

Abstract
Proteins function in the crowded cellular environments with high salt concentrations, thus facing tremendous challenges of misfolding/aggregation which represents a pathological hallmark of aging and an increasing spectrum of human diseases. Recently, intrinsically disordered regions (IDRs) were recognized to drive liquid–liquid phase separation (LLPS), a common principle for organizing cellular membraneless organelles (MLOs). ATP, the universal energy currency for all living cells, mysteriously has concentrations of 2–12 mM, much higher than required for its previously‐known functions. Only recently, ATP was decoded to behave as a biological hydrotrope to inhibit protein LLPS and aggregation at mM. We further revealed that ATP also acts as a bivalent binder, which not only biphasically modulates LLPS driven by IDRs of human and viral proteins, but also bind to the conserved nucleic‐acid‐binding surfaces of the folded proteins. Most unexpectedly, ATP appears to act as a hydration mediator to antagonize the crowding‐induced destabilization as well as to enhance folding of proteins without significant binding. Here, this review focuses on summarizing the results of these biophysical studies and discussing their implications in an evolutionary context. By linking triphosphate with unique hydration property to adenosine, ATP appears to couple the ability for establishing hydrophobic, π‐π, π‐cation and electrostatic interactions to the capacity in mediating hydration of proteins, which is at the heart of folding, dynamics, stability, phase separation and aggregation. Consequently, ATP acquired a category of functions at ~mM to energy‐independently control protein homeostasis with diverse mechanisms, thus implying a link between cellular ATP concentrations and protein‐aggregation diseases.
Keywords: adenosine triphosphate, crowding‐induced destabilization, FUS, intrinsically disordered proteins, liquid–liquid phase separation, NMR spectroscopy, protein aggregation, protein homeostasis, SARS‐CoV‐2, TDP‐43
1. INTRODUCTION
Proteins are the most important functional players for all forms of life, which are linear heteropolymers composed of 20 α‐amino acids. In his Nobel Lecture, Anfinsen stated “the native conformation is determined by the totality of interatomic interactions and hence by the amino acid sequence, in a given environment”. 1 Now it becomes established that based on amino acid sequences, proteins encoded by higher eukaryotic genomes can in fact be grouped into two major categories: random and high‐complexity (I of Figure 1(a)), and non‐random and low‐complexity (II of Figure 1(a)) sequences. Only a portion of the first category can spontaneously fold into unique three‐dimensional structures but the mechanisms still remain not completely understood, particularly for the role of protein hydration despite exhaustive studies. 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 On the other hand, many proteins in the second category remain highly disordered but are functional (Figure 1(a)), thus designated as intrinsically disordered proteins (IDPs), and ~ 44% of human proteins contain intrinsically disordered regions (IDRs) of >30 amino acids. 9 , 10
FIGURE 1.
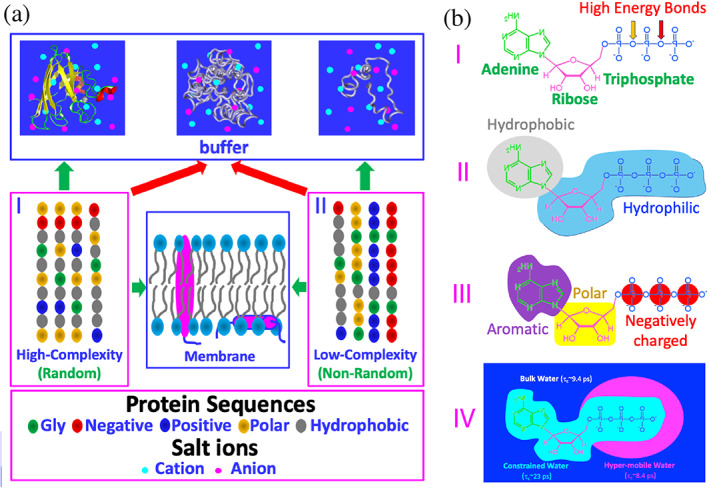
Sequence‐structure relationship of proteins and structure–function relationship of ATP. (a) Based on the sequences as represented by five types of amino acids, proteins can be classified into high‐complexity or random (I), and low‐complexity or non‐random (II) sequences. A portion of proteins of high‐complexity sequence can fold into uniquely‐defined structures soluble in vivo with high concentrations of salts (~150 mM), while many of proteins of low‐complexity sequence remain intrinsically disordered. Interestingly, a large amount of proteins of both high‐ and low‐complexity sequences appear to be aggregation‐prone or even insoluble in vivo. Furthermore, ~30% proteins from both sequences can fold in the membrane environments. (b) ATP has unique structural properties and thus may act as: (I) energy currency by hydrolysis of high energy bonds; (II) biological hydrotrope with the presence of hydrophobic adenine and hydrophilic ribose and triphosphate; (III) bivalent binder by the aromatic purine ring and highly negatively charged triphosphate chain; and (IV) hydration mediator resulting from its unique hydration structure previously derived from the results of microwave dielectric spectroscopy, which was modeled to contain “constrained water” with dielectric relaxation time (τc) of ~23 ps, as well as “hyper‐mobile water” with τc of ~8.4 ps, even smaller than that of bulk water (9.4 ps)
Recently, in addition to the classic functions, IDRs have been identified to drive liquid–liquid phase separation (LLPS) by multivalent and dynamic interactions at concentrations much lower than those required for the folded proteins such as lysozyme. 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 LLPS has been recognized as a common principle for organizing cellular membraneless organelles (MLOs), as well as hypothesized as a key mechanism for the formation of the protocells. Initially LLPS was described by the Flory‐Huggins theory developed for the phase separation of polymers. 21 Very recently, a stickers‐and‐spacers model has been proposed, in which the groups participating in attractive interactions are defined as “stickers”, while the regions that do not significantly contribute to attractive interactions are considered to be “spacers”. 16
Intriguingly, many proteins in both categories are prone to aggregation or even insoluble in cells or salted aqueous solution (Figure 1(a)), which is not only problematic for protein research and industry applications, but also associated with aging and an increasing spectrum of human disease, particularly neurodegenerative diseases including Parkinson's disease (PD), Alzheimer's disease (AD), Huntington's disease (HD), spinocerebellar ataxias (SCA), amyotrophic lateral sclerosis (ALS). 8 , 9 , 19 , 22 , 23 , 24 , 25 IDR‐rich proteins are particularly prone to aggregation in the phase separated state because protein concentrations in droplets are 50–300 fold higher than those in the surrounding environments. As such, LLPS might further exaggerate into aggregation or amyloid fibrillation, thus jumping from physiology to pathology.
2. WATER, SALT AND CELLULAR CROWDING
Where there is water, there is life. 26 , 27 , 28 , 29 Water is not just a passive scaffold but also has many active roles in molecular biology. The absolutely essential role of water for life is associated with its many abnormal properties which are universally believed to be irreplaceable by another single molecule system. 26 , 27 , 28 , 29 , 30 , 31 , 32 Very unexpectedly, in 2005 we found that insoluble proteins including the most hydrophobic transmembrane fragment in nature could be all solubilized in unsalted water for high‐resolution biophysical studies. 9 , 30 , 31 , 32 Since then, this has been extensively observed on other insoluble proteins, 33 , 34 , 35 and in particular the total protein extract of human cells including membrane proteins were demonstrated to be soluble in unsalted water. 35 These results logically suggest that proteins are so designed to be intrinsically soluble in unsalted water and salt ions play a central role in controlling protein aggregation by the interplay of the complex interactions including electrostatic screening, altering the electrostatic property of proteins and perturbing the protein hydration. 9 , 30 , 31 , 32 , 36 , 37 , 38 Furthermore, anions were shown to selectively bind both folded and intrinsically disordered proteins at physiologically relevant concentrations, highlighting that the electrostatic property of proteins in the presence of salts is a critical determinant for solubility and aggregation particularly at high protein concentrations. 37 , 38
Cellular environments are extremely crowded, and it was estimated that ~5–40% of the volume of a living cell is occupied by various biological macromolecules including proteins, nucleic acids, polysaccharides with a total up to 80–400 mg/ml. 39 , 40 , 41 , 42 , 43 , 44 , 45 The cellular crowding is anticipated to significantly impact folding, dynamics, stability and aggregation of proteins, but the exact mechanisms appear to be complex. Initially the macromolecular crowding was considered to enhance stability of proteins due to the excluded‐volume effects. 40 , 41 , 42 , 43 , 44 , 45 However, further studies indicated that the crowding may also destabilize the protein stability because of the presence of various non‐specific interactions. 42 , 43 , 44 , 45 In particular, accumulating results suggest that the hydration property of proteins appears to be critical for protein folding, dynamics, stability, phase separation and aggregation but is currently underappreciated due to the extreme challenge in experimentally and computationally characterizing protein hydration. 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50
2.1. ATP functions beyond “high energy bonds”
Adenosine triphosphate (ATP) is a unique nucleotide well‐known as the “biological fuel” for all living cells. By hydrolysis of high energy bonds (I of Figure 1(b)), ATP provides energy to drive a myriad of biological processes, including muscle contraction, nerve impulse propagation, protein homeostasis, chemical synthesis and signaling transduction. 51 Mysteriously, although all known ATP‐dependent proteins/enzymes require only micromolar concentrations, the cellular concentrations of ATP are very high, which range from 2 to 12 mM depending on cell types. So a fundamental question arises: why do cells invest so much energy to maintain such high ATP concentrations? Previously, it was hypothesized that the free‐energy gradient between ATP and ADP/AMP is required and consequently a ~ 50‐fold ATP/ADP ratio is necessary to drive ATP‐dependent reactions. 51
Only recently, it was decoded that at concentrations >5 mM, ATP acts as a biological hydrotrope to dissolve LLPS and aggregates/fibrils of proteins. 52 , 53 This new role was proposed to result from the amphiphilic property of ATP, which is composed of the hydrophobic adenine and polar triphosphate (II of Figure 1(b)). Briefly, the aromatic purine ring of ATP was proposed to be clustered over hydrophobic patches of protein liquid droplets or aggregates, while its triphosphate chain strongly interacts with bulk water, thus dissolving LLPS and aggregates. 52 , 53
By NMR visualization of the residue‐specific interaction of ATP with intrinsically disordered domains (IDDs) of FUS, we found that ATP in fact could biphasically modulate the RGG‐rich domain which has no hydrophobic residues and is incapable of phase separation by itself: induction of LLPS at low ATP concentrations and dissolution at high concentrations. 54 , 55 The results led to the proposal that ATP also functions to modulate LLPS by acting as a bivalent binder specifically targeting Arg/Lys residues (III of Figure 1(b)). ATP was also shown to bivalently bind the conformation‐dependent pockets of the folded proteins and inhibit their amyloid fibrillation. 56 , 57 , 58 , 59 , 60
Very recently, we found that even without significant binding, ATP can efficiently antagonize the crowding‐induced destabilization 43 , 44 , 45 as well as enhance the folding and inhibit aggregation. 61 To the best of our knowledge, ATP appears to achieve these functions by acting as a hydration mediator (IV of Figure 1(b)). Indeed, previously by high‐resolution microwave dielectric spectroscopy, it was fitted out that around the triphosphate chain of ATP, in addition to the constrained water with dielectric relaxation time longer than that of bulk water, there exists an anormal layer of water which was modeled to be “hyper‐mobile” water with dielectric relaxation time even shorter than that of bulk water. 62
This review is only focused on summarizing the biophysical studies of the novel energy‐independent functions of ATP at mM and subsequently discussing their implications and future challenges.
2.2. ATP biphasically modulates LLPS of FUS
The 526‐residue FUS is an RNA‐binding protein (RBP) which functions by binding a large array of nucleic acids. 63 FUS also undergoes LLPS to participate in the formation of stress granules (SGs) composed of RBPs and nucleic acids in response to environmental stresses. On the other hand, its aggregation has been extensively identified to be associated with a large spectrum of neurodegenerative diseases, including Amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia (FTLD). 19 , 22 , 63 , 64 , 65 As shown in Figure 2(a), FUS consists of the N‐terminal domain (NTD) composed of QGSY‐rich prion‐like domain (PLD) over 1–165 and RGG‐rich region (RGG1) over 166–267; middle RNA‐recognition motif (RRM) over 282–371, and C‐terminal domain (CTD) over 371–526 containing RGG2, zinc finger (ZnF), and RGG3. Strikingly, except for RRM and ZnF domains, the rest (~75%) of the FUS sequence are all intrinsically disordered. FUS owns two types of IDRs with distinctive compositions (Figure 2(b)): while PLD is rich in polar residues including Tyr but completely absent of Arg/Lys, RGG−/RG‐rich regions are abundant in Arg and Lys residues, whose side chains are significantly different but both positively‐charged in physiological pH (Figure 2(c)).
FIGURE 2.
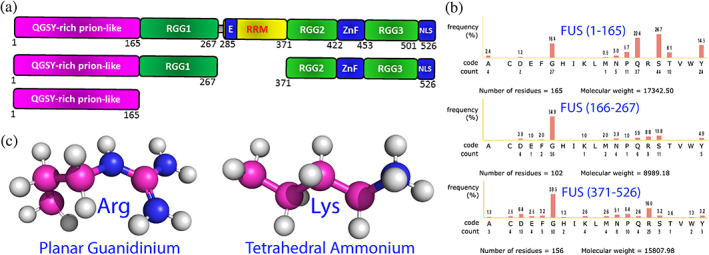
Intrinsically‐disordered domains of FUS and sidechain structures of Arg/Lys. (a) Schematic representation of the domain organization of FUS and its differentially dissected domains. (b) Amino acid compositions of three intrinsically disordered regions of FUS. (C) Stick‐and‐ball models of the side chains of Arg and Lys residues showing the planar guanidinium cation of arginine and tetrahedral ammonium cation of lysine
To delineate the mechanism for LLPS of FUS, FUS was dissected into differential combinations of domains and the interactions between different IDRs were shown to drive its LLPS. 54 , 55 Noticeably, regardless of whether ZnF is folded or not, CTD with 25 Arg and 4 Lys was incapable of LLPS by itself even at 1 mM in various buffers at different pH and salt concentrations. 54 However, ATP, RNA and single‐stranded DNA (ssDNA) including the telomeric ssDNA (TssDNA) and 24‐mer poly(dT) (T24) were identified to biphasically modulate its LLPS: induction at low concentrations but dissolution at high concentrations 54 , 55 as exemplified by the results with ATP (Figure 3(a)).
FIGURE 3.
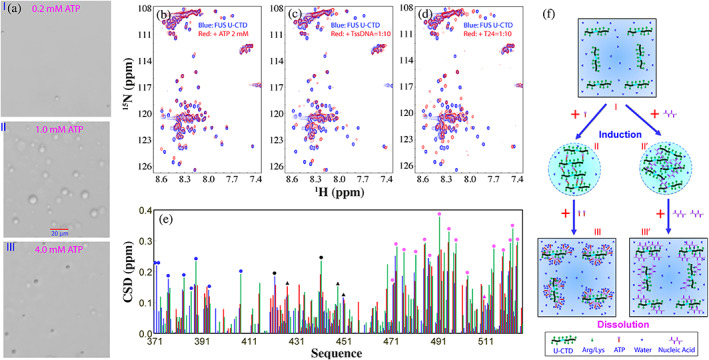
ATP biphasically modulates LLPS of FUS CTD. (a) DIC images of dynamic droplets formed by FUS CTD in the presence of ATP at different concentrations. (b) HSQC spectra of CTD with the zinc finger (ZnF) unfolded (U‐CTD) in the absence (blue) and in the presence of ATP at 2 mM (red). (c) HSQC spectra of U‐CTD in the absence (blue) and in the presence of TssDNA at a ratio of 1:10 (red). (d) HSQC spectra of U‐CTD in the absence (blue) and in the presence of T24 at a ratio of 1:10 (red). (e) Chemical shift difference (CSD) of FUS U‐CTD in the presence of ATP at 2 mM (blue) as well as TssDNA (red) and T24 (green) at a ratio of 1:10. Filled circles are used for indicating the locations of Arg, while triangles indicate Lys. Blue is for those within RGG2, black for those in the unfolded ZnF, and purple for those within RGG3. (f) A speculative model to rationalize the specific binding of ATP and ssDNA to Arg/Lys residues within FUS U‐CTD to induce LLPS at low concentrations but to dissolve at high concentrations
Residue‐specific interactions of FUS CTD with ATP (Figure 3(b)), TssDNA (Figure 3(c)) and T24 (Figure 3(d)) have been visualized by NMR HSQC spectroscopy which is particularly powerful in detecting very weak interactions. Detailed analysis revealed that ATP and two oligonucleic acids induced the very similar shift patterns of HSQC peaks, in which HSQC peaks of Arg and Lys are significantly shifted (Figure 3(e)). On the other hand, the molar ratios of TssDNA and T24 needed to biphasically modulate LLPS of FUS CTD are much lower than that of ATP. The results led to the proposal that ATP and oligonucleic acids share the similar mechanism to biphasic modulate LLPS of CTD mainly by specifically targeting Arg/Lys residues (Figure 3(f)).
To corroborate this proposal, the effects of ATP and nucleic acids were further examined on PLD and entire NTD. Previously, FUS PLD without any Arg/Lsy residue was shown to undergo LLPS only in the presence of the crowding‐inducing reagent or at very high concentrations. 18 , 65 Unexpectedly, upon inclusion of the residues 166–267 containing no hydrophobic residues but 9 Arg and 4 Lys residues (Figure 4(a)), NTD suddenly could undergo LLPS at very low concentrations. To understand the underlying mechanism, NMR relaxation data were acquired and analyzed to gain insights into the backbone dynamics of NTD. 55 The obtained spectral densities at three frequencies revealed: (1) NTD is similarly disordered over the whole sequence; and (2) the backbone dynamics of PLD are highly similar in the isolated form and in the context of the entire NTD (Figure 4(b)). This set of results strongly suggests that the significantly‐enhanced capacity of FUS NTD in LLPS is not due to the conformational or/and dynamic change of PLD in the presence of RGG‐rich 166–261 (Figure 4(a)), but because of introduction of Arg/Lys residues which could additionally established π‐π and π‐cation interactions between the PLD aromatic residues and RGG1 Arg/Lys residues.
FIGURE 4.
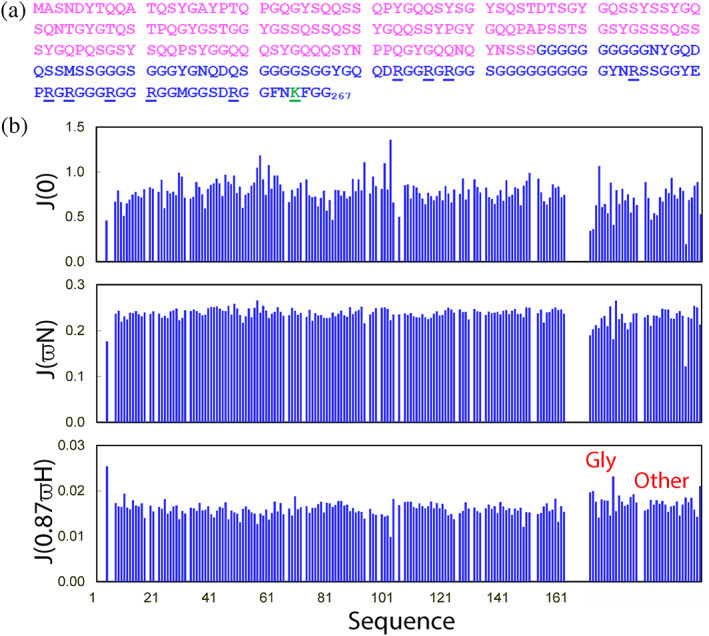
NMR backbone dynamics of FUS NTD (1–267) on the ps–ns timescale. (a) Sequence of FUS NTD with PLD colored in purple and RGG1 in blue. Arg residues are underlined, while one Lys residue is colored in green and underlined. (b) Spectral densities at J(0), J(ωN), and J(0.87ωH) of FUS NTD calculated from the 15N backbone relaxation data as measured at 800 MHz
FUS PLD was titrated with ATP up to 20 mM but no induction of LLPS was observed as well as no significant binding was detected, indicating that ATP has no significant binding to PLD residues including aromatic residues. 49 However, as for NTD capable of phase separation by itself, the addition of ATP induced the monotonic dissolution of the droplets (Figure 5(a)) as TssDNA and T24. 55 Intriguingly, NMR characterization revealed an unexpected phenomenon that the increase of ATP concentrations induced the disappearance of HSQC peaks of NTD (Figure 5(b)), and at 10 mM of ATP, most HSQC peaks became too weak to be detected. 54 , 55 By contrast, although TssDNA and T24 also could induce the same monotonic dissolution as ATP but at the much lower concentrations, both of them only trigged the shifts/disappearance of HSQC peaks of several Arg residues within RGG1. 55 Strikingly, the extensively disappeared HSQC peaks induced by ATP could be restored by additionally adding TssDNA (Figure 5(c)). In particular, the HSQC spectrum of NTD in the presence of both ATP and TssDNA is highly superimposable to that in the presence of only TssDNA at the same molar ratio (Figure 5(d)). The results led to the proposal of a model (Figure 5(e)): (1) ATP and oligonucleic acids dissolved the droplets of NTD by binding to the same set of RGG1 Arg residues to disrupt their dynamic interaction with PLD aromatic residues, but the binding affinity of oligonucleic acids are much higher than that of ATP. (2) Upon exceedingly binding to ATP, NTD appears to further assemble into large oligomers while the binding of oligonucleic acids trigger no assembly of such oligomers. The distinctive effects of ATP and oligonucleic acids are most likely due to the presence of the triphosphate chain in ATP, which owns the unique hydration property (IV of Figure 1(b)), thus responsible for driving the assembly of NTD into large oligomers with HSQC peaks undetectable. 55
FIGURE 5.
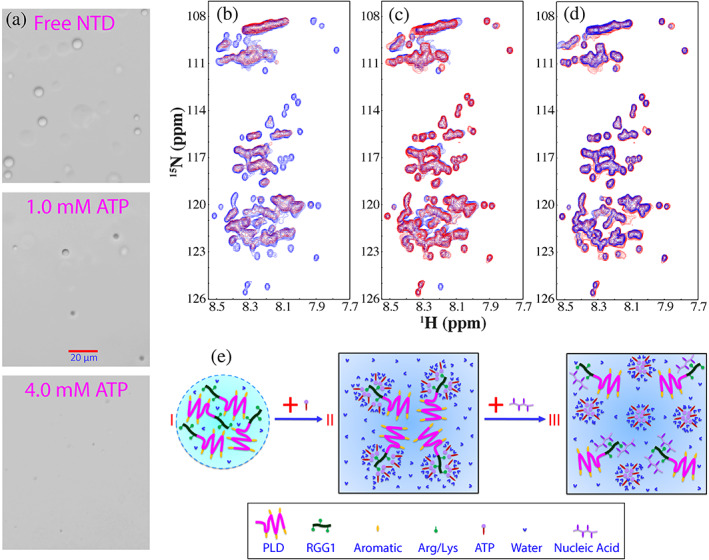
The modulation of LLPS of FUS NTD by ATP and ssDNA. (a) DIC images of dynamic droplets formed by FUS NTD in the presence of ATP at different concentrations. (b) HSQC spectra of FUS NTD in the absence (blue) and in the presence of ATP at 3 mM (red). (c) HSQC spectra of FUS NTD in the absence (blue), and in the presence of ATP at 3 mM with an extra addition of telomeric ssDNA (TssDNA) at 60 μM (red). (d) HSQC spectra of FUS NTD in the presence of TssDNA at a ratio of 1:5 (blue) and in the presence of both ATP at 3 mM and TssDNA at 60 μM (red). (e) A speculative model to illustrate: (1) FUS NTD undergoes LLPS resulting from the π‐cation or/and π‐π interactions between aromatic residues in PLD and Arg in RRG1; (2) the specific binding of ATP to Arg within RGG1 leads to dissolution of LLPS, which, however, can be displaced by ssDNA due to the much higher binding affinity of ssDNA
2.3. ATP biphasically modulate LLPS of TDP‐43 PLD
TDP‐43 is another RNA‐binding protein whose aggregation represents a common pathological hallmark of most cases of ALS, as well as other major neurodegenerative diseases, including Alzheimer's (AD), Parkinson's (PD), frontotemporal dementia (FTD), and Huntington's (HD) diseases. 8 , 18 , 19 , 20 , 22 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 As shown in Figure 6(a), 414‐residue TDP‐43 consists of the folded N‐terminal 67 , 68 and two RNA recognition motif (RRM) domains, 69 as well as the C‐terminal PLD domain over the residues 267–414. Intriguingly, TDP‐43 PLD hosts almost all ALS‐causing mutations although previous NMR studies indicate that TDP‐43 PLD is intrinsically disordered. 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 On the other hand, TDP‐43 PLD has the composition very different from those of FUS PLD, which contains 5 Arg and 1 Lys, as well as a unique hydrophobic region 311–343, which was identified to be the key driver for LLPS by the dynamic dimerization/oligomerization. 73 , 74 , 78
FIGURE 6.
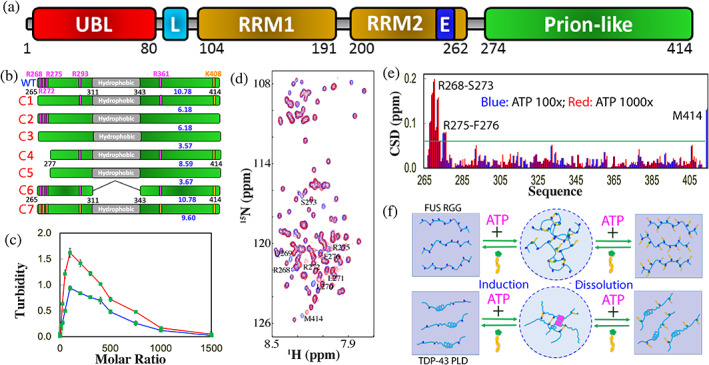
ATP biphasically modulates LLPS of TDP‐43 PLD. (a) Domain organization of TDP‐43. (b) Diagrams showing the wild‐type TDP‐43 PLD and its six mutants. (c) Turbidity curves of PLD at pH 7.0 (red) and pH 5.5 (green) in the presence of ATP at different molar ratios. (d) HSQC spectra of TDP‐43 PLD in the absence (blue) and in the presence of ATP at a molar ratio of 1:1000 (red). (e) CSD of TDP‐43 PLD in the presence of ATP at 1:100 (blue) and 1:1000 (red) with the significantly‐perturbed residues labeled. (f) Speculative models for ATP to biphasically modulate LLPS by “Direct Mechanism” (upper) or “Indirect Mechanism” (lower), which needs the additional driving force such as the dimerization‐induced formation of the helix (pink rectangles) as previously proposed (Reference 73)
Very recently, the effect of ATP on LLPS and its molecular interaction have been systematically studied 71 on TDP‐43 PLD and its seven mutants (Figure 6(b)). At lower protein concentrations, no LLPS was detected, but upon adding ATP, TDP‐43 PLD was induced to phase separate with the highest turbidity values at 1:100 (PLD:ATP) (Figure 6(c)). Further addition of ATP led to reduction of turbidity. DIC visualization showed that only upon adding ATP, the dynamic droplets were formed, and further addition of ATP led to dissolution of the droplets, which were completely dissolved at 1:1000.
Subsequently the molecular interaction of ATP with TDP‐43 PLD was monitored by HSQC spectroscopy (Figure 6(d)). Very unexpectedly, although ATP contains both hydrophobic adenine and highly negatively charged triphosphate, it only induces the shift of a small set of HSQC peaks even up to 1:1000 (Figure 6(e)). The residues with significant shifts are clustered over N‐terminal three Arg residues, and the shifting process is largely saturated at 1:100 (PLD:ATP). Moreover, seven mutants (Figure 6(b)) were constructed which include C1 with Arg268, Arg272 and Arg275 mutated to Ala. ATP also biphasically modulated LLPS of C1 as observed on WT. On the other hand, NMR characterization showed that at 1:100 only HSQC peak of Met414 showed a large shift, while the residues clustered over N‐terminal three Arg no longer showed any significant shifts. The results indicate that the peak shift of the N‐terminal residues other than three Arg is due to the perturbation by the binding of ATP to neighboring three Arg residues and unambiguously reveal that ATP only significantly bind Arg residues within IDRs.
To better characterize the interaction of ATP with Arg, the mutant C7 was generated with all five Arg residues mutated to Lys, which only has the slightly smaller pI (9.6) than that (10.78) of WT (Figure 6(b)). Very interestingly, C7 could undergo LLPS even without ATP, while the addition of ATP only induced the slight increase of turbidity with the highest at 1:300. Further addition of ATP led to a slow decrease of turbidity and the droplet number, but only at 1:3000 the droplets became completely dissolved. NMR characterization revealed that ATP induced the shift of a small set of peaks. Detailed analysis revealed that C7 has the overall shift pattern very similar to that of WT with the significantly shifted residues including Lys268, Leu270‐Lys272, Lys275‐Gly277, Ser407‐Lys408 and Met414. Examination of the chemical shift tracings of the significantly‐shifted peaks of WT and mutants disclosed that the shifting of HSQC peaks of WT, C4 and C6 residues were largely saturated at 1:100, while the shifting of all C7 residues remained unsaturated even at 1:500 (protein:ATP). This clearly indicates that the binding affinity of ATP to Arg is much higher than that to Lys, most likely because the purine ring of ATP can establish both π‐cation and π‐π stacking interactions with the delocalized planar guanidinium cation of Arg but only π‐cation interaction with the tetrahedral ammonium cation of Lys (Figure 2(c)). This observation is in general consistent with the previous results that the aromatic rings of aromatic residues (18) and RNA (82) interact with Arg more tightly than with Lys.
Interestingly, the mutant C6 with the deletion of the hydrophobic region 311–343 (Figure 6(b)) no longer has the capacity in phase separation even with addition of ATP to 1:1500, although ATP was found to bind to the same Arg residues with the similar affinity as detected by NMR. This suggests that two distinctive mechanisms exist to underlie the biphasic modulation of LLPS of FUS RGG‐rich domain and TDP‐43 PLD respectively. Briefly, as illustrated by Figure 6(f), for 156‐residue FUS RGG‐rich domain with 25 Arg residues, the multiple bivalent bindings of ATP to Arg is sufficient to directly induce LLPS by forming large and dynamic complexes which manifest as dynamic droplets, thus designated as “direct mechanism”. 55 By contrast, for 150‐residue TDP‐43 PLD with only 5 Arg residues, the ATP‐binding to Arg is insufficient to solely drive LLPS, which thus needs to coordinate other diving forces, particularly the dimerization/oligomerization of the hydrophobic region to indirectly induce LLPS, thus designated as “indirect mechanism”. For both mechanisms, the exceedingly binding of ATP not only disrupts the ATP‐linked dynamic complexes, but also introduces the long‐range electrostatic repulsive interaction, thus leading to dissolution of dynamic droplets as well as inhibition of further aggregation (Figure 6(f)). Therefore, as a highly‐charged molecule ATP can impose two different electrostatic effects onto IDRs: alternation of the conformation‐specific electrostatic property by specific and relatively‐tight binding to Arg, and non‐specific electrostatic screening effect common to all charge molecules such as NaCl.
Recently, a neuronal cell death mechanism was established which is initiated by LLPS of TDP‐43 independent of conventional SGs. 66 Briefly, TDP‐43 may undergo LLPS by itself to form liquid droplets capable of recruiting other proteins and leading to cell death. Neurons have ATP concentrations of ~3 mM while TDP‐43 is only ~1 μM, 51 , 79 thus with a molar ratio of ~1:3000. As such, TDP‐43 is largely bound with ATP over its PLD Arg residues as well as RRM1 domain. 52 Additionally, ATP also has the hydrotropic capacity in dissolving LLPS of RNA‐binding proteins at high concentrations. 52 , 53 Altogether, TDP‐43 is inhibited for the LLPS in the neuronal cytoplasm. Only under pathological or/and aging conditions with significantly increased TDP‐43 or/and reduced ATP concentrations, TDP‐43 PLD will drive LLPS which may even be even enhanced by the bivalent binding with ATP.
Intriguingly, with respect to the role of Arg/Lys, in FUS they behave as the major stickers for driving LLPS. 18 , 55 By contrast, in TDP‐43 PLD they serve as inhibitors for maintaining the reversibility of LLPS and preventing the exaggeration of LLPS into aggregation. This implies that the role in LLPS of a certain amino acid is highly context‐dependent. Moreover, LLPS and its exaggeration into aggregates are controlled by a delicate network composed of “stickers” and “spacers” as well as “inhibitors”. These results also bear the unprecedented implication that in addition to the covalent Arg methylation, 80 , 81 , 82 , 83 ATP can act as a universal and specific regulator for most, if not all, R‐containing IDRs by binding to Arg to alter their physicochemical properties, conformations, dynamics, LLPS, assembly and aggregation. Remarkably, the number of the proteins with R‐containing IDRs should be much larger than that of RG/RGG‐rich domains which were found in >1,700 human proteins. 81 , 82 , 83
2.4. ATP appears to be exploited by SARS‐CoV‐2 to promote its life cycle
Viruses have no ability to generate ATP 84 and thus it is of great interest to explore whether ATP can bind and modulate LLPS of the viral proteins. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) is the etiologic agent of the ongoing pandemic, 85 which already led to >127 millions of infections and > 2.7 millions of deaths. Its nucleocapsid (N) protein is not only the key component of the packed viral genome, but also a key candidate for vaccine development. Very recently, the N protein was identified to phase separate as induced by nucleic acids. 86 , 87 , 88 , 89 , 90 419‐residue N protein is an RNA‐binding protein, composed of the folded N‐terminal domain (NTD) for binding RNA 91 and C‐terminal domain (CTD) for dimerization, 92 as well as three IDRs (Figure 7(a)). Very recently, the effect of ATP was assessed on its LLPS with or without 24‐mer poly(dA) (A24) which was used to mimic nucleic acids. 90 As shown in Figure 7(b) N protein at 20 μM has a turbidity of only 0.08 and showed no droplets as imaged by DIC. However, upon adding ATP, the turbidity increased and reached the highest at 1:200 (N‐protein:ATP), and further addition of ATP led to the decrease of the turbidity. DIC revealed that dynamic droplets could be observed upon adding ATP at 1:25 and at 1:100, many droplets were formed with the diameters of some of ~1 μm (Figure 7(c)). However, further increase of ATP concentrations led to the reduction of droplet numbers, and at 1:500, all droplets were dissolved. This set of results clearly indicate that as observed on human FUS and TDP‐43, ATP also biphasically modulates LLPS of the viral N protein.
FIGURE 7.
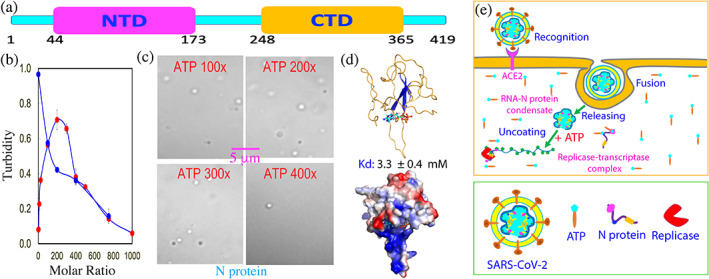
ATP appears to be exploited by SARS‐CoV‐2 to promote its life cycle. (a) Domain organization of SARS‐CoV‐2 nucleocapsid (N) protein composed of the N‐terminal domain (NTD) for RNA‐binding and C‐terminal domain (CTD) for dimerization, as well as three intrinsically disordered regions (IDRs). (b) Turbidity curves of N protein at 20 μM without (red circles) and with the pre‐existence of A24 at 1:0.5 (blue circles) upon addition of ATP at different ratios. (c) DIC images of N protein in the presence of ATP at different ratios. (d) The lowest‐energy docking model of the ATP‐RBD complex with ATP in ball‐and‐stick, and RBD in ribbon or in electrostatic potential surface. (e) A proposed scheme to illustrate that ATP might be hijacked by SARS‐CoV‐2 to promote its life cycle by facilitating the initial uncoating of the gRNA‐Nprotein condensate and subsequent localizing to replicase‐transcriptase complex
Furthermore, A24 was also able to biphasically modulate LLPS of N protein as ATP, but the ratios required to induce and dissolve droplets were much lower than those of ATP. Addition of A24 even at 1:0.5 resulted in a turbidity value of 0.97 (Figure 1(c)) as well as the formation of dynamic droplets with the diameter of some even close to ~2 μm. Interestingly, additional addition of ATP into this sample led to the monotonic reduction of the turbidity, as well as dissolution of the droplets as imaged by DIC. At 1:750, the droplets were completely disappeared. The results strongly imply that ATP and A24 most likely target the same sites of N protein for biphasic modulation, similar to what were previously observed on human FUS 55 and TDP‐43. 71 As such, ATP and A24 appear to biphasically modulate LLPS of the SARS‐CoV‐2 N proteins by the bivalent interaction with Arg/Lys residues within its three IDRs. However, A24 has much higher capacity than ATP because A24 can achieve multiple bivalent interactions with Arg/Lys residues of IDRs which consequently has the much higher affinity. 93 Due to the low numbers of Arg/Lys within three IDRs of N protein, ATP appears to modulate its LLPS likely by the indirect mechanism, as proposed for TDP‐43 PLD (Figure 6(f)). This thus explain the reports that the dimerization domain of N protein is essential for its RNA‐induced LLPS. 86 , 87 , 88 , 89 Moreover, ATP was also identified to specifically bind its NTD, or RNA‐binding domain (RBD) and with NMR‐derived constraints, the ATP‐BRD complex was constructed in which ATP specifically bind the conserved RNA‐binding surface of the N protein RBD (Figure 7(d)).
Therefore, ATP appears to be hijacked by SARS‐CoV‐2 to promote its life cycle although SARS‐CoV‐2 cannot generate ATP by itself. As illustrated in Figure 7(e), immediately after the infection, SARS‐CoV‐2 will release its gRNA‐Nprotein condensate into infected cell, which is tightly packed into the gel‐like state. 86 As one infected cell might only have one to several copies of the condensate, the ratio between ATP and Nprotein/gRNA is very high. Consequently at this stage ATP acts to facilitate the uncoating of the condensate. However, once new copies of the viral RNA or/and N protein are synthesized by the host cell machinery, the ratios will reduce, and consequently ATP may enhance LLPS of the mixture of the viral gRNA and N protein together with the host cell replicases to form replicase‐transcriptase complexes. Finally, after all components needed for assembling new virions are synthesized, the ratios between ATP and Nprotein/gRNA will be further reduced and therefore a large population of N protein becomes unbound with ATP. As such, the ATP‐unbound RBD/CTD become available for recognizing the specific package signal sites of gRNA to initiate the packing of gRNA and N proteins into virions.
The critical role of LLPS in the life cycle implies that to disrupt LLPS may represent a promising strategy to discover/design anti‐SARS‐CoV‐2 drugs. Hydroxychloroquine (HCQ) has been shown to own promising potential in clinically combating SARS‐CoV‐2 but the underlying mechanisms still remain largely elusive. 94 , 95 , 96 In particular, so far, all action sites were proposed on the host cells, and no specific viral target protein has been experimentally identified. Very recently, by use of DIC microscopy and NMR spectroscopy, HCQ has been decoded to specifically bind to both NTD and CTD of SARS‐CoV‐2 N protein to inhibit their interactions with nucleic acids, as well as to disrupt its LLPS induced by ssDNA. 97 The results suggest that HCQ may achieve its anti‐SARS‐CoV‐2 activity partly by directly interfering in the viral life cycle. The study not only provides a structural basis for the anti‐SARS‐CoV‐2 activity of HCQ, but also indicates that SARS‐CoV‐2 N protein and its LLPS represent key targets for further optimization and development of anti‐SARS‐CoV‐2 drugs.
2.5. ATP specifically binds the folded nucleic‐acid‐binding domains
RNA‐binding proteins such as FUS, TDP‐43 and hnRNPA1 also contain the folded RNA‐recognition motif (RRM) domains which participate in pathological aggregation or amyloid fibrillation. 56 , 57 , 58 , 69 , 98 , 99 So it is of significant interest to assess whether they can bind ATP. Recently, ATP was first identified to specifically bind FUS RRM with a Kd of 3.8 mM 56 to a pocket within the conserved surface for RRM to bind various nucleic acids (Figure 8(a)). Extensive studies with ADP, AMP and triphosphate further revealed that both adenine and triphosphate chain of ATP are critical for the specific binding. 56 Intriguingly, although this binding showed no significant alternation of the thermal stability of FUS RRM, it is sufficient to inhibit its amyloid fibrillation likely by kinetically blocking the dynamic opening of the folded structure to expose the hydrophobic core for the pathological assembly into amyloid fibrils. 56 , 98
FIGURE 8.
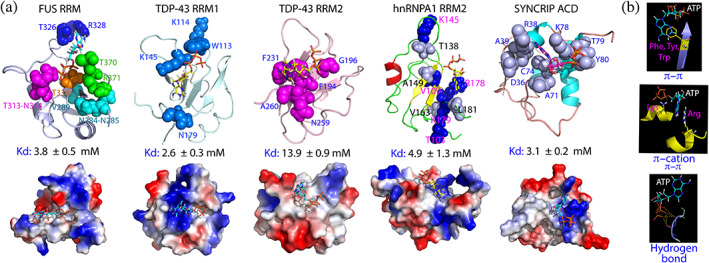
Structures of the ATP‐protein complexes with Kd at ~ mM. (a) NMR‐derived structures of ATP in complex with FUS RRM, TDP‐43 RRM1 and RRM2, hnRNPA1 RRM2 as well as the acidic domain (AcD) of Syncrip respectively in ribbon (upper) and electrostatic potential surface (lower). The significantly perturbed residues derived from NMR HSQC titrations are displayed in spheres and labeled. (b) The major types of the ATP‐protein interactions identified in these complexes
Subsequently ATP has been shown to bind the tandem RRM domains of TDP‐43 and hnRNPA1. 57 , 58 , 99 The results reveal that ATP is capable of binding to the pockets within the conserved nucleic‐acid‐binding surfaces of both RRM1 and RRM2 of TDP‐43 but with very differential affinity (Kd of 2.6 mM for RRM1 and 13.9 mM for RRM2). For TDP‐43 RRM domains, ATP could increase their thermal stability to different degree and inhibit amyloid fibrillation. 57 Moreover, the ALS‐causing D169G mutation of TDP‐43 RRM1 at the back of the ATP‐binding pocket is able to reduce the binding affinity of ATP. 58 On the other hand, ATP only binds the hnRNPA1 RRM2 domain with Kd of 4.9 mM while has no complete binding pocket on the hnRNPA1 RRM1 domain. 99
So an interesting question arise: whether ATP can bind other nucleic‐acid‐binding domains without the RRM fold. To this end, ATP was found to bind the N‐terminal acidic domain (AcD) of 623‐residue SYNCRIP with Kd of 3.1 mM, 59 which has no sequence and structure homology to those of RRM but is a cryptic RNA binding domain with an all‐helix fold (Figure 8(a)). Very recently, ATP was also deciphered to specifically bind NTD of SARS‐CoV‐2 N protein 90 with Kd of 3.3 mM to the surface for recognizing the viral gRNA (Figure 7(d)). This domain not only represents the first viral protein capable of binding ATP at mM, but also adopts a structural fold very different from those of RRM and AcD.
Kinases constitute the largest superfamily of the classic ATP‐binding proteins. 100 , 101 , 102 So far, 555 members of the human kinases have been identified, which include a main class of 497 eukaryotic kinases (ePKs) and 58 atypical kinases (aPKs). Previous studies revealed that the human kinase superfamily, regardless of being ePKs and aPKs, all shares the overall structural fold with the high binding affinity to ATP. 100 , 101 , 102 The results discussed here decode that ATP can also specifically bind RRM, AcD and SARS‐CoV‐2 NTD of diverse folds with very weak binding affinity, which thus prevented further determination of the complex structures by classic X‐ray crystallography or NMR spectroscopy. Nevertheless, with the constraints derived from NMR titrations, six complexes of ATP with human and viral protein domains with Kd at ~ mM have been constructed (Figure 7(d), 8(a)). Despite adopting three distinctive folds, they are all nucleic‐acid‐binding domains and their ATP complexes share three common features: (1) the ATP binding pockets are all located within the conserved surface for binding various nucleic acids. (2) the binding affinities are very weak, with Kd values of ~mM. (3) both adenine and triphosphate chain of ATP are essential to establish the bivalent binding. Briefly, the purine ring of ATP have been shown to establish π‐π interactions with the aromatic rings of Phe, Tyr and Trp as well as π‐π or/and π‐cation interactions with the side chains of Arg/Lys, while the triphosphate chain of ATP is involved in establishing electrostatic interactions including hydrogen bonds with protein sidechain or/and backbone atoms (Figure 8(b)). Noticeably, π‐π interaction between the purine ring of ATP and the rings of aromatic amino acids appears to be weaker than π‐π and π‐cation interaction between the purine ring of ATP and Arg sidechains as evident by the result that the affinity of ATP to TDP‐43 RRM2 is lower than that to TDP‐43 RRM1. In the ATP‐RRM2 complex, the purine ring of ATP is sandwiched by the aromatic rings of two Phe residues to establish π‐π interactions while in the ATP‐RRM1 complex, the purine ring of ATP contacts the side chain of Arg to establish π‐π and π‐cation interaction. 57
2.6. ATP antagonizes the crowding‐induced destabilization and enhances folding
Eye lens is one of the most crowded cells with protein concentrations up to 400 mg/ml in human and > 1,000 mg/ml in fish lens. 103 , 104 , 105 , 106 , 107 , 108 , 109 , 110 , 111 The major ocular proteins are crystallins, which consist of three types (α, β and γ), accounting for ~90% of the lens proteins. Owing to the absence of any repair and recycling systems for the proteins in lens, crystallins need to sustain for the whole life‐span. As such, the solubility and stability of the lens crystallins are extremely critical for their functions. While α‐crystallins functions as molecular chaperones, β‐ and γ‐crystallins belong to the βγ‐crystallin superfamily, which are evolutionarily selected for their high solubility, stability and refractive indices to construct the eye lens characteristic of transparency and refractive index gradient. However, genetic mutation may lead to aggregation of crystallins of either type, thus resulting in cataract which accounts for 48% of blindness worldwide. 103 , 104 , 105 , 106 , 107 , 108 , 109 , 110 , 111
The 178‐residue γS‐crystallin is the most abundant structural protein in the cortex of the lens, which adopts the four Greek key fold organized into the N‐ and C‐domains with four cataract‐causing mutation identified: namely G18V, D26G, S39C and V42M (Figure 9(a)). 109 Although γS‐crystallin was decoded to have weak attractive intermolecular interactions, it remains monomeric even at very high concentrations. A variety of biophysical studies indicates that the attractive interaction is independent of salt concentrations, thus most unlikely being electrostatic. The unusual stability of γS‐crystallin was thus proposed to result from its unique hydration shell and the G18V mutation appears to disrupt the hydration shell. 109 , 110 , 111
FIGURE 9.
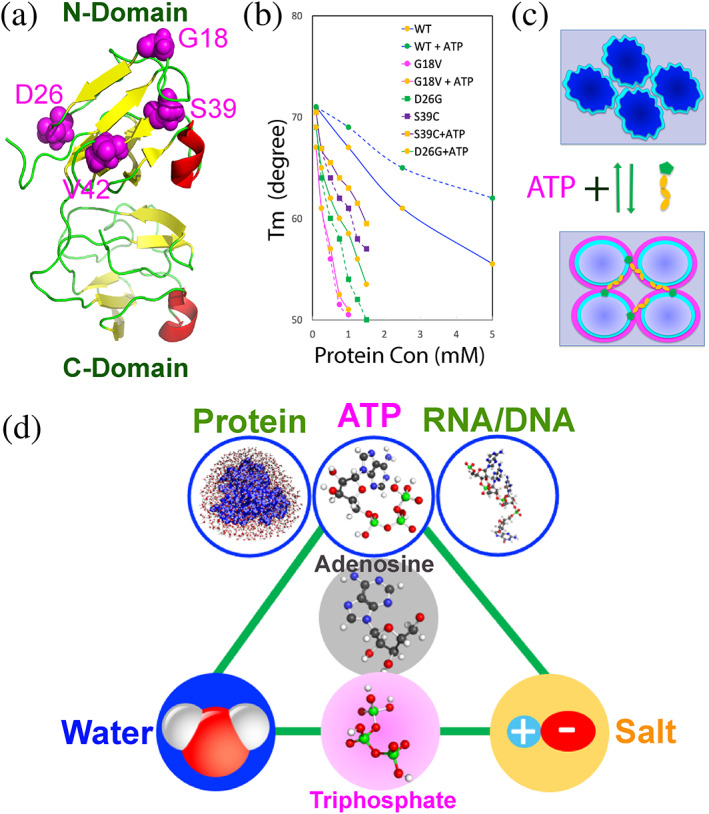
ATP controls protein homeostasis by coupling various capacities to hydration. (a) Three structure of human γS‐crystallin with four cataract‐causing mutation residues in spheres and labeled. (b) Concentration‐dependence curves of melting temperature (Tm) of WT, G18V, D26G and S39C γS‐crystallins without and with ATP at 20 mM. (c) A speculative model to illustrate that ATP antagonizes the crowding‐induced destabilization by targeting protein hydration. Under the extremely‐crowded condition, the hydration shell (cyan) is disrupted/twisted. Upon the presence of ATP, the disrupted/twisted hydration shell (cyan) is restored, and the additional “hyper‐mobile” hydration shell (pink) might be formed. (d) ATP appears to couple features of water, salt and nucleic acids, and consequently acquires the integrated capacity in controlling protein homeostasis and regulating the communication between proteins and nucleic acids in cells. The molecules connected by green lines appear to share the similar components or/and properties
Mysteriously, vertebrate lens is a metabolically quiescent organ but still maintains ATP concentrations of 3–7 mM, which is even higher than that in neurons. 51 , 104 So what is role of ATP in lens? Very recently, the effects of ATP were investigated on the conformation, self‐association, thermal stability and interaction with α‐crystallin for the wild‐type (WT) and four cataract‐causing mutants of human γS‐crystallin at various protein concentration. 43 , 44 , 45 The results reveal: (1) WT γS‐crystallin remains to be soluble and monomeric even at ~100 mg/ml, while the four mutants became prone to aggregation at concentrations >40 mg/ml. (2) The crowding induces a significant decrease of the thermal stability. Although at low protein concentration (100 μM), WT and four mutants only have small differences of the melting temperatures (Tm), the cataract‐causing mutations significantly speed up the destabilization at high protein concentration (Figure 9(b)). (3) ATP at ratios up to 1:200 showed no significant binding as well as no detectable effect on the conformations of WT and four mutants. (4) ATP also showed no significant interference in the interaction of WT/four γS‐crystallin mutants with α‐crystallin. (5) Unexpectedly, however, ATP is able to antagonize the crowding‐induced destabilization of WT even at 1:1. Intriguingly, the antagonizing effects of ATP became weaker for the mutants and in particular ATP loses antagonizing effect on the G18V mutant, whose hydration shell was biophysically characterized to be disrupted. 110
The antagonizing effect of ATP appears not to result from electrostatic effect, as NaCl and triphosphate have no such effect. In the human lens, the concentration of γS‐crystallin is >5 mM while the ATP concentrations are only ~5 mM. As such, one γS‐crystallin molecule can only averagely interacts with one ATP molecule. In this regard, it was proposed that ATP antagonizes the crowding‐induced destabilization of γS‐crystallin most likely by mediating its hydration shell (Figure 9(c)). Briefly, for γS‐crystallin under the crowding conditions, the shape/thickness or/and dynamics of the hydration shell might be largely disrupted. 110 , 111 However, upon the presence of ATP, the purine ring of ATP might transiently interact with the hydrophobic patches of the protein to facilitate its triphosphate chain to interact with the hydration shell. As the triphosphate chain of ATP has unique “hyper‐mobile” water, the altered shape/thickness or/and dynamics of the hydration shell might be partially restored, thus manifesting as the capacity of ATP to antagonize the crowding‐induced destabilization. As cataract‐causing mutations disrupt the hydration shell to different degree, ATP thus shows differential antagonizing effects on the crowding‐induced destabilization for WT and four mutants (Figure 9(b)). Therefore, even without significant binding, ATP could still function in lens to antagonize the crowding‐induced destabilization of γS‐crystallin to prevent its aggregation. This may partly rationalize the observation that cataractogenesis is age‐related as the concentration of ATP is constantly declining upon aging.
Recently, ATP was also found to be capable of inducing protein folding with very high efficiency for two ALS‐causing mutants of the well‐folded human profilin‐1 (hPFN1) and superoxide dismutase 1 (hSOD1). 61 , 112 , 113 , 114 Very unexpectedly, even without significant binding, ATP at 1:4 is sufficient to induce the complete conversion of the unfolded population into the folded state of C71G‐hPFN1 which has the co‐existence of the folded and unfolded states exchanging at 11.7 Hz. 61 , 112 On the other hand, ATP at 1:8 induces the folding of the highly unfolded state of hSOD1. 61 , 113 , 114 Moreover, the effects of ATP and 11 related molecules were successfully visualized by NMR directly on the folding and aggregation of the ALS‐causing mutants, which include ATPP, AMP‐PCP, AMP‐PNP, ADP, AMP, Adenosine, triphosphate, diphosphate, phosphate and NaCl. Most unexpectedly, the capacity of ATP in inducing folding has been decoded to come from triphosphate, which, however, is able to trigger severe aggregation of two ALS‐causing mutants with significant exposure of hydrophobic patches. Amazingly, upon joining with adenosine to form ATP, the ability of triphosphate to trigger aggregation is shielded. In particular, only ATP has three integrated abilities: to simultaneously induce folding, inhibit aggregation and increase thermal stability, that are absent in all analogues including ATPP, AMP‐PCP and AMP‐PNP. 61
Interestingly, the crosstalk might exist between LLPS and misfolded proteins. 115 Briefly, ALS‐causing hPFN1 mutants C71G‐, G118V‐ and E117G‐PFN1 have differential capacity in disrupting dynamics of liquid droplets formed by FUS NTD. 115 One possible mechanism might be that upon destabilization and misfolding, the residues of hPFN1 such as aromatic or/and Arg/Lys become more dynamic or/and exposed, thus accessible for interacting with FUS NTD residues that are critical for rendering the dynamics of liquid droplets.
3. PERSPECTIVES AND CHALLENGES
Proteins in cells function in either folded or intrinsically disordered forms under the crowded environments with high salt concentrations (~150 mM), thus facing tremendous challenge of misfolding and aggregation, although they are intrinsically soluble in unsalted water. 9 , 30 , 31 , 32 , 33 , 34 , 35 To tackle the challenge, universal mechanisms need to be evolved particularly in the protocells where the ATP‐energy‐dependent chaperone systems had not developed. 116 For the emergence of life, two key components are also essential: the molecules encoding/storing genetic information and the biological fuel providing energy to drive various biological processes. Nature solved the two problems by selecting phosphorus as a key building element for living organisms. 117 , 118 , 119 , 120 , 121 , 122 Indeed, phosphate esters not only provide the extremely stable backbone for RNA and DNA, but out of them ATP emerged as the universal energy currency by hydrolysis of its high energy bonds, which is thermodynamically favorable but kinetically controlled. So Nobelist Alexander Todd remarked: “Where there is life, there is phosphorus”. 117
The emerging results reviewed here imply that nature selected phosphorus also because ATP can energy‐independently control protein homeostasis at mM by manifesting as a biological hydrotrope, a bivalent binder and a hydration mediator (Figure 1(b)). The multifaced roles of ATP appear to critically depend on the presence of triphosphate, which was proposed to be the central intermediate in prebiotic chemistry. 117 , 118 , 119 , 120 Previously, inorganic polyphosphates were identified to function as a primordial chaperone in single‐cell organisms, but the underlying mechanism remains elusive. 121 , 122 The results discussed here suggest that polyphosphates might share the same mechanism of triphosphate to generally enhance protein folding by mediating the protein hydration. However, due to being highly charged, by electrostatic screening triphosphate is also able to severely trigger aggregation of unfolded and partially‐folded proteins with considerable exposure of hydrophobic patches. Amazingly, however, by linking the anionic triphosphate with the unique hydration property to adenosine containing the aromatic purine ring (Figure 9(d)), the aggregation‐triggering ability of triphosphate is largely shielded. This also implies the existence of the interaction between triphosphate and adenosine in ATP. Most unexpectedly, ATP appears to couple the ability for establishing hydrophobic, π‐π, π‐cation, and electrostatic interactions with proteins to the capacity in mediating the hydration of proteins (Figure 9(d)). Consequently, ATP acquires the integrated capacity in energy‐independently controlling protein homeostasis simultaneously with diverse mechanisms, because the hydration is at the heart of protein folding, dynamics, stability, phase separation and aggregation. Therefore, ATP appears to play an irreplaceable role in promoting the transition from the protocells to modern cells for Origin of Life.
Noticeably, ATP owns the basic features of nucleotides for constructing RNA and DNA, thus providing the foundation for the observations that ATP and nucleic acids not only specifically bind to the conformation‐dependent pockets of the folded proteins, but also to Arg/Lys residues within IDRs. However, ATP generally has the binding affinity much lower than that of nucleic acids. In this context, ATP with the high concentrations might be required for cells to regulate the communication between nucleic acids and proteins (Figure 9(d)). For example, the majority of cellular MLOs such as P‐body and SGs are composed of both IDR‐rich proteins and nucleic acids. While these IDR‐rich proteins such as FUS and TDP‐43 functionally participate in forming MLOs by driving LLPS through the dynamic interaction between their Arg/Lys residues within IDRs and nucleic acids, they also have the intrinsic capacity in LLPS by themselves even in the absence of nucleic acids. Nevertheless, as recently decrypted for TDP‐43, 66 their phase separation could be further exaggerated into the pathological consequence. As such, by maintaining sufficiently high ATP concentrations, cells own the ability to inhibit the pathological phase separation of these IDR‐rich proteins in the absence of nucleic acids. This might explain two puzzling observations: (1) while a large number of proteins have been shown to have the intrinsic ability to phase separate by themselves, only limited MLOs have been identified in cells. (2) Aggregation of proteins including TDP‐43 becomes dramatically provoked after a certain age, which might partly result from the reduction of cellular ATP concentrations upon aging.
In the future, it is of great interest to decipher the biological consequences of the altered properties of proteins due to the presence of ATP at mM, part of which have been biophysically characterized in vitro as presented here. As ATP appears to emerge in probiotic chemistry, it is thus anticipated that the energy‐independent effects of ATP at mM have been extensively integrated into various cellular processes during evolution. Indeed, ATP appears to be exploited by SARS‐CoV‐2 to promote its life cycle although viruses have no ability to generate ATP. Nevertheless, this task might be extremely challenging because: (1) ATP already has very high background concentrations in all living cells and is engaged in a plethora of biological processes, many of which are essential. (2) The energy‐independent functions of ATP are overlapped or even coupled with its energy‐dependent roles, as well as with other specific cellular regulations on protein homeostasis. (3) a measurable property of proteins might be the sum of the interplay of many interactions which could be modulated by ATP. For example, even protein solubility is a result of the sophisticated interplay of protein electrostatic properties, conformations, dynamics and hydration, all of which might be simultaneously modulated by ATP through diverse mechanisms. Indeed, an in vitro characterization of the effects of ATP on protein solubility showed an unusual complexity on a proteome‐scale. 123 A study on in vivo effects of ATP on solubility and aggregation revealed the coupling of ATP‐energy‐independent and ATP‐energy‐dependent processes. 124
Even for the in vitro biophysical studies, significant challenges exist at two levels. At technic level, more methods need to be developed to quantitatively characterize the dynamic interactions of ATP with proteins at atomic/residue‐specific resolution. At fundamental level, as the multifaceted capacities of ATP root in its interaction with water, it thus appears that only if we have a significant breakthrough in experimentally and computationally understanding the abnormality of water, we will be able to decode the core mechanisms for ATP to energy‐independently control protein homeostasis.
AUTHOR CONTRIBUTIONS
Jianxing Song: Conceptualization; formal analysis; funding acquisition; investigation; resources; writing‐original draft; writing‐review & editing.
ACKNOWLEDGEMENT
The study is supported by Ministry of Education of Singapore (MOE) Tier 1 Grant R‐154‐000‐B45‐114 and R‐154‐000‐B92‐114 to Jianxing Song. The author is grateful to the contribution of all lab members.
Song J. Adenosine triphosphate energy‐independently controls protein homeostasis with unique structure and diverse mechanisms. Protein Science. 2021;30:1277–1293. 10.1002/pro.4079
Funding information Ministry of Education of Singapore, Grant/Award Number: R‐154‐000‐B92‐114
REFERENCES
- 1. Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. [DOI] [PubMed] [Google Scholar]
- 2. Baldwin RL. New directions in the study of peptide h‐bonds and peptide solvation. Adv Protein Chem. 2005;72:ix–xi. [DOI] [PubMed] [Google Scholar]
- 3. Rose GD, Fleming PJ, Banavar JR, Maritan A. A backbone‐based theory of protein folding. Proc Natl Acad Sci U S A. 2006;103:16623–16633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dill KA, Bromberg S, Yue K, et al. Principles of protein folding–a perspective from simple exact models. Protein Sci. 1995;4:561–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baase WA, Liu L, Tronrud DE, Matthews BW. Lessons from the lysozyme of phage T4. Protein Sci. 2010;19:631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Powers ET, Deechongkit S, Kelly JW. Backbone‐backbone H‐bonds make context‐ dependent contributions to protein folding kinetics and thermodynamics: Lessons from amide‐to‐Ester mutations. Adv Protein Chem. 2005;72:39–78. [DOI] [PubMed] [Google Scholar]
- 7. Kim DE, Gu H, Baker D. The sequences of small proteins are not extensively optimized for rapid folding by natural selection. Proc Natl Acad Sci U S A. 1998;95:4982–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. [DOI] [PubMed] [Google Scholar]
- 9. Song J. Environment‐transformable sequence‐structure relationship: A general mechanism for proteotoxicity. Biophys Rev. 2018;10:503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wright PE, Dyson HJ. Intrinsically unstructured proteins: Reassessing the protein structure‐function paradigm. J Mol Biol. 1999;293:321–331. [DOI] [PubMed] [Google Scholar]
- 11. van der Lee R, Buljan M, Lang B, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. André C, Dumetz 1, Aaron M, et al. Protein phase behavior in aqueous solutions: Crystallization, liquid‐liquid phase separation, gels, and aggregates. Biophys J. 2008;94:570–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brangwynne CP, Eckmann CR, Courson DS, et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. [DOI] [PubMed] [Google Scholar]
- 14. Hyman AA, Weber CA. Ju ¨licher F. Liquid‐liquid phase separation in biology. Annu Rev Cell Dev Biol. 2014;30:39–58. [DOI] [PubMed] [Google Scholar]
- 15. Brangwynne CP, Tompa P, Pappu RV. Polymer physics of intracellular phase transitions. Nature Phys. 2015;11:899–904. [Google Scholar]
- 16. Choi JM, Holehouse AS, Pappu RV. Physical principles underlying the complex biology of intracellular phase transitions. Annu Rev Biophys. 2020;49:107–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: Organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18:285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang J, Choi JM, Holehouse AS, et al. Molecular grammar underlying the driving forces for phase separation of prion‐like RNA binding proteins. Cell. 2018;174:688–699.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: From genes to mechanism. Nature. 2016;539:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poudyal RR, Cakmak FP, Keating CD, Bevilacqua PC. Physical principles and extant biology reveal roles for RNA‐containing Membraneless compartments in origins of life chemistry. Biochemistry. 2018;57:2509–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Flory PJ. Thermodynamics of high polymer solutions. J Chem Phys. 1942;10:51–61. [Google Scholar]
- 22. Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lindner AB, Demarez A. Protein aggregation as a paradigm of aging. Biochim Biophys Acta. 1790;2009:980–996. [DOI] [PubMed] [Google Scholar]
- 24. Tannous P, Zhu H, Nemchenko A, et al. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. 2008;117:3070–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu M, Hodish I, Haataja L, et al. Proinsulin misfolding and diabetes: Mutant INS gene‐induced diabetes of youth. Trends Endocrinol Metab. 2010;21:652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Finney JL. Water? What's so special about it! Philos Trans Roy Soc Lond B: Biol Sci. 2004;359:1145–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ball P. Water: Water – An enduring mystery. Nature. 2008;452:291–292. [DOI] [PubMed] [Google Scholar]
- 28. Ball P. Water as an active constituent in cell biology. Chem Rev. 2008;108:74–108. [DOI] [PubMed] [Google Scholar]
- 29. Chaplin M. Do we underestimate the importance of water in cell biology? Nat Rev Mol Cell Biol. 2006;7:861–866. [DOI] [PubMed] [Google Scholar]
- 30. Li M, Liu J, Ran X, et al. Resurrecting abandoned proteins with pure water: CD and NMR studies of protein fragments solubilized in salt‐free water. Biophys J. 2006;91:4201–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song J. Insight into “insoluble proteins” with pure water. FEBS Letts. 2009;583:953–959. [DOI] [PubMed] [Google Scholar]
- 32. Song J. Why do proteins aggregate? “Intrinsically insoluble proteins” and “dark mediators” revealed by studies on “insoluble proteins” solubilized in pure water. F1000Research. 2013;2:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Delak K, Harcup C, Lakshminarayanan R, et al. The tooth enamel protein, porcine ame‐ logenin, is an intrinsically disordered protein with an extended molecular configuration in the monomeric form. Biochemistry. 2009;48:2272–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aguado‐Llera D, Goormaghtigh E, De Geest N, et al. The basic helix‐loop‐helix region of human neurogenin 1 is a monomeric natively unfolded protein which forms a “fuzzy” complex upon DNA binding. Biochemistry. 2010;49:1577–1589. [DOI] [PubMed] [Google Scholar]
- 35. Futami J, Fujiyama H, Kinoshita R, et al. Denatured mammalian protein mixtures exhibit unusually high solubility in nucleic acid‐free pure water. PLoS One. 2014;9:e113295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baldwin RL. How Hofmeister ion interactions affect protein stability. Biophys J. 1996;4:2056–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miao L, Qin H, Koehl P, Song J. Selective and specific ion binding on proteins at physiologically‐relevant concentrations. FEBS Lett. 2011;585:3126–3132. [DOI] [PubMed] [Google Scholar]
- 38. Laue T. Charge matters. Biophys Rev. 2016;8:287–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moran U, Phillips R, Milo R. SnapShot: Key numbers in biology. Cell. 2010;141:1262–1262. [DOI] [PubMed] [Google Scholar]
- 40. Zimmerman SB, Minton AP. Macromolecular crowding: Biochemical, biophysical, and physiological consequences. Annu Rev Biophys Biomol Struct. 1993;22:27–65. [DOI] [PubMed] [Google Scholar]
- 41. Ellis RJ. Macromolecular crowding: Obvious but underappreciated. Trends Biochem Sci. 2001;26:597–604. [DOI] [PubMed] [Google Scholar]
- 42. Inomata K, Ohno A, Tochio H, et al. High‐resolution multi‐dimensional NMR spectroscopy of proteins in human cells. Nature. 2009;458:106–109. [DOI] [PubMed] [Google Scholar]
- 43. He Y, Kang J, Song J. ATP antagonizes the crowding‐induced destabilization of the human eye‐lens protein γS‐crystallin. Biochem Biophys Res Commun. 2020;526:1112–1117. [DOI] [PubMed] [Google Scholar]
- 44. He Y, Kang J, Song J. Cataract‐causing G18V eliminates the antagonization by ATP against the crowding‐induced destabilization of human γS‐crystallin. Biochem Biophys Res Commun. 2020;530:554–560. [DOI] [PubMed] [Google Scholar]
- 45. He Y, Kang J, Song J. ATP differentially antagonizes the crowding‐induced destabilization of human γS‐crystallin and its four cataract‐causing mutants. Biochem Biophys Res Commun. 2020;533:913–918. [DOI] [PubMed] [Google Scholar]
- 46. Baldwin RL. Dynamic hydration shell restores Kauzmann's 1959 explanation of how the hydrophobic factor drives protein folding. Proc Natl Acad Sci U S A. 2014;111:13052–12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levy Y, Onuchic JN. Water mediation in protein folding and molecular recognition. Annu Rev Biophys Biomol Struct. 2006;35:389–415. [DOI] [PubMed] [Google Scholar]
- 48. Fogarty AC, Duboué‐Dijon E, Sterpone F, et al. Distinct role of hydration water in protein misfolding and aggregation revealed by fluctuating thermodynamics analysis. Acc Chem Res. 2015;48:956–965. [DOI] [PubMed] [Google Scholar]
- 49. Laage D, Elsaesser T, Hynes JT. Water dynamics in the hydration shells of biomolecules. Chem Rev. 2017;117:10694–10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mazza MG, Stokely K, Pagnotta SE, et al. More than one dynamic crossover in protein hydration water. Proc Natl Acad Sci U S A. 2011;108:19873–19878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leningher A. Principles of biochemistry. New York: W. H. Freeman and Company, 2005. [Google Scholar]
- 52. Patel A, Malinovska L, Saha S, et al. ATP as a biological hydrotrope. Science. 2017;356:753–756. [DOI] [PubMed] [Google Scholar]
- 53. Rice AM, Rosen MK. ATP controls the crowd. Science. 2017;356:701–702. [DOI] [PubMed] [Google Scholar]
- 54. Kang J, Lim L, Song J. ATP enhances at low concentrations but dissolves at high concentrations liquid‐liquid phase separation (LLPS) of ALS/FTD‐causing FUS. Biochem Biophys Res Commun. 2018;504:545–551. [DOI] [PubMed] [Google Scholar]
- 55. Kang J, Lim L, Lu Y, Song J. A unified mechanism for LLPS of ALS/FTLD‐causing FUS as well as its modulation by ATP and oligonucleic acids. PLoS Biol. 2019;17:e3000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kang J, Lim L, Song J. ATP binds and inhibits the neurodegeneration‐associated fibrillization of the FUS RRM domain. Commun Biol. 2019;2:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dang M, Kang J, Lim L, et al. ATP is a cryptic binder of TDP‐43 RRM domains to enhance stability and inhibit ALS/AD‐associated fibrillation. Biochem Biophys Res Commun. 2020;522:247–253. [DOI] [PubMed] [Google Scholar]
- 58. Dang M, Song J. ALS‐causing D169G mutation disrupts the ATP‐binding capacity of TDP‐43 RRM1 domain. Biochem Biophys Res Commun. 2020;524:459–464. [DOI] [PubMed] [Google Scholar]
- 59. He Y, Kang J, Lim L, Song J. ATP binds nucleic‐acid‐binding domains beyond RRM fold. Biochem Biophys Res Commun. 2020;522:826–831. [DOI] [PubMed] [Google Scholar]
- 60. Wang L, Lim L, Dang M, Song J. A novel mechanism for ATP to enhance the functional oligomerization of TDP‐43 by specific binding. Biochem Biophys Res Commun. 2019;514:809–814. [DOI] [PubMed] [Google Scholar]
- 61. Kang J, Lim L, Song J. ATP induces protein folding, inhibits aggregation and antagonizes destabilization by effectively mediating water‐protein‐ion interactions, the heart of protein folding and aggregation. BioRxiv. 2020. 10.1101/2020.06.21.163758. [DOI] [Google Scholar]
- 62. Mogami G, Wazawa T, Morimoto N, et al. Hydration properties of adenosine phosphate series as studied by microwave dielectric spectroscopy. Biophys Chem. 2011;154:1–7. [DOI] [PubMed] [Google Scholar]
- 63. Schwartz JC, Cech TR, Parker RR. Biochemical properties and biological functions of FET proteins. Annu Rev Biochem. 2015;84:355–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Han TW, Kato M, Xie S, et al. Cell‐free formation of RNA granules: Bound RNAs identify features and components of cellular assemblies. Cell. 2012;149:768–779. [DOI] [PubMed] [Google Scholar]
- 65. Burke KA, Janke AM, Rhine CL, Fawzi NL. Residue‐by‐residue view of in vitro FUS granules that bind the C‐terminal domain of RNA polymerase II. Mol Cell. 2015;60:231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gasset‐Rosa F, Lu S, Yu H, et al. Cytoplasmic TDP‐43 De‐mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP‐43, and cell death. Neuron. 2019;102:339–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Qin H, Lim L, Wei Y, Song J. TDP‐43 N terminus encodes a novel ubiquitin‐ like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA, Proc. Natl Acad Sci U S A. 2014;111:18619–18624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Afroz T, Hock EM, Ernst P, et al. Functional and dynamic polymerization of the ALS‐linked protein TDP‐43 antagonizes its pathologic aggregation. Nat Commun. 2017;8:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lukavsky PJ, Daujotyte D, Tollervey JR, et al. Molecular basis of UG‐rich RNA recognition by the human splicing factor TDP‐43. Nat Struct Mol Biol. 2013;20:1443–1449. [DOI] [PubMed] [Google Scholar]
- 70. Lim L, Wei Y, Lu Y, Song J. ALS‐causing mutations significantly perturb the self‐assembly and interaction with nucleic acid of the intrinsically disordered prion‐like domain of TDP‐43. PLoS Biol. 2016;14:e1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dang M, Lim L, Kang J, Song J. ATP regulates TDP‐43 pathogenesis by specifically binding to an inhibitory component of a delicate network controlling LLPS. BioRxiv. 2020. 10.1101/2020.10.08.330829. [DOI] [Google Scholar]
- 72. Cao Q, Boyer DR, Sawaya MR, et al. Cryo‐EM structures of four polymorphic TDP‐43 amyloid cores. Nat Struct Mol Biol. 2019;26:619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Conicella AE, Zerze GH, Mittal J, Fawzi NL. ALS mutations disrupt phase separation mediated by a‐helical structure in the TDP‐43 low‐ complexity C‐terminal domain. Structure. 2016;24:1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schmidt HB, Rohatgi R. In vivo formation of vacuolated multi‐phase compartments lacking membranes. Cell Rep. 2016;16:1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li HR, Chiang WC, Chou PC, et al. TAR DNA‐binding protein 43 (TDP‐43) liquid– Liquid phase separation is mediated by just a few aromatic residues. J Biol Chem. 2018;293:6090–6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Babinchak WM, Haider R, Dumm BK, et al. The role of liquid–liquid phase separation in aggregation of the TDP‐43 low‐complexity domain. J Biol Chem. 2019;294:6306–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang L, Kang J, Lim L, Wei Y, Song J. TDP‐43 NTD can be induced while CTD is significantly enhanced by ssDNA to undergo liquid‐liquid phase separation. Biochem Biophys Res Commun. 2018;499:189–195. [DOI] [PubMed] [Google Scholar]
- 78. Lin Y, Zhou X, Kato M, et al. Redox mediated regulation of an evolutionarily conserved cross‐β structure formed by the TDP43 low complexity domain. Proc Natl Acad Sci U S A. 2020;117:28727–28734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Maharana S, Wang J, Papadopoulos DK, et al. RNA buffers the phase separation behavior of prion‐like RNA binding proteins. Science. 2018;360:918–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Guccione E, Richard S. The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol. 2019;20:642–657. [DOI] [PubMed] [Google Scholar]
- 81. Thandapani P, O'Connor TR, Bailey TL, Richard S. Defining the RGG/RG motif. Mol Cell. 2013;50:613–623. [DOI] [PubMed] [Google Scholar]
- 82. Rajyaguru P, Parker R. RGG motif proteins: Modulators of mRNA functional states. Cell Cycle. 2012;11:2594–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chong PA, Vernon RM, Forman‐Kay JD. RGG/RG motif regions in RNA binding and phase separation. J Mol Biol. 2018;430:4650–4665. [DOI] [PubMed] [Google Scholar]
- 84. Wessner DR. The origins of viruses. Nat Edu. 2010;3:37. [Google Scholar]
- 85. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lu S, Ye Q, Singh D, et al. The SARS‐CoV‐2 Nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane‐associated M protein. BioRxiv. 2020. 10.1101/2020.07.30.228023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Savastano A, de Opakua AI, Rankovic M, et al. Nucleocapsid protein of SARS‐CoV‐2 phase separates into RNA‐rich polymerase‐containing condensates. Nat Commun. 2020;11:6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Perdikari TM, Murthy AC, Ryan VH, et al. SARS‐CoV‐2 nucleocapsid protein phase‐separates with RNA and with human hnRNPs. EMBO J. 2020;39:e106478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Carlson CR, Asfaha JB, Ghent CM, et al. Phosphoregulation of phase separation by the SARS‐CoV‐2 N protein suggests a biophysical basis for its dual functions. Mol Cell. 2020;80:1092–1103.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dang M, Li Y, Song J. ATP biphasically modulates LLPS of SARS‐CoV‐2 nucleocapsid protein and specifically binds its RNA‐binding domain. Biochem Biophys Res Commun. 2021;94:50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dinesh DC, Chalupska D, Silhan J, et al. Structural basis of RNA recognition by the SARS‐CoV‐2 nucleocapsid phosphoprotein. PLoS Pathog. 2020;16:e1009100–e1009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zinzula L, Basquin J, Bohn S, et al. High‐resolution structure and biophysical characterization of the nucleocapsid phosphoprotein dimerization domain from the Covid‐19 severe acute respiratory syndrome coronavirus 2. Biochem Biophys Res Commun. 2021;538:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Song J, Ni F. NMR for the design of functional mimetics of protein‐protein interactions: One key is in the building of bridges. Biochem Cell Biol. 1998;76:177–188. [DOI] [PubMed] [Google Scholar]
- 94. Maisonnasse P, Guedj J, Contreras V, et al. Hydroxychloroquine use against SARS‐CoV 2 infection in non‐human primates. Nature. 2020;585:584–587. [DOI] [PubMed] [Google Scholar]
- 95. Roldan EQ, Biasiotto G, Magro P, Zanella I. The possible mechanisms of action of 4‐aminoquinolines (chloroquine/hydroxychloroquine) against Sars‐Cov‐2 infection (COVID‐19): A role for iron homeostasis? Pharmacol Res. 2020;158:104904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Satarker S, Ahuja T, Banerjee M, et al. Hydroxychloroquine in COVID‐19: Potential mechanism of action against SARS‐CoV‐2. Curr Pharmacol Rep. 2020;24:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Dang M, Song J. Structural basis of anti‐SARS‐CoV‐2 activity of hydroxychloroquine: Specific binding to NTD/CTD and disruption of LLPS of N protein. BioRxiv. 2021. 10.1101/2021.03.16.435741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lu Y, Lim L, Song J. RRM domain of ALS/FTD‐causing FUS characteristic of irreversible unfolding spontaneously self‐assembles into amyloid fibrils. Sci Rep. 2017;7:1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dang M, Li Y, Song J. Tethering‐induced destabilization and ATP‐binding for tandem RRM domains of ALS‐causing TDP‐43 and hnRNPA1. Sci Rep. 2021;11:1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Buchanan SG, Hendle J, Lee PS, et al. SGX523 is an exquisitely selective, ATP‐competitive inhibitor of the MET receptor tyrosine kinase with antitumor activity in vivo. Mol Cancer Ther. 2009;8:3181–3190. [DOI] [PubMed] [Google Scholar]
- 101. Scheeff ED, Bourne PE. Structural evolution of the protein kinase‐like superfamily. PLoS Comput Biol. 2005;1:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kanev GK, de Graaf C, de Esch IJP, et al. The landscape of atypical and eukaryotic protein kinases. Trends Pharmacol Sci. 2019;40:818–832. [DOI] [PubMed] [Google Scholar]
- 103. Ghosh KS, Chauhan P. Crystallins and their complexes. Subcell Biochem. 2019;93:439–460. [DOI] [PubMed] [Google Scholar]
- 104. Greiner JV, Glonek T. Hydrotropic function of ATP in the crystalline lens. Exp Eye Res. 2020;190:107862. [DOI] [PubMed] [Google Scholar]
- 105. Tardieu A, Vérétout F, Krop B, et al. Protein interactions in the calf eye lens: Interactions between beta‐crystallins are repulsive whereas in gamma‐crystallins they are attractive. Eur Biophys J. 1992;21:1–12. [DOI] [PubMed] [Google Scholar]
- 106. Petitt P, Edwards ME, Forciniti D. A small‐angle neutron scattering study of gamma‐crystallins near their isoelectric point. Eur J Biochem. 1997;243:415–421. [DOI] [PubMed] [Google Scholar]
- 107. Mills IA, Flaugh SL, Kosinski‐Collins MS, et al. Folding and stability of the isolated Greek key domains of the long‐lived human lens proteins gammaD‐ crystallin and gammaS‐crystallin. Protein Sci. 2007;16:2427–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhao H, Chen Y, Rezabkova L, et al. Solution properties of γ‐crystallins: Hydration of fish and mammal γ‐crystallins. Protein Sci. 2014;23:88e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kingsley CN, Brubaker WD, Markovic S, et al. Preferential and specific binding of human αB‐crystallin to a cataractrelated variant of γS‐crystallin. Structure. 2013;21:2221–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Huang KY, Kingsley CN, Sheil R, et al. Stability of protein‐specific hydration Shell on crowding. J Am Chem Soc. 2016;138:5392–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Harada R, Sugita Y, Feig M. Protein crowding affects hydration structure and dynamics. J Am Chem Soc. 2012;134:4842–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lim L, Kang J, Song J. ALS‐causing profilin‐1‐mutant forms a non‐native helical structure in membrane environments. Biochim Biophys Acta Biomembr. 1859;2017:2161–2170. [DOI] [PubMed] [Google Scholar]
- 113. Lim L, Lee X, Song J. Mechanism for transforming cytosolic SOD1 into integral membrane proteins of organelles by ALS‐causing mutations. Biochim Biophys Acta. 1848;2015:1–7. [DOI] [PubMed] [Google Scholar]
- 114. Lim L, Song J. SALS‐linked WT‐SOD1 adopts a highly similar helical conformation as FALS‐causing L126Z‐SOD1 in a membrane environment. Biochim Biophys Acta. 1858;2016:2223–2230. [DOI] [PubMed] [Google Scholar]
- 115. Kang J, Lim L, Song J. Misfolded proteins share a common capacity in disrupting LLPS organizing membrane‐less organelles. BioRxiv. 2018. 10.1101/317347. [DOI] [Google Scholar]
- 116. Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. [DOI] [PubMed] [Google Scholar]
- 117. Todd AR. In: Nakamura K, Kageyama M, Oshima T, editors. Where there's life there's phosphorus. Japan: Science and Society Press, 1981; p. 275. [Google Scholar]
- 118. Westheimer FH. Why nature chose phosphates. Science. 1987;235:1173–1178. [DOI] [PubMed] [Google Scholar]
- 119. Bowler MW, Cliff M, Walthoand JP, Blackburn GM. Why did nature select phosphate for its dominant roles in biology ? New J Chem. 2010;34:784–794. [Google Scholar]
- 120. Kamerlin SC, Sharma PK, Prasad RB, et al. Why nature really chose phosphate. Q Rev Biophys. 2013;46:1–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Gray MJ, Wholey WY, Wagner NO, et al. Polyphosphate is a primordial chaperone. Mol Cell. 2014;53:689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Rao NN, Gómez‐García MR, Kornberg A. Inorganic polyphosphate: Essential for growth and survival. Annu Rev Biochem. 2009;78:605–647. [DOI] [PubMed] [Google Scholar]
- 123. Sridharan S, Kurzawa N, Werner T, et al. Proteome‐wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat Commun. 2019;10:1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hayes MH, Peuchen EH, Dovichi NJ, Weeks DL. Dual roles for ATP in the regulation of phase separated protein aggregates in Xenopus oocyte nucleoli. Elife. 2018;7:e35224. [DOI] [PMC free article] [PubMed] [Google Scholar]