

Abstract
Obesity increases the risk for breast cancer and is associated with poor outcomes for cancer patients. A variety of rodent models have been used to investigate these relationships; however, key differences in experimental approaches, as well as unique aspects of rodent physiology lead to variability in how these valuable models are implemented. We combine expertise in the development and implementation of preclinical models of obesity and breast cancer to disseminate effective practices for studies that integrate these fields. In this review, we share, based on our experience, key considerations for model selection, highlighting important technical nuances and tips for use of preclinical models in studies that integrate obesity with breast cancer risk and progression. We describe relevant mouse and rat paradigms, specifically highlighting differences in breast tumor subtypes, estrogen production, and strategies to manipulate hormone levels. We also outline options for diet composition and housing environments to promote obesity in female rodents. While we have applied our experience to understanding obesity-associated breast cancer, the experimental variables we incorporate have relevance to multiple fields that investigate women’s health.
Keywords: obesity, adipose tissue, preclinical model, breast cancer, thermoneutrality, diet, ovariectomy, menopause, carcinogen, tumor subtype
Introduction
Obesity has become a worldwide epidemic, with nearly 70% of adults in the United States and 52% worldwide identified as either overweight or obese (1–3). There are many negative health consequences associated with obesity, including an increased risk of breast cancer and poorer prognosis, particularly in postmenopausal women with estrogen dependent tumors (4–9). One limitation of research in this field has been the difficulty of developing preclinical models that accurately reflect the physiology of tumors that develop in humans. Thus, the goal of this paper is to provide insight into our experience in the development and implementation of rodent models that combine obesity and simulated menopause with the development of hormone-receptor (HR) positive mammary/breast tumors. The techniques that we describe for promoting excess adiposity can, however, be applied to any cancer that is associated with obesity, including endometrial, colon, pancreatic, and kidney cancers (10).
There are many research groups who have done tremendous work to optimize and improve preclinical models of breast cancer. Throughout this review we will touch on several such areas and direct the reader towards relevant publications that describe this work in detail. Our contribution to this field is incorporation of the relevant variable of obesity (increased adiposity) into these models. In the rat, we have been successful in establishing models with intact immune systems, while in mice we have done so in both immune-compromised and -competent animals.
Many of the tips, tools, considerations, and caveats that we describe can be applied to other existing models of breast cancer. In the current review, we will highlight key features and considerations as shown in Figure 1. We aim to provide examples based on our personal experience with these models and highlight considerations for reproducing and/or incorporation of these techniques for the development of new models. Together we hope that this will provide the basis for other investigators to further contribute to our knowledge of the complex relationship between obesity and breast cancer.
Figure 1. Considerations for preclinical modeling of obesity and breast cancer.
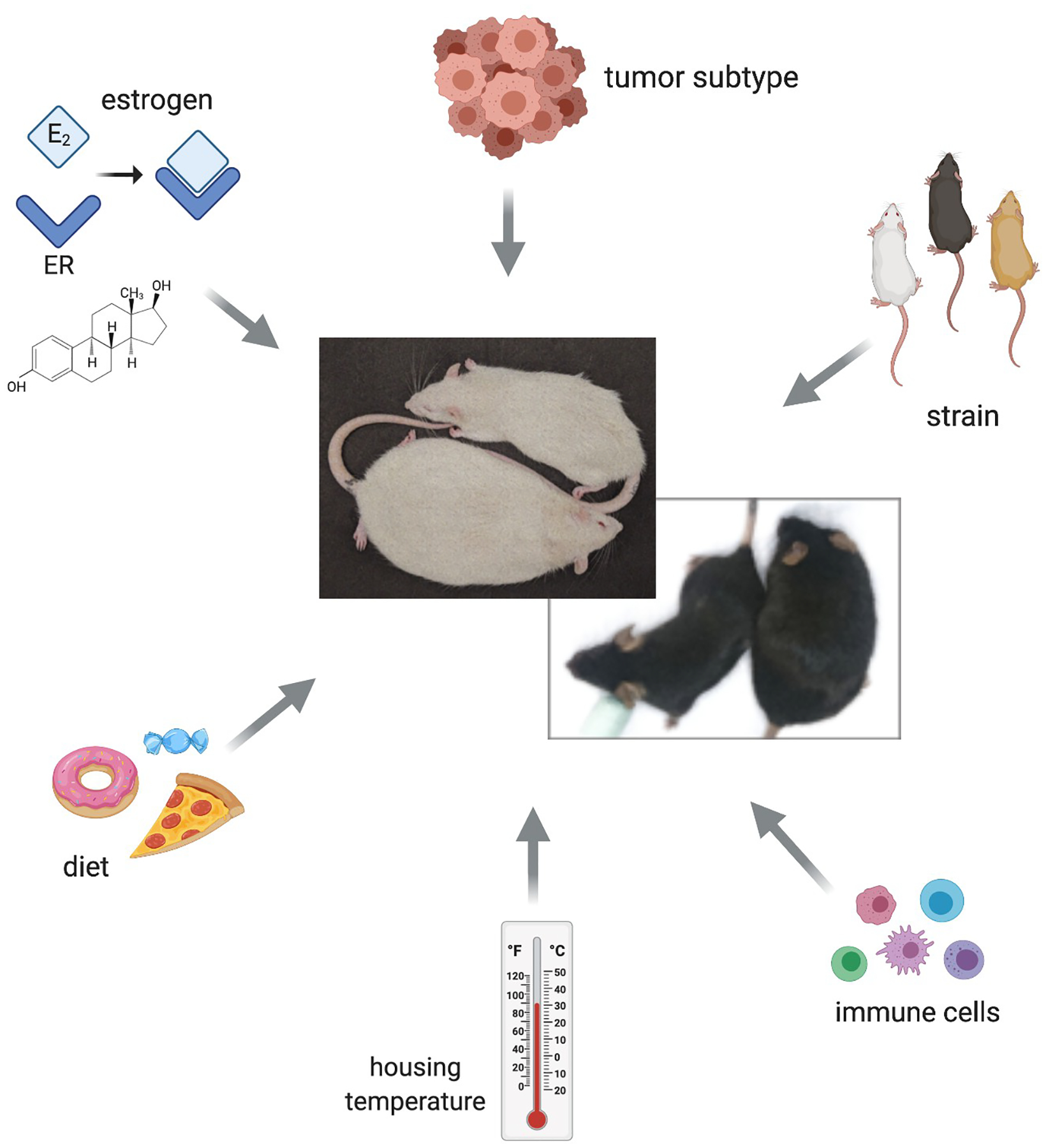
Each factor, including the presence of estrogen, tumor subtype, rodent strain, immune function, housing temperature, and diet influences and/or is influenced by obesity. Representative images of Lean and Obese female rats (top; white) and mice (bottom; black) derived from these models are shown.
1. Models of Hormone Dependent Tumors
a. Breast Cancer Subtypes
It is well established that breast cancer is not one disease; rather, it occurs as distinct subtypes that display inter- and intra-tumoral heterogeneity. Clinically, tumors are analyzed and subtyped based on immunohistochemistry and/or in situ hybridization for expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) (11). Presence or absence of these receptors is currently used to dictate treatment. Over the past decade, scientists have used gene expression profiling to classify breast cancers by their molecular (intrinsic) subtypes, including normal-like, luminal A, luminal B, HER2-enriched, and basal (12, 13). Although not identical to the clinical subtypes, there are similarities between ER+/PR+ and luminal A/B tumors, HER2/ERBB2+ and HER2-enriched, and TN and basal cancers in terms of frequency, gene expression profiles, and prognoses (12, 13). The tumor subtypes and their association with obesity are as follows:
HER-2 / ERBB2 Positive (HER2+):
Approximately 15% of breast tumors have amplification or overexpression of the human epidermal growth factor receptor 2 and are referred to as HER2+ (14). The HER2+ subtype is treated with targeted therapy that includes monoclonal antibodies or small molecule inhibitors and the 5-year survival for stage I HER2+ breast cancer is approximately 94% (14). Overall, most studies have failed to report an association between obesity and development of HER2+ tumors; however, a recent study has shown that bariatric surgery was associated with reduced HER2+ breast cancers (15) and emerging evidence suggests that obesity may be correlated with worse overall survival in patients with HER2+ metastatic disease (16). Thus, the link between obesity and HER2+ cancers warrants further investigation.
Triple Negative (TN):
Tumors that lack ER, PR and HER2 are classified as triple negative (TN) (11, 14). These represent approximately 15% of all diagnoses and have a poorer disease free and overall survival compared with HR- and HER2+ tumors. For stage I TN tumors, the 5-year survival is approximately 85% (17). Because they lack the receptor targets that are present in the other subtypes, TN breast cancers are treated with chemotherapy (14). Obesity is associated with an increased risk for TN tumors in premenopausal women, but may not associate with overall- or disease-free survival in this population (6, 18).
Hormone Receptor Positive (HR+):
Approximately 70% of tumors have >1% cells positive for the estrogen receptor (ER) and/or progesterone receptor (PR) and are referred to as ER/PR+ or hormone receptor (HR) positive (11, 14, 19). Obesity is associated with an increased risk for HR+ breast cancer, particularly after menopause (6). Patients with HR+ breast cancer are often treated with anti-estrogen therapies, such as the selective estrogen receptor modulator tamoxifen or one of several aromatase inhibitors. The prescribed treatment depends in part on the patient’s age and menopausal status, as well as the stage and grade of the tumor. HR+ breast cancers have a favorable prognosis, with a 5-year disease-free survival for patients diagnosed with stage I tumors of 99% (14). However, the relatively long latency of tumor recurrence remains a clinical challenge (20). Women with obesity are more likely to experience HR+ cancer recurrence after treatment with the aromatase inhibitor anastrozole (21), suggesting the possibility that the excess estrogens associated with obesity may not be completely suppressed by standard doses (22, 23). However, clinical studies have found no added benefit to tumor response with high versus low aromatase inhibitor treatment (24, 25), and a recent study found that increasing the dose of letrozole, a more potent aromatase inhibitor than anastrozole (26), did not further reduce circulating estrogen levels in women with a body mass index (BMI) > 25 kg/m2 (27). Together, these data indicate that factors instead of or in addition to estrogen may drive breast cancer growth in the context of obesity, which is an important consideration when designing preclinical models of this disease.
b. Modeling HR+ Tumors
One of the greatest limitations in developing preclinical models to study the interaction between obesity and breast cancer has been to model HR+ tumors in a postmenopausal setting. This is particularly important given the strong association between obesity and breast cancer risk after menopause, where HR+ tumors are the predominant subtype. Despite this need, it has been notoriously difficult to generate models where grafted human tumors retain expression of the estrogen receptor (ER) after circulating estrogens are removed. Additionally, there are relatively few mouse models of mammary tumorigenesis that produce ER+ cancers (28). While we have not fully overcome all of these challenges, we have made significant strides in working to improve many aspects of these models, thus improving our ability to study the complex relationship between obesity and breast cancer in the postmenopausal setting.
The field of breast cancer research has a wide variety of model systems to use for scientific and preclinical studies. These include xenograft models that use patient-derived tumors (PDX) or established human cell lines, syngeneic mouse mammary tumor cell lines, genetically engineered/transgenic mice, and chemical carcinogen-induced models. Each system has benefits and limitations.
Chemical Carcinogen Models
The use of chemical carcinogens to induce tumor formation is not new; this approach was pioneered for development of mammary tumors by Dr. Charles Brenton Huggins (reviewed in (29)) and has been used for more than 70 years. One of the benefits of this approach is that it allows for induction of tumors in immune competent animals. The two most common carcinogens used to induce mammary tumors are 1-methyl-1-nitrosourea (MNU) and 7,12-Dimethylbenzathracene (DMBA). Work from Henry Thompson’s lab has significantly expanded our knowledge regarding the use of carcinogens for tumor formation, and they have detailed the procedure for both MNU and DMBA elsewhere (30, 31). While some researchers have argued that carcinogen induces variability in tumor incidence (32), such studies have given multiple injections of MNU; administration of a single dose of MNU (either i.v. or i.p) has been shown to be highly specific and reproducible for induction of mammary carcinomas (33). Work by Thompson and colleagues has been instrumental in detailing the timing, doses, and administration of carcinogen needed to induce mammary tumor formation in a reproducible manner (30). By following these procedures, we have similarly found MNU to be a reliable and consistent means of inducing mammary tumor formation in rats (34–39). The tumors produced also have the benefit of reflecting clinical disease with respect to the proportion of tumors that are intraductal, the progression of histologic stages from hyperplasia to in situ to invasive, and hormone receptor positivity (40, 41). While we have not done so, MNU will also produce mammary tumors in mice; however, the dose of MNU needed will differ from that in the rat.
The technical aspects of preparing and administering MNU to rats are all described in detail in (30), and adhering to these guidelines will increase the specificity and reproducibility of tumor development. Briefly, key considerations critical to the success of this model, as well as our approach include:
-
Source and Handling of carcinogen
There are several reputable sources for MNU, and some that are less reliable. MNU is sensitive to temperature, light, and humidity. It should be stored at temperatures below −10°C, under conditions where exposure to light and moisture are minimized. This includes shipping on dry ice. Ash Stevens (Detroit Michigan) was previously a preferred source for MNU (30), however, it is no longer producing this compound. Similarly, in recent years the chemical repository at the National Cancer Institute has not been able to provide sufficient volumes of carcinogen for studies. For the past several years, we have obtained MNU from MRI Global (Kansas City, Missouri) and found this to be a reliable source of the agent.
-
Preparation and Administration of MNU; Concentration of MNU
The dose of carcinogen will influence tumor incidence and multiplicity achieved. Studies generally use doses ranging from 10 to 50 mg/kg body weight (30, 42). 50 mg/kg body weight was reported to induce tumors in 100% of rats (41). Our unpublished data would suggest that higher doses may lead to an increased number of off-target effects.
We have consistently administered MNU at a dose of 50 mg/kg body weight, by i.p. injection. In our hands, and as reported by Thompson, this dose results in >80% of rats developing palpable tumors by 6 months (34–36, 38, 39). Although MNU can be delivered intravenously, subcutaneously, or intraperitoneally, the i.p. route has been reported to generate the most consistent tumor response (30). This increased reliability has been attributed to better accuracy in the delivery with this approach. We prepare MNU as described (30). For large cohorts of rats, we prepare several small vials of MNU which are solubilized in acidified saline (pH <5.0, final concentration 14 mg/mL) so that the powered carcinogen is dissolved immediately prior to injecting the animals. We ensure that MNU is used within 1h of addition of saline to the powered agent, although in most cases we prepare small enough batches that the solubilized MNU does not sit for longer than 40 minutes.
-
Age of Rat at Time of Administration
Early work by Huggins with DMBA found that the ideal window of sensitivity for carcinogen induction of mammary tumors was 50 to 65 days of age, and thus this range was often also used for MNU studies. This age range corresponds to the rapid ductal and alveolar epithelial proliferation taking place in the mammary gland during puberty. The carcinogenic insult to the mammary gland is diminished when delivered after 65 days of age (for both MNU and DMBA), thus carcinogen must be administered prior to this age.
Thompson and colleagues have performed dose-response studies with MNU, demonstrating no statistical differences in tumor incidence, number, or latency in rats injected with 50 mg MNU/kg body weight at 28, 35 or 42 days of age (42). Thus, when merging MNU-induced tumors with models of obesity, we have chosen to use the ~55 ± 2 days of age time frame, with a dose of 50mg/kg body weight, to ensure that almost all rats will develop a tumor, but will do so in a time frame that allows them to also develop diet-induced obesity before moving into the “menopausal” phase of our studies. In our experience, a single injection of MNU given at this time does not alter weight gain or the development of obesity in these animals.
Transgenic and Syngeneic Models
Transgenic mouse models of breast cancer rely on mammary-specific promoters (e.g. mouse mammary tumor virus, MMTV; whey acidic protein, WAP) to drive expression of various oncogenes in the mammary ductal and alveolar epithelial cells (43, 44). Examples include (but are not limited to) the MMTV-Neu line, which expresses the rat HER2/ERBB2 proto-oncogene, the MMTV-Polyomavirus Middle T antigen (PyMT), which produces numerous rapidly growing tumors in young mice, and the MMTV-c-myc, which encodes a transcription factor that is often mutated in human breast cancer (43, 44). Mouse mammary tumor cell lines, such as the mouse-derived syngeneic transplant collection (MDST) (45) have been established that can be grafted orthotopically to study tumor progression. Syngeneic mammary tumor lines are established cancer cells, which precludes the investigation of the early stages of tumorigenesis in these models.
The use of transgenic mouse models and grafted syngeneic lines to study breast cancer has benefits such as the relatively short disease course compared to humans and the wide availability of molecular tools to interrogate DNA, RNA, and protein. Importantly, tumors grow in the presence of intact immune systems, which is critical for understanding many aspects of cancer biology (46). The variability in obesity predisposition among mouse strains should be considered when choosing a transgenic mouse model. While it is commonly thought that BALB/c mice are obesity-resistant, at least one recent study has shown that a high fat diet increased body weight, body fat, and circulating leptin levels in BALB/c mice, and this was associated with increased metastasis of 4T1 breast cancer cells in this model (47). Several transgenic breast cancer models are on the FVB background, and we have found that these females are also obesity-prone on a high fat diet using the methods described below (E.A. Wellberg and S.M. Anderson, unpublished data). A complete analysis of obesity predisposition in various transgenic mouse models of breast cancer would be a great benefit to the scientific community.
One main limitation of using mouse models to study human breast cancer is that, with few exceptions, most mouse mammary tumors lack ER either completely, or only express it very early in tumorigenesis (48), despite having luminal molecular signatures (49). Some exceptions to this include the prolactin transgenic mouse model (NRL-PRL), which produces tumors that express ER, as do cell lines derived from tumors in this line (50, 51). Another model, which conditionally expresses the SV40 large T Antigen (TAg) and ERα, under control of an MMTV-driven tetracycline-responsive transactivator produces mammary adenocarcinomas, salivary tumors, and lymphomas (52). The mammary tumors can be grafted into immune-compromised mice, where they grow larger in the presence versus absence of E2 (52). A different group reported the generation of mammary cancer cell lines from a spontaneous tumor that formed in a STAT1-null mouse (53). Two lines from this mouse model, called SSM2 and SSM3, are reported to express ERα and PR, and to grow either in ovary-intact females or in the presence of supplemental E2 (53). The SSM3 cells are sensitive to the ER downregulator, Fulvestrant, while the SSM2 cells are resistant (53). When implanted into a mouse from the same background as the original tumor donor, these cells may offer the ability to study ER+ breast cancer progression in the presence of a full immune system. Another consideration when studying mammary development or breast cancer in rodents is that the morphology of the gland and surrounding stroma have key distinctions (54). Human terminal duct lobular units are typically more developed even before pregnancy than the mouse terminal end bud or side branches. In addition, human breasts can have loose intralobular stroma and dense interlobular stroma, while mouse mammary stroma has a predominance of adipocytes (54, 55). Finally, when using transgenic mouse models it should be noted that distant metastases are mainly found in the lungs, whereas humans develop metastases is lung, liver, bone, brain, and other tissues (56).
Human Cell Lines and Patient Derived Xenografts (PDX)
An alternative to transgenic or grafted mouse mammary tumor cells is the use of grafted established human breast cancer cells or patient-derived tumor tissues (57). There is a variety of human breast cancer cells that capture the clinical and molecular subtypes, including luminal/ER+ (MCF7, ZR75.1, T47D), HER2-enriched/HER2+ (BT474, SkBR3), and basal/TN (MDA-MB-231, BT20). Human breast cancer cells can be grown orthotopically in the inguinal mammary fat pads or as intraductal tumors (28, 58), which can support more relevant and sustained expression of ER (58). Basic and preclinical scientists have relied heavily upon these tools to elucidate mechanisms of breast cancer progression, metastasis, drug resistance, and to test the roles of individual genes and pharmacological therapies.
More recently, several institutions have in parallel developed banks of patient-derived breast cancer tissues that can be grafted into immune-compromised mice, referred to as patient-derived xenografts (PDX; reviewed in depth in (57, 59)). These PDX tumors are often propagated in NOD-SCID-IL2Rγ (NSG) mice, where small pieces of tumor (~1 mm × 2 mm) are implanted into the inguinal mammary fat pad using a 10-gauge trochar. Recently, novel cell lines have been derived from some PDX tumors, offering an even wider variety of resources for scientists to rigorously study breast cancer (60). The benefit of using these resources is that they recapitulate the heterogeneity found within and between human breast tumors, and notably, they represent the most commonly diagnosed subtype, HR+. The main limitation of using human tissues in mice is the risk of graft rejection. These cells and tumors will only grow in immune-compromised hosts, which limits their use in studies investigating the role of inflammation or immunity in the obesity or cancer process. One solution to overcome this limitation is the use of humanized mice. This can be accomplished by humanizing the mammary fat pad, where human stromal fibroblasts are injected along with human breast cancer epithelial cells, or by repopulating the immune system with human PBMCs (reviewed in (57)). While possible, this approach is expensive, and is not easily accomplished without assistance from those with significant experience with these techniques. Importantly, it is unclear how humanizing the immune system of mice will impact obesity predisposition and whole-body metabolism.
Each model system has benefits and limitations, but valuable information about breast cancer initiation, progression, and therapy response has been obtained using all of the paradigms described above. Overall, investigators must carefully consider the goals of their experiments and choose the approach and model system that allows these goals to be met.
2. Modeling Obesity in Rats and Mice
a. Rodent Models of Obesity
Defining and measuring obesity in rodents
There are several approaches to promote obesity in rodents; however, not all are amenable to studies that are combined with models of breast cancer. Importantly, there is not a standardized definition of obesity for rats and mice like there is for humans. To address this, work from the Hursting lab has attempted to characterize obesity criteria in mice that are reflective of the body mass index (BMI) cut points used in women. They generated female C57Bl/6 mice with a range of body fat percentages by feeding diets with varying fat content (up to 60%kCal fat) and energy densities (61). They then compared the % body fat of mice following 10 weeks on each diet to the % body fat of women within each BMI category, using data from the National Health and Nutrition Examination Survey (NHANES) III study (62). Using these criteria, they defined lean (calorie restricted), overweight (control diet-fed), and obese (DIO-fed) non-ovariectomized female mice as having percent body fat ranges of <26%, 26%−33%, and >37%, respectively. For mice OVX’d at 5-weeks of age and then fed the same diets, the percent body fat ranges for mice on the lean, overweight and obesity-inducing diets were <29%, 34%−40%, and >45% respectively (61). Our approach has been similar, although we have generally use the word “obesity” to describe an excess of adiposity, relative to a lean control group, and have used HF diets with slightly lower fat content (maximum of 46% kCal fat), which could affect adipose deposition. Further, while Hursting’s group identified these % adiposity at the end of their study, we have generally used cut points earlier in a study time-course to prospectively categorize rodents as lean or obese before performing additional study interventions. Thus, in our studies, obesity has generally been identified as >20% body fat for adult female mice and rats prior to OVX. However, it can vary between cohorts and depending on the age at which body fat is measured. Adiposity then increases over time, with a significant increase in body fat deposition after OVX resulting in rodents than are upwards of 40% body fat. Excess adiposity is often accompanied by fasting insulin and/or glucose that are significantly higher than levels measured in LF fed controls; although, in animals that are all consuming the same HF diet, this difference in insulin and/or glucose may not always be apparent. The transdisciplinary field of obesity and cancer research would greatly benefit from adopting a standard approach to measuring and categorizing adiposity.
We most often measure body composition using quantitative magnetic resonance (qMR; EchoMRI, Houston TX), which is a rapid, non-invasive way to obtain fat mass, lean mass, as well as free and total body water content. While this is fast (<3 minutes per animal) and performed without anesthesia, it is limited in that it does not allow for measurements of regional fat distribution. Dual energy X-ray absorptiometry (DEXA) has the benefit that it allows for measures of regional adiposity and bone density; however, DEXA is time-intensive, requires placing animals under anesthesia, and requires more specialized equipment than available for many investigators. For investigators without either of these machines, total fat mass can be approximated by excising and weighing the subcutaneous (mammary), gonadal, retroperitoneal, and/or mesenteric fat pads at the time of sacrifice and expressing them as a percent of total body mass. Further, this method can be used to supplement qMR data to gain insight into fat distribution at the end of a study.
Available obesity models
Preclinical models of obesity can be categorized as either 1) genetic models – single gene mutation, 2) diet-induced models, or 3) selective inbreeding of diet-induced models (37). At least seven single-gene mutations that lead to obesity have been identified in rodents, including those affecting leptin or the leptin receptor. These mutations give rise to monogenic forms of obesity with an extreme phenotype (63–65) and have proven valuable in understanding the mechanisms and contributions of specific factors involved in energy homeostasis and body weight regulation. Obesity in humans involves complex interactions between genes and the environment that converge to affect multiple tissues and cells in the body. Together these interactions favor the accumulation of excess adipose tissue, and obesity does not result from changes to a single gene. Thus, using a diet-induced approach to promote obesity may increase the physiological relevance of cancer studies and strengthen the ability to translate findings to humans.
Separating contributions of diet composition, adiposity, and metabolic dysfunction
Recent studies suggest that metabolic health may be an important contributor to obesity-associated cancer, with the highest risks for breast cancer diagnosis occurring in women who are classified as “metabolically unhealthy” and as obese based on BMI (8, 66, 67). In addition, a low dietary fat pattern associates with decreased incidence of death after breast cancer (68–71). Based on these observations, it may be desirable to dissociate the contributions of underlying metabolic dysfunction or diet composition from adiposity in preclinical cancer studies. Our approach has been to use semi-purified diets purchased from a reputable company, which supports reproducibility across studies and offers the advantage that investigational agents can be blended directly into the diet at the time of manufacturing, as needed. There are several stock diets available, or researchers can create custom diets tailored to individual study needs. Because there is considerable variability in the amount and source of fat, carbohydrate, and protein, the reproducibility of results within and across laboratories and institutions depends on investigators reporting the specific formulations and catalog numbers of all diets used. Regardless of the diet chosen, we encourage researchers to avoid the use of “chow” as a control. Chow provided by most animal facilities varies significantly across batches, institutions, and seasons, making a semi-purified low-fat diet a better choice to maintain lean rodents. The compositions of low, moderate, and high fat diets with and without sucrose that we have commonly used in our rodent studies are shown in Table 1.
Table 1.
Diet numbers and composition used in mouse and rat obesity studies.
| MOUSE | RAT | |||||||
|---|---|---|---|---|---|---|---|---|
| DIET | Low Fat Low Sugar (LFLS) | Low Fat High Sugar (LFHS) | High Fat High Sugar (HFHS) | High-Fat (HF) | ||||
| Product # | D11092101 | D19043002 | D15031601 | D12344 | ||||
| (Research Diets) | g % | kcal % | g % | kcal % | g % | kcal % | g % | kcal % |
| Protein | 20.3 | 20.8 | 20.3 | 20.8 | 24.0 | 20.8 | 24.6 | 20.8 |
| Carbohydrate | 66.0 | 67.7 | 66.0 | 67.7 | 45.2 | 39.2 | 39.7 | 33.1 |
| Fat | 5.0 | 11.5 | 5.0 | 11.5 | 20.5 | 40.0 | 24.6 | 46.1 |
| Total | 100.0 | 100.0 | 100.0 | 100.0 | ||||
| kCal/gram | 3.90 | 3.90 | 4.61 | 4.80 | ||||
| Ingredient | grams | kcal | grams | kcal | grams | kcal | grams | kcal |
| Casein, Lactic | 200 | 800 | 200 | 800 | 200 | 800 | 200 | 800 |
| DL-Methionine | 3 | 12 | 3 | 12 | 3 | 12 | 3 | 12 |
| Corn Starch | 531 | 2124 | 278 | 1110 | - | - | 163 | 650 |
| Maltodextrin | 119 | 476 | 80 | 320 | 80 | 320 | 150 | 600 |
| Sucrose | - | - | 293 | 1170 | 293 | 1170 | - | - |
| Cellulose BW200 | 50 | - | 50 | - | 50 | - | 50 | - |
| Milk Fat (Butter) | 11 | 99 | 11 | 99 | 134 | 1209 | ||
| Corn Oil | 39 | 351 | 39 | 351 | 39 | 351 | 200 | 1800 |
| Minerals Mix S10001 | 35 | - | 35 | - | 35 | - | 35 | - |
| Vitamin Mix V10001 | 10 | 40 | 10 | 40 | 10 | 40 | 10 | 40 |
| Choline Bitartrate | 2 | - | 2 | - | 2 | - | 2 | - |
| Cholesterol | - | - | - | - | - | - | - | - |
| Total | 1000.0 | 3902.0 | 1000.0 | 3902.0 | 845.8 | 3902.1 | 812.5 | 3902.0 |
Bold values indicate key differences in the fat and sucrose content of the diets, as described in the text
b. Diet Selection
Mouse Studies
In mice, we have used diets with varying levels of fat and sugar to produce the physiological response(s) desired for our studies (72). Details of typical diets that we have used can be found in Table 1. Generally, we would expect that any diet containing at least 40% kcal from fat (such as the HF diet we use in rats; described below) should induce obesity in mice (regardless of the fat source), especially if combined with thermoneutral housing (described below). However, based on specific outcomes of interest in our ongoing studies, we have specifically used a high-fat high-sugar (HFHS) diet with 40% kCal fat with butter as the primary fat source, with additional ~30% kCal from sucrose (72). Milk fat (butter) provides a variety of fatty acids and also includes small amounts of cholesterol; however, it is likely that several fat sources, such as corn oil or lard, would result in a similar obesity phenotype. The low-fat diet we typically use contains 11% kCal from fat, with no added sucrose. We have also included a low-fat/high sucrose (LFHS) diet in preliminary studies (11% kCal fat; 30% kCal sucrose; data below) to separate the effects of sucrose-induced insulin resistance from the excess adiposity induced by high fat/high sucrose (HFHS). Studies performed using the HFHS diet, with LFLS as a control lead to reproducible fat gain in HFHS-fed compared to LFLS-fed females on the C57Bl/6 background, with ~50% of HFHS-fed mice achieving a higher body fat percentage than LF-fed mice irrespective of sucrose contents. In contrast, only ~20% of NSG females develop excess body fat on a HFHS diet at thermoneutrality (72).
Using these three diets, and also taking advantage of the distribution of body fat percentage across the HFHS-fed group, it is possible to generate several metabolic phenotypes that can be informative for mammary development and/or breast cancer studies. Mice fed either a LFLS or LFHS diet have similar body fat content of <20%, while HFHS-fed females average ~ 26% body fat (Figure 2A). The HFHS mice can be separated into two groups, one that is obesity-resistant (OR) and one that is obesity-prone (OP). The resulting body fat content is ~16% in the OR and ~39% in the OP (Figure 2B). Although the LFHS-fed and OR females maintain a body composition similar to LFLS-fed mice, their fasting insulin, glucose, and resulting HOMA-IR are elevated (Figures 2C–E). This allows the investigation of breast cancer in response to impaired glucose tolerance and hyperinsulinemia but in the absence of excess adiposity. In some cases, it may be desirable to keep the diet constant and investigate the effects of varying body fat. In the HFHS-fed group, the OR and OP phenotypes have different body fat contents, but have similarly elevated insulin, glucose, and HOMA-IR compared to LFLS-fed mice (Figure 2B–E).
Figure 2. Diet-induced separation of obesity and metabolic dysfunction in mice.
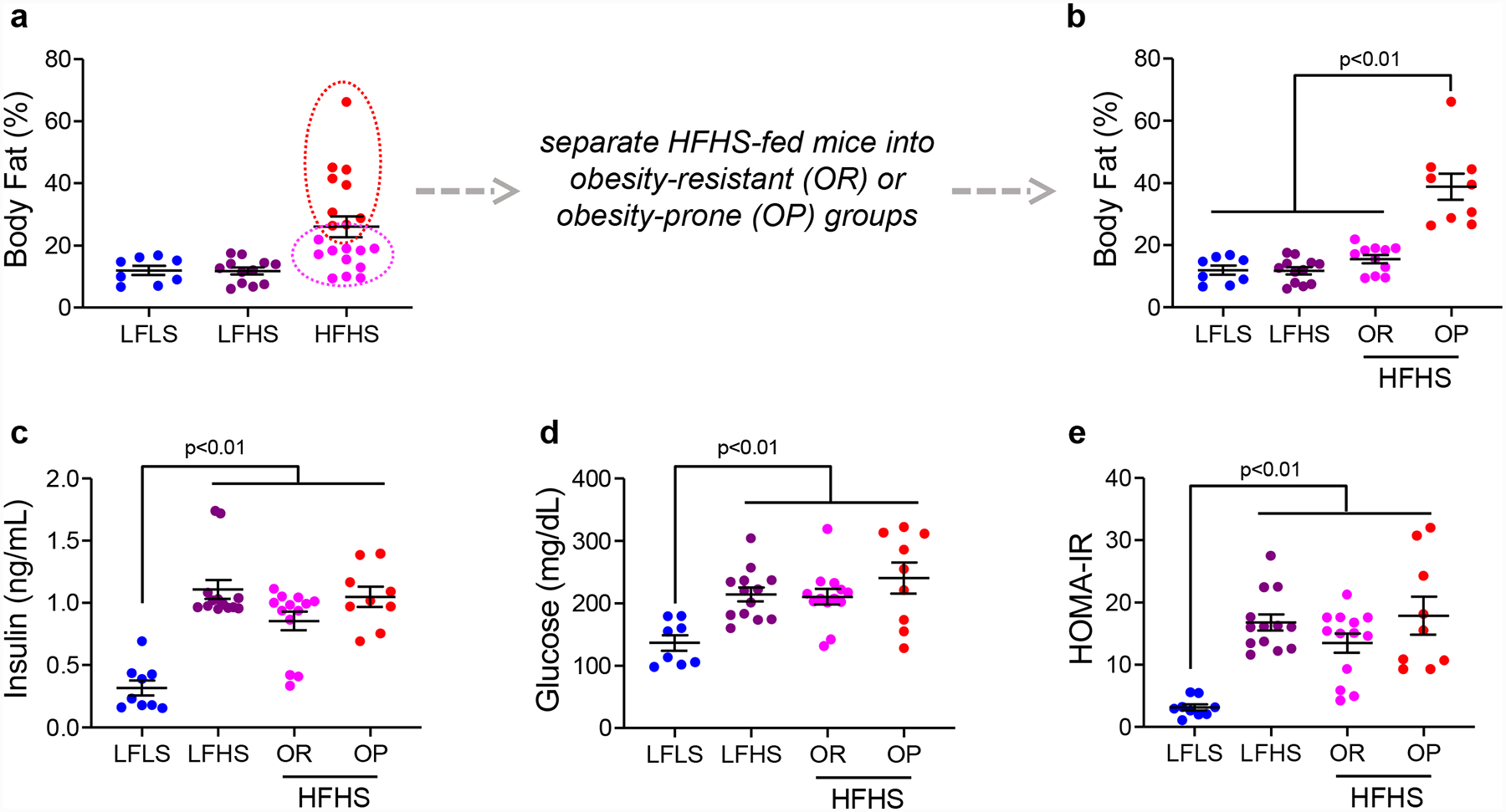
(A) Body fat percentage measured by qMR in Rag1-null C57Bl/6 mice fed low-fat/low-sucrose (LFLS), low-fat/high-sucrose (LFHS), or high-fat/high-sucrose (HFHS) diets. Note the variability in body fat across the HFHS group. (B) Body fat percentage in LFLS, LFHS, and HFHS fed mice, showing the obesity resistant (OR) and obesity prone (OP) groups separately. (C) Fasting insulin and (D) glucose measured in mice on LFLS, LFHS, or HFHS diets. (E) HOMA-IR calculated from fasting insulin and glucose measures in mice on LFLS, LFHS, or HFHS diets. Bars represent mean and SEM. Data were analyzed by unpaired t-test.
Rat Studies: OR/OP Model
We take a similar, but slightly different approach in rat studies. The details of this approach (the OR/OP model of obesity) has evolved over the past two decades, based on early work in male rat; historical details and female-specific aspects of this model are reviewed elsewhere (37), and the timeline is summarized at the end of this review. Female Wistar rats are ordered to arrive at our facility at 5 weeks of age (±2d, with a known date of birth). Because the development of obesity requires that we limit physical activity, animals are individually housed in wire-bottom caging for the duration of the study. These cages also allow for the measurement of food intake and spill throughout the study. All rats are provided ad libitum access to a 46% fat diet (Research Diets, #12344; Table 1). Consumption of the HF diet begins as soon as possible upon arrival, subject to the facility and regulations on quarantine period. Our early studies in rats often relied on 60% fat in the diet; however, over the past decade we have shifted to using high fat diets that are more reflective of human consumption (40–50% kcal; Table 1) [34, 35, 37], with corn oil as the fat source. In our rat studies, we typically do not include sucrose as a portion of the carbohydrate component. By carefully selecting diets, we are able to consider and isolate variables that contribute to the obesity-cancer relationship, potentially strengthening the translational impact of cancer studies.
The response of Wistar rats to this obesogenic environment (HF diet + limited physical activity) is varied (Figure 3A–B). We take advantage of this variability to select rats that are resistant or prone to obesity (OR and OP, respectively; Figure 3B), and have found that they reliably mature into lean and obese adults. Unlike male rats, where this propensity for obesity can be easily identified as early as 5–6 weeks of age, selection of these phenotypes is more complex in females (37). In our experience, percent body fat at 14–18 weeks of age is a more reliable predictor of adult adiposity (37). Often, we select those in the bottom 30–40% as OR, and those in the top 30–40% as OP, although these ratios can be altered as needed based on study design. Further, we try to stratify based on natural breaks in the data, which sometimes results in slightly unbalanced groups. Figure 3C shows example data from a large study of approximately 300 rats. Selection of the OR and OP phenotype occurred using % body fat (qMR) at 14 weeks of age (Figure 3B). In this example, the bottom 36% and top 32% were selected as OR and OP, respectively, with the middle group moved to other studies. We then assessed body composition in these same animals 8 weeks later (~22 weeks of age). As shown in Figure 3C, obese animals had higher body weight, fat mass, and lean mass compared to their lean counterparts. Animals then underwent surgical ovariectomy (OVX; further described below), which induced significant weight gain in both the lean and obese groups (Figure 3D). This selection of lean and obese phenotypes and OVX-induced weight gain is reproducible, and we have seen similar results in several of our previously published studies of females using this model, both with (34–37, 39) and without tumors (73–75).
Figure 3. Diet-induced separation of obesity prone and resistant rats.
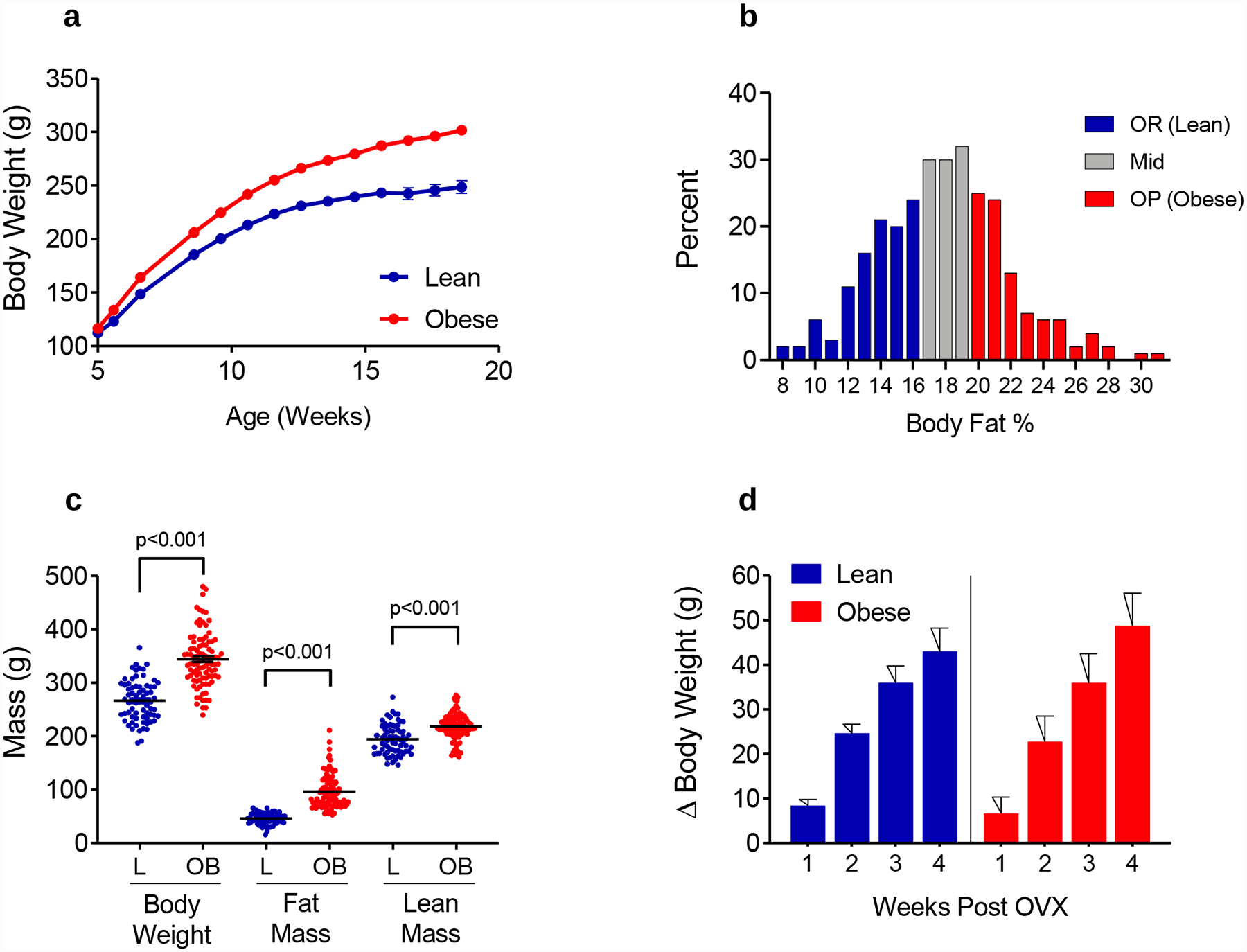
(A) Body weight for lean and obese rats all on the same HF diet (46% kcal fat) demonstrates the differential predisposition to obesity in the outbred Wistar strain. Graph represents weekly body weights from 288 rats (mean +/− SEM) based on classification as obesity resistant (OR) or obesity-prone (OP) using % body fat at 18 weeks of age. (B) Frequency distribution of % Body Fat at 18 weeks of age (N=288 rats). Color coding shows selection of OR (n=105 rats; 36%) and OP (n=91 rats; 32%) from the entire cohort; Middle Tertile animals (n=92 rats; 32%) were moved to other studies. (C) Body Composition in mature Lean and Obese Rats (mean = 22 weeks of age). (D) Loss of circulating hormones following OVX induces rapid weight gain over approximately 3–4 weeks in both lean and obese rats, with no difference in the cumulative weight gained between lean and obese groups. All data is presented as mean and SEM.
c. Housing Temperature
It can be challenging to promote obesity or fat gain in female mice, particularly if they have intact ovaries. This can be even more difficult in immune-compromised mice that lack T-cells, B-cells, and even NK cells and macrophages. To overcome these challenges, we use thermoneutrality. While the perfect housing temperature for mice is widely debated (76–80), and may depend on the sex, strain, age, or mass of the animal (80–82), it is clear that ambient vivarium temperatures cause chronic cold stress. While animal housing rooms with temperatures 20–23°C are comfortable for humans, mice housed at these temperatures can experience adaptive thermogenesis (83–86). The thermoneutral zone is the temperature range in which the metabolic rate is at resting levels and no adjustments (e.g. shivering, panting) are necessary to maintain a stable core body temperature (81). For rats, this was reported to be ~25°C–31°C (87) and for mice, it’s reported to be ~26°C–34°C (80) with the recommendation of ~ 28°C–31°C based on robust metabolic phenotyping data (76, 77). We take two approaches to raise temperatures in our rats and mice; however, it should be noted that warming the mice is critical for the reproducible development of obesity in our studies, but it may not be necessary in rats. For the majority of our rat studies, juvenile Wistar females arrive at 5–6 weeks of age and are singly housed at ambient temperature (~23°C) in a standard vivarium. After an acclimation period of 2–3 days, females are placed on HFD (34, 37). MNU is administered at 55 ± 2 days of age, and rats are monitored weekly for palpable tumor formation. Once tumors reach a predefined size, rats are moved to a climate-controlled satellite room for anti-cancer interventions. The temperature of this room is maintained at 25–26°C; however, we have observed robust weight gain in female rats at standard vivarium temperatures.
To create the diet-induced obesity xenograft (DIOX) mouse model, we use the following approach. Female Rag1-null mice on the C57Bl/6 background (Jackson Labs #002216) arrive at ~5–6 weeks of age and are group housed (5 mice per cage) in a vivarium at ambient temperature. Mice are randomized based on body mass to either HFHS or LFLS diets and are rearranged so that they remain group housed. Mice remain on diet for ~2 weeks before they are moved to static cages placed upon warming blankets. To our knowledge, there are no published studies that suggest the timing of diet and warming influences adiposity development; however, our protocol relies on administering diet for a short time prior to commencing thermoneutrality.
When mice are approximately 8 weeks old, the cages are placed on blankets with warm circulating water. Figure 4A shows a typical cage set up. Cages are positioned such that half of the cage is directly on the blanket and half of the cage is directly on the rack. This allows the internal cage temperature to rise but gives the mice the option to choose the warmer or cooler side of the cage. It has been reported that mice do not prefer to spend all of their time at ~30°C and will choose a cooler temperature particularly at night when they are active (76, 77). Mice remain in cages on warming blankets for the duration of all tumor studies.
Figure 4. Effect of thermoneutral housing on body composition in Rag1-null C57Bl/6 female mice.
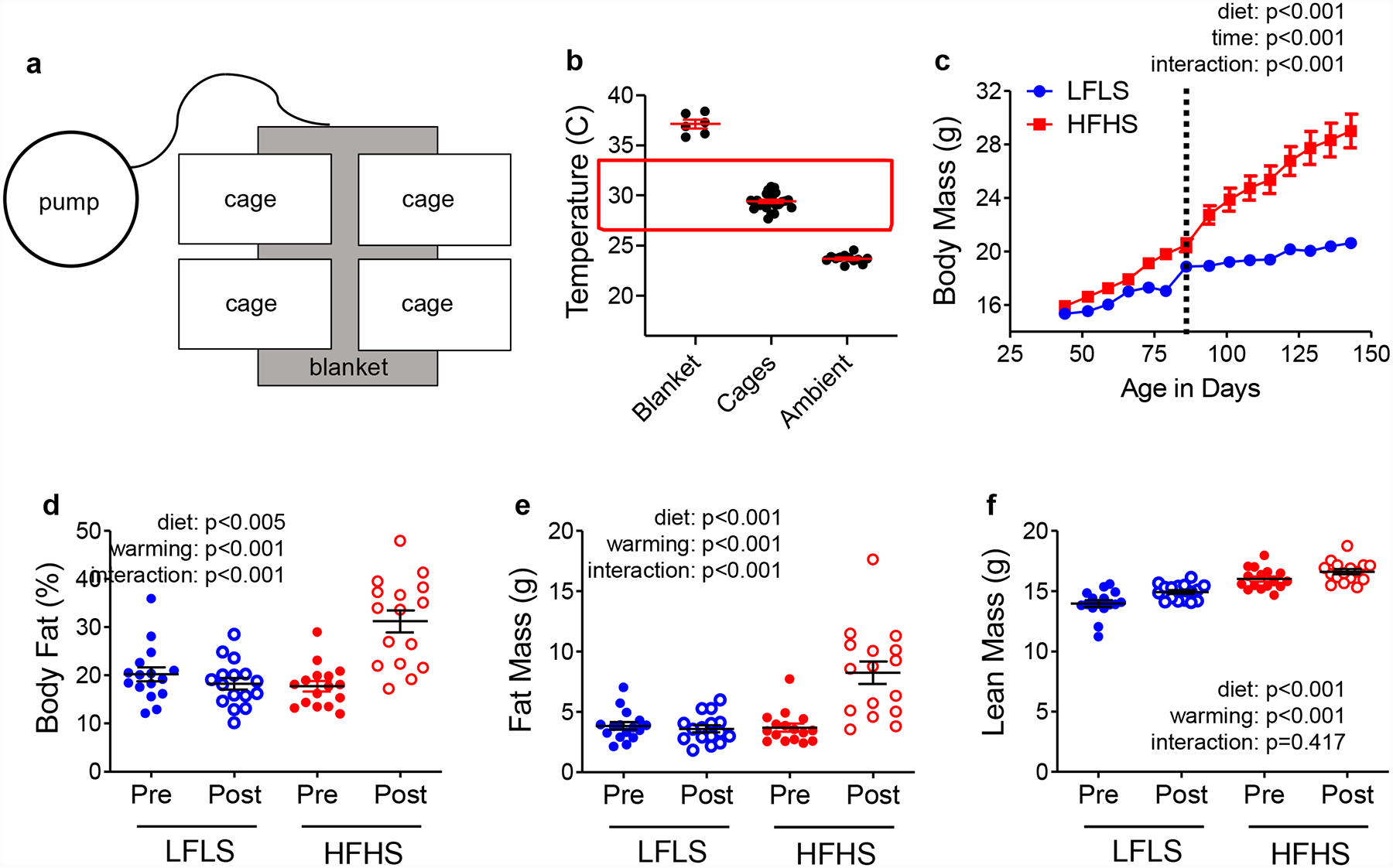
(A) diagram of cage placement on warm water blankets. Each cage is housed half on, and half off, the blanket. (B) Temperatures of the blanket surface, the bottom of the warmed cage, and the bottom of cages housed at ambient vivarium temperature, measured with an infrared thermometer. The red box indicates the mouse thermoneutral zone. (C) Body mass of LFLS and HFHS Rag1-null females. The vertical line indicates the initiation of thermoneutral housing; N=16 per group. (D) Body fat percentage, (E) Fat mass, and (F) lean mass measured before (pre) and after (post) warming with qMR in mice fed LFLS or HFHS diets; N=16 per group. Data were analyzed by 2-way ANOVA testing for main effects of diet or time (panel c) or diet and warming (panels d-f) and for an interaction.
The warm water pump has four temperature settings: 10°C, 35°C, 38°C, and 42°C. In our studies, we select the highest setting (42°C) and also select continuous circulation. We used infrared thermometers to measure the temperature of the floors of cages housed on or off the warming blankets and the temperature of the blanket itself (Figure 4B) (72). We found the temperatures of the blankets, the floors of the cages housed on blankets, and the cages at ambient temperatures averaged 37.1°C, 29.4°C, and 23.7°C respectively (72) and Figure 4B. The average temperature of cages housed on warming blankets falls within the recommended temperature zone of ~28°C-30°C (76, 77). Within a few days of being placed on warming blankets, HFHS-fed but not LFLS-fed females begin to gain weight (Figure 4C, vertical line). In LFLS fed mice, warming does not affect body fat percentage (Figure 4D), nor does it affect fat or lean mass (Figure 4E–F). In contrast, warming increases body fat (Figure 4D–E) but does not increase lean mass in HFHS-fed mice (Figure 4F). Similar effects of warming are seen on body mass and composition in wild type C57Bl/6 females (Figure 5). HFHS-fed but not LFLS-fed gain more weight after warming compared to LFLS-fed mice (Figure 5A). Overall, compared to LFLS-fed females, HFHS-fed females housed for >6 weeks on warming blankets have a higher percentage of body fat (Figure 5B), nearly twice as much fat (Figure 5C), and slightly greater lean mass (Figure 5D).
Figure 5. Effect of thermoneutral housing on body composition in wild type C57Bl/6 females.
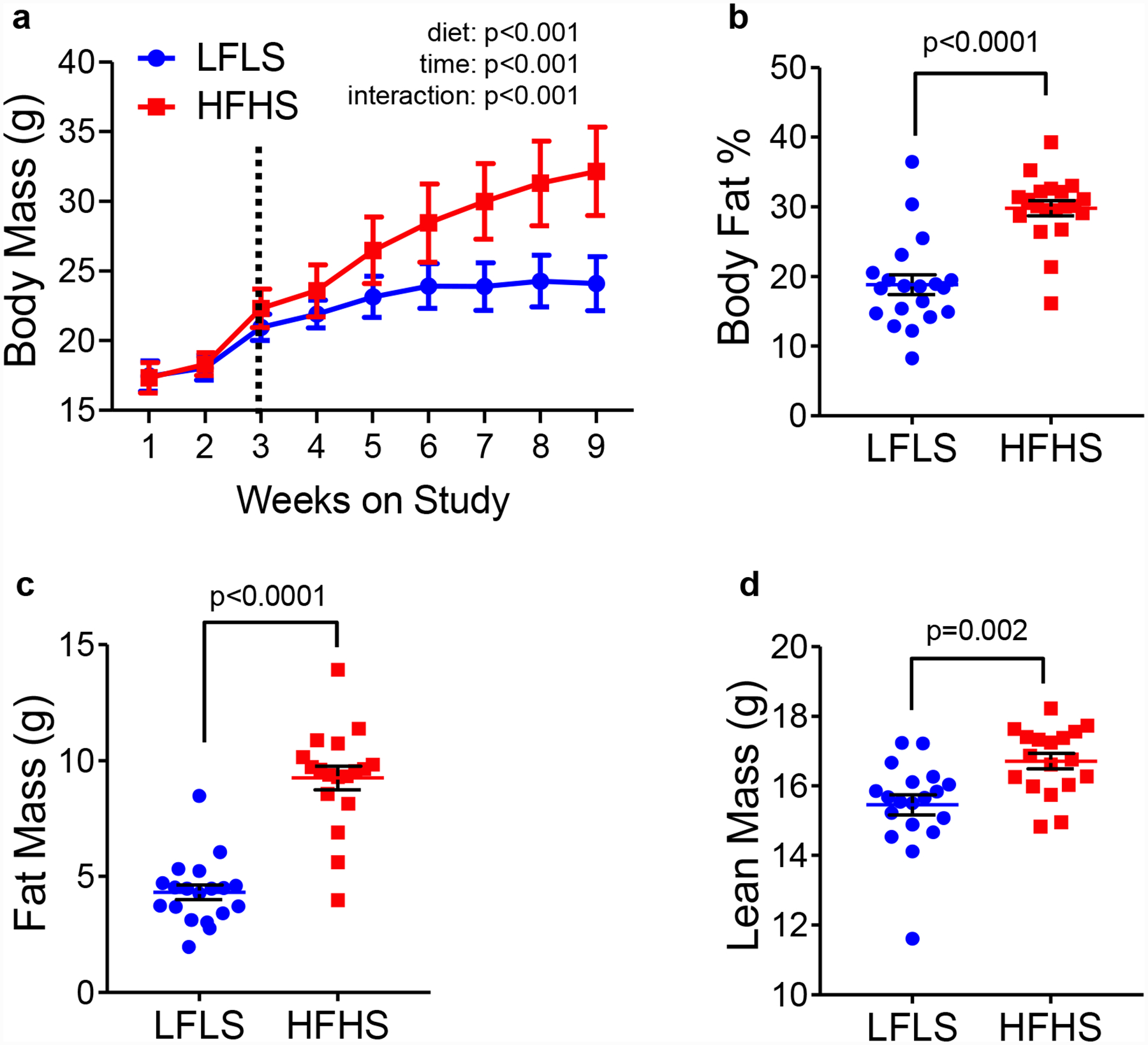
(A) Body mass of LFLS and HFHS wild type C57Bl/6 females. The vertical line indicates the initiation of thermoneutral housing. (B) Body fat percentage in mice fed LFLS or HFHS diets measured after 9 weeks on study via qMR. Fat mass (C) and lean mass (D) in g are shown. N=19 LFLS, N=18 HFHS. Data were analyzed by 2-way ANOVA (panel a) testing for main effects of diet and time, or by unpaired t-tests (panels b-d).
We chose temperatures that supported robust and reproducible adipose gain in outbred Wistar rats (Figure 3 and [35, 53–55]), in immune compromised Rag1-null C57Bl/6 mice (Figure 4), and (72), and in wild type C57Bl/6 mice (Figure 5). We have applied the same methods to the NSG strain, which is widely used as a PDX recipient; however, only ~20% of mice are susceptible to obesity compared to ~50% of Rag1-null C57Bl/6 females (72). While the exact temperature at which mice should be housed is debated, one important consideration is whether the results of any preclinical study translate to humans (88). In our studies, we strive to confirm that the mechanisms we identify during obesity-associated breast cancer progression in rodents are also seen in breast cancer patients (35, 72), thus we feel the approaches to promote obesity are valid.
3. Modeling Menopause
a. Surgical ovariectomy (OVX)
Surgical ovariectomy (OVX) is a well-established approach to create a pseudo-postmenopausal state. While it is not a perfect reflection of the postmenopausal condition, it does have many correlates that make it an appropriate model for menopause. Like menopause in humans, OVX induces a transient, predictable period of weight gain and increase in adiposity (73, 89, 90) and in the context of breast cancer, we have also shown that OVX in the rat model causes the adverse effects of obesity on tumor promotion to emerge (34–36, 39). Using a surgical approach to model menopause allows the investigator to control the timing of menopause induction so that post-menopausal weight gain and tumor progression can be monitored in a time dependent fashion. While it may seem desirable to OVX rodents at an early age to shorten study timelines (and therefore cost), we highly recommend that investigators wait to perform this procedure until animals have reached maturity. We typically OVX mice when they are at least 12 weeks old, and rats when they are at least 18 weeks of age. These time point are used as they reflect the time at which weight gain generally begins to taper off, which suggests that animals are no longer depositing lean mass and are therefore considered fully mature (37)
b. Chemical Toxicant
An alternative approach to model menopause is the use of a chemical toxicant, such as 4-vinylcyclohexene diepoxide (VCD). VCD induces apoptosis of primordial follicles in the ovary without affecting other peripheral tissues, eventually leading to ovarian failure (menopause), as reviewed in (91, 92). While this approach more closely mimics the natural human progression through perimenopause into menopausal, is requires repeated daily injections of VCD and the loss of ovarian function is variable, occurring over weeks or months depending on the protocol used. This would make it difficult to follow timelines in studies investigating the menopause-induced changed in obesity, energy balance, and metabolism on tumor outcomes such as those of related to obesity and postmenopausal breast cancer; however, it may more accurately simulate the gradual loss of ovarian function observed in women. In our studies where we aim to isolate the metabolic changes that occur following estrogen loss, surgical ovariectomy provides a more appropriate and logistically feasible approach.
c. Estrogen supplementation & withdrawal for ER+ tumors
For many of these mouse models, the growth of estrogen-responsive tumors requires that animals be supplemented with estrogens, in the form of E2 or conjugated estrogens, at least briefly for tumors to establish. Obviously, this becomes problematic when attempting to model the postmenopausal condition, where these hormone-responsive tumors grow in the absence of ovarian production of hormones.
Most human ER+ breast cancer cell lines or PDX tumors require supplemental E2 to sustain initial growth in mice. This can be accomplished with several approaches. The most common is the subcutaneous pellet. E2-containing pellets can be made by mixing powdered E2 with cellulose at the desired ratio and packing the mixture into ~0.8 cm pieces of silastic tubing that are sealed on both ends with adhesive. Beeswax hormone-containing pellets can similarly be made with the desired E2 levels and implanted subcutaneously with a trochar, or pellets can be purchased (Innovative Research of America) and are available in a range of doses and delivery durations. One drawback of using pellets is the tendency for them to promote urine retention and potentially severe cystitis (93), but this may be avoided by reducing the levels of E2 (94). Another approach to deliver E2 is by diluting a stock solution into the drinking water (57, 95–97), which we now use in Rag1-null and wild type C57Bl/6 female mice. Supplementation of OVX’d C57Bl/6 female mice with 0.5 μM oral E2 decreases gonadal fat mass (Figure 6A), increases uterine mass (Figure 6B), and increases the percent of mammary epithelial cells that express the ER target, PR (Figure 6C). While the pellets may result in circulating E2 levels that decrease over time, the administration of oral E2 achieves relatively constant circulating hormone levels in the animal. Wild type C57Bl/6 females supplemented with E2 maintain a steady body mass over time, which we recently reported in a preprint (98), but HFHS-females were still heavier than LFLS-fed females. Likewise, HFHS-fed Rag1-null females supplemented with E2 are heavier and have greater body fat than LFLS-females, and these parameters increase after E2 withdrawal (72). For studies where it may be desirable to mimic the cyclical nature of hormone levels, such as that achieved by oral administration of hormone-replacement therapy in women, a novel approach using E2 delivered in a bolus of hazelnut spread (such as Nutella) has been described (99).
Figure 6. Effect of oral E2 administration in mice.
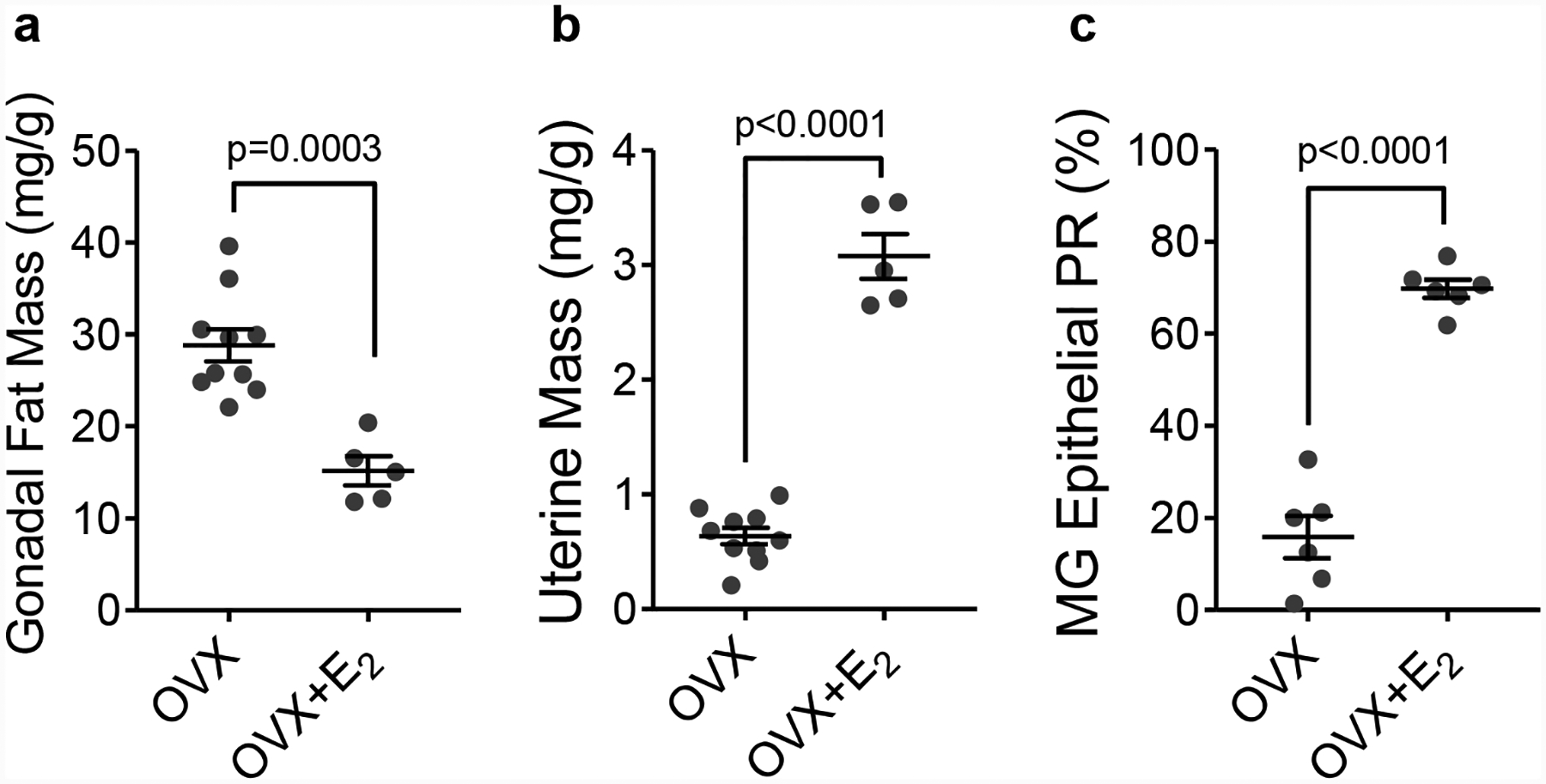
(A) Gonadal fat pad mass and (B) uterine mass expressed as mg per g body mass, and (C) percentage of mammary epithelial cells positive for the progesterone receptor (PR) by IHC in OVX mice or OVX mice supplemented with 0.5 μM E2 in drinking water. For a and b, N=10 OVX and N=5 OVX+E2. For c N=6 OVX and N=6 OVX+E2. Data were analyzed by unpaired t-test.
4. General Study Timelines
General timelines, showing key features of both mouse and rat models are shown in Figure 7. Female mice (Figure 7A) are given semi-purified diets and housed at thermoneutrality for the duration of the study. Mature mice are OVX’d, supplemented with E2 and implanted with tumors. Once tumors are established and reach the desired size, mice are either maintained on E2 or E2 is withdrawn. E2 withdrawal associates with a period of weight gain and elevated insulin resistance, which we have shown is tumor promotional in some cases (72). Juvenile female rats (Figure 7B) are given semi-purified diet and tumors are initiated with MNU. Tumors emerge and grow as the animals mature, and rats are OVX’d based on a defined tumor burden. As we see in mice, the ~3 week period following OVX is characterized by weight gain and insulin resistance, and supports tumor progression particularly in obesity-prone (obese) females (35–37, 39).
Figure 7. General Overview of Models.
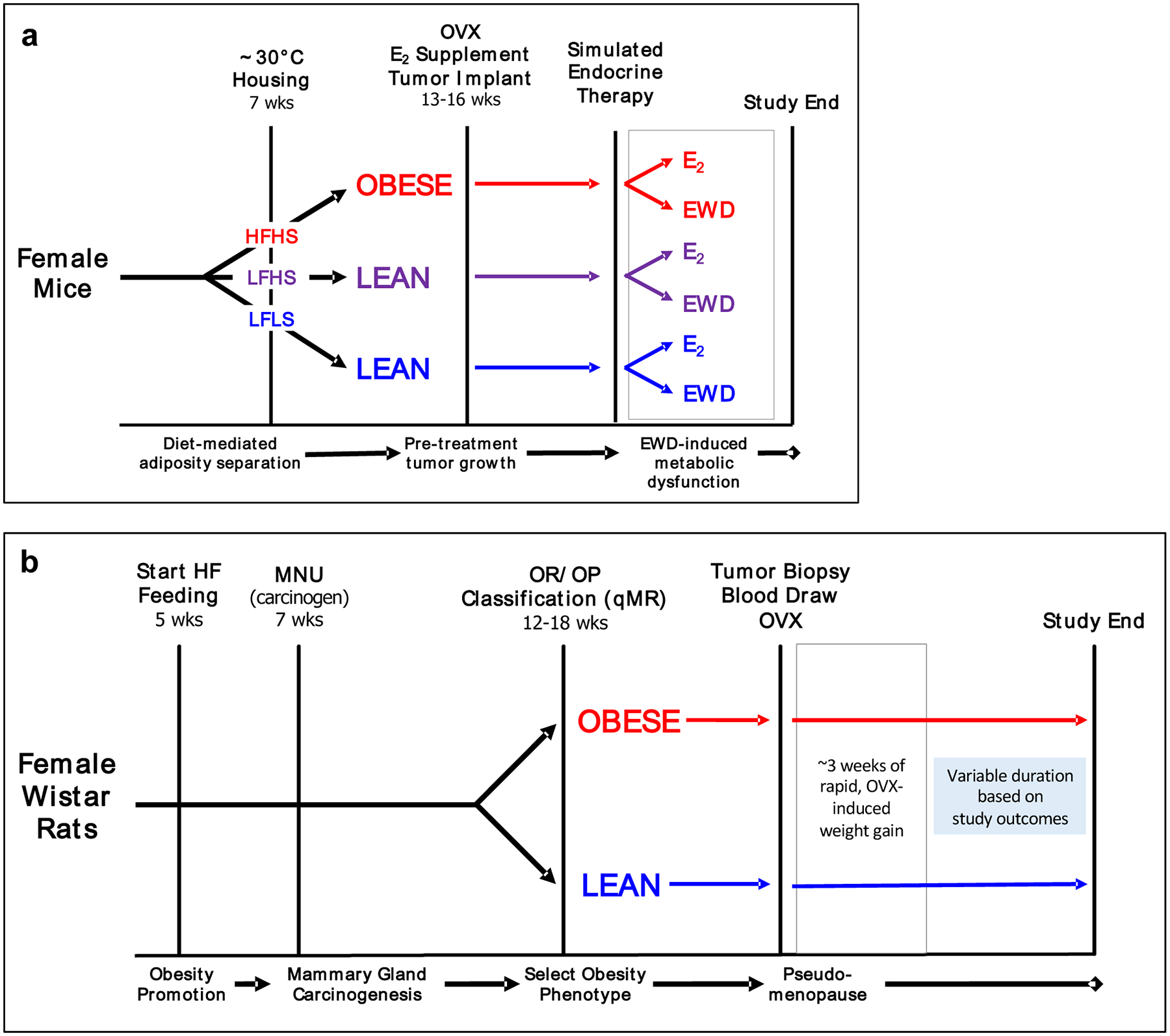
Key features of studies done in (A) mice and (B) rats are shown. The grey box in each diagram represents the period following estrogen loss when weight gain and insulin resistance increase. OVX-ovariectomy; EWD-estrogen withdrawal; LFLS-low fat/low sucrose; LFHS-low fat/high sucrose; HFHS-high fat/high sucrose.
Because tumors are carcinogen-induced, additional tumors will develop in many of the animals, allowing for the study of new tumor development in the postOVX period. Depending on the desired study outcome, rats can be maintained in the post-OVX period for up to 20 weeks, unless tumor burden requires an earlier endpoint.
5. Application to Other Paradigms
The approaches described in this paper have evolved through the efforts of numerous research teams and reflect our experiences with these models. Our studies have applied these techniques to breast cancer investigations; however, it is likely that similar approaches could be used to study mammary development and differentiation. We hope that by sharing some of the nuances and intricacies of these techniques, other investigators will be able to apply these approaches to reproduce our findings and also to their own projects on breast cancer and mammary development, further expanding the toolbox available for future studies. As a research community, continuing to publish the details of these models and changes made as they evolve over time will strengthen our ability to answer important questions with increased rigor and reproducibility, which we hope will ultimately increase the clinical applicability of our findings.
Acknowledgments:
Development of these models was a team effort, and we are incredibly grateful to the members of the ‘Fat Rat’ team, who were instrumental in developing and characterizing these models. This includes: Drs. Paul MacLean, Pepper Schedin, Ann Thor, Steven Anderson, and members of each of their labs. We are equally grateful to the skilled research technicians who ensured the success of the many studies in mice and rats that contributed to this work. Summary figures were created using BioRender.com.
Funding: This work was supported by the NIH R00CA169430 (Giles), R01CA241156 (Wellberg), R01CA164166 (MacLean), Colorado Nutrition Obesity Research Center Metabolic Phenotyping Core and Pilot Grant Program P30DK48520, TREC Training Workshop R25CA203650 (Giles and Wellberg), KL2TR002534 (Wellberg), the Komen Foundation CCR17483321 (Wellberg), and seed grants from the University of Colorado’s Center for Women’s Health Research (Giles and Wellberg)
Footnotes
Conflicts of interest/Competing interests: The authors declare that they have no conflict of interest.
Ethics approval: All animal work was approved by Texas A&M and/or University of Colorado Institutional Animal Care and Use Committees.
Consent to participate: Not applicable
Consent for publication: (include appropriate statements)
Availability of data and material: The data generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability: Not applicable
REFERENCES
- 1.Flegal KM, Kruszon-Moran D, Carroll MD, Fryar CD, Ogden CL. Trends in Obesity Among Adults in the United States, 2005 to 2014. JAMA. 2016;315(21):2284–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of Obesity Among Adults and Youth: United States, 2015–2016. NCHS Data Brief. 2017(288):1–8. [PubMed] [Google Scholar]
- 3.Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA. 2014;311(8):806–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan DSM, Vieira AR, Aune D, Bandera EV, Greenwood DC, McTiernan A, et al. Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Ann Oncol. 2014;25(10):1901–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neuhouser ML, Aragaki AK, Prentice RL, Manson JE, Chlebowski R, Carty CL, et al. Overweight, Obesity, and Postmenopausal Invasive Breast Cancer Risk: A Secondary Analysis of the Women’s Health Initiative Randomized Clinical Trials. JAMA Oncol. 2015;1(5):611–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matthews SB, Thompson HJ. The Obesity-Breast Cancer Conundrum: An Analysis of the Issues. Int J Mol Sci. 2016;17(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen L, Cook LS, Tang MT, Porter PL, Hill DA, Wiggins CL, et al. Body mass index and risk of luminal, HER2-overexpressing, and triple negative breast cancer. Breast Cancer Res Treat. 2016;157(3):545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park YM, White AJ, Nichols HB, O’Brien KM, Weinberg CR, Sandler DP. The association between metabolic health, obesity phenotype and the risk of breast cancer. Int J Cancer. 2017;140(12):2657–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Picon-Ruiz M, Morata-Tarifa C, Valle-Goffin JJ, Friedman ER, Slingerland JM. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA Cancer J Clin. 2017;67(5):378–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Azvolinsky A Cancer risk: the fat tissue-BMI-obesity connection. J Natl Cancer Inst. 2014;106(4):dju100. [DOI] [PubMed] [Google Scholar]
- 11.Howlader N, Altekruse SF, Li CI, Chen VW, Clarke CA, Ries LA, et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst. 2014;106(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. [DOI] [PubMed] [Google Scholar]
- 13.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waks AG, Winer EP. Breast Cancer Treatment: A Review. JAMA. 2019;321(3):288–300. [DOI] [PubMed] [Google Scholar]
- 15.Heshmati K, Harris DA, Rosner B, Pranckevicius E, Ardestani A, Cho N, et al. Association of Bariatric Surgery Status with Reduced HER2+ Breast Cancers: a Retrospective Cohort Study. Obes Surg. 2019;29(4):1092–8. [DOI] [PubMed] [Google Scholar]
- 16.Krasniqi E, Pizzuti L, Barchiesi G, Sergi D, Carpano S, Botti C, et al. Impact of BMI on HER2+ metastatic breast cancer patients treated with pertuzumab and/or trastuzumab emtansine. Real-world evidence. J Cell Physiol. 2020;235(11):7900–10. [DOI] [PubMed] [Google Scholar]
- 17.Chavez-MacGregor M, Mittendorf EA, Clarke CA, Lichtensztajn DY, Hunt KK, Giordano SH. Incorporating Tumor Characteristics to the American Joint Committee on Cancer Breast Cancer Staging System. Oncologist. 2017;22(11):1292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torres-de la Roche LA, Steljes I, Janni W, Friedl TWP, De Wilde RL. The Association between Obesity and Premenopausal Breast Cancer According to Intrinsic Subtypes - a Systematic Review. Geburtshilfe Frauenheilkd. 2020;80(6):601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28(16):2784–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N Engl J Med. 2017;377(19):1836–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sestak I, Distler W, Forbes JF, Dowsett M, Howell A, Cuzick J. Effect of body mass index on recurrences in tamoxifen and anastrozole treated women: an exploratory analysis from the ATAC trial. J Clin Oncol. 2010;28(21):3411–5. [DOI] [PubMed] [Google Scholar]
- 22.Folkerd EJ, Dixon JM, Renshaw L, A’Hern RP, Dowsett M. Suppression of plasma estrogen levels by letrozole and anastrozole is related to body mass index in patients with breast cancer. J Clin Oncol. 2012;30(24):2977–80. [DOI] [PubMed] [Google Scholar]
- 23.Ligibel JA, Winer EP. Aromatase inhibition in obese women: how much is enough? J Clin Oncol. 2012;30(24):2940–2. [DOI] [PubMed] [Google Scholar]
- 24.Buzdar AU, Jones SE, Vogel CL, Wolter J, Plourde P, Webster A. A phase III trial comparing anastrozole (1 and 10 milligrams), a potent and selective aromatase inhibitor, with megestrol acetate in postmenopausal women with advanced breast carcinoma. Arimidex Study Group. Cancer. 1997;79(4):730–9. [PubMed] [Google Scholar]
- 25.Jonat W, Howell A, Blomqvist C, Eiermann W, Winblad G, Tyrrell C, et al. A randomised trial comparing two doses of the new selective aromatase inhibitor anastrozole (Arimidex) with megestrol acetate in postmenopausal patients with advanced breast cancer. Eur J Cancer. 1996;32A(3):404–12. [DOI] [PubMed] [Google Scholar]
- 26.Dixon JM, Renshaw L, Young O, Murray J, Macaskill EJ, McHugh M, et al. Letrozole suppresses plasma estradiol and estrone sulphate more completely than anastrozole in postmenopausal women with breast cancer. J Clin Oncol. 2008;26(10):1671–6. [DOI] [PubMed] [Google Scholar]
- 27.Elliott MJ, Ennis M, Pritchard KI, Townsley C, Warr D, Elser C, et al. Association between BMI, vitamin D, and estrogen levels in postmenopausal women using adjuvant letrozole: a prospective study. NPJ Breast Cancer. 2020;6:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozdemir BC, Sflomos G, Brisken C. The challenges of modeling hormone receptor-positive breast cancer in mice. Endocr Relat Cancer. 2018;25(5):R319–R30. [DOI] [PubMed] [Google Scholar]
- 29.Welsch CW. Host factors affecting the growth of carcinogen-induced rat mammary carcinomas: a review and tribute to Charles Brenton Huggins. Cancer Res. 1985;45(8):3415–43. [PubMed] [Google Scholar]
- 30.Thompson HJ. Methods for the Induction of Mammary Carcinogenesis in the Rat Using either 7,12-Dimethylbenz[α]anthracene or 1-Methyl-1-Nitrosourea. In: Ip M.M. ABB, editor. Methods in Mammary Gland Biology and Breast Cancer Research. Boston, MA: Springer; 2000. p. 19–29. [Google Scholar]
- 31.Thompson HJ, McGinley JN, Rothhammer K, Singh M. Rapid induction of mammary intraductal proliferations, ductal carcinoma in situ and carcinomas by the injection of sexually immature female rats with 1-methyl-1-nitrosourea. Carcinogenesis. 1995;16(10):2407–11. [DOI] [PubMed] [Google Scholar]
- 32.Perse M, Cerar A, Injac R, Strukelj B. N-methylnitrosourea induced breast cancer in rat, the histopathology of the resulting tumours and its drawbacks as a model. Pathol Oncol Res. 2009;15(1):115–21. [DOI] [PubMed] [Google Scholar]
- 33.McCormick DL, Adamowski CB, Fiks A, Moon RC. Lifetime dose-response relationships for mammary tumor induction by a single administration of N-methyl-N-nitrosourea. Cancer Res. 1981;41(5):1690–4. [PubMed] [Google Scholar]
- 34.MacLean PS, Giles ED, Johnson GC, McDaniel SM, Fleming-Elder BK, Gilman KA, et al. A surprising link between the energetics of ovariectomy-induced weight gain and mammary tumor progression in obese rats. Obesity (Silver Spring). 2010;18(4):696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giles ED, Wellberg EA, Astling DP, Anderson SM, Thor AD, Jindal S, et al. Obesity and overfeeding affecting both tumor and systemic metabolism activates the progesterone receptor to contribute to postmenopausal breast cancer. Cancer Res. 2012;72(24):6490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giles ED, Jindal S, Wellberg EA, Schedin T, Anderson SM, Thor AD, et al. Metformin inhibits stromal aromatase expression and tumor progression in a rodent model of postmenopausal breast cancer. Breast Cancer Res. 2018;20(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giles ED, Jackman MR, MacLean PS. Modeling Diet-Induced Obesity with Obesity-Prone Rats: Implications for Studies in Females. Front Nutr. 2016;3:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Checkley LA, Rudolph MC, Wellberg EA, Giles ED, Wahdan-Alaswad RS, Houck JA, et al. Metformin Accumulation Correlates with Organic Cation Transporter 2 Protein Expression and Predicts Mammary Tumor Regression In Vivo. Cancer Prev Res (Phila). 2017;10(3):198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wellberg EA, Checkley LA, Giles ED, Johnson SJ, Oljira R, Wahdan-Alaswad R, et al. The Androgen Receptor Supports Tumor Progression After the Loss of Ovarian Function in a Preclinical Model of Obesity and Breast Cancer. Horm Cancer. 2017;8(5–6):269–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson HJ, McGinley JN, Wolfe P, Singh M, Steele VE, Kelloff GJ. Temporal sequence of mammary intraductal proliferations, ductal carcinomas in situ and adenocarcinomas induced by 1-methyl-1-nitrosourea in rats. Carcinogenesis. 1998;19(12):2181–5. [DOI] [PubMed] [Google Scholar]
- 41.Thompson HJ, Adlakha H, Singh M. Effect of carcinogen dose and age at administration on induction of mammary carcinogenesis by 1-methyl-1-nitrosourea. Carcinogenesis. 1992;13(9):1535–9. [DOI] [PubMed] [Google Scholar]
- 42.Thompson HJ, Adlakha H. Dose-responsive induction of mammary gland carcinomas by the intraperitoneal injection of 1-methyl-1-nitrosourea. Cancer Res. 1991;51(13):3411–5. [PubMed] [Google Scholar]
- 43.Hennighausen L Mouse models for breast cancer. Oncogene. 2000;19(8):966–7. [DOI] [PubMed] [Google Scholar]
- 44.Hutchinson JN, Muller WJ. Transgenic mouse models of human breast cancer. Oncogene. 2000;19(53):6130–7. [DOI] [PubMed] [Google Scholar]
- 45.Federico L, Chong Z, Zhang D, McGrail DJ, Zhao W, Jeong KJ, et al. A murine preclinical syngeneic transplantation model for breast cancer precision medicine. Sci Adv. 2017;3(4):e1600957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, et al. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol. 2016;13(4):228–41. [DOI] [PubMed] [Google Scholar]
- 47.Evangelista GCM, Salvador PA, Soares SMA, Barros LRC, Xavier F, Abdo LM, et al. 4T1 Mammary Carcinoma Colonization of Metastatic Niches Is Accelerated by Obesity. Front Oncol. 2019;9:685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirma NB, Tekmal RR. Transgenic mouse models of hormonal mammary carcinogenesis: advantages and limitations. J Steroid Biochem Mol Biol. 2012;131(3–5):76–82. [DOI] [PubMed] [Google Scholar]
- 49.Usary J, Darr DB, Pfefferle AD, Perou CM. Overview of Genetically Engineered Mouse Models of Distinct Breast Cancer Subtypes. Curr Protoc Pharmacol. 2016;72:14 38 1–14 38 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jallow F, Brockman JL, Helzer KT, Rugowski DE, Goffin V, Alarid ET, et al. 17beta-Estradiol and ICI182,780 Differentially Regulate STAT5 Isoforms in Female Mammary Epithelium, With Distinct Outcomes. J Endocr Soc. 2018;2(3):293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, Schuler LA. Prolactin induces ERalpha-positive and ERalpha-negative mammary cancer in transgenic mice. Oncogene. 2003;22(30):4664–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tilli MT, Frech MS, Steed ME, Hruska KS, Johnson MD, Flaws JA, et al. Introduction of estrogen receptor-alpha into the tTA/TAg conditional mouse model precipitates the development of estrogen-responsive mammary adenocarcinoma. Am J Pathol. 2003;163(5):1713–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chan SR, Vermi W, Luo J, Lucini L, Rickert C, Fowler AM, et al. STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast Cancer Res. 2012;14(1):R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cardiff RD, Wellings SR. The comparative pathology of human and mouse mammary glands. J Mammary Gland Biol Neoplasia. 1999;4(1):105–22. [DOI] [PubMed] [Google Scholar]
- 55.Fleming JM, Long EL, Ginsburg E, Gerscovich D, Meltzer PS, Vonderhaar BK. Interlobular and intralobular mammary stroma: genotype may not reflect phenotype. BMC Cell Biol. 2008;9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dankort DL, Muller WJ. Transgenic models of breast cancer metastasis. Cancer Treat Res. 1996;83:71–88. [DOI] [PubMed] [Google Scholar]
- 57.Dobrolecki LE, Airhart SD, Alferez DG, Aparicio S, Behbod F, Bentires-Alj M, et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 2016;35(4):547–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sflomos G, Dormoy V, Metsalu T, Jeitziner R, Battista L, Scabia V, et al. A Preclinical Model for ERalpha-Positive Breast Cancer Points to the Epithelial Microenvironment as Determinant of Luminal Phenotype and Hormone Response. Cancer Cell. 2016;29(3):407–22. [DOI] [PubMed] [Google Scholar]
- 59.Matthews SB, Sartorius CA. Steroid Hormone Receptor Positive Breast Cancer Patient-Derived Xenografts. Horm Cancer. 2017;8(1):4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Finlay-Schultz J, Jacobsen BM, Riley D, Paul KV, Turner S, Ferreira-Gonzalez A, et al. New generation breast cancer cell lines developed from patient-derived xenografts. Breast Cancer Res. 2020;22(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nunez NP, Perkins SN, Smith NC, Berrigan D, Berendes DM, Varticovski L, et al. Obesity accelerates mouse mammary tumor growth in the absence of ovarian hormones. Nutr Cancer. 2008;60(4):534–41. [DOI] [PubMed] [Google Scholar]
- 62.Chumlea WC, Guo SS, Kuczmarski RJ, Flegal KM, Johnson CL, Heymsfield SB, et al. Body composition estimates from NHANES III bioelectrical impedance data. Int J Obes Relat Metab Disord. 2002;26(12):1596–609. [DOI] [PubMed] [Google Scholar]
- 63.Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, et al. Animal models of obesity and diabetes mellitus. Nat Rev Endocrinol. 2018;14(3):140–62. [DOI] [PubMed] [Google Scholar]
- 64.Speakman J, Hambly C, Mitchell S, Krol E. The contribution of animal models to the study of obesity. Lab Anim. 2008;42(4):413–32. [DOI] [PubMed] [Google Scholar]
- 65.Tschop M, Heiman ML. Overview of rodent models for obesity research. Curr Protoc Neurosci. 2002;Chapter 9:Unit 9 10. [DOI] [PubMed] [Google Scholar]
- 66.Gunter MJ, Xie X, Xue X, Kabat GC, Rohan TE, Wassertheil-Smoller S, et al. Breast cancer risk in metabolically healthy but overweight postmenopausal women. Cancer Res. 2015;75(2):270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kabat GC, Kim MY, Lee JS, Ho GY, Going SB, Beebe-Dimmer J, et al. Metabolic Obesity Phenotypes and Risk of Breast Cancer in Postmenopausal Women. Cancer Epidemiol Biomarkers Prev. 2017;26(12):1730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chlebowski RT, Aragaki AK, Anderson GL, Thomson CA, Manson JE, Simon MS, et al. Low-Fat Dietary Pattern and Breast Cancer Mortality in the Women’s Health Initiative Randomized Controlled Trial. J Clin Oncol. 2017;35(25):2919–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chlebowski RT, Anderson GL, Manson JE, Prentice RL, Aragaki AK, Snetselaar L, et al. Low-Fat Dietary Pattern and Cancer Mortality in the Women’s Health Initiative (WHI) Randomized Controlled Trial. JNCI Cancer Spectr. 2018;2(4):pky065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chlebowski RT, Aragaki AK, Anderson GL, Simon MS, Manson JE, Neuhouser ML, et al. Association of Low-Fat Dietary Pattern With Breast Cancer Overall Survival: A Secondary Analysis of the Women’s Health Initiative Randomized Clinical Trial. JAMA Oncol. 2018;4(10):e181212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chlebowski RT, Aragaki AK, Anderson GL, Pan K, Neuhouser ML, Manson JE, et al. Dietary Modification and Breast Cancer Mortality: Long-Term Follow-Up of the Women’s Health Initiative Randomized Trial. J Clin Oncol. 2020;38(13):1419–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wellberg EA, Kabos P, Gillen AE, Jacobsen BM, Brechbuhl HM, Johnson SJ, et al. FGFR1 underlies obesity-associated progression of estrogen receptor-positive breast cancer after estrogen deprivation. JCI Insight. 2018;3(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Giles ED, Jackman MR, Johnson GC, Schedin PJ, Houser JL, MacLean PS. Effect of the estrous cycle and surgical ovariectomy on energy balance, fuel utilization, and physical activity in lean and obese female rats. Am J Physiol Regul Integr Comp Physiol. 2010;299(6):R1634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sherk VD, Jackman MR, Giles ED, Higgins JA, Foright RM, Presby DM, et al. Prior weight loss exacerbates the biological drive to gain weight after the loss of ovarian function. Physiol Rep. 2017;5(10):e13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sherk VD, Jackman MR, Higgins JA, Giles ED, Foright RM, Presby DM, et al. Impact of Exercise and Activity on Weight Regain and Musculoskeletal Health Post-Ovariectomy. Med Sci Sports Exerc. 2019;51(12):2465–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fischer AW, Cannon B, Nedergaard J. The answer to the question “What is the best housing temperature to translate mouse experiments to humans?” is: thermoneutrality. Mol Metab. 2019;26:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ganeshan K, Chawla A. Warming the mouse to model human diseases. Nat Rev Endocrinol. 2017;13(8):458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Keijer J, Li M, Speakman JR. What is the best housing temperature to translate mouse experiments to humans? Mol Metab. 2019;25:168–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Keijer J, Li M, Speakman JR. To best mimic human thermal conditions, mice should be housed slightly below thermoneutrality. Mol Metab. 2019;26:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Speakman JR, Keijer J. Not so hot: Optimal housing temperatures for mice to mimic the thermal environment of humans. Mol Metab. 2012;2(1):5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gordon CJ. Thermal physiology of laboratory mice: Defining thermoneutrality. Journal of Thermal Biology. 2012;37(8):654–85. [Google Scholar]
- 82.Morris EM, Noland RD, Allen JA, McCoin CS, Xia Q, Koestler DC, et al. Difference in Housing Temperature-Induced Energy Expenditure Elicits Sex-Specific Diet-Induced Metabolic Adaptations in Mice. Obesity (Silver Spring). 2020;28(10):1922–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cui X, Nguyen NL, Zarebidaki E, Cao Q, Li F, Zha L, et al. Thermoneutrality decreases thermogenic program and promotes adiposity in high-fat diet-fed mice. Physiol Rep. 2016;4(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hylander BL, Repasky EA. Thermoneutrality, Mice, and Cancer: A Heated Opinion. Trends Cancer. 2016;2(4):166–75. [DOI] [PubMed] [Google Scholar]
- 85.Ravussin Y, LeDuc CA, Watanabe K, Leibel RL. Effects of ambient temperature on adaptive thermogenesis during maintenance of reduced body weight in mice. Am J Physiol Regul Integr Comp Physiol. 2012;303(4):R438–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stemmer K, Kotzbeck P, Zani F, Bauer M, Neff C, Muller TD, et al. Thermoneutral housing is a critical factor for immune function and diet-induced obesity in C57BL/6 nude mice. Int J Obes (Lond). 2015;39(5):791–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Szymusiak R, Satinoff E. Maximal REM sleep time defines a narrower thermoneutral zone than does minimal metabolic rate. Physiol Behav. 1981;26(4):687–90. [DOI] [PubMed] [Google Scholar]
- 88.Neff EP. A point on thermoneutrality for mice. Lab Animal. 2020;49(6):169-. [Google Scholar]
- 89.Rogers NH, Perfield JW 2nd, Strissel KJ, Obin MS, Greenberg AS. Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology. 2009;150(5):2161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Witte MM, Resuehr D, Chandler AR, Mehle AK, Overton JM. Female mice and rats exhibit species-specific metabolic and behavioral responses to ovariectomy. Gen Comp Endocrinol. 2010;166(3):520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brooks HL, Pollow DP, Hoyer PB. The VCD Mouse Model of Menopause and Perimenopause for the Study of Sex Differences in Cardiovascular Disease and the Metabolic Syndrome. Physiology (Bethesda). 2016;31(4):250–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Van Kempen TA, Milner TA, Waters EM. Accelerated ovarian failure: a novel, chemically induced animal model of menopause. Brain Res. 2011;1379:176–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pearse G, Frith J, Randall KJ, Klinowska T. Urinary retention and cystitis associated with subcutaneous estradiol pellets in female nude mice. Toxicol Pathol. 2009;37(2):227–34. [DOI] [PubMed] [Google Scholar]
- 94.Dall G, Vieusseux J, Unsworth A, Anderson R, Britt K. Low Dose, Low Cost Estradiol Pellets Can Support MCF-7 Tumour Growth in Nude Mice without Bladder Symptoms. J Cancer. 2015;6(12):1331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eyre R, Alferez DG, Spence K, Kamal M, Shaw FL, Simoes BM, et al. Patient-derived Mammosphere and Xenograft Tumour Initiation Correlates with Progression to Metastasis. J Mammary Gland Biol Neoplasia. 2016;21(3–4):99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Levin-Allerhand JA, Sokol K, Smith JD. Safe and effective method for chronic 17beta-estradiol administration to mice. Contemp Top Lab Anim Sci. 2003;42(6):33–5. [PubMed] [Google Scholar]
- 97.Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res. 2007;13(13):3989–98. [DOI] [PubMed] [Google Scholar]
- 98.Scalzo RL, Foright RM, Hull SE, Knaub LA, Johnson-Murguia S, Kinanee F, et al. Breast cancer endocrine therapy exhausts adipocyte progenitors promoting weight gain and glucose intolerance. BioRxiv The Preprint Server for Biology. 2020. [Google Scholar]
- 99.Strom JO, Theodorsson A, Ingberg E, Isaksson IM, Theodorsson E. Ovariectomy and 17beta-estradiol replacement in rats and mice: a visual demonstration. J Vis Exp. 2012(64):e4013. [DOI] [PMC free article] [PubMed] [Google Scholar]