

Abstract
Myeloid master regulator CCAAT enhancer-binding protein alpha (C/EBPα) is deregulated by multiple mechanisms in leukemia. Inhibition of C/EBPα function plays pivotal roles in leukemogenesis. While much is known about how C/EBPα orchestrates granulopoiesis, our understanding of molecular transformation events, the role(s) of cooperating mutations and clonal evolution during C/EBPα deregulation in leukemia remains elusive. In this review, we will summarize the latest research addressing these topics with special emphasis on CEBPA mutations. We conclude by describing emerging therapeutic strategies to restore C/EBPα function.
INTRODUCTION
Hematopoiesis is the process by which multipotent hematopoietic stem cells (HSCs) develop into mature blood cells through a highly organized hierarchy of successive differentiation events. Transcription factors play a central role in the multi-layered regulation of hematopoiesis. Perturbations in transcription factor activity result in a differentiation block, lineage infidelity and transformation events, which lead to leukemia.1
The C/EBP family of transcription factors consists of six members—C/EBPα, C/EBPβ, C/EBPγ, C/EBPδ, C/EBPζ and C/EBPε.2 C/EBPα, the founding member of C/EBP family, was initially characterized in adipogenesis and later found to be expressed in multiple tissues, including the liver, lung, skin, mammary glands and hematopoietic cells, acting as a tumor suppressor. Studies on C/EBPs led to the discovery of the basic region leucine zipper (bZIP) and the basic helix-loop-helix (bHLH) classes of transcription factors.3,4 Research on C/EBPα has contributed immensely to our understanding of transcription factor function in lineage specificity, cell cycle control by transcription factors, the transcriptional program of differentiation, and the molecular basis for transformation in cancer.1,5
C/EBPα FUNCTION IN HEMATOPOIETIC DIFFERENTIATION
Development of HSC to various lineages is the result of two fundamental processes: self-renewal and differentiation. Both these processes are orchestrated by transcription factors by complex mechanisms.1 The intron-less CEBPA gene encodes a mRNA that can translate into two major forms—42 kD C/EBPα-p42 and 30 kD C/EBPα-p30 based on alternate translation initiation codons.6 Cells regulate the p42/p30 ratio at translation initiation by mTOR signaling and C/EBPα-p30 dominantly inhibits the C/EBPα-p42 function when the ratio of p42/p30 is less than one.7 C/EBPα-p30 lacks two N-terminal transactivation domains that are unique to C/EBPα-p42, which play a central role in the inhibition of E2F transcription factors.8,9
Conditional deletion of Cebpa in mouse displays lack of granulocytes with a specific block in the common myeloid progenitor (CMP) to granulocyte-monocyte progenitor (GMP) step, underlining the central role of C/EBPα in granulopoiesis.10 C/EBPα regulates myeloid differentiation by upregulating myeloid-specific genes and by blocking myeloid cell proliferation. Well characterized examples of genes regulated by C/EBPα in granulopoiesis include transcription factors (PU.1,11 c-Jun,12 c-Myc,13 SOX4(ref 14) and E2F9), growth factor receptors (granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor), primary granule proteins (myeloperoxidase), secondary granule proteins (lactoferin)15 and microRNAs (miR-223,16,17 miR-34a18 and miR-30c19). Even though C/EBPα is widely considered as a master regulator of granulopoiesis, reports suggest it is important in monocyte/macrophage differentiation,20,21 suggesting the role of C/EBPα in lineage commitment is context dependent.
Even though C/EBPα is expressed at low levels in HSC, it negatively regulates HSC self-renewal.10 Ye et al.22 reported that C/EBPα controls maintenance of adult HSC quiescence, but not the survival of HSCs. Loss of C/EBPα establishes a fetal transcriptional program in adult HSCs and enhances HSC proliferation by de-repressing N-Myc.22 The function of C/EBPα in HSCs was re-evaluated recently by Hasemann and colleagues.23 Their study demonstrated that C/EBPα positively regulates HSC self-renewal, protects adult HSCs from apoptosis and maintains their quiescent state.23 These studies demonstrated opposite views for the role of C/EBPα in HSC function. In these studies, the timing of C/EBPα excision could have resulted in disparate findings. In most of the experiments in Ye’s study,22 donor cells were transplanted into a recipient mouse followed by pIpC-mediated deletion. Meanwhile, in experiments that evaluated HSC function in Hasemann’s study,23 donor cells had undergone pIpC-mediated deletion followed by transplantation. So, loss of C/EBPα (before transplantation) may have compromised HSC engraftment during transplantation, resulting in lower reconstitution, which was interpreted as a competitive disadvantage in the Hasemann’s study.23 This also explains why CebpaΔ/Δ HSCs in that study do not exhibit a proliferative advantage and eventually underwent apoptosis. In support of this possibility, C/EBPα directly regulates CXC chemokine receptor 4 (CXCR4), one of the major regulators for homing, engraftment, and quiescence of HSCs in the bone marrow niche,24,25 and is downregulated in AML samples with CEBPA mutations.26
The discrepancies in C/EBPα-mediated HSC function are similar to previous studies, which investigated RUNX1 function in HSCs. While Growny et al.27 reported that RUNX1-deficient HSCs display impaired chimerism in competitive transplantation assays, they did not observe defective LT-HSC activity; Ichikawa et al.,28,29 showed that in their experimental settings RUNX1 deletion does not impair chimerism in competitive transplantation assays and is inversely correlated with LT-HSC activity. Collectively, the timing of C/EBPα excision and whether it affected engraftment of bone marrow cells in transplantation experiments could have resulted in varying interpretations of the role of C/EBPα in HSCs. Another explanation for the different views of C/EBPα function in HSCs is the timing of analysis (5–7 days after pIpC in most of the experiments in Ye’s study versus ⩾ 16 weeks after pIpC in Hasemann’s study).
The above-mentioned studies bring our attention to the unexplored role of C/EBPα in HSCs, which is compromised in leukemia (discussed below).
C/EBPα AND CELL CYCLE CONTROL
In addition to acting as a classical transcription factor, C/EBPα blocks cell cycle progression30 with mechanisms that vary from cell type to cell type (reviewed in Nerlov et al.31). In hematopoiesis, inhibition of E2F transcription factors have been widely accepted as the mechanism for C/EBPα-mediated growth arrest.9,32,33 We have showed that C/EBPα inhibits E2F by directly regulating two microRNAs miR-223 and miR-34a, which target E2F1 and E2F3 respectively.17,18 In line with these findings, acute myeloid leukemia (AML) with C/EBPα deregulation is associated with decreased activity of miR-223 and miR-34a. Mouse models of Cebpa mutants suggests loss of cell cycle regulation by C/EBPα plays a major role in leukemogenesis.34,35
C/EBPα DEREGULATION IN AML EXCLUDING MUTATION
The fundamental characteristics of leukemia are block of differentiation in a particular lineage, leading to enhanced proliferation and cell survival resulting from inactivation of tumor suppressors and/or activation of oncogenes. The first report of C/EBPα deregulation was shown by RUNX1/ETO, the product of the t(8;21) translocation.36 RUNX1/ETO blocks C/EBPα expression by inhibiting positive autoregulation of the CEBPA promotor37 and by directly binding to the +42 kb enhancer,38 which is regulated by RUNX1 in granulopoiesis.39,40 CBFβ-SMMHC fusion protein found in AML patients with inv(16) inhibits C/EBPα by upregulating calreticulin, which binds to GC-rich regions in C/EBPα mRNA.41 Treatment of inv(16) cells with AI-10-49, a CBFβ-SMMHC inhibitor can reactivate C/EBPα expression in inv(16) cells.42 Inhibition of C/EBPα by calreticulin is also found in RUNX1-MDS-EVI1 translocation t(3;21).43
BCR-ABL, the product of the t(9;22) translocation in chronic myeloid leukemia upregulates poly(rC)-binding protein hnRNP-E2, inhibiting C/EBPα translation by binding to C/EBPα mRNA.44 The BCR-ABL inhibitor Imatinib downregulates hnRNP-E2 and restores C/EBPα protein levels and granulocyte differentiation. C/EBPα serine 21 phosphorylation by ERK1/2 and cyclin-dependent kinase (CDK1) in FLT3 activating mutation is another mechanism for C/EBPα disruption in AML.45,46 Acetylation of C/EBPα at lysine residues K298 and K302 by general control non-derepressible 5 (GCN5) impairs C/EBPα DNA-binding ability and compromises C/EBPα transcriptional activity.47 Also, RARα-PLZF found in t(11;17) inhibits C/EBPα by protein interaction and recruitment of histone Deacetylase1 to target loci.48 In addition to leukemic oncogenes mentioned above, another layer of C/EBPα deregulation in AML is by miR-182, which binds to CEBPA 3′-untranslated region, thereby blocking C/EBPα translation.49 C/EBPα binds to miR-182 promotor and inhibits its expression during granulopoiesis. AML samples with CEBPA mutations display elevated levels of miR-182, suggesting C/EBPα-miR-182 feedback loop is critical for proper C/EBPα regulation.
Epigenetic modifications of the CEBPA promotor have been found as another major mechanism for C/EBPα inactivation in AML. PMR-RARα, the leukemic fusion protein observed in acute promyelocytic leukemia with t(15;17) deregulates C/EBPα accompanied by DNA methylation.50,51 CEBPA distal promotor methylation associated with decreased C/EBPα expression has been reported in 38% in normal karyotype AML.52,53 Taken together, these findings show that deregulation of C/EBPα function acts as a major step for myeloid leukemia development. Multiple mechanisms for inhibiting C/EBPα function in leukemia are shown in Figure 1.
Figure 1.
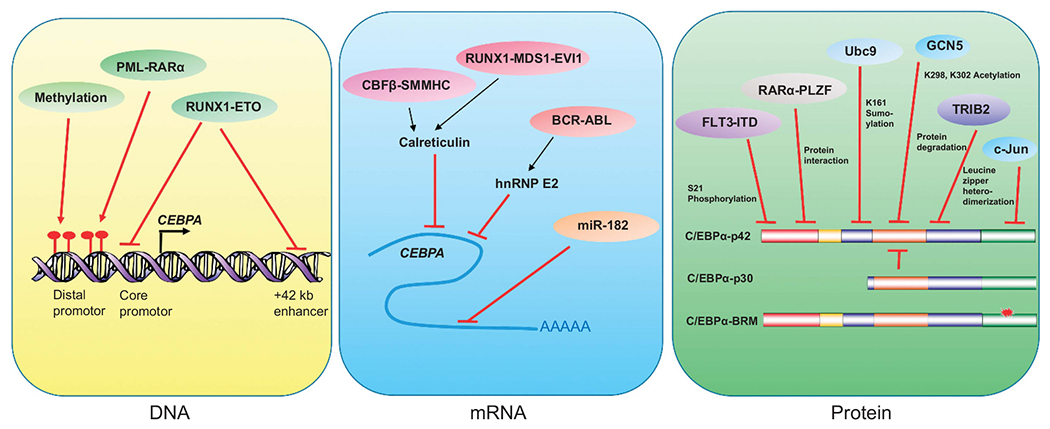
Multiple mechanisms for C/EBPα inhibition in leukemia. Genetic, epigenetic, transcriptional, translational and post-translational mechanisms of C/EBPα inactivation, which result in block in granulocytic differentiation and leukemia.
CEBPα MUTATIONS IN AML
The role of C/EBPα as a tumor suppressor is underlined by the discovery that CEBPA is mutated in AML.54,55 CEBPA is mutated in around 11% of AML patients.56 The mutations reported in CEBPA are point mutations at a bZIP (mentioned as C/EBPα-BRM) and a frame-shift mutation at N-terminus, resulting in a 30 kD isoform of C/EBPα (C/EBPα-p30).54 CEBPA mutations are mostly bi-allelic with one mutation located in N-terminus and other mutation in C-terminus. Absence of homozygous N- or C-terminal mutations suggest that the two mutations possess different functions in leukemogenesis. Studies from multiple laboratories have shown that C/EBPα-p30 is the founder mutation, followed by point mutations in the basic region of C/EBPα. Patients undergoing AML relapse retain the same mutation pattern in both CEBPA alleles in comparison to diagnosis, suggesting CEBPA mutations are pre-leukemic molecular events in the transformation processes.57,58 C/EBPα-p42, mutant forms of C/EBPα observed in AML and their functional characteristics are depicted in Figure 2a.
Figure 2.
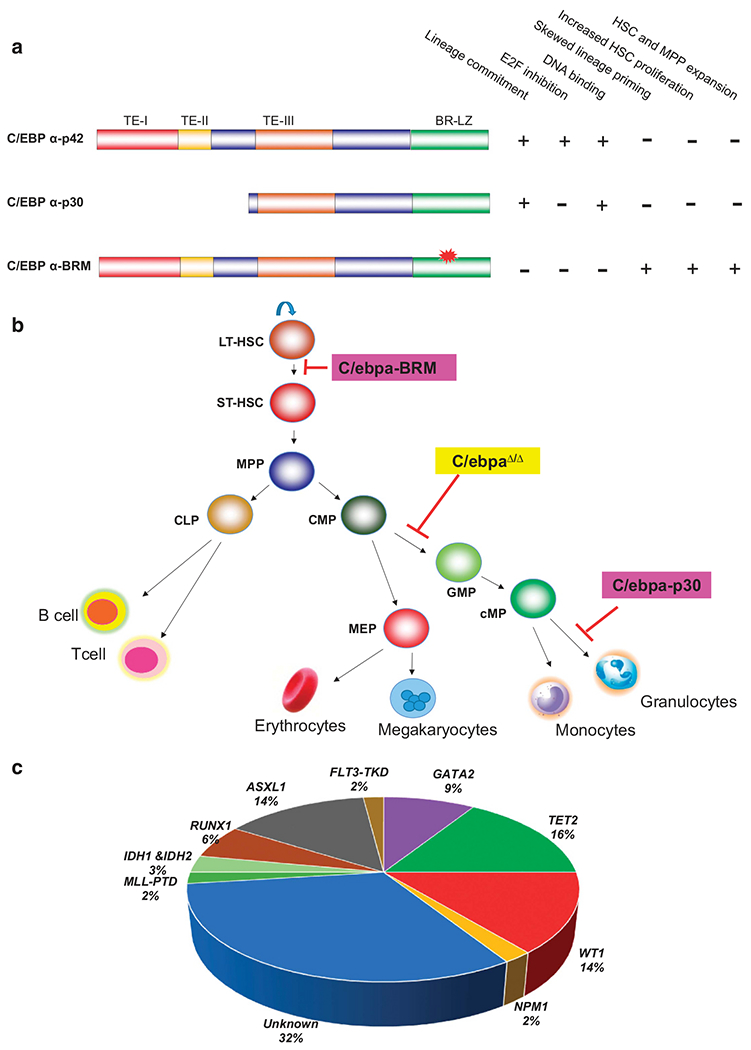
Characteristics of CEBPA mutations in AML. (a) Biological roles of C/EBPα-p42 and C/EBPα mutant forms. Three transactivation elements (TE-I, TE-II and TE-III) and a basic-region leucine zipper (BR-LZ) are depicted. C/EBPα-BRM, C/EBPα basic region mutant. (b) Distinct differentiation blocks observed during C/EBPα deletion and CEBPA mutations. (c) Common mutations associated with bi-allelic CEBPA mutations (prepared based on Fasan et al.,56). CLP, common lymphoid progenitors; cMP, committed myeloid progenitors; CMP, common myeloid progenitors; GMP, granulocyte/macrophage progenitors; LT-HSC, long-term hematopoietic stem cells; MEP, megakaryocyte/erythroid progenitors; MPP, multi-potential progenitors; ST-HSC, short-term hematopoietic stem cells.
CEBPα MUTATIONS AND LEUKEMIA-INITIATING CELLS
Leukemia-initiating cells (LIC) can originate from hematopoietic stem cells or progenitors with an altered self-renewal function during the transformation process. Proof of CEBPA mutations activating self-renewal in committed myeloid progenitor cells in a genetic model was provided by Nerlov and colleagues.34 By using a non-conditional mouse model for Cebpa-p30, they demonstrated that C/ebpa-p30 is able to induce differentiation to committed myeloid progenitors (cMP). This basal differentiation provided by Cebpa-p30, not the loss of expression of Cebpa-p42, can act as a platform for induction of myeloid leukemia. Mouse models for Cebpa basic region mutants display increased proliferation of long-term HSCs (LT-HSC) and skewed lineage programing of HSCs, resulting in pre-malignant expansion of multipotent progenitors (MPP).35 In addition, this study demonstrated that combining the N- and C-terminal Cebpa mutations in mouse models accelerate leukemia development in comparison to a single mutation, reproducing the bi-allelic mutation pattern seen in patients with CEBPA mutations. These two seminal works bring several key points to the field. (1) Loss of C/EBPα-mediated cell cycle control is instrumental in the transformation process of myeloid leukemia. (2) Leukemic stem cells (LSC) in CEBPA mutations do not display features of HSC or MPP, rather a c-kit+Mac-1+ committed myeloid progenitor observed for mixed lineage leukemia (MLL).59 (3) Even though expressed at low levels, C/EBPα plays a major role in HSC lineage priming for downstream myeloid commitment. (4) N-terminal and C-terminal mutations of CEBPA have different functions ,which are instrumental in the leukemic transformation. Collectively, these findings suggest mutations of CEBPA can convert HSCs to a pre-leukemic state and a downstream progenitor compartment can act as the target for secondary mutations, triggering transformation. Patterns of differentiation block by C/EBPα mutants in comparison to C/EBPα deletion are shown in Figure 2b.
C/EBPα MUTANTS AND MECHANISM OF ACTION
Several models have been suggested for the mechanism of action of C/EBPα-p30. The first model suggested C/EBPα-p30 hetero-dimerize with C/EBPα-p42 and inhibit DNA binding to C/EBPα-p42. This possibility seems less likely, as C/EBPα-p30 with mutations in the heterodimerization domain, which cannot bind C/EBPα-p42, can still block granulocyte differentiation.60 However, it is clear that C/EBPα-p30 can inhibit C/EBPα-p42 DNA binding, at least on certain targets.54 Another potential mechanism for the C/EBPα-p30--mediated differentiation block is differential regulation of target genes by C/EBPα-p30. An earlier report suggested C/EBPα-p30 has quite distinct functional properties compared with the C/EBPα-p42 in the lung.61 We have reported that C/EBPα-p30, but not C/EBPα-p42, upregulates PIN1 as well as UBC9, which in turn blocks C/EBPα-p42 function by multiple mechanisms in AML with CEBPA mutations62,63 Trib2 blocks C/EBPα by proteasome-dependent degradation and is overexpressed in AML with CEBPA mutations.64 Even though both C/EBPα-p42 and C/EBPα-p30 bind to the Trib2 promotor, they have opposite effect in Trib2 transcription, suggesting differential effects in transcription.65 Since both C/EBPα-p42 and C/EBPα-p30 have the same DNA-binding motif, a mechanistic explanation for the selective binding of C/EBPα-p30 to target promotors remains unclear. One possibility is that C/EBPα-p42 and C/EBPα-p30 proteins recruit different protein complexes to target promotors, which can have distinct transcriptional signatures. Careful molecular studies are needed to determine C/EBPα-p30 mediated transcriptional regulation.
CEBPα MUTATIONS AND COOPERATING MUTATIONS
Cancer, including leukemia, results from sequential accumulation of multiple mutations in a specific cell lineage. Gene-expression studies indicated that bi-allelic; but not mono-allelic CEBPA mutant samples have distinctive gene-expression profiles66 that can be predicted by a unique gene signature.67 The major cooperating molecular abnormalities reported in CEBPA mutations are mutations in GATA2, TET2, ASXL1, WT1, RUNX1 and FLT3.56,68 Recent reports show mutations in JAK-STAT pathway members such as CSF3R and STAT5B are frequent in AML with CEBPA mutations.69,70 The most common mutations associated with bi-allelic CEBPA mutations and their frequencies are shown in Figure 2c. Even though the N-terminal mutation, resulting in C/EBPα-p30, is known as the founding mutations, our understanding of secondary transformation events in CEBPA mutations is limited. A recent study showed that internal tandem duplications in the Flt3 gene in combination with CEBPA mutation result in expansion of the MPP compartment and generation of leukemia-initiating GMPs.71 This study demonstrated that Flt3 mutants cooperate with CEBPA mutant to induce leukemia in mice. Further studies are needed to elucidate the function of other cooperating events in CEBPA mutations, its impact on leukemic stem cell activity and determine if C/EBPα mutants play a role in the acquisition of secondary mutations.
GERMLINE CEBPα MUTATION AND CLONAL ORIGIN
Germline CEBPA mutations are relatively rare in AML.72 A recent report found that germline CEBPA mutations occur primarily within the N-terminal domain, with acquired (somatic) mutations preferentially targeting the C-terminal.73 This study also found that GATA2 mutations are the most common secondary events in germline CEBPA mutations. Molecular sequencing of diagnostic and relapsed AML samples with germline CEBPA mutations showed leukemic clones that were distinct from the diagnostic AML, suggesting that the recurrences are a second primary leukemia rather than a relapse. Detailed analysis of clonal evolution at relapse showed presence of a novel leukemic clone as well as a sub-clone with mutations in components of DNA hydroxylmethylation such as WT1 and TET2, respectively. This study highlighted that CEBPA germline mutations may influence the acquisition and/or selection of cooperating mutations, which may be governed by genetic factors.
REQUIREMENT OF A MYELOID PLATFORM FOR LEUKEMIA INITIATION
Conditional deletion of C/EBPα in mice show a block of myeloid differentiation, but do not develop leukemia.10 Whether C/EBPα per se is required for leukemogenesis was recently debated. Using a transplantation model for CebpaΔ/Δ bone marrow cells transduced with MLL-AF9 retrovirus, Ohlsson and colleagues, reported that C/EBPα is required for the initiation of MLL-driven AML and suggested C/EBPα may collaborate with MLL-ENL.74 In a related area, Collins and colleagues showed that C/EBPα is essential for the proliferation of Hoxa9/Meis1 transformed cells and that deficiency of C/EBPα displays enhanced survival in primary murine models of Hoxa9/Meis1-induced leukemia.75 These two papers suggested C/EBPα either collaborate with oncogenes and/or provide enhanced proliferation to leukemic cells, in multiple contexts. These findings have recently been challenged by Ye and colleagues.76 The study reported that even though C/EBPα deletion results in lack of leukemia development, it can be reverted by inducing myeloid differentiation by cytokines such as GM-CSF and IL-3. This suggests myeloid differentiation rather than the presence of C/EBPα is the requirement for leukemia development.76 Multiple factors could be contributing to the discrepancy in the interpretation of these results. Ye study shows that compared to CMPs, GMPs provide more accessible genomic architecture for oncogenes, suggesting a lack of GMPs could be preventing leukemic transformation in the Ohlsson study. Reduced expression of Meis1 and Hoxa9 oncogenes in Cebpa deleted MLL-ENL cells, reported by Ohlsson study, could be due to the fact that expression patterns of genes can vary according to the bone marrow compartment and the C/EBPα deletion results in complete block towards GMP. Leukemic cells with RUNX1-ETO and CBFb-SMMHC oncoproteins require wild-type RUNX1 for proliferation and survival, even though both oncoproteins block RUNX1 function.77,78 So, once transformed MLL leukemic cells could be addicted to C/EBPα, which would be instrumental in the proliferation and survival reported by Collins study. Collectively, these studies show that to initiate myeloid leukemic transformation a differentiation program towards GMP is required, and once the transformation happens, these cells are addicted to key myeloid transcription factors.
THERAPY FOR C/EBPα DEREGULATION IN LEUKEMIA
Being the master regulator in granulopoiesis, restoration of normal C/EBPα levels warrants induction of granulocytic differentiation in leukemic stem and progenitor cells. Several studies demonstrated that restoration of C/EBPα function results in abrogation of leukemia and better survival in leukemic mice.79,80 2-Cyano-3, 12-dioxooleana-1, 9-dien-28-oic acid (CDDO), a terpenoid has been shown to increase C/EBPα-p42 protein levels and induces granulocyte differentiation.81 MEK1/2 inhibitor CI-1040 inhibits hnRNP-E2, which results in enhanced C/EBPα protein levels and granulocytic differentiation.82 MLN518, a Flt3 tyrosine kinase inhibitor, can induce granulocytic differentiation by inhibiting phosphorylation of C/EBPα on serine 21.45 Silencing SOX4, a transcription factor overexpressed in bi-allelic Cebpa mutation could rescue myeloid differentiation in the humanized mouse model for CEBPA mutation, suggesting SOX4 as a potential therapeutic target.14 Another study of the gene signature in CEBPA dysfunctional AML suggested responsiveness to histone deacetylase inhibitors.83 High Sensitivity of JAK inhibitors suggest another promising avenue for improved therapeutic strategies for bi-allelic CEBPA mutant AML.69 A recent study showed C/EBPα-p30-dependent inhibition of myeloid differentiation required direct protein–protein interaction with Wdr5, and small molecule-mediated inhibition of this interaction affected the viability of human primary samples with an N-terminal CEBPA mutation.84 Further studies are needed to find therapeutic values of this inhibitor. Discovery of targeted therapeutic approaches such as small molecule-mediated inhibitors that can target C/EBPα-p30 oncogenic function will have great therapeutic value in the treatment of CEBPA mutations.
FUTURE DIRECTIONS
Genetic analyses have provided in-depth knowledge to our understanding of leukemia. Significant progress has been made in elucidating transcriptional control in hematopoiesis. However, our understanding of molecular pathogenesis for C/EBPα deregulation in leukemia is limited. Several key questions remain unanswered. (1) What are the molecular mechanisms behind leukemic transformation events during C/EBPα deregulation? (2) How does C/EBPα-p30 selectively bind to target promotors? (3) What are the genes regulated by C/EBPα-p30 that play key roles in self-renewal, survival and proliferation? (4) What are the pre-leukemic events that define the pattern for acquisition of secondary mutations? (5) How does clonal evolution of CEBPA mutations regulate treatment outcomes in AML? Future studies addressing these questions will provide novel insights, which can be translated to therapeutic approaches.
ACKNOWLEDGEMENTS
JAP is the recipient of a Scholar Award from American Society of Hematology, Young Investigator Award from Alex’s Lemonade Stand Foundation for Childhood Cancer and a Discovery Grant from Lauri Strauss Leukemia Foundation. This work was supported by grants to DGT (a STaR Investigator Award; a RCE Core grant; a Tier 3 RNA Biology Center grant MOE2014-T3-1-006 from the NRF and MOE, Singapore; and grants R35CA197697 from the NCI/NIH and P01HL131477 from the NHLBI/NIH) and to GB (grants BE 2042/12-1, BE 2042/7-1 and GRK1591 from DFG, German Research Foundation; grants DJCLS R 11/17, 17R/2016, R15/18, R12/31 from Deutsche Jose Carreras Leukämie-Stiftung e.V. and grants 2013.153.1 and 2015.093.1 from Wilhelm Sander Stiftung).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol 2007; 7: 105–117. [DOI] [PubMed] [Google Scholar]
- 2.Johnson PF. Molecular stop signs: regulation of cell-cycle arrest by C/EBP transcription factors. J Cell Sci 2005; 118(Pt 12): 2545–2555. [DOI] [PubMed] [Google Scholar]
- 3.Landschulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science 1988; 240: 1759–1764. [DOI] [PubMed] [Google Scholar]
- 4.Landschulz WH, Johnson PF, McKnight SL. The DNA binding domain of the rat liver nuclear protein C/EBP is bipartite. Science 1989; 243: 1681–1688. [DOI] [PubMed] [Google Scholar]
- 5.McKnight SL. McBindall–a better name for CCAAT/enhancer binding proteins? Cell 2001; 107: 259–261. [DOI] [PubMed] [Google Scholar]
- 6.Lin FT, MacDougald OA, Diehl AM, Lane MD. A 30-kDa alternative translation product of the CCAAT/enhancer binding protein alpha message: transcriptional activator lacking antimitotic activity. Proc Natl Acad Sci USA 1993; 90: 9606–9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calkhoven CF, Muller C, Leutz A. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev 2000; 14: 1920–1932. [PMC free article] [PubMed] [Google Scholar]
- 8.Slomiany BA, D’Arigo KL, Kelly MM, Kurtz DT. C/EBPalpha inhibits cell growth via direct repression of E2F-DP-mediated transcription. Mol Cell Biol 2000; 20: 5986–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porse BT, Pedersen TA, Xu X, Lindberg B, Wewer UM, Friis-Hansen L et al. E2F repression by C/EBPalpha is required for adipogenesis and granulopoiesis in vivo. Cell 2001; 107: 247–258. [DOI] [PubMed] [Google Scholar]
- 10.Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity 2004; 21: 853–863. [DOI] [PubMed] [Google Scholar]
- 11.Kummalue T, Friedman AD. Cross-talk between regulators of myeloid development: C/EBPalpha binds and activates the promoter of the PU.1 gene. J Leuk Biol 2003; 74: 464–470. [DOI] [PubMed] [Google Scholar]
- 12.Rangatia J, Vangala RK, Treiber N, Zhang P, Radomska H, Tenen DG et al. Downregulation of c-Jun expression by transcription factor C/EBPalpha is critical for granulocytic lineage commitment. Mol Cell Biol 2002; 22: 8681–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johansen LM, Iwama A, Lodie TA, Sasaki K, Felsher DW, Golub TR et al. c-Myc is a critical target for C/EBPalpha in granulopoiesis. Mol Cell Biol 2001; 21: 3789–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Alberich-Jorda M, Amabile G, Yang H, Staber PB, Di Ruscio A et al. Sox4 is a key oncogenic target in C/EBPalpha mutant acute myeloid leukemia. Cancer Cell 2013; 24: 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W, Wang X, Ward AC, Touw IP, Friedman AD. C/EBPalpha and G-CSF receptor signals cooperate to induce the myeloperoxidase and neutrophil elastase genes. Leukemia 2001; 15: 779–786. [DOI] [PubMed] [Google Scholar]
- 16.Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005; 123: 819–831. [DOI] [PubMed] [Google Scholar]
- 17.Pulikkan JA, Dengler V, Peramangalam PS, Peer Zada AA, Muller-Tidow C, Bohlander SK et al. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood 2010; 115: 1768–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pulikkan JA, Peramangalam PS, Dengler V, Ho PA, Preudhomme C, Meshinchi S et al. C/EBPalpha regulated microRNA-34a targets E2F3 during granulopoiesis and is down-regulated in AML with CEBPA mutations. Blood 2010; 116: 5638–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katzerke C, Madan V, Gerloff D, Brauer-Hartmann D, Hartmann JU, Wurm AA et al. Transcription factor C/EBPalpha-induced microRNA-30c inactivates Notch1 during granulopoiesis and is downregulated in acute myeloid leukemia. Blood 2013; 122: 2433–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahl R, Walsh JC, Lancki D, Laslo P, Iyer SR, Singh H et al. Regulation of macrophage and neutrophil cell fates by the PU.1:C/EBPalpha ratio and granulocyte colony-stimulating factor. Nat Immunol 2003; 4: 1029–1036. [DOI] [PubMed] [Google Scholar]
- 21.Wang D, D’Costa J, Civin CI, Friedman AD. C/EBPalpha directs monocytic commitment of primary myeloid progenitors. Blood 2006; 108: 1223–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye M, Zhang H, Amabile G, Yang H, Staber PB, Zhang P et al. C/EBPa controls acquisition and maintenance of adult haematopoietic stem cell quiescence. Nat Cell Biol 2013; 15: 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hasemann MS, Lauridsen FK, Waage J, Jakobsen JS, Frank AK, Schuster MB et al. C/EBPalpha is required for long-term self-renewal and lineage priming of hematopoietic stem cells and for the maintenance of epigenetic configurations in multipotent progenitors. PLoS Genet 2014; 10: e1004079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nie Y, Han YC, Zou YR. CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med 2008; 205: 777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006; 25: 977–988. [DOI] [PubMed] [Google Scholar]
- 26.Kuo YY, Hou HA, Chen YK, Li LY, Chen PH, Tseng MH et al. The N-terminal CEBPA mutant in acute myeloid leukemia impairs CXCR4 expression. Haematologica 2014; 99: 1799–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 2005; 106: 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ichikawa M, Asai T, Saito T, Seo S, Yamazaki I, Yamagata T et al. AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med 2004; 10: 299–304. [DOI] [PubMed] [Google Scholar]
- 29.Ichikawa M, Goyama S, Asai T, Kawazu M, Nakagawa M, Takeshita M et al. AML1/Runx1 negatively regulates quiescent hematopoietic stem cells in adult hematopoiesis. J Immunol 2008; 180: 4402–4408. [DOI] [PubMed] [Google Scholar]
- 30.Umek RM, Friedman AD, McKnight SL. CCAAT-enhancer binding protein: a component of a differentiation switch. Science 1991; 251: 288–292. [DOI] [PubMed] [Google Scholar]
- 31.Nerlov C C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer 2004; 4: 394–400. [DOI] [PubMed] [Google Scholar]
- 32.D’Alo F, Johansen LM, Nelson EA, Radomska HS, Evans EK, Zhang P et al. The amino terminal and E2F interaction domains are critical for C/EBP alpha-mediated induction of granulopoietic development of hematopoietic cells. Blood 2003; 102: 3163–3171. [DOI] [PubMed] [Google Scholar]
- 33.Porse BT, Bryder D, Theilgaard-Monch K, Hasemann MS, Anderson K, Damgaard I et al. Loss of C/EBP alpha cell cycle control increases myeloid progenitor proliferation and transforms the neutrophil granulocyte lineage. J Exp Med 2005; 202: 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirstetter P, Schuster MB, Bereshchenko O, Moore S, Dvinge H, Kurz E et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell 2008; 13: 299–310. [DOI] [PubMed] [Google Scholar]
- 35.Bereshchenko O, Mancini E, Moore S, Bilbao D, Mansson R, Luc S et al. Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPalpha mutant AML. Cancer Cell 2009; 16: 390–400. [DOI] [PubMed] [Google Scholar]
- 36.Westendorf JJ, Yamamoto CM, Lenny N, Downing JR, Selsted ME, Hiebert SW. The t(8;21) fusion product, AML-1-ETO, associates with C/EBP-alpha, inhibits C/EBP-alpha-dependent transcription, and blocks granulocytic differentiation. Mol Cell Biol 1998; 18: 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pabst T, Mueller BU, Harakawa N, Schoch C, Haferlach T, Behre G et al. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat Med 2001; 7: 444–451. [DOI] [PubMed] [Google Scholar]
- 38.Avellino R A myeloid-specific gene-dosage regulator for cebpa expression in myeloid cells is commonly targeted by onco-proteins in AML. Blood 2014; 124: 2205. [Google Scholar]
- 39.Cooper S, Guo H, Friedman AD. The +37kb cebpa enhancer is critical for cebpa myeloid gene expression and contains functional sites that bind SCL, GATA2, C/EBPalpha, PU.1, and additional Ets factors. PloS One 2015; 10: e0126385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Avellino R, Havermans M, Erpelinck C, Sanders MA, Hoogenboezem R, van de Werken HJ et al. An autonomous CEBPA enhancer specific for myeloid-lineage priming and neutrophilic differentiation. Blood 2016; 127: 2991–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Helbling D, Mueller BU, Timchenko NA, Schardt J, Eyer M, Betts DR et al. CBFB-SMMHC is correlated with increased calreticulin expression and suppresses the granulocytic differentiation factor CEBPA in AML with inv(16). Blood 2005; 106: 1369–1375. [DOI] [PubMed] [Google Scholar]
- 42.Illendula A, Pulikkan JA, Zong H, Grembecka J, Xue L, Sen S et al. Chemical biology. A small-molecule inhibitor of the aberrant transcription factor CBFbeta-SMMHC delays leukemia in mice. Science 2015; 347: 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Helbling D, Mueller BU, Timchenko NA, Hagemeijer A, Jotterand M, Meyer-Monard S et al. The leukemic fusion gene AML1-MDS1-EVI1 suppresses CEBPA in acute myeloid leukemia by activation of Calreticulin. Proc Natl Acad Sci USA 2004; 101: 13312–13317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perrotti D, Cesi V, Trotta R, Guerzoni C, Santilli G, Campbell K et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet 2002; 30: 48–58. [DOI] [PubMed] [Google Scholar]
- 45.Radomska HS, Basseres DS, Zheng R, Zhang P, Dayaram T, Yamamoto Y et al. Block of C/EBP alpha function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J Exp Med 2006; 203: 371–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radomska HS, Alberich-Jorda M, Will B, Gonzalez D, Delwel R, Tenen DG. Targeting CDK1 promotes FLT3-activated acute myeloid leukemia differentiation through C/EBPalpha. J Clin Invest 2012; 122: 2955–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bararia D, Kwok HS, Welner RS, Numata A, Sarosi MB, Yang H et al. Acetylation of C/EBPalpha inhibits its granulopoietic function. Nat Commun 2016; 7: 10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Girard N, Tremblay M, Humbert M, Grondin B, Haman A, Labrecque J et al. RARalpha-PLZF oncogene inhibits C/EBPalpha function in myeloid cells. Proc Natl Acad Sci USA 2013; 110: 13522–13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wurm AA, Zjablovskaja P, Kardosova M, Gerloff D, Brauer-Hartmann D, Katzerke C et al. Disruption of the C/EBPalpha-miR-182 balance impairs granulocytic differentiation. Nat Commun 2017; 8: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guibal FC, Alberich-Jorda M, Hirai H, Ebralidze A, Levantini E, Di Ruscio A et al. Identification of a myeloid committed progenitor as the cancer-initiating cell in acute promyelocytic leukemia. Blood 2009; 114: 5415–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santana-Lemos BA, de Lima Lange AP, de Lira Benicio MT, Jose TD, Lucena-Araujo AR, Krause A et al. The CEBPA gene is down-regulated in acute promyelocytic leukemia and its upstream promoter, but not the core promoter, is highly methylated. Haematologica 2011; 96: 617–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fasan A, Alpermann T, Haferlach C, Grossmann V, Rollr A, Kohlmann A et al. Frequency and prognostic impact of CEBPA proximal, distal and core promoter methylation in normal karyotype AML: a study on 623 cases. PloS One 2013; 8: e54365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Musialik E, Bujko M, Kober P, Grygorowicz MA, Libura M, Przestrzelska M et al. Comparison of promoter DNA methylation and expression levels of genes encoding CCAAT/enhancer binding proteins in AML patients. Leuk Res 2014; 38: 850–856. [DOI] [PubMed] [Google Scholar]
- 54.Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet 2001; 27: 263–270. [DOI] [PubMed] [Google Scholar]
- 55.Gombart AF, Hofmann WK, Kawano S, Takeuchi S, Krug U, Kwok SH et al. Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein alpha in myelodysplastic syndromes and acute myeloid leukemias. Blood 2002; 99: 1332–1340. [DOI] [PubMed] [Google Scholar]
- 56.Fasan A, Haferlach C, Alpermann T, Jeromin S, Grossmann V, Eder C et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia 2014; 28: 794–803. [DOI] [PubMed] [Google Scholar]
- 57.Shih LY, Liang DC, Huang CF, Wu JH, Lin TL, Wang PN et al. AML patients with CEBPalpha mutations mostly retain identical mutant patterns but frequently change in allelic distribution at relapse: a comparative analysis on paired diagnosis and relapse samples. Leukemia 2006; 20: 604–609. [DOI] [PubMed] [Google Scholar]
- 58.Tiesmeier J, Czwalinna A, Muller-Tidow C, Krauter J, Serve H, Heil G et al. Evidence for allelic evolution of C/EBPalpha mutations in acute myeloid leukaemia. BrJ Haematol 2003; 123: 413–419. [DOI] [PubMed] [Google Scholar]
- 59.Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006; 442: 818–822. [DOI] [PubMed] [Google Scholar]
- 60.Niebuhr B, Iwanski GB, Schwieger M, Roscher S, Stocking C, Cammenga J. Investigation of C/EBPalpha function in human (versus murine) myelopoiesis provides novel insight into the impactofCEBPA mutations in acute myelogenous leukemia (AML). Leukemia 2009; 23: 978–983. [DOI] [PubMed] [Google Scholar]
- 61.Wang C, Chen X, Wang Y, Gong J, Hu G. C/EBPalphap30 plays transcriptional regulatory roles distinct from C/EBPalphap42. Cell Research 2007; 17: 374–383. [DOI] [PubMed] [Google Scholar]
- 62.Pulikkan JA, Dengler V, Peer Zada AA, Kawasaki A, Geletu M, Pasalic Z ef al. Elevated PIN1 expression by C/EBPalpha-p30 blocks C/EBPalpha-induced granulocytic differentiation through c-Jun in AML. Leukemia 2010; 24: 914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Geletu M, Balkhi MY, Peer Zada AA, Christopeit M, Pulikkan JA, Trivedi AK ef al. Target proteins of C/EBPalphap30 in AML: C/EBPalphap30 enhances sumoylation of C/EBPalphap42 via up-regulation of Ubc9. Blood 2007; 110: 3301–3309. [DOI] [PubMed] [Google Scholar]
- 64.Keeshan K, He Y, Wouters BJ, Shestova O, Xu L, Sai H ef al. Tribbles homolog 2 inactivates C/EBPalpha and causes acute myelogenous leukemia. Cancer Cell 2006; 10: 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rishi L, Hannon M, Salome M, Hasemann M, Frank AK, Campos J et al. Regulation of Trib2 by an E2F1-C/EBPalpha feedback loop in AML cell proliferation. Blood 2014; 123: 2389–2400. [DOI] [PubMed] [Google Scholar]
- 66.Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. The N Engl J Med 2004; 350: 1617–1628. [DOI] [PubMed] [Google Scholar]
- 67.Taskesen E, Bullinger L, Corbacioglu A, Sanders MA, Erpelinck CA, Wouters BJ et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood 2011; 117: 2469–2475. [DOI] [PubMed] [Google Scholar]
- 68.Greif PA, Dufour A, Konstandin NP, Ksienzyk B, Zellmeier E, Tizazu B et al. GATA2 zinc finger 1 mutations associated with biallelic CEBPA mutations define a unique genetic entity of acute myeloid leukemia. Blood 2012; 120: 395–403. [DOI] [PubMed] [Google Scholar]
- 69.Lavallee VP, Krosl J, Lemieux S, Boucher G, Gendron P, Pabst C et al. Chemo-genomic interrogation of CEBPA mutated AML reveals recurrent CSF3R mutations and subgroup sensitivity to JAK inhibitors. Blood 2016; 127: 3054–3061. [DOI] [PubMed] [Google Scholar]
- 70.Maxson JE, Ries RE, Wang YC, Gerbing RB, Kolb EA, Thompson SL et al. CSF3R mutations have a high degree of overlap with CEBPA mutations in pediatric AML. Blood 2016; 127: 3094–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reckzeh K, Bereshchenko O, Mead A, Rehn M, Kharazi S, Jacobsen SE et al. Molecular and cellular effects of oncogene cooperation in a genetically accurate AML mouse model. Leukemia 2012; 26: 1527–1536. [DOI] [PubMed] [Google Scholar]
- 72.Pabst T, Eyholzer M, Haefliger S, Schardt J, Mueller BU. Somatic CEBPA mutations are a frequent second event in families with germline CEBPA mutations and familial acute myeloid leukemia. J Clin Oncol 2008; 26: 5088–5093. [DOI] [PubMed] [Google Scholar]
- 73.Tawana K, Wang J, Renneville A, Bodor C, Hills R, Loveday C et al. Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood 2015; 126: 1214–1223. [DOI] [PubMed] [Google Scholar]
- 74.Ohlsson E, Hasemann MS, Willer A, Lauridsen FK, Rapin N, Jendholm J et al. Initiation of MLL-rearranged AML is dependent on C/EBPalpha. J Exp Med 2014; 211: 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Collins C, Wang J, Miao H, Bronstein J, Nawer H, Xu T et al. C/EBPalpha is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis. Proc Natl Acad Sci USA 2014; 111: 9899–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ye M, Zhang H, Yang H, Koche R, Staber PB, Cusan M et al. Hematopoietic differentiation is required for initiation of acute myeloid leukemia. Cell Stem Cell 2015; 17: 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ben-Ami O, Friedman D, Leshkowitz D, Goldenberg D, Orlovsky K, Pencovich N et al. Addiction of t(8;21) and inv(16) acute myeloid leukemia to native RUNX1. Cell Rep 2013; 4: 1131–1143. [DOI] [PubMed] [Google Scholar]
- 78.Mandoli A, Singh AA, Prange KH, Tijchon E, Oerlemans M, Dirks R et al. The hematopoietic transcription factors RUNX1 and ERG prevent aml1-eto oncogene overexpression and onset of the apoptosis program in t(8;21) AMLs. Cell Rep 2016; 17: 2087–2100. [DOI] [PubMed] [Google Scholar]
- 79.Truong BT, Lee YJ, Lodie TA, Park DJ, Perrotti D, Watanabe N et al. CCAAT/Enhancer binding proteins repress the leukemic phenotype of acute myeloid leukemia. Blood 2003; 101: 1141–1148. [DOI] [PubMed] [Google Scholar]
- 80.Lee YJ, Jones LC, Timchenko NA, Perrotti D, Tenen DG, Kogan SC. CCAAT/enhancer binding proteins alpha and epsilon cooperate with all-trans retinoic acid in therapy but differ in their antileukemic activities. Blood 2006; 108: 2416–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koschmieder S, D’Alo F, Radomska H, Schoneich C, Chang JS, Konopleva M et al. CDDO induces granulocytic differentiation of myeloid leukemic blasts through translational up-regulation of p42 CCAAT enhancer binding protein alpha. Blood 2007; 110: 3695–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chang JS, Santhanam R, Trotta R, Neviani P, Eiring AM, Briercheck E et al. High levels of the BCR/ABL oncoprotein are required for the MAPK-hnRNP-E2 dependent suppression of C/EBPalpha-driven myeloid differentiation. Blood 2007; 110: 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liss A, Ooi CH, Zjablovskaja P, Benoukraf T, Radomska HS, Ju C et al. The gene signature in CCAAT-enhancer-binding protein alpha dysfunctional acute myeloid leukemia predicts responsiveness to histone deacetylase inhibitors. Haematologica 2014; 99: 697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, Avellino R et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat Chem Biol 2015; 11: 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]