

SUMMARY
The zinc finger transcription factor SALL4 is highly expressed in embryonic stem cells, downregulated in most adult tissues, but reactivated in many aggressive cancers. This unique expression pattern makes SALL4 an attractive therapeutic target. However, whether SALL4 binds DNA directly to regulate gene expression is unclear, and many of its targets in cancer cells remain elusive. Here, through an unbiased screen of protein binding microarray (PBM) and cleavage under targets and release using nuclease (CUT&RUN) experiments, we identify and validate the DNA binding domain of SALL4 and its consensus binding sequence. Combined with RNA sequencing (RNA-seq) analyses after SALL4 knockdown, we discover hundreds of new SALL4 target genes that it directly regulates in aggressive liver cancer cells, including genes encoding a family of histone 3 lysine 9-specific demethylases (KDMs). Taken together, these results elucidate the mechanism of SALL4 DNA binding and reveal pathways and molecules to target in SALL4-dependent tumors.
In Brief
In this paper, Kong et al. elucidate the DNA binding mechanisms of the transcription factor SALL4 and an epigenetic pathway that it regulates. Due to its important role in driving aggressive cancers, better understanding of SALL4 function will lead to strategies to target this protein in cancer.
Graphical Abstract
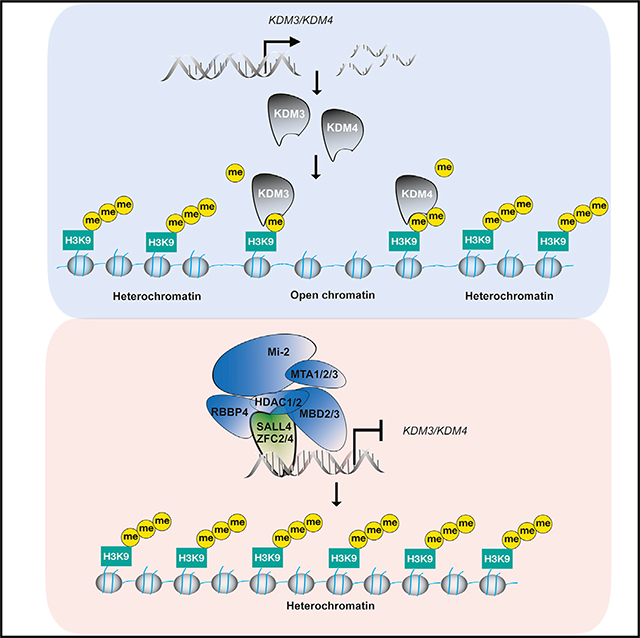
INTRODUCTION
SALL4 is a nuclear factor that plays an important role in embryonic development (Elling et al., 2006; Sakaki-Yumoto et al., 2006; Zhang et al., 2006). Normally, SALL4 is downregulated in most adult tissues except germ cells (Chan et al., 2017; Hobbs et al., 2012; Yamaguchi et al., 2015) and hematopoietic stem cells (Gao et al., 2013b). However, it is dysregulated in hematopoietic pre-leukemias and leukemias (Gao et al., 2013a; Ma et al., 2006). SALL4 is also reactivated in a significant fraction of almost all solid tumors, including lung cancer, endometrial cancer, germ cell tumors, and hepatocellular carcinomas (Li et al., 2015; Miettinen et al., 2014; Yong et al., 2013, 2016). This unique expression pattern demonstrates that SALL4 can be a potential link between pluripotency and cancer and thus targeted therapeutically with limited side effects. Accordingly, SALL4-positive liver cancers share a similar gene expression signature to that of fetal liver tissues and are associated with a more aggressive cancer phenotype, drug resistance, and worse patient survival (Oikawa et al., 2013; Yong et al., 2013).
Despite its important roles in pluripotency and association with certain types of cancers, it is still unclear how SALL4 functions as a transcription factor. SALL4 has two main isoforms, namely, the full-length SALL4A and a spliced variant, SALL4B (Tatetsu et al., 2016). SALL4A has four zinc finger clusters (ZFCs), of which three contain either a pair or a trio of C2H2-type zinc fingers, which are thought to confer nucleic-acid-binding activity (Al-Baradie et al., 2002). However, these clusters are scattered throughout the linear polypeptide sequence, and it is not known which ZFC of SALL4 is responsible for DNA binding. Demonstrating their functional importance, SALL4 ZFC with either missense or frameshift mutations are frequently found in patients with Okihiro Syndrome (Borozdin et al., 2004; Kohlhase et al., 2003; Miertus et al., 2006; Terhal et al., 2006), which is proposed to be a result of impaired SALL4-dependent transcription. Furthermore, immunomodulatory drug (IMiD)-mediated SALL4 degradation through the Cullin RING E3 ubiquitin ligase complex, CUL4-RBX1-DDB1-CRBN (CRL4CRBN), depends on its zinc finger amino acid sequences that show species-specific selectivity (Donovan et al., 2018; Matyskiela et al., 2018). Despite evidence of their functional importance, it is not known whether SALL4 binds DNA through its ZFCs directly or, if so, which ZFC is responsible for binding. It is also unclear what consensus sequence SALL4 prefers. Finally, although SALL4 has been shown to function as a transcriptional repressor by recruiting the nucleosome remodeling and histone deacetylase complex (NuRD) (Gao et al., 2013a; Lu et al., 2009; Yong et al., 2013), many of its target genes and downstream pathways have yet to be elucidated. Its association with the NuRD has led to the hypothesis that SALL4 may play a role in global chromatin regulation. However, its direct involvement with heterochromatin or euchromatin has yet to be determined (Böhm et al., 2007; Kim et al., 2017; Sathyan et al., 2011).
Here, we have used an unbiased screen to discover that SALL4 binds an AT-rich motif through its C-terminal ZFC. These results were further confirmed using a recently developed method of targeted in situ genome-wide profiling (cleavage under targets and release using nuclease [CUT&RUN]) (Skene et al., 2018) to identify true SALL4 binding sites in liver cancer cells. These experiments, coupled with RNA sequencing (RNA-seq) after SALL4 knockdown (KD), allowed us to unveil SALL4’s transcriptional regulation of a family of histone 3 lysine 9-specific demethylases (KDMs 3/4), through which it can regulate the chromatin landscape in cancer cells. Understanding its mechanism as a transcription factor can thus provide new insight of how SALL4-dependent pathways can be targeted in therapeutic approaches.
RESULTS
SALL4 Binds DNA through an AT-Rich Motif
In order to identify the SALL4 consensus binding sequence(s), we used the universal protein binding microarray (PBM) technology (Berger et al., 2006) to conduct an unbiased analysis of all possible DNA sequences to which SALL4 binds. Using this method, we discovered that SALL4 prefers to bind an AT-rich sequence with little degeneracy: AA[A/T]TAT[T/G][A/G][T/A] (Figure 1A; see also Table S1), in which the WTATB in the center of the motif represents the core sequence. In addition, this sequence is highly specific compared with other AT-rich sequences on the array (Figure S1A), and the control FLAG peptide alone does not bind this sequence (Figure S1B). Along its linear polypeptide sequence, the full-length SALL4 protein (SALL4A) has three C2H2-type ZFCs (ZFC2–ZFC4) either in pairs or in a triplet, as well as one C2HC-type zinc finger. Zinc finger motifs are frequently associated with nucleic acid binding (Struhl, 1989); however, it is not known which ZFC of SALL4 is responsible for its DNA binding activity. Therefore, we deleted two of the clusters individually and generated SALL4A mutants that lack either their ZFC2 or ZFC4 domains, hereafter referred to as AΔZFC2 and AΔZFC4, and repeated the PBM experiments. Interestingly, the AΔZFC2 mutant was unaffected compared to the wild-type (WT) SALL4A protein in DNA binding specificity, but the AΔZFC4 mutant was unable to bind the AT-rich consensus motif (Figures 1B and 1C), suggesting that ZFC4 is responsible for sequence recognition. SALL4 also has a shorter isoform resulting from alternative splicing, SALL4B, which shares only the ZFC4 domain with SALL4A. Supporting our finding that ZFC4 is the DNA sequence recognition domain of SALL4, we confirmed that WT SALL4B also binds the specific AT-rich motif, but the SALL4BΔZFC4 mutant lacking this domain does not (Figures S1C and S1D). To validate our PBM results, we performed electrophoretic mobility shift assays (EMSAs) with two randomly picked oligos on the PBM chip, both containing the AT-rich consensus binding site. These assays demonstrated that SALL4 could shift biotinylated oligos containing the WT AT-rich sequence but not those with the probe sequence randomly scrambled (Figure 1D; see also Figure S2A). Furthermore, anti-FLAG or anti-SALL4 antibodies were able to diminish or super-shift the signal, the latter through binding to and slowing down the electrophoretic mobility of the SALL4-DNA complex, while mouse immunoglobulin G (IgG) isotype control could not (Figure 1E, compare lanes 4 and 5 to lane 2; Figure 1F, compare lanes 4–7 to 2). This finding demonstrated that the binding event was highly specific to SALL4 and could not be attributed to any other proteins co-purified with FLAG-tagged SALL4.
Figure 1. Discovery of a Novel SALL4 DNA-Binding Motif.

(A–C) DNA sequence motifs bound by WT SALL4A (A), a mutant lacking the 2nd zinc finger cluster (AΔZFC2) (B), or a mutant lacking the 4th ZFC (AΔZFC4) (C), discovered in universal PBM assays. The color bars above the position weighted matrices indicate the linear structure of SALL4, and the ZFCs are denoted by black ovals.
(D) EMSA showing SALL4A shifts the AT-rich motif-containing oligos (lanes 1–3) but not when the motif is scrambled (lanes 4–6, gel cut for clarity); UC, unlabeled competitor probes.
(E) SALL4-DNA complex is super-shifted by a SALL4 monoclonal antibody (lane 4) but not by mouse IgG isotype control (lane 5); mAb, SALL4 mouse antibody (Santa Cruz EE-30).
(F) EMSA showing that SALL4-DNA complex was reduced or super-shifted in the presence of FLAG or SALL4 antibodies; rAb-1, SALL4 rabbit antibody (Cell Signaling D16H12); rAb-2, SALL4 rabbit antibody (Abcam ab57577). Lanes 8–10 show that the AΔZFC3 mutant binds to the same WT sequence, whereas binding by AΔZFC2 and AΔZFC4 mutants is abrogated. All EMSA reactions contain poly dI:dC competitor to reduce background binding.
(G) Isothermal titration calorimetry (ITC) experiments showing purified SALL4 ZFC4 (amino acids 864–929) binds DNA oligos containing the WT WTATB motif (top) and not when the motif was mutated (bottom). All EMSA and ITC oligo sequences can be found in the Key Resources Table.
Next, we performed EMSA experiments with AΔZFC2 and AΔZFC4 mutants described above along with a SALL4A mutant lacking ZFC3 (AΔZFC3). Although AΔZFC3 can still bind WT oligos (Figure 1F, compare lanes 9 to 2), suggesting ZFC3 is not involved in DNA binding, the AΔZFC4 mutant was unable to bind the oligos (Figure 1F, compare lanes 10 to 2), suggesting that deleting ZFC4 completely abrogated SALL4 DNA binding ability. In addition, deletion of ZFC2 had impaired DNA binding, suggesting that ZFC2 contributes to the ability of ZFC4 to bind to DNA in the context of SALL4A (Figure 1F, compare lanes 8 to 2). These results were consistent with our observation that the smaller SALL4B isoform lacking ZFCs 2 and 3 appears to bind DNA less strongly than the A isoform (Figure S2B, compare lanes 6–9 for SALL4B to lanes 2–5 for SALL4A).
To further validate the SALL4 DNA binding domain, we performed isothermal titration calorimetry (ITC) experiments with purified ZFC4 domain of SALL4 with either WT probes containing the AT-rich motif or mutated probes with only the core motif changed. ITC experiments demonstrated that although SALL4’s C-terminal ZFC4 domain can bind WT probes with a Kd of 6.4 μM for probe 1 and 6.88 μM for probe 2, it cannot bind mutated probes (Figure 1G; see also Figure S2C). Results from these experiments supported our PBM and EMSA findings of a specific SALL4 motif that is AT rich, as well as the importance of SALL ZFC4 domain in DNA sequence recognition and binding.
The SALL4 Motif Is Enriched in CUT&RUN Binding Experiments
Given the in vitro results demonstrating that SALL4 binds to an AT-rich DNA motif, we sought to determine if it binds a similar motif in cells. Previously, we performed SALL4 chromatin immunoprecipitation sequencing (ChIP-seq) experiments in human cells and found they were challenging because SALL4 is located in the chromatin fractions that can be difficult to sonicate (Figure S2D). Furthermore, our previous method of interrogating the chromatin fraction still required cross-linking, although it did not generate many SALL4 peaks or yield a SALL4 motif (Liu et al., 2018a). Here, we took advantage of the availability of a highly specific antibody against human SALL4 (Cell Signaling Technology, clone D16H12, lot 2) and performed the CUT&RUN assay (Skene et al., 2018), which is an in situ profiling of protein-DNA binding that eliminates the cross-linking step and generates reads with low background and more precise localization.
We chose SNU398 liver cancer cells to identify endogenous SALL4 binding sites genome wide because (1) these cells have high SALL4 expression compared with other cancer cells (Figure S2E) and SALL4 ChIP data are available in these cells (Liu et al., 2018a); and (2) SALL4 is required for the viability of a large fraction of hepatocellular carcinoma cells, while serving as a biomarker for worse prognosis in liver cancer (Oikawa et al., 2013; Yong et al., 2013).
Three separate CUT&RUN experiments using SNU398 cell nuclei revealed SALL4 binds over 11,200 common peaks genome wide, at least 2-fold above isotype control IgG peak enrichment (representative track shown in Figure 2A; see also Table S2). Furthermore, SALL4 peaks were distributed 33.8% intergenic, 30% intronic, and 21% within the proximal promoter (Figure 2B) and can be annotated near 4,364 genes. On average, the reads are about 80–100 bp long, allowing for better identification of SALL4 binding motif. Consequently, when a de novo motif search was performed on SALL4 peaks, we observed a significant enrichment of the AT-rich motif with the core WTATB motif that was independently identified from PBM experiments (Figure 2C; see also Figure S3A). This motif was present in similar percentages of peaks in all three SALL4 CUT&RUN replicates through both de novo and direct searches using motif 2 (Figure 2D).
Figure 2. SALL4 CUT&RUN Showing Its Binding Genome wide in Liver Cancer Cells.

(A) Representative genomic tracks of three SALL4 CUT&RUN replicates (rep) and their isotype rabbit IgG control experiments; scale is 0–25.
(B) The genomic distribution of SALL4 CUT&RUN peaks.
(C) The top five HOMER motifs from de novo analysis of three CUT&RUN reps with their respective p values, resulting in the two composite motifs shown on right.
(D) Bar graph showing the percentage of peaks containing Motif 2 in de novo (black bars) and direct (gray bars) analyses from SALL4 CUT&RUN reps.
Discovery of New SALL4 Gene Targets in Liver Cancer Cells
The SALL4 target genes in liver cancer that are associated with transcriptional and chromatin regulation are not well defined. Furthermore, it has been shown that SALL4 can act like a transcriptional activator and/or repressor dependent on the cellular context (Gao et al., 2013a; Li et al., 2013; Lu et al., 2009; Ma et al., 2006; Young et al., 2014; Zhang et al., 2006). To understand how SALL4 DNA binding affects the expression of its downstream target genes, we performed RNA-seq at 72 h after SALL4 KD (Figures S3B and S3C) in biological duplicates. We then compared the RNA-seq results to our CUT&RUN peaks and found that among 2,695 significantly differentially expressed genes (red circles in Figure 3A; see also Table S3), 430 genes had annotated SALL4 CUT&RUN peaks (totaling 1,192 peaks annotated by their proximity to the nearest transcriptional start site; Figure 3B; see also Table S4), suggesting they were directly regulated by SALL4. When de novo motif search was performed on the peaks, the core WTATB motif was among the top hits (Figure S3D).
Figure 3. RNA-seq Data Revealed Direct SALL4 Target Genes.

(A) Volcano plot showing genes that are down- or upregulated significantly after SALL4 KD with log2 fold change (FC) represented on the x axis; red circles denote differentially expressed genes with false discovery rate (FDR) of <0.05.
(B) Number of differentially expressed genes after SALL4 KD (2,695) as well as those with annotated SALL4 peaks nearby (430) (Tables S3 and S4).
(C) Bar graph representing number of up- and downregulated genes after SALL4 KD (Table S3) and their Gene Ontology (GO) molecular pathway analysis focusing on GO 0140110.
(D) Volcano plot from (A) with genes encoding KDM proteins labeled; green circles denote genes containing SALL4 CUT&RUN peaks; the size of the circles corresponds to log2FC in expression.
(E) Quantitative real-time PCR analysis of two SALL4 direct targets 40 h after SALL4 KD, summarized from either 3 or 4 independent experiments (primer sequences found in Table S5); SCR, scrambled shRNA control.
(F) EMSA showing that SALL4 binds KDM3A promoter region containing the WT AT-rich motif but not the mutated sequence (Key Resources Table)
For genes with SALL4 CUT&RUN peaks and that are differentially expressed after KD, 240 were upregulated (repressed by SALL4) and 190 were downregulated (activated by SALL4). When Gene Ontology analysis was performed, we found that one of the most differentially represented molecular pathways that was repressed by SALL4 was transcription regulation, accounting for 11.1% of the upregulated genes after SALL4 KD, compared to 3.1% of downregulated genes after SALL4 KD (Figure 3C; see also Table S4, columns G–I). These categories of SALL4-repressed genes included those encoding chromatin modifiers such as KDM3A and several family of transcription factors such as Forkhead (FOXA1 and FOXO1), BCL (BCL11A, BCL11B, and BCL6), and KLF (KLF10 and KLF12), as well as TBX5. Many of these targets were not previously reported in liver cancer cells (Liu et al., 2018a), which demonstrates the sensitivity of the CUT&RUN technique. Taken together, these genome-wide binding assays have confirmed the bona fide SALL4 motif we found in vitro.
SALL4 Regulates the Expression of Histone Demethylases
SALL4 can interact with the NuRD (Lu et al., 2009; Figure S3E), and we have previously shown that blocking SALL4’s transcriptional repressive function by interrupting its interaction with NuRD was an effective therapeutic approach in liver cancer cells (Gao et al., 2013a; Liu et al., 2018a). Furthermore, SALL4 has been shown to localize in and regulate chromatin in cells (Böhm et al., 2007; Chan et al., 2017; Hobbs et al., 2012; Kim et al., 2017; Xiong et al., 2016). Therefore, we focused on chromatin-associated genes that were upregulated after SALL4 KD, as well as potential direct targets identified by CUT&RUN. One of the upregulated genes, KDM3A, encoded a histone 3 lysine 9 (H3K9)-specific demethylase (Gray et al., 2005), as well as several other members of the KDM demethylase family (Figure 3D; see also Figures S4A and S4B). We validated the RNA-seq data by performing qPCR after SALL4 KD (Figure 3E), and we validated the binding by ChIP-qPCR with primers targeting the SALL4 binding site at the KDM3A promoter (Figure S4C). Then, we used the KDM3A peak to design probes for EMSA experiments. We found that SALL4A binds double-stranded oligos containing the SALL4 motif, but not when this motif was mutated (Figure 3F).
In order to confirm the importance of SALL4 binding on KDM3A gene expression, we further used CRISPR-Cas9 to disrupt one of the SALL4 motifs in its binding site in the KDM3A promoter. After sorting cells that had successful CRISPR targeting through GFP expression present on the lentiviral vector, we collected mRNA and performed qPCR analysis. We observed a two-fold increase in expression of KDM3A in cells with deleted SALL4 binding site compared to mock-transfected control cells (Figure S4D).
Our combined analyses of RNA-seq and CUT&RUN data, in addition to EMSA and gene editing experiments, demonstrated that SALL4 directly regulates a subset of chromatin modifying genes in cancer cells, raising the possibility that SALL4 could regulate the global chromatin landscape of cells.
SALL4 and Heterochromatin
It has been shown previously that SALL4 KD in liver cancer cells led to cell death (Yong et al., 2013), and we confirmed that cells could not survive after prolonged loss of SALL4 expression by KD by two different short hairpin RNAs (shRNAs) (Figure S4E). However, at an earlier time point (40 h post-viral transduction), SALL4 KD was highly efficient (Figure S4E, and detected by GFP expression from the shRNA lentiviral plasmid), and yet, no significant cell death was observed, as GFP and DAPI double-positive cells did not increase until 72 h post-transduction (Figure S4F). This observation allows a window to assess true SALL4-dependent cellular functions with fewer potential secondary effects.
Because of SALL4’s known association with the NuRD and its regulation of the KDM3/4 pathway (Figure 3D), we sought to ascertain whether there were any changes in the global chromatin landscape. We first confirmed our RNA-seq data by showing that KDM3A protein was upregulated upon SALL44 KD at the early time point of 40 h after transduction (Figure 4A). Given that KDM3A is known to demethylate H3K9me2/3 (Gray et al., 2005), we found that although total histone 3 levels were unchanged, there was a marked reduction of global H3K9me2/3 levels after SALL4 KD (Figure 4A).
Figure 4. SALL4 and Heterochromatin.

(A) Western blotting of SNU398 liver cancer cell lysates at 40 h after SALL4 KD with gel cut for clarity; molecular weight marker is indicated on the right (left panel); Kd, kilodalton; summary of western blot densitometry from two separate SALL4 KD experiments of H3K9me2/3 protein expression (black bars) and KDM3A expression (gray bars) (right panel).
(B) Western blotting of SNU398 cell lysates collected after either control DMSO or pomalidomide (Pom; 10 μM) treatments at the indicated time points.
(C) Immunofluorescence staining of HP1 of SNU398 liver cancer cells transduced with SCR control or two shRNAs against SALL4; white scale bar denotes 33 μm; DAPI, 4’,6-diamidino-2-phenylindole DNA stain.
In addition to shRNA-mediated KD of SALL4 expression, we used a second approach to pharmacologically delete SALL4. It has been shown that as a neo-substrate of CRL4CRBN, SALL4 can be induced to degrade by treatment with IMiDs, such as thalidomide (Donovan et al., 2018; Matyskiela et al., 2018). Therefore, we treated SNU398 liver cancer cells with a thalidomide analog, pomalidomide, for 6, 12, or 24 h and collected protein lysates. We observed robust SALL4 degradation as soon as 6–12 h after treatment, at which points H3K9me2/3 marks were already diminished significantly, whereas total histone 3 levels remained unchanged (Figure 4B). We further confirmed SALL4’s importance in heterochromatin regulation by performing immunofluorescence for HP1α protein after SALL4 KD. HP1 binds H3K9me2/3 and is a hallmark of heterochromatin (Zeng et al., 2010). Again, using two shRNAs targeting SALL4 (Figure S4G), we observed substantial reduction of HP1 staining in the nuclei of KD cells compared to SCR control shRNA-targeted cells (Figure 4C).
DISCUSSION
Although SALL4 has been referred to as a transcriptional regulator, biochemical evidence of direct DNA binding has been scant. Its ZFCs have been individually tested in EMSA assays and shown to preferentially bind 5-hydroxymethylcytosine, and thus, SALL4 was proposed to stabilize the Tet2-DNA interaction during DNA demethylation in murine embryonic stem cells (ESCs) (Xiong et al., 2016). However, the role of SALL4 DNA binding to regulate specific gene expression is still unclear. Here, we discovered an AT-rich SALL4 binding motif by PBM assays and confirmed this finding by EMSA and ITC assays. In addition, through biochemical approaches, we demonstrated that the fourth ZFC of SALL4 is responsible for DNA recognition and binding. Our DNA motif was further supported by CUT&RUN assays, which resulted in much smaller DNA fragments than previous ChIP-seq experiments, allowing for better de novo motif discovery. In all, we used four separate biochemical or cellular assays to demonstrate SALL4 binds a unique AT-rich motif through its fourth ZFC.
Our novel CUT&RUN results in cancer cells, coupled with RNA-seq data from SALL4 KD in the same cells, identified hundreds of genes that SALL4 directly regulates. Of note, many of these targets are involved in transcriptional regulation or chromatin modification. We found that SALL4 binds and represses members of the KDM family of genes, resulting in changes in the methylation status of H3K9 and chromatin, as assessed by staining with HP1. Our identification of the link between SALL4 KD and decreased heterochromatin marks presents a previously undescribed potential mechanism by which SALL4 acts as a regulator of global chromatin landscape. One of the few adult tissues in which SALL4 is expressed is the spermatogonial progenitor cells, where it antagonizes PLZF transcription factor function to drive cellular differentiation (Hobbs et al., 2012). Interestingly, KDM3A is highly expressed in post-meiotic male germ cells to regulate expression of protamine in spermatids (Okada et al., 2007). It remains to be seen whether SALL4 directly represses KDM3A in the testes, which would suggest that the SALL4-KDM3A connection discovered here is applicable to other progenitor cell tissues. KDM3A has also been shown to be important in maintaining self-renewal property of ESCs (Kuroki et al., 2018). Therefore, the SALL4/KDM pathway in embryonic development, spermatogenesis, and adult cancer should be examined more closely, wherein SALL4 expression is potentially important to prevent premature overexpression of KDM3A. An unbiased shRNA screen found that KDM3A promotes epithelial cell apoptosis through activating pro-apoptotic BNIP3 genes (Pedanou et al., 2016). Therefore, SALL4 regulation of the KDM pathway may contribute to its regulation of both heterochromatin and cell death.
The recent discoveries of SALL4 as a IMiD-dependent neo-substrate of CRL4CRBN, which promotes its degradation, have demonstrated that SALL4A ZFC2 can be targeted (Donovan et al., 2018; Matyskiela et al., 2018). In contrast, SALL4B, lacking this ZFC, is not affected. We have observed that thalidomide does not affect growth of SALL4+ cancer cells, suggesting that SALL4B is required for SALL4-mediated tumorigenesis, further supported by the observation that mice overexpressing SALL4B develop acute myeloid leukemia (Ma et al., 2006). Because SALL4B lacks the degron-containing ZFC2, it is imperative to understand how we can target its ZFC4, which is shared by both isoforms, in order to degrade this protein completely. Knowing the consensus DNA sequence SALL4 ZFC4 prefers will facilitate solving the structure of this domain by X-ray crystallography because DNA-bound SALL4 may be at its most stable conformation.
Overall, our findings contribute to further understanding SALL4 function in cancer cells and the underlying molecular mechanisms, thereby uncovering novel therapeutic approaches in SALL4-positive cancers.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
The lead contact for this manuscript is Daniel G.Tenen (daniel.tenen@nus.edu.sg)
Materials availability
Further information and requests for resources such as recombinant DNA plasmids generated in this study should be directed to and will be fulfilled by the lead contact
Data and code availability
The datasets and code utilized in this study are available at GEO: GSE136332 and on GitHub: https://github.com/mbassalbioinformatics/CnRAP.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
SNU398 and SNU387 hepatocellular carcinoma cells (ATCC) were cultured in RPMI media with 10% fetal bovine serum (FBS, GIBCO). HEK293T and HeLa cells were cultured in DMEM media with 10% FBS.
METHOD DETAILS
Protein binding microarray (PBM)
SALL4 proteins were purified using M2 FLAG agarose beads (Sigma) from nuclear extracts of HEK293T cells that were transfected with FLAG-tagged SALL4. “Universal” all 10-mer double-stranded oligonucleotide arrays in 8 × 60K, GSE format (Agilent Technologies; AMADID #030236) were used to perform PBM experiments following previously described experimental protocols (Berger and Bulyk, 2009; Berger et al., 2006). Each SALL4A WT or mutant protein was assayed in PBM at 600nM. The PBM scan images were obtained using a GenePix 4000A Microarray Scanner (Molecular Devices).
Electrophoretic mobility shift assay
Biotinylated probes were designed based on PBM data and obtained from Integrated DNA Technology. Oligonucleotide annealing was performed by heating mixed oligonucleotides to 95 degrees for 5 min, and slowly cooled in a water bath (initially 70 degrees) overnight. SALL4 proteins (purified as described for PBM) were premixed with unlabeled probes or appropriate antibodies for 20 min at 4 degrees, then mixed with labeled probes and incubated for 20 min at room temperature. Binding buffers B1 and B2 containing poly dIdC blocking DNA, binding buffer C1, and stabilization buffer D (Active Motif) were used. Free DNA and protein-DNA complexes were run for 2 h in the cold room in 6% polyacrylamide gels in tris-borate-EDTA, then transferred onto nylon membranes (0.45um pore), and visualized via streptavidin-HRP according to manufacturer’s instructions (Thermo Fisher). 1ug of antibody against SALL4 (Santa Cruz EE-30) or isotype control IgG (Santa Cruz) was pre-incubated with the proteins, before incubation with the DNA probes. All EMSA probe sequences can be found in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Sall4 | Santa Cruz | Cat# EE-30; RRID: AB_1129262 |
| SALL4 | Abcam | Cat# Ab29112; RRID: AB_777810 |
| SALL4 | Cell signaling | Cat# 8459 |
| Normal mouse IgG | Santa Cruz | Cat# SC-2025; RRID: AB_737182 |
| Normal Rabbit IgG | Abcam | Cat# Ab171870; RRID: AB_2687657 |
| Actin | Sigma | Cat# A2066; RRID: AB_476693 |
| H3K9Me3 (D4W1U) | Cell Signaling | Cat# 13969: RRID: AB_2798355 |
| Histone 3 | Abcam | Cat# Ab1791: RRID: AB_302613 |
| FLAG (M2) | Sigma | Cat# F3165: RRID: AB_259529 |
| HDAC1 | Cell Signaling | Cat# 2062 |
| HP1 | Abcam | Cat# Ab77256: RRID: AB_1523784 |
| KDM3A | Abcam | Cat# Ab80598 |
| Bacterial and Virus Strains | ||
| BL21 (DE3) | Novagen | Cat# 69450 |
| Chemicals | ||
| EMSA Buffer B1 | Active Motif | Cat# 37480 |
| EMSA Buffer B2 | Active Motif | Cat# 37481 |
| EMSA Buffer C1 | Active Motif | Cat# 37484 |
| EMSA Buffer D | Active Motif | Cat# 37488 |
| FBS | Sigma | Cat# F2442 |
| RPMI | Thermo Fisher | Cat# 11875119 |
| DMEM | Thermo Fisher | Cat# 11965118 |
| OptiMEM | Thermo Fisher | Cat# 31985070 |
| Trypsin-EDTA (0.25%) | Thermo Fisher | Cat# 25200114 |
| iScript | BIO-RAD | Cat# 1708891 |
| IQ SYBR Green supermix | BIO-RAD | Cat# 1708882 |
| FLAG-M2 beads | Sigma | Cat# A2220 |
| Glutathione Sepharose | GE Healthcare | Cat# GE17-0756-01 |
| Concanavalin A beads | Bangs Laboratories | Cat# BP531 |
| proteinA-micrococcal nuclease | This paper | |
| DAPI | Sigma | Cat# 5087410001 |
| Vectashield antifade mounting medium | Vector Laboratories | Cat# H-1000-10 |
| TransIT-LT1 | Mirus | Cat# MIR2300 |
| Digitonin | Sigma | Cat# D141 |
| Critical Commercial Assays | ||
| TruSeq stranded mRNA Kit | Illumina | Cat# RS-122-21001 |
| NEBNext Ultra II DNA Library prep kit | NEB | Cat# M0541 |
| NEBNext Multiplex Oligos for Illumina (index primers set 1 | NEB | Cat# E7335 |
| LightShift Chemiluminescent EMSA kit | Thermo Fisher | Cat# 20148 |
| Deposited Data | ||
| CUT&RUN and RNA-seq data | Gene Expression Omnibus | GSE136332 |
| Experimental Models: Organisms/Strains | ||
| SNU-398 | ATCC | Cat# CRL-2233 |
| SNU-387 | ATCC | Cat# CRL-2237 |
| HEK293T | ATCC | Cat# CRL-1573 |
| HeLa | ATCC | Cat# CCL-2 |
| Oligonucleotides | ||
| PBM EMSA WT oligo 1: GTGAAAAAAAATATTAACGTACAGCGGGGAGGCGGC | This paper | N/A |
| PBM EMSA Mutant oligo 1: AAAAGCGCAGGCATTAAAGGTATACGTGTGAAAAGA | This paper | N/A |
| PBM EMSA WT oligo 2: TTAAGCAGAAATATTACGGTCTCCGGATTTGGCGCT | This paper | N/A |
| PBM EMSA Mutant oligo 2: ATTTACAACAGGCCAGAAGTTCTTTGGCTTATCCAT | This paper | N/A |
| ITC WT oligo 1: GAGTTATTAATG | This paper | N/A |
| ITC Mutant oligo 1: GAGTCGCTAATG | This paper | N/A |
| ITC WT oligo 2: GATAAATATTTG | This paper | N/A |
| ITC Mutant oligo 2: GATAAACGCTTG | This paper | N/A |
| KDM3A EMSA WT oligo: TCTTCATTTATCCTTCAAAA | This paper | N/A |
| KDM3A EMSA Mutant oligo: TCTTCTTTTAACCTTCAAAA | This paper | N/A |
| RT-qPCR, ChIP-qPCR primers, shRNA/CRISPR target regions | This paper | Table S5 |
| Software and Algorithms | ||
| GenePix Pro v7.2 | Molecular Devices | https://mdc.custhelp.com/app/answers/detail/a_id/18792/~/genepix%E2%AE-pro-7-microarray-acquisition-%26-analysis-software-download-page |
| Masliner | Dudley et al., 2002 Berger et al., 2006 | http://arep.med.harvard.edu/masliner/supplement.htm |
| Universal PBM Analysis Suite | Berger and Bulyk, 2009 | http://thebrain.bwh.harvard.edu/PBMAnalysisSuite/indexSep2017.html |
| Enologos | Dobin et al., 2013 | http://www.benoslab.pitt.edu/cgi-bin/enologos/enologos.cgi |
| Trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| BWA | Li et al., 2009 | http://bio-bwa.sourceforge.net/ |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Stampy | Lunter and Goodson, 2011 | https://www.rdm.ox.ac.uk/research/lunter-group/lunter-group/stampy |
| deepTools | Ramírez et al., 2014 | https://deeptools.readthedocs.io/en/develop/ |
| Bedtools | Quinlan and Hall, 2010; Kent et al., 2010 | https://bedtools.readthedocs.io/en/latest/ |
| SEACR | Meers et al., 2019 | https://github.com/FredHutch/SEACR |
| ChIPSeeker | Yu et al., 2015 | https://guangchuangyu.github.io/software/ChIPseeker/ |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| BBMap | https://sourceforge.net/projects/bbmap/ | |
| BamCoverage | Ramírez et al., 2014 | https://deeptools.readthedocs.io/en/develop/content/tools/bamCoverage.html |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| RseQC | Wang et al., 2012 | http://rseqc.sourceforge.net/ |
| htseq-count | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/ |
| EnhancedVolcano | https://github.com/kevinblighe/EnhancedVolcano | |
| ImageJ | https://imagej.nih.gov/ij/ | |
| MEME-ChIP | Bailey et al., 2009 | http://meme-suite.org/tools/meme-chip |
Isothermal titration calorimetry (ITC)
The DNA fragments encoding SALL4 ZFC4 (residues 864–929) cloned into modified pGEX-4T1 vector (GE Healthcare) with a tobacco etch virus (TEV) cleavage site after the GST tag. All the proteins were expressed in Escherichia coli BL21 (DE3) cells (Novagen), purified using glutathione Sepharose (GE healthcare), and cleaved by TEV protease overnight at 4 degrees to remove the GST tag. The cleaved protein was further purified by size-exclusion chromatography on a HiLoad 16/60 Superdex 75 column (GE healthcare), dialyzed with buffer C (20 mM Tris-HCl (pH 7.5), 150 mM NaCl), and concentrated for subsequent experiments. ITC assays were carried out on a Microcal PEAQ-ITC instrument (Malvern) at 25 degrees. The titration protocol consisted of a single initial injection of 1 μl, followed by 19 injections of 2 μl of the protein (concentration: 0.5–1mM) into the sample cell containing double stranded DNA oligos (concentration: 20 μM).
Cleavage under targets and release using nucleases (CUT&RUN)
A detailed protocol can be found on protocol.io from the Henikoff lab (https://doi.org/10.17504/protocols.io.mgjc3un; Skene et al., 2018). Briefly, 2 million cell nuclei were immobilized on Concanavalin A beads after washing. SALL4 (CST D16H12) or H3K9me2/3 (Cell Signaling D4W1U) antibodies, or normal rabbit IgG (Cell Signaling DA1E) were incubated with the nuclei overnight in the presence of 0.02% digitonin at 4 degrees. The next day, 700ng/mL of proteinA-micrococcal nuclease (pA-Mnase purified in house with vector from Addgene 86973, protocol from Schmid et al., 2004) were incubated with the nuclei at 4 degrees for an hour. After washing, the tubes were placed in heat blocks on ice set to 0 degrees, CaCl2 (1mM) was added and incubated for 30 min before 2× Stop buffer containing EDTA was added. DNA was eluted by heat and high-speed spin, then phenol-chloroform extracted. Qubit was used to quantify purified DNA and Bioanalyzer (2100) traces were run to determine the size of the cleaved products. NEBNext Ultra II DNA library prep kit (NEB E7645) was used to make the libraries according to Liu et al.’s protocol, outlined on protocols.io (https://doi.org/10.17504/protocols.io.wvgfe3w; Liu et al., 2018b). Pair-end (42bp) Illumina sequencing was performed on the bar-coded and amplified libraries.
Lentivirus-mediated gene expression knockdown and western blotting
Two shRNAs targeting SALL4 were previously described (Gao et al., 2013b): shSALL4–1 and shSALL4–2 (Table S5); both target exon 2 of SALL4 mRNA. A pLL.3 vector containing the shRNA and GFP were transfected into HEK293T cells using TransIT-Lenti (Mirus). Viruses were collected at 48 h and 72 h post-transfection. After cell debris was filtered out with 45micron syringe filters, viral supernatants were spun at 20,000 RPM at 4 degrees for 2 h, and re-suspended in RPMI media (GIBCO). The viral titer was calculated by serial dilution and transduction of HeLa cells. MOI of 2 were used for these cells. Transduction was performed with polybrene (8ug/mL) and spinning at 70 g at room temperature for an hour. SNU398 cells were >90% GFP positive starting at 40 h post-transduction. At either 40 or 72 h after transduction, cells were either counted with trypan blue exclusion method or collected and stained with DAPI nuclear stain. GFP and DAPI+ dead cells were counted using a Canto II flow cytometer (BD Biosciences). Western blotting was performed by running the collected cell lysates in a 4%–20% gradient tris-glycine SDS-PAGE gel, transferred onto methylcellulose, and blotted with antibodies raised against SALL4 (Santa Cruz, clone EE30), Actin (Sigma, clone AC-74), total histone 3 (Cell Signaling Technology, clone D1H2), di/tri-methyl histone 3 lysine 9 (CST, D4W1U), tri-methyl histone 3 lysine 9 (EMD/Millipore, catalog 07–442), or KDM3A (Abcam, catalog ab80598).
RNA-seq
RNA was extracted by Trizol in three biological replicates 72 h after SALL4 KD and libraries were made following manufacturer’s instructions (Illumina). Pair-end Illumina sequencing was performed on the bar-coded and amplified libraries.
Co-immunoprecipitation
Cells were lysed with RIPA lysis buffer and sonicated with a microtip sonicator at 90% duty, 15 bursts. The lysates were incubated with SALL4 antibody (same as ChIP) over night at 4 degrees, followed by 6hr incubation with protein A/G beads at 4 degrees. After washing, the beads were boiled in 2× SDS sample buffer containing beta-mercaptoethanol and the supernatant was separated in Tris-glycine gels. Western blotting was performed with SALL4 antibody (EE30, Santa Cruz Biotechnology) and HDAC1/2 antibodies (Cell Signaling 8349).
Immunofluorescence staining
40 h after SALL4 KD with shRNA 1 and 2, SNU398 liver cancer cells were fixed with 4% paraformaldehyde in PBS for 15 min. Fixed cells were permeabilized with 0.1% Triton-x in PBS, washed with PBS containing 0.1% Tween-20, and blocked with 3% bovine serum albumin (BSA). Primary antibodies against HP1 (Abcam ab77256) or SALL4 (Abcam ab57577) were incubated with cells overnight at 4 degrees, in PBS containing 0.3% BSA and 0.1% Tween-20. The next day, cells were washed with Tween-20/PBS, incubated with secondary anti-goat or anti-rabbit antibodies conjugated to Alexa Fluorophore 594 at room temperature for an hour. Cells were washed and stained with DAPI DNA stain for 5 min and mounted with Vectashield mounting medium. Images were taken with a confocal microscope (Zeiss LSM710) with the same settings for all samples.
QUANTIFICATION AND STATISTICAL ANALYSIS
PBM analysis
PBM image data were processed using GenePix Pro v7.2 to obtain signal intensity data for each spot. The data were then further processed by using Masliner software (v1.02) (Berger et al., 2006; Dudley et al., 2002) to combine scans from different intensity settings, increasing the effective dynamic range of the signal intensity values. If a dataset had any negative background-subtracted intensity (BSI) values (which can occur if the region surrounding a spot is brighter than the spot itself), consistent pseudocounts were added to all BSI values such that they all became nonnegative. All BSI values were normalized using the software for spatial de-trending providing in the Universal PBM Analysis Suite (Berger and Bulyk, 2009). Motifs were derived using the Seed-and-Wobble algorithm, and Enologos was used to generate logos from PWMs, as previously described (Berger and Bulyk, 2009; Berger et al., 2006).
ITC analysis
Data obtained from ITC assays were fitted to one-site binding model via the MicroCal PEAQ-ITC analysis software provided by the manufacturer and the oligonucleotide sequences can be found in the Key Resources Table.
CUT&RUN analysis
Detailed data analysis combining Henikoff (Skene et al., 2018) and Orkin (Liu et al., 2018b) labs’ pipelines can be found on github (https://github.com/mbassalbioinformatics/CnRAP). Briefly, raw fastq files were trimmed with Trimmomatic v0.36 (Bolger et al., 2014) in pair-end mode. Next, the kseq trimmer developed by the Orkin lab was run on each fastq file. BWA (v0.7.17-r1188) (Li and Durbin, 2009) was first run in “aln” mode on a masked hg38 genome downloaded form UCSC to create *.sai files; then BWA was run in “sampe” mode with the flag “-n 20” on the *.sai files. Afterward, Stampy (v1.0.32) (Lunter and Goodson, 2011) was in “—sensitive” mode. Next, using SAMtools (v1.5) (Li et al., 2009), bam files were sorted (“sort - | 0 –O bam”), had read pair mates fixed (“fixmate”), and indexed (“index”). Bam coverage maps were generated using bamCoverage from the deepTools suite (v2.5.7) (Ramírez et al., 2014). The same procedure was run to align fastq files to a masked Saccharomyces Cerevisiae v3 (sacCer3) genome for spike-in control DNA, also downloaded form UCSC. A normalization factor was determined for each hg38 aligned replicate based on the corresponding number of proper-pairs aligned to the sacCer3 genome, as recommended in the Henikoff pipeline, this was calculated as follow: normalization factor = 10,000,000#”proper_pairs’2. Next, from the hg38 aligned bam files, “proper-paired” reads were extracted using SAMtools with the output piped into Bedtools (Quinlan and Hall, 2010), producing BED files of reads that have been normalized to the number of reds aligned to the sacCer3 genome. BedGraphs of these files were generated as intermediary fiels to facilitate generation of BigWig coverage maps using the bedGraphToBigWig tool from UCSC (v4) (Kent et al., 2010). For peak calling, the recently developed SEACR (v.1.1) (Meers et al., 2019)was utilized and run in “relaxed’ mode to produce peak files as the BED files used were already normalized to the number of yeast spike-in reads. Subsequent peak file columns were re-arranged to facilitate motif discovery using HOMER (v4.10) (Heinz et al., 2010). Peaks were annotate using the R package ChIPSeeker (v.1.20.0) (Yu et al., 2015). Overlapping peak subsets within 3kb of each other were generated using mergePeaks.py from the HOMER suite (Heinz et al., 2010). Peak positions for those that are common to all three replicates and at least 2-fold above IgG control can be found in Table S2. Heatmap of SALL4-bound genes encoding lysine demethylases was generated using the R package pheatmap (https://cran.r-project.org/web/packages/pheatmap/index.html, Raivo Kolde. Pheatmap under R 3.6.2.
RNA-seq analysis
Raw fastq files had optical duplicates removed using clumpify form BBMap (https://sourceforge.net/projects/bbmap/). Next, adapt trimming was performed using BBDuk (from BBMap) and reads were trimmed using trimmomatic (Bolger et al., 2014). After read cleanup, reads were aligned to hg38 genome using STAR (Dobin et al., 2013). BamCoverage (Ramírez et al., 2014) maps were generated using default parameters and read distributions were calculated using read_distribution.py from the RseQC suite of tools (Wang et al., 2012). Counts tables were generated using htseq-count (Anders et al., 2015). The fold change was plotted as a volcano plot using EnhancedVolcano (https://github.com/kevinblighe/EnhancedVolcano) with FDR cut-off of 0.05. Quantitative real-time PCR for selected targets were performed with primer sequences found Table S5.
Integration of CUT&RUN and RNA-seq data were accomplished by first annotating the CUT&RUN peaks based on their genomic location with respect to their nearest transcriptional start site (TSS). Then we looked for matching gene names between our annotated CUT&RUN peaks and the list of differentially expressed genes from the RNA-seq analysis.
Motif search
De novo motif search was performed using both HOMER (Heinz et al., 2010) with the flags “-size given –mask –S 50” and MEME-ChIP (Bailey et al., 2009) with the flags “-drene-m 50 –meme-nmotifs 50.” For directed motif search wherein we searched for the abundance of our motif of interest (Motif 2 in Figure 3C) in called CUT&RUN peaks, HOMER was utilized with a calculated motif position weight matrix and the flags “-find pos_weight_matrix.motif.”
Supplementary Material
Highlights.
Transcription factor SALL4 binds an AT-rich DNA sequence
The C-terminal zinc finger cluster of SALL4 is responsible for DNA binding
SALL4 negatively regulates genes encoding histone 3 lysine 9-specific demethylases
SALL4 regulates heterochromatin through its repression of KDM genes
ACKNOWLEDGMENTS
This work was supported by a Pathology Research Microgrant from Brigham and Women’s Hospital. In addition, this work was supported by the National Institutes of Health (NIH) grant number T32 HL066987 to N.R.K., grant numbers P01 CA66996 and HL131477 to D.G.T., and grant number R01 HG003985 to M.L.B. This work was further supported by the National Cancer Institute grant number R35 CA197697 to D.G.T.; the National Heart, Lung, and Blood Institute grant number P01 HL095489 to L.C.; the Leukemia and Lymphoma Society grant number P-TRP-5855-15 to L.C.; and Xiu Research Fund to L.C. This work was also supported by the Singapore Ministry of Health’s National Medical Research Council under its Singapore Translational Research (STaR) Investigator Award and by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence Initiative.
We thank Bee Hui Liu for sharing data, Steve Gisselbrecht for assistance with PBM data analysis, and Yanzhou Zhang for discussion of the paper.
Footnotes
DECLARATIONS OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108574.
REFERENCES
- Al-Baradie R, Yamada K, St Hilaire C, Chan W-M, Andrews C, McIntosh N, Nakano M, Martonyi EJ, Raymond WR, Okumura S, et al. (2002). Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet 71, 1195–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, and Bulyk ML (2009). Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat. Protoc 4, 393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW 3rd, and Bulyk ML (2006). Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol 24, 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm J, Kaiser FJ, Borozdin W, Depping R, and Kohlhase J (2007). Synergistic cooperation of Sall4 and Cyclin D1 in transcriptional repression. Biochem. Biophys. Res. Commun 356, 773–779. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borozdin W, Wright MJ, Hennekam RCM, Hannibal MC, Crow YJ, Neumann TE, and Kohlhase J (2004). Novel mutations in the gene SALL4 provide further evidence for acro-renal-ocular and Okihiro syndromes being allelic entities, and extend the phenotypic spectrum. J. Med. Genet 41, e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A-L, La HM, Legrand JMD, Mäkelä J-A, Eichenlaub M, De Seram M, Ramialison M, and Hobbs RM (2017). Germline Stem Cell Activity Is Sustained by SALL4-Dependent Silencing of Distinct Tumor Suppressor Genes. Stem Cell Reports 9, 956–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan KA, An J, Nowak RP, Yuan JC, Fink EC, Berry BC, Ebert BL, and Fischer ES (2018). Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. eLife 7, e38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley AM, Aach J, Steffen MA, and Church GM (2002). Measuring absolute expression with microarrays with a calibrated reference sample and an extended signal intensity range. Proc. Natl. Acad. Sci. USA 99, 7554–7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elling U, Klasen C, Eisenberger T, Anlag K, and Treier M (2006). Murine inner cell mass-derived lineages depend on Sall4 function. Proc. Natl. Acad. Sci. USA 103, 16319–16324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Dimitrov T, Yong KJ, Tatetsu H, Jeong H-W, Luo HR, Bradner JE, Tenen DG, and Chai L (2013a). Targeting transcription factor SALL4 in acute myeloid leukemia by interrupting its interaction with an epigenetic complex. Blood 121, 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Kong NR, Li A, Tatetu H, Ueno S, Yang Y, He J, Yang J, Ma Y, Kao GS, et al. (2013b). SALL4 is a key transcription regulator in normal human hematopoiesis. Transfusion 53, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SG, Iglesias AH, Lizcano F, Villanueva R, Camelo S, Jingu H, Teh BT, Koibuchi N, Chin WW, Kokkotou E, and Dangond F (2005). Functional characterization of JMJD2A, a histone deacetylase- and retinoblastoma-binding protein. J. Biol. Chem. 280, 28507–28518. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs RM, Fagoonee S, Papa A, Webster K, Altruda F, Nishinakamura R, Chai L, and Pandolfi PP (2012). Functional antagonism between Sall4 and Plzf defines germline progenitors. Cell Stem Cell 10, 284–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Xu S, Xiong L, Yu L, Fu X, and Xu Y (2017). SALL4 promotes glycolysis and chromatin remodeling via modulating HP1α-Glut1 pathway. Oncogene 36, 6472–6479. [DOI] [PubMed] [Google Scholar]
- Kohlhase J, Schubert L, Liebers M, Rauch A, Becker K, Mohammed SN, Newbury-Ecob R, and Reardon W (2003). Mutations at the SALL4 locus on chromosome 20 result in a range of clinically overlapping phenotypes, including Okihiro syndrome, Holt-Oram syndrome, acro-renal-ocular syndrome, and patients previously reported to represent thalidomide embryopathy. J. Med. Genet 40, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroki S, Nakai Y, Maeda R, Okashita N, Akiyoshi M, Yamaguchi Y, Kitano S, Miyachi H, Nakato R, Ichiyanagi K, et al. (2018). Combined Loss of JMJD1A and JMJD1B Reveals Critical Roles for H3K9 Demethylation in the Maintenance of Embryonic Stem Cells and Early Embryogenesis. Stem Cell Reports 10, 1340–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Jiao Y, Yong KJ, Wang F, Gao C, Yan B, Srivastava S, Lim GSD, Tang P, Yang H, et al. (2015). SALL4 is a new target in endometrial cancer. Oncogene 34, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Yang Y, Gao C, Lu J, Jeong H-W, Liu BH, Tang P, Yao X, Neuberg D, Huang G, et al. (2013). A SALL4/MLL/HOXA9 pathway in murine and human myeloid leukemogenesis. J. Clin. Invest 123, 4195–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu BH, Jobichen C, Chia CSB, Chan THM, Tang JP, Chung TXY, Li J, Poulsen A, Hung AW, Koh-Stenta X, et al. (2018a). Targeting cancer addiction for SALL4 by shifting its transcriptome with a pharmacologic peptide. Proc. Natl. Acad. Sci. USA 115, E7119–E7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, et al. (2018b). Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Jeong H-W, Kong N, Yang Y, Carroll J, Luo HR, Silberstein LE, Yupoma, and Chai L (2009). Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS One 4, e5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G, and Goodson M (2011). Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 21, 936–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Cui W, Yang J, Qu J, Di C, Amin HM, Lai R, Ritz J, Krause DS, and Chai L (2006). SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice. Blood 108, 2726–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela ME, Couto S, Zheng X, Lu G, Hui J, Stamp K, Drew C, Ren Y, Wang M, Carpenter A, et al. (2018). SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol 14, 981–987. [DOI] [PubMed] [Google Scholar]
- Meers MP, Tenenbaum D, and Henikoff S (2019). Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin 12, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miertus J, Borozdin W, Frecer V, Tonini G, Bertok S, Amoroso A, Miertus S, and Kohlhase J (2006). A SALL4 zinc finger missense mutation predicted to result in increased DNA binding affinity is associated with cranial midline defects and mild features of Okihiro syndrome. Hum. Genet 119, 154–161. [DOI] [PubMed] [Google Scholar]
- Miettinen M, Wang Z, McCue PA, Sarlomo-Rikala M, Rys J, Biernat W, Lasota J, and Lee Y-S (2014). SALL4 expression in germ cell and non-germ cell tumors: a systematic immunohistochemical study of 3215 cases. Am. J. Surg. Pathol 38, 410–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Kamiya A, Zeniya M, Chikada H, Hyuck AD, Yamazaki Y, Wauthier E, Tajiri H, Miller LD, Wang XW, et al. (2013). Sal-like protein 4 (SALL4), a stem cell biomarker in liver cancers. Hepatology 57, 1469–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Scott G, Ray MK, Mishina Y, and Zhang Y (2007). Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature 450, 119–123. [DOI] [PubMed] [Google Scholar]
- Pedanou VE, Gobeil S, Tabariè s S, Simone TM, Zhu LJ, Siegel PM, and Green MR (2016). The histone H3K9 demethylase KDM3A promotes anoikis by transcriptionally activating pro-apoptotic genes BNIP3 and BNIP3L. eLife 5, e16844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). Deep-Tools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaki-Yumoto M, Kobayashi C, Sato A, Fujimura S, Matsumoto Y, Takasato M, Kodama T, Aburatani H, Asashima M, Yoshida N, and Nishinakamura R (2006). The murine homolog of SALL4, a causative gene in Okihiro syndrome, is essential for embryonic stem cell proliferation, and cooperates with Sall1 in anorectal, heart, brain and kidney development. Development 133, 3005–3013. [DOI] [PubMed] [Google Scholar]
- Sathyan KM, Shen Z, Tripathi V, Prasanth KV, and Prasanth SG (2011). A BEN-domain-containing protein associates with heterochromatin and represses transcription. J. Cell Sci 124, 3149–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Durussel T, and Laemmli UK (2004). ChIC and ChEC; genomic mapping of chromatin proteins. Mol. Cell 16, 147–157. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Struhl K (1989). Helix-turn-helix, zinc-finger, and leucine-zipper motifs for eukaryotic transcriptional regulatory proteins. Trends Biochem. Sci 14, 137–140. [DOI] [PubMed] [Google Scholar]
- Tatetsu H, Kong NR, Chong G, Amabile G, Tenen DG, and Chai L (2016). SALL4, the missing link between stem cells, development and cancer. Gene 584, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhal P, Rösler B, and Kohlhase J (2006). A family with features overlapping Okihiro syndrome, hemifacial microsomia and isolated Duane anomaly caused by a novel SALL4 mutation. Am. J. Med. Genet. A 140, 222–226. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Xiong J, Zhang Z, Chen J, Huang H, Xu Y, Ding X, Zheng Y, Nishinakamura R, Xu G-L, Wang H, et al. (2016). Cooperative Action between SALL4A and TET Proteins in Stepwise Oxidation of 5-Methylcytosine. Mol. Cell 64, 913–925. [DOI] [PubMed] [Google Scholar]
- Yamaguchi YL, Tanaka SS, Kumagai M, Fujimoto Y, Terabayashi T, Matsui Y, and Nishinakamura R (2015). Sall4 is essential for mouse primordial germ cell specification by suppressing somatic cell program genes. Stem Cells 33, 289–300. [DOI] [PubMed] [Google Scholar]
- Yong KJ, Gao C, Lim JSJ, Yan B, Yang H, Dimitrov T, Kawasaki A, Ong CW, Wong K-F, Lee S, et al. (2013). Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N. Engl. J. Med 368, 2266–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong KJ, Li A, Ou W-B, Hong CKY, Zhao W, Wang F, Tatetsu H, Yan B, Qi L, Fletcher JA, et al. (2016). Targeting SALL4 by entinostat in lung cancer. Oncotarget 7, 75425–75440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JJ, Kjolby RAS, Kong NR, Monica SD, and Harland RM (2014). Spalt-like 4 promotes posterior neural fates via repression of pou5f3 family members in Xenopus. Development 141, 1683–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, and He Q-Y (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zeng W, Ball AR Jr., and Yokomori K (2010). HP1: heterochromatin binding proteins working the genome. Epigenetics 5, 287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tam W-L, Tong GQ, Wu Q, Chan H-Y, Soh B-S, Lou Y, Yang J, Ma Y, Chai L, et al. (2006). Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat. Cell Biol 8, 1114–1123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets and code utilized in this study are available at GEO: GSE136332 and on GitHub: https://github.com/mbassalbioinformatics/CnRAP.