

Abstract
Purpose:
To investigate the toxicity profile and establish an optimal dosing schedule of zotiraciclib with temozolomide (TMZ) in patients with recurrent high-grade astrocytoma.
Experimental Design:
This two-stage phase 1 trial determined the maximum tolerated dose (MTD) of zotiraciclib combined with either dose-dense (Arm1) or metronomic (Arm2) TMZ using a Bayesian Optimal Interval design; then a randomized cohort-expansion compared the progression-free survival rate at 4 month (PFS4) of the two arms for an efficient determination of a TMZ schedule to combine with zotiraciclib at MTD. Pharmacokinetics (PK) and pharmacogenomic (PG) profiling were included. Patient-reported outcome was evaluated by longitudinal symptom burden.
Results:
Fifty-three patients were enrolled. Dose-limiting toxicities were neutropenia, diarrhea, elevated liver enzymes, and fatigue. MTD of zotiraciclib was 250mg in both arms and thus selected for the cohort expansion. Dose-dense TMZ plus zotiraciclib (PSF4 40%) compared favorably with metronomic TMZ (PFS4 25%). Symptom burden worsened at Cycle 2 but stabilized by Cycle 4 in both arms. A significant decrease in absolute neutrophil count and neutrophil reactive oxygen species production occurred 12–24 hours after an oral dose of zotiraciclib but both recovered by 72 hours. PK/PG analyses revealed that the CYP1A2_5347T>C (rs2470890) polymorphism was associated with higher AUCinf value.
Conclusions:
Zotiraciclib combined with TMZ is safe in patients with recurrent high-grade astrocytomas. Zotiraciclib-induced neutropenia can be profound but mostly transient, warranting close monitoring rather than treatment discontinuation. Once validated, polymorphisms predicting drug metabolism may allow personalized dosing of zotiraciclib.
Keywords: Astrocytoma, glioblastoma, CDK9 inhibitor, zotiraciclib, clinical trial
Introduction
Brain tumors are challenging to treat, largely due to their biological features including a high level of heterogeneity and complicated resistance mechanisms (1,2). Only a small percentage of drugs investigated through clinical trials become an established therapy, underscoring the importance of preclinical investigations where promising drug candidates can be studied, thereby increasing the chance for successful drug development. Glioblastoma, which accounts for half of malignant primary brain tumors, has an extremely poor prognosis. Only a quarter of patients survive 2 years after their initial diagnosis, despite the aggressive treatments (3). As the disease progresses, there are few treatment options, and the survival time is counted by weeks, suggesting a large unmet need for developing effective therapies for recurrent glioblastoma (4).
Zotiraciclib (TG02) is a pyrimidine-based multi-kinase inhibitor with inhibitory effects on cyclin-dependent kinases (CDKs) (5). It has high potency to inhibit CDK9 with an IC50 of 3nM. CDK9 is a serine/threonine kinase with a catalytic core of the positive transcription elongation factor and it is critical for stimulating transcription elongation through RNA polymerase II (6–8). Clinical experiences of zotiraciclib, which is orally administered, have been mostly from hematological malignancies (9,10). We had performed extensive preclinical studies to investigate zotiraciclib as a single agent and in combination with temozolomide (TMZ) in both in vivo and in vitro preclinical glioma models (11). Our preclinical studies demonstrated that in addition to suppressing transcriptional process through CDK9 inhibition, zotiraciclib also decreased the cellular ATP production by suppressing glycolysis and causing mitochondrial dysfunction, leading to cell death in glioblastoma but not in normal cells. A synergistic effect with TMZ was found in both TMZ-sensitive and resistant glioblastoma models. The survival benefit was demonstrated in an in vivo orthotopic glioblastoma mouse model, where a pharmacodynamic experiment demonstrated a suppression of CDK9 activity in tumor tissues of mice treated with zotiraciclib, suggesting that zotiraciclib penetrates the blood-brain barrier. These preclinical findings suggest that zotiraciclib targets multiple survival pathways in glioblastoma and synergistically decreases cellular energy production with TMZ, representing a promising therapeutic strategy in glioblastoma. Based on these preclinical findings, we launched a phase 1 clinical trial to investigate zotiraciclib combined with TMZ in recurrent glioblastoma and anaplastic astrocytoma (NCT02942264). Here we report the results of this phase 1 study.
Methods
Study design
This phase 1 trial comprised a dose-escalation and a cohort expansion phase, where it concurrently optimized the dose and schedule of the combination of zotiraciclib with TMZ. The primary objective was to assess the safety and preliminary anti-tumor activities to establish the optimal dose-schedule for zotiraciclib combined with TMZ, with dose-limiting toxicity (DLT) as the primary safety endpoint. The determination of progression-free survival rate at 4 month (PFS4) and patient-reported outcomes (PROs) were secondary endpoints.
Patients were equally randomized to the dose-dense (DD) TMZ (Arm1) or metronomic (MN) TMZ (Arm2) arms, each combined with zotiraciclib at 4 dose-levels (150mg, 200mg, 250mg, 300mg). A Bayesian Optimal Interval (BOIN) design was employed to establish the maximum tolerated dose (MTD) of zotiraciclib in combination within each TMZ schedule arm (12). After the MTD was established in both arms, patients were randomized to receive a combination regimen using the established MTDs for each combination (DD or MN TMZ) until the total number of patients treated at each MTD reached 18. This included patients treated at the MTD during the dose escalation. Utilizing a “Pick the Winner” design, whereby the better performing arm would be the non-inferior, we tested the two TMZ dosing schedules in combination with zotiraciclib. The PFS4, rather than the traditional PFS6, was used as a prespecified landmark to facilitate a rapid determination of a potentially better TMZ dosing schedule to combine with zotiraciclib for the future studies.
Patients
All patients were enrolled at the Center for Cancer Research of the National Cancer Institute (NCI), Bethesda, Maryland, USA. Patients were eligible if they were older than 18 years with a recurrent, histologically confirmed anaplastic astrocytoma (WHO grade III, intact 1p/19q chromosome), or glioblastoma/gliosarcoma (WHO grade IV), and no more than two prior relapses. Additional eligibility criteria included a Karnofsky performance score (KPS) of 60 or higher and adequate bone marrow, hepatic and renal functions. Pregnant or nursing women were excluded because of the unknown effects of zotiraciclib on fetus or infant. Due to zotiraciclib being primary metabolized by cytochrome P450 (CYP)1A2 and 3A4, strong inducers or inhibitors of CYP1A2 or 3A4 were prohibited. PROs were assessed in English speaking patients who were not cognitively impaired to self-report.
The study was conducted in accordance with the ethics principles of the Declaration of Helsinki. The study protocol was reviewed and approved by the NCI Institutional Review Board. Written informed consent was obtained from all study participants.
Treatment
TMZ was administered at 125mg/m2 daily on days 1–7 and 15–21 orally in DD TMZ arm and 50mg/m2 daily in MN TMZ arm. Patients received zotiraciclib 200mg/day orally on days 1, 12, 15, and 26 on a 28-day cycle, after a single initial dose given 3 days prior to cycle 1/day1. As detailed in the protocol (Supplementary Materials), this dosing schedule was designed primarily based on previous experiences of zotiraciclib and carfilzomib in hematological malignancy and the anticipations of more toxicities in patients receiving zotiraciclib and TMZ (10). In addition, the dosing schedule reduced the number of times patients had to take both medications on the same day, avoiding overlapping toxicities from both drugs. The dose escalation and de-escalation of zotiraciclib in each arm were guided by the BOIN design, with the starting dose of 200mg.
After the MTD of zotiraciclib was determined in both arms, a cohort expansion was initiated, and patients were randomized to receive zotiraciclib at MTD combined with DD TMZ in Arm1or MN TMZ in Arm2. The treatment was planned for 12 cycles unless patient had progressive disease, unacceptable toxicities, pregnancy, or withdrew from the study. All patients received premedication to prevent nausea, vomiting and diarrhea before and after each dose of zotiraciclib.
Study Assessment
Physical exams, toxicity evaluation, and laboratory exams, including complete blood count (CBC) with differential, chemistry panel, pregnancy test and EKG, were performed at the baseline and before each cycle, with an additional CBC with differential performed on day 14 of each treatment cycle. Brain MRIs with and without gadolinium contrast were completed at the baseline and every 2 cycles of treatment to determine the treatment response based on the Response Assessment in Neuro-Oncology (RANO) Criteria (13). The same dose of corticosteroid for at least 5 days was required before each MRI.
All toxicities were graded based on the Common Terminology Criteria for Adverse Events (CTCAE Version 4.0). Patients in the MTD finding phase were evaluable for assessment of toxicity to define DLT, which was defined as any of the following treatment-related adverse events (AEs) in the first 4 weeks of treatment: grade 4 neutropenia ≥ 5 days; febrile neutropenia; grade 4 thrombocytopenia ≥2 days or grade 3 thrombocytopenia with bleeding; grade 3 or 4 anemia ≥ 2 days; grade 3 or 4 non-hematological toxicities except for diarrhea responds to treatment within 2 days; nausea, vomiting, fatigue that respond to treatments within 7 days and deep venous thrombosis. During the entire phase 1 study, if an adverse event occurred wherein both TMZ and zotiraciclib required dose de-escalation, the Principal Investigator had the discretion to dose-reduce one or both study drugs with the consideration of known side effects in an effort to better define attribution.
The MD Anderson Symptom Inventory-Brain Tumor Module (MDASI-BT) was administered at baseline and the time of imaging (14). The MDASI-BT consists of 22 symptoms rated on a scale of 0 (not present) to10 (as bad as you can image) to indicate the severity of each symptom at its worst in the last 24 hours. Factor groupings of symptoms associated with the disease or treatment have been identified and were used for this analysis. The MDASI-BT also assesses how much symptoms interfere with different aspects of a patient’s life in the last 24 hours. These interference items are general activity, mood, work, relations with other people, walking, and enjoyment of life. The interference items are also measured on a 0 (“Did not interfere”) to 10 (“Interfered completely”) scale.
Exploratory Assessment
Patients enrolled in the cohort expansion phase participated in pharmacokinetics (PK), pharmacogenetics (PG), and neutrophil analysis. Blood samples were drawn prior to the first dose on cycle 1 day −3 (C1D-3) and at 1, 2, 4, 12, 24, 48, and 72-hour post-dosing.
Blood samples collected at all time points were tested for CBC with differential and blood smear. Blood cell images were captured and analyzed by CellaVision instrumentation. Samples collected at 0, 24 and 72 hours were analyzed for neutrophil chemotaxis, reactive oxygen species (ROS) production and neutrophil cell surface markers. Briefly, the in vitro chemotaxis was measured using an imaging system (EZ-TAXIScan) with either N-formylmethionyl-leucyl-phenylalanine (fMLF) or C5a as chemoattractant as described previously (15). Neutrophil ROS production was measured by luminol-enhanced chemiluminescence, using either fMLF, opsonized zymosan or phorbol myristate acetate (PMA) as stimuli (15). The area under the curve (AUC) of the luminescence response was calculated as the measure of ROS production. The expression of neutrophil surface markers (IgG1, CD11b, CD16, CD18, CD66b, 7D5, IgG2a, CD62L) was analyzed using flow cytometry (15). A panel of cytokines of the plasma samples was measured using a human 30-plex cytokine array (Meso Scale Discovery).
In parallel to neutrophil analysis, PK was characterized by using the samples obtained at the same time points. Plasma concentrations of zotiraciclib were measured using a validated liquid chromatography-tandem mass spectrometry assay. The parameters of PK analysis were AUC from time zero to the time of the final quantifiable sample (AUClast), or extrapolated to infinity AUCinf, calculated using the linear-up/log-down trapezoidal method; the maximum plasma concentration (Cmax); and half-life (T1/2), determined by the terminal elimination rate constant (λz).
One baseline blood sample per patient was analyzed for the genotype of the most relevant drug metabolizing enzymes and transporters from the genomic DNA. DNA was analyzed on a PharmacoScan™ (ThermoFisher) genotyping platform. Patient exposures (AUCinf) were compared with different genotype groups for each polymorphism in CYP1A2 and CYP3A4, which metabolize zotiraciclib (16).
Statistical Analysis
The BOIN design was employed to guide the dose escalation and establish the MTD in each arm, with the target DLT rate of 0.35 and patients treated in a cohort size of 3. The dose escalation stops for MTD determination when the number of patients treated at any dose reached 12 or the maximum sample size of 24 per arm.
Descriptive statistics were used to summarize the demographics and clinical characteristics of all patients. All AEs were tabulated by grade and dose levels. The determination of MTD was based on the treatment related AEs occurred during the DLT evaluation period. The AEs in the cohort expansion were used for overall toxicity assessment. For the cohort expansion, the response rate was summarized using a 95% binomial confidence interval, and PFS and OS were estimated using the Kaplan-Meier method.
A Jonckheere-Terpstra trend test was preferred over a Kruskal-Wallis test for the comparison of AUC of zotiraciclib in groups with different genotypes. However, both were performed.
The ROS luminescence response of freshly isolated neutrophils from each patient to each stimulus was plotted and compared to the response of freshly isolated neutrophils isolated from healthy volunteers using Student’s t-test. Cytokine levels at baseline and peak time were compared using paired t-test.
Descriptive statistics were used to summarize the mean severity of MDASI-BT symptom subscales and symptom interference over time and by dose groups. Linear mixed models for every MDASI-BT symptom subscales and interference were fitted to determine if there isstatistically significant difference between the treatment arms over time. These analyses were performed using all available data and were compared using data from patients who completed baseline, cycle 2, and cycle 4 (Completers).
Results
Patients
Between December 2016 and December 2019, fifty-three patients (40 in MTD finding phase and 13 in cohort expansion) were enrolled to the study (Figure 1). The patient characteristics in both arms are comparable as shown in Table 1. Because this study was initiated before the most recent revision of WHO Classification of Tumors of CNS (17), we used the 2016 classification (18).
Figure 1.
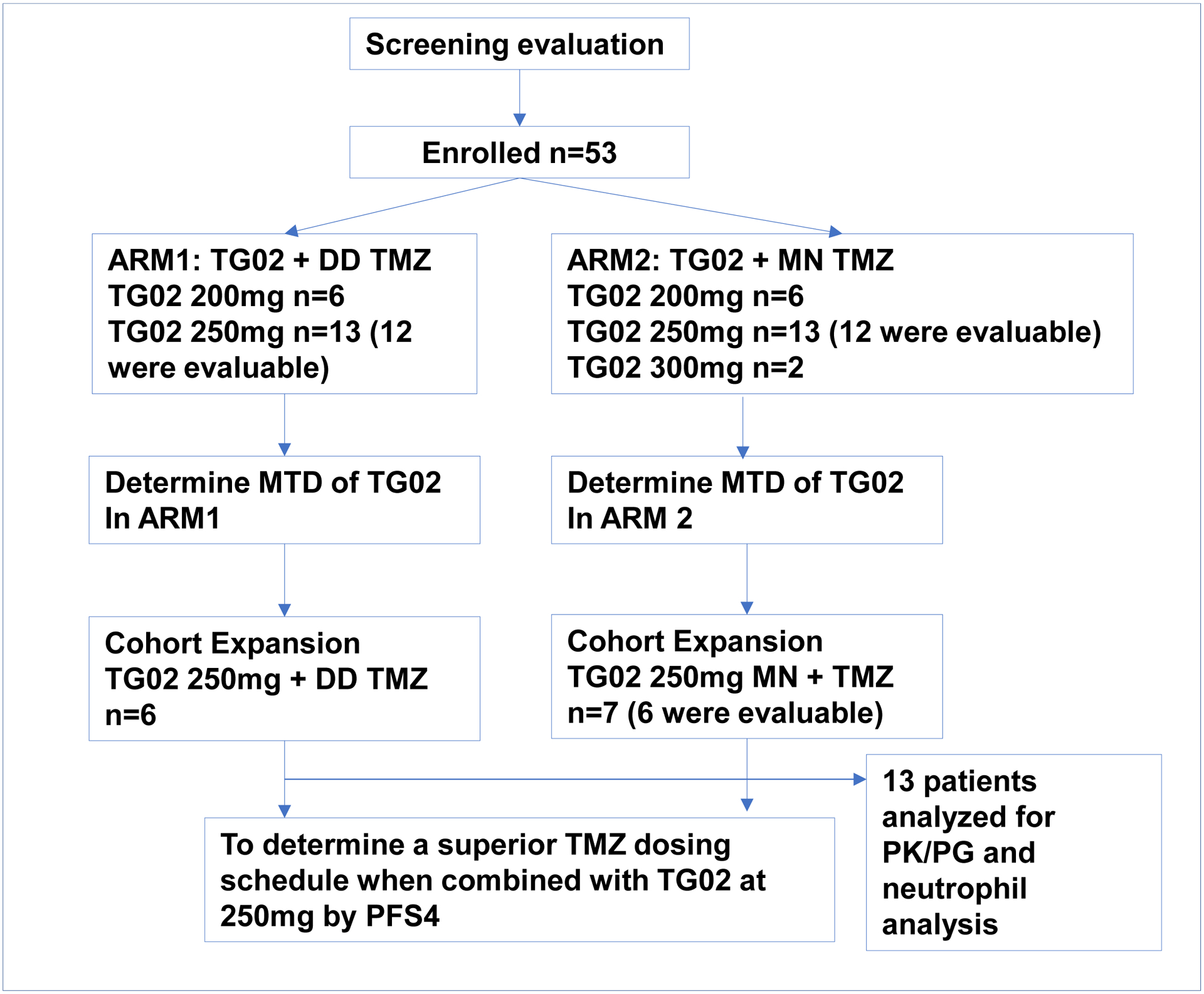
CONSORT Diagram
Table 1.
Patient Characteristics
| All Patients n=53 |
ARM 1 (DD) n=25 |
ARM 2 (MN) n=28 |
|
|---|---|---|---|
| Median Age, years (range) | 52.3 (20.9–76.4) | 50.8 (21.3–75.9) | 55.5 (20.9–76.4) |
| Female/male | 13/40 | 6/19 | 7/21 |
| Median KPS (range) | 90 (70–100) | 90 (70–100) | 90 (70–100) |
| Tumor type at screening* | |||
| Glioblastoma, IDH-wildtype | 35 | 16 | 19 |
| Glioblastoma, IDH-mutant | 4 | 0 | 4 |
| Glioblastoma, NOS | 1 | 1 | 0 |
| Anaplastic astrocytoma, IDH-mutant | 9 | 6 | 3 |
| Anaplastic astrocytoma, IDH-wildtype | 2 | 2 | 0 |
| Diffuse midline glioma, H3K27M-mutant | 2 | 0 | 2 |
| 1st/2nd recurrence | |||
| 1st relapse | 38 | 18 | 20 |
| 2nd relapse | 15 | 7 | 8 |
| IDH mutation status** | |||
| IDH-mutant | 13 | 6 | 7 |
| IDH-wildtype | 39 | 18 | 21 |
| MGMT promoter status*** | |||
| methylated | 24 | 10 | 14 |
| unmethylated | 28 | 14 | 14 |
| Prior brain tumor therapies | |||
| Concurrent XRT/TMZ Followed by adjuvant TMZ | 50 | 23 | 27 |
| XRT and TMZ in sequential | 3 | 2 | 1 |
| Others**** | 18 | 10 | 8 |
Note:
Tumor type at screening is based on “The 2016 World Health Organization Classification of the Tumor of the Central Nervous System”.
IDH mutation status is not available in one patient due to lack of enough tumor material.
MGMT promoter methylation status is not available due to lack of enough tumor material in one case.
Other therapies include clinical trial therapies, re-irradiation, TTF (Tumor Treating Field). All 18 patients who received these therapies also received concurrent XRT/TMZ followed by adjuvant TMZ as well.
Abbreviations: DD, dose-dense; MN, metronomic; KPS, Karnofsky performance status; GBM, glioblastoma; AA, anaplastic astrocytoma; IDH, isocitrate dehydrogenase; TMZ, temozolomide; XRT, radiation therapy.
Safety
As shown in Table 2, the DLT rate is proportional to the dosage of zotiraciclib in both arms. The non-hematologic DLTs included fatigue, elevated liver enzymes and diarrhea, while neutropenia was the only hematologic DLT. Two patients were enrolled to dose-level II and received zotiraciclib at 300mg, one patient developed severe AEs, including febrile neutropenia, thrombocytopenia, and hepatic dysfunction, which led to an intensive care unit admission. Interestingly, an other patient received zotiraciclib at 300mg tolerated the medication well other than mild fatigue, suggesting an individual variation in zotiraciclib metabolism. Nevertheless, the dose of 300mg zotiraciclib was eliminated without further testing due to the safety concern. Among all 38 DLT-evaluable patients, 11 (28.9%) patients experienced DLT.
Table 2.
Dose Limiting Toxicity Analysis
| Dose level | DLT | DLT Rate (%) | Description | |
|---|---|---|---|---|
| ARM 1 (DD) | D 0 200mg | 1/6 | 16.7 | Gr 3 diarrhea |
| D I 250mg | 3/12 | 25 | G4 neutropenia G3 ALT elevation G3 fatigue |
|
| ARM 2 (MN) | D 0 200mg | 1/6 | 16.7 | Dose reduction due to G3 neutropenia |
| D I 250mg | 5/12 | 41.7 | G3 ALT elevation G3 fatigue Dose reduction due to G3 neutropenia |
|
| D II 300mg | 1/2 | 50 | G4 neutropenia G4 ALT elevation G4 AST elevation |
Abbreviations: DD, dose-dense; MN, metronomic; D, dose level; DLT, dose limiting toxicity; Gr, grade; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
The AEs continued to be collected during the cohort expansion. Table S1 summarizes all treatment-related AEs at any grade that occurred among all 53 patients. Across all zotiraciclib dose-levels and different TMZ dosing schedules, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) elevation, diarrhea, fatigue and nausea were common non-hematologic AEs, mostly occurring at grade 1–2 level. ALT elevation was the most common grade 3–4 non-hematologic AE, having occurred in 11 (20.8%) patients, though only one case was likely attributed to zotiraciclib alone. Five (9.4%) patients developed grade 3 fatigue with 3 of the 5 likely attributed to zotiraciclib alone. Grade 3 diarrhea was found in 3 (5.6%) patients, each caused by zotiraciclib alone. Most hematologic toxicities were grades 1–2. There were 13 (24.5%) treatment-related grade 4 neutropenia; four occurred after zotiraciclib alone, of which two resolved to grade 1 within one day. Among these 13 patients who had grade 4 neutropenia, 9 of them recovered to grade 2 or less in no more than 3 days; two recovered to grade 3 by 3 days. These observations compelled us to further investigate the functional consequences of the transient neutropenia.
Outcomes
As of July 30, 2020, all 50 evaluable patients were off study treatment; other than 2 patients (4%) lost-to-follow up, 41 patients (82%) were deceased and 7 patients (14%) are still alive. Although most of the patients (72%) were off treatment due to disease progression, 5 patients (10%) completed the study’s entire treatment course. It was encouraging to observe 2 patients had partial response as the best response while they were on treatment. Both patients had IDH-wild type glioblastoma, one had unmethylated MGMT, suggesting the treatment activity in TMZ refractory glioblastoma (Figure S1).
In dose-escalation phase, 250mg of zotiraciclib was established as the MTD in both arms. Following cohort expansion, a total of 36 patients were treated for at least one cycle of zotiraciclib at MTD with either DD or MN TMZ. PFS4 was 0.40 (95% CI: 0.17, 0.63) in Arm 1 (DD TMZ) and 0.25 (95% CI: 0.08, 0.47) in Arm 2 (MN TMZ). The DD TMZ combined with 250mg zotiraciclib was selected as the optimal dose and schedule for future studies. There was no significant difference of overall PFS in each arm at MTD as shown in Figure S2. Subset analysis demonstrated that the PFS in patients with IDH-mutant gliomas was 171 days versus 74 days in IDH-wild type tumors (p=0.015, HR 2.3 95% CI 1.02–5.25) (Figure S3).
AE of Special Interest-Early Onset and Transient Neutropenia
CBC with differential demonstrated a consistent decrease in the absolute neutrophil count (ANC) between 12–24 hours after an oral dose of zotiraciclib and recovered to their baselines by 72 hours (Figure 2A). The nadir of ANC occurred within a few hours after the Tmax of zotiraciclib and recovered as its plasma concentration fell below the detectable level (Figure 2B). A transient decrease in the count of other blood cells, such as platelet or red blood cells, was not observed. The morphology of neutrophils captured by CellaVision, illustrated in Figure S4, was reviewed by certified hematology staff. A significant drop was observed in the absolute number of mature, segmented neutrophils but not in bands, the immature neutrophils at 12 hours (Figure S5), suggesting a loss of mature neutrophils, perhaps to the marginated pool, or into the organs, such as lungs, etc., without a concomitant release of immature neutrophils from the bone marrow. Defects in the chemotaxis of neutrophils, which may cause severe infections, were investigated. Zotiraciclib was not found to alter the fMLF-induced nor C5a-induced chemotaxis as demonstrated in the video files in supplementary materials. The release of ROS from neutrophils is an important component in the bactericidal repertoire of neutrophils. While ROS production stimulated by fMLF and opsonized zymosan was significantly reduced at 24 hours post zotiraciclib treatment, ROS production stimulated by PMA was not reduced (Figure 2C and 2D). Interestingly, the loss of ROS production to fMLF and opsonized zymosan was transient, and completely recovered within 72 hours. The expression of the surface antigens on neutrophils, including IgG1, CD11b, CD16, CD18, CD66b, 7D5, IgG2a and CD62L was unremarkable. To explore the potential etiology for indirect effects on neutrophils, a cytokine array study of the plasma sample from patients’ blood was conducted. We observed an increase in the plasma levels of several cytokines that often exceeded the normal range (defined as the mean±2SD, n=114 healthy volunteers). The peak level of IL-8 coincided temporally with the ANC nadir and may reflect an impact neutrophil biology (Figure 3). Several other cytokines (IL-6, MIP-1α, MIP-1β, IP-10, and MCP-1) exhibited increased plasma levels that peaked at 24 hours, suggesting that zotiraciclib therapy may be associated with transient immune activation. More importantly, the elevated level of these cytokines recovered to the normal range at 72 hours post-treatment.
Figure 2.
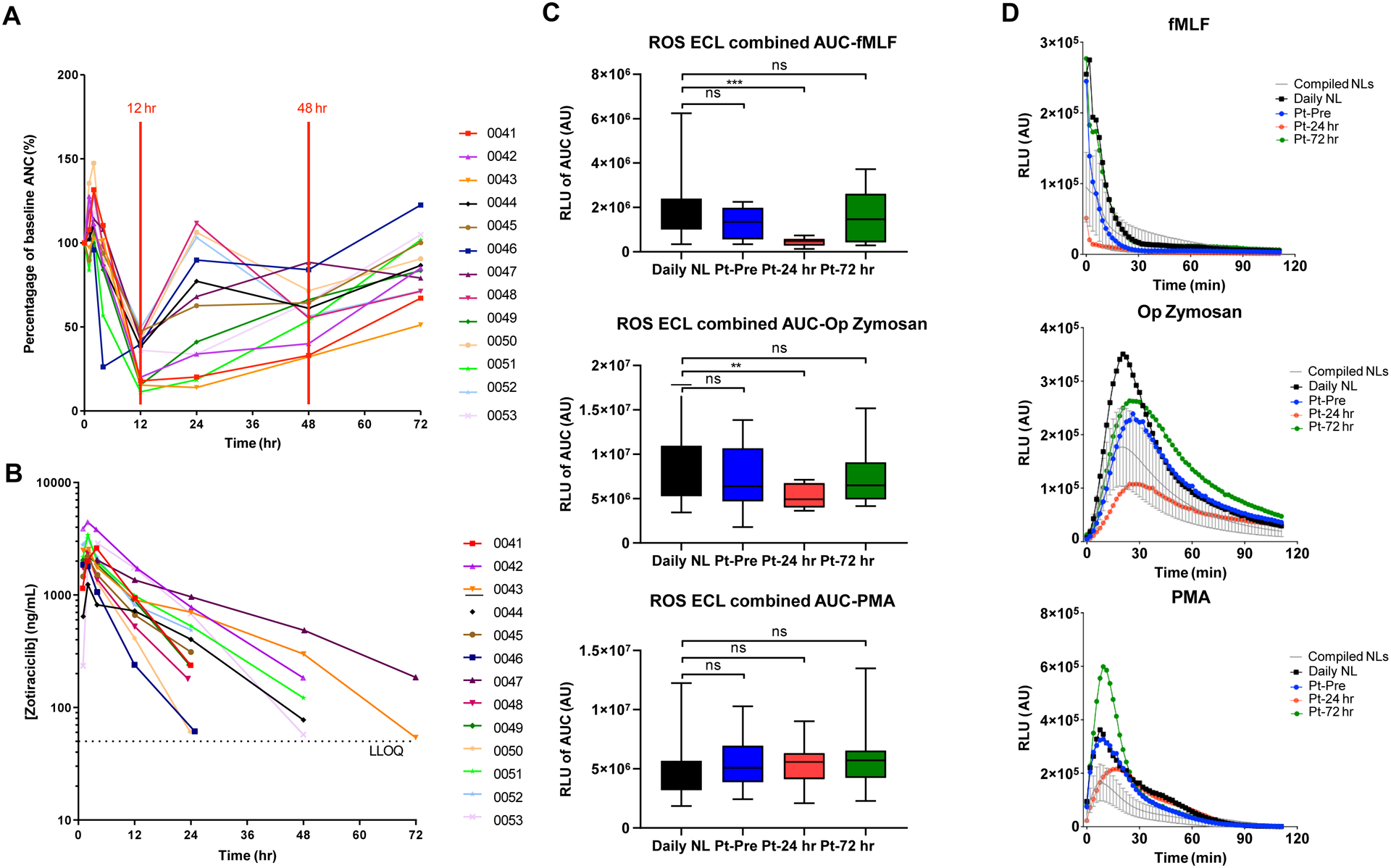
Neutrophil and PK analysis. A Plot of the percentage of baseline absolute neutrophil count over time in each patient (n=13). B Serum concentration of zotiraciclib over time after treatment in each patient (n=13). C Neutrophil ROS production was measured by luminol-enhanced chemiluminescence (ECL), using fMLF, zymosan and phorbol myristate acetate (PMA) as stimuli. The area under the curve (AUC) was calculated as the measure of ROS production. Data are expressed as the mean ± SD. Black bar: daily normal neutrophil; blue bar: patient baseline; red bar: 24hr after treatment; green bar: 72hr after treatment. ns, not significant, ** p<0.01, *** p< 0.001. (C) Changes of fMLF (top panel), op-zymosan (middle panel), and PMA-induced ROS production at baseline (blue lines), 24 hours (red lines) and 72 hours (green lines) after treatment in a representative case compared to the normal neutrophils (black lines).
Figure 3.
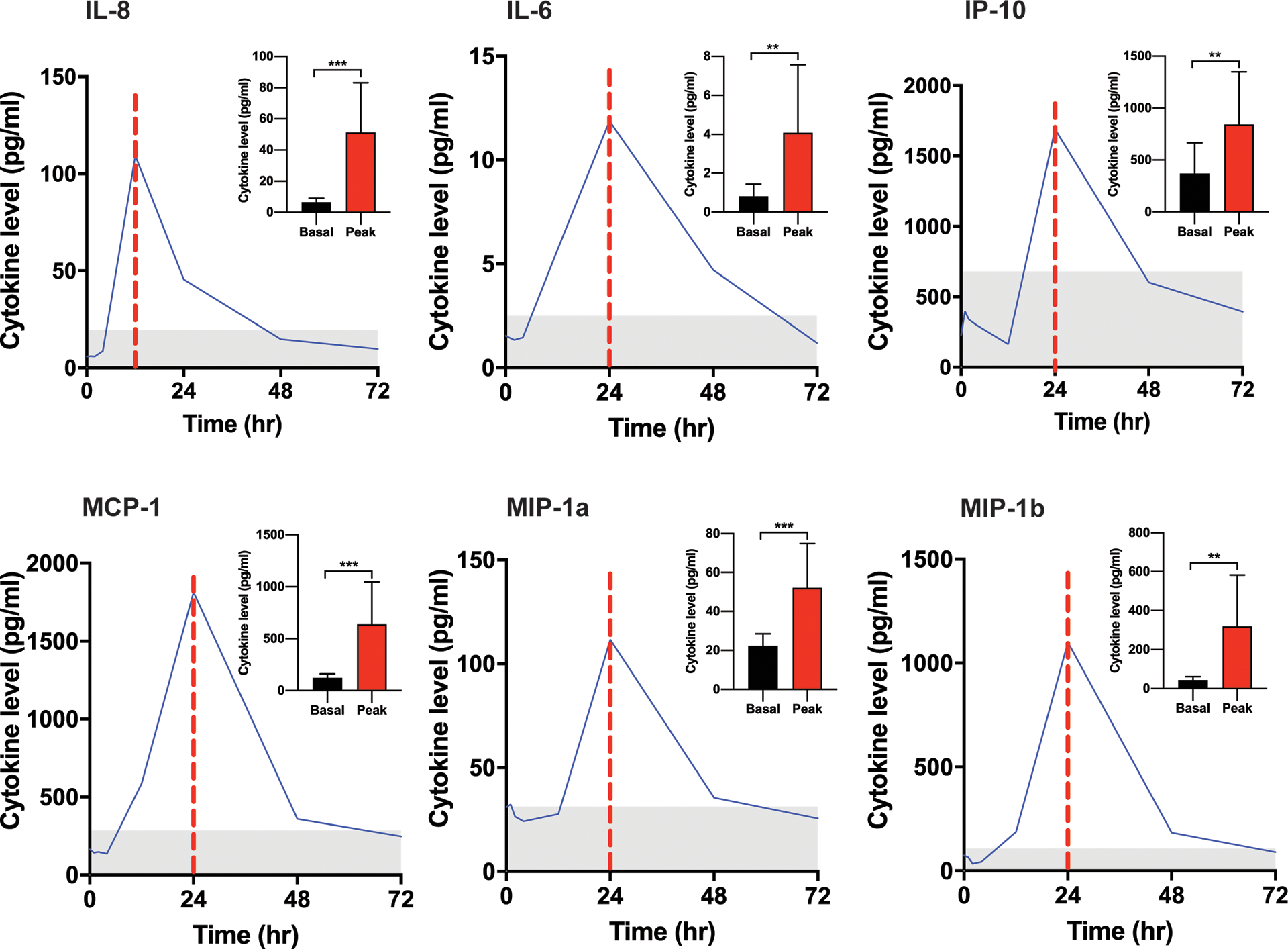
Cytokine analysis in patients received an oral dose of zotiraciclib at 250mg. Cytokine level of a representative study subject versus time points from 0–72 hours after zotiraciclib dosing is plotted to demonstrate the temporal change of the cytokine release. The vertical dotted red line indicates the peak time of each cytokine release. The gray area represents the normal range defined as the mean ± 2 standard deviation that was generated from 114 healthy volunteer. The mean level of cytokines at baseline and peak time from entire cohort of patients are summarized in the insert in each plot (n=13). Cytokine level at peak time was compared with baseline level using paired t-test. n=13. * p<0.05, ** p<0.01, *** p<0.001.
Pharmacokinetic and Pharmacogenetics
Blood samples obtained from all 13 patients in cohort expansion were analyzed for PK and PG. Following apparent first order absorption, zotiraciclib appeared to follow a mono-exponential elimination (Figure S6). Individual concentration versus time profiles for all 13 patients are depicted in Figure 2A. A non-compartmental analysis of plasma concentration versus time data was used to determine PK parameters for each patient. While the general pattern of absorption and elimination was consistent across patients, the PK parameters varied among patients (range 31–52%CV) (Table S2). No significant difference was observed in PK parameters between patients enrolled to different arms (Figure S7). Additionally, no significant correlation was found between apparent oral clearance or volume of distribution and body weight or age (Figure S8).
All 13 patients in the cohort expansion had PG data available. While several genetic variants were analyzed (n=17 in CYP1A2, n=25 in CYP3A4), inter-individual variation was only observed for more than one patient at five loci: CYP1A2 5347T>C (N516N; rs2470890), CYP1A2*1D (rs35694136), CYP1A2*1F (rs762551), CYP3A4_20230G>A (rs2242480), CYP3A4_−392A>G (rs2740574). The mean AUCINF of zotiraciclib was lower in CYP1A2 5347T/T genotype carriers (23.4 hr*ug/mL, n=5) than in those carrying the 5347C/T (31.7 hr*ug/mL, n=6) and 5347T/T (51.6 hr*ug/mL, n=2) genotypes, respectively (p=0.044, Figure 4), which is consistent with the hypothesis that rs2470890 “C” allele affects the zotiraciclib metabolism by reducing the CYP1A2 expression. The 4 remaining polymorphisms demonstrated expected genotype-dependent changes in zotiraciclib AUC (Figure S9). However, due to low numbers of samples, a statistical analysis could not be completed.
Figure 4.
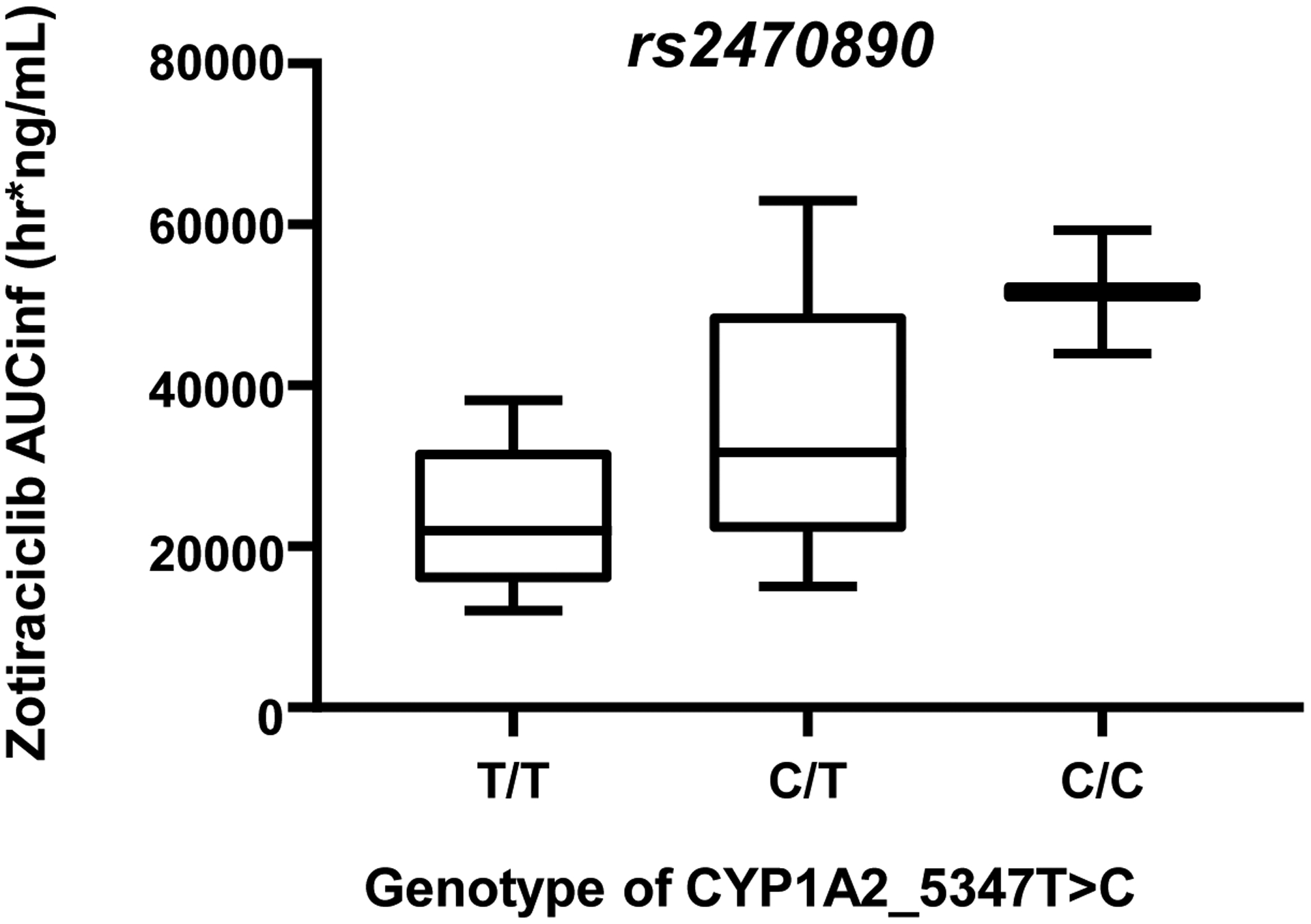
AUCinf of zotiraciclib in patients have homozygous loss (C/C, n=2), heterozygous (C/T n=6) and wild type (T/T, n=5) of CYP1A-5347>C, a variant is suspected to be loss of function of CYP1A2. p=0.044 by Jonckeere-Terpstra trend test.
Patient-Reported Outcomes
Of the 50 evaluable patients, the compliance rate of completing MDASI-BT were 98%, 89% and 100% at the baseline, cycle 2 and cycle 4, respectively. As shown in Table S3, overall symptom burden was 1.5 (SD=1.3) at baseline. Of the symptom factors, the affective factor was rated the highest. Overall interference at baseline was rated 2.3 (SD=2.6) with activity-related interference rated the highest. Among patients who received treatment through cycle 4, the overall symptom burden at baseline was 1.3 (SD=1.1) and overall interference was 2.1 (SD=2.8).
A longitudinal evaluation using data from all available patients reveals that overall symptom burden and interference worsen during cycle 2 but remained the same from cycle 2 to cycle 4 (Table S3). However, no significant statistical differences on any of the symptom subscales and interference between treatment arms were found. Analyses of the data using completers yielded similar results.
Discussion
Development of a promising therapeutic approach in refractory malignant gliomas remains challenging. Here we report a randomized phase 1 clinical trial investigating the toxicity and tolerability of a novel drug, zotiraciclib, in combination with TMZ in patients with recurrent glioblastoma and anaplastic astrocytoma.
Our preclinical studies were compelling and strongly supported the clinical investigation of zotiraciclib with TMZ (11). While zotiraciclib is also under investigation as a single agent in newly diagnosed glioblastoma (NCT03224104), the synergistic effects and alternative dosing schedule of TMZ support the clinical investigation of combined therapy in recurrent disease. Furthermore, zotiraciclib and TMZ synergistically induced gliomas cell death regardless of MGMT expression levels, suggesting a role in treating TMZ-resistant gliomas. Knowing most patients would have already received the standard dosing of TMZ (150mg-200mg/m2 on days 1–5 of a 28 day-schedule) as first-line therapy, alternative schedules were considered. The 7-days on/7-days off and metronomic are the most evaluated schedules, which have shown promising clinical activities in recurrent glioblastomas (19,20).
This trial simultaneously optimized the dosing and schedule of the combination of zotiraciclib and TMZ using the BOIN dose-finding design. This approach minimized the chance of exposing patients to subtherapeutic or overly toxic doses, and has better operating characteristics than the 3+3 design (21,22). Furthermore, the use of a randomization schema in the phase 1 design enabled the objective comparison of two different TMZ dosing schedules while using results from patients enrolled on the dose-finding as well as the expansion component. We prespecified PFS4 as a landmark to facilitate the rapid determination of a potentially better TMZ schedule to combine with zotiraciclib for the phase 2 study, not to establish that one schedule is truly better than the other, given the small sample size. It is possible that the PFS at a specific landmark (e.g., 4 months) is not fully consistent with the trend of the overall PFS. In our trial, the PFS of the two schedules are mostly comparable, which means that choosing either schedule for the phase 2 is probably reasonable.
In this phase 1 study, extensive investigations were done to understand the toxicity and tolerability profile of the combined treatment in the study patients. Longitudinal measures of symptom burden support that after an early increase in symptom burden, supportive measures lead to either improvement or stabilization of symptoms. Although DD TMZ with higher zotiraciclib dose seems to be associated with worsening symptom compared to other treatment arms, no statistically significant differences were found, likely due to small sample size.
A zotiraciclib-induced neutropenia with an unusual pattern was observed, which typically developed within 24 hours after treatment and recovered within 72 hours in patients. Although it reported that CDK inhibitors may induce neutrophil apoptosis through CDK7 and CDK9 inhibition (23), we did not observe a similar effect of zotiraciclib. In vitro experiments using neutrophils from healthy volunteers demonstrated that zotiraciclib has no effects on neutrophil viability and morphology within a range of concentration (10–200 nM) (data not shown). Zotiraciclib also had little effects on fMLF-induced chemotaxis and expression of surface markers in neutrophils from healthy volunteers, suggesting that an indirect effect of zotiraciclib may be attributed to the transient neutropenia. To ensure patient safety, we performed an in-depth neutrophil analysis along with a concurrent determination of zotiraciclib PK to potentially correlate with the drug metabolism and neutrophil functions as part of the clinical trial. The response of neutrophils to PMA, including induced ROS production, bypasses many of the physiologic receptor-mediated signaling pathways required for response to fMLF and opsonized zymosan (24,25). PMA-induced ROS production following zotiraciclib was found to be intact, suggesting that the potential of ROS production remained following the treatment. The ROS production induced by fMLF and opsonized zymosan was found to be decreased at 24 hours, coinciding with the nadir of ANC. However, it recovered by 72 hours, suggesting a transient loss of signaling responses, rather than a decrease in ROS production capacity. Neutrophil chemotaxis and surface marker expression were found to be unaffected in the neutrophils isolated from patients treated with zotiraciclib. Furthermore, despite the profound drop in peripheral blood neutrophil count, there were no instances of febrile neutropenia. This may be partially explained by the associated cytokine release with zotiraciclib treatment which may partially mediate its indirect effects on neutrophils. Although more studies are needed to fully uncover the causes of the transient neutropenia, our extensive neutrophil studies demonstrate that zotiraciclib causes a significant but transient neutropenia, which does not warrant immediate drug discontinuation in most cases.
To understand the individual variance observed in PK parameters and treatment related toxicities, we performed a PG study to interrogate possible genomic polymorphisms or variants in genes coding the drug metabolizing enzymes or drug transporters (26,27). Genetic variations in CYPs genes or their protein products are known to cause variabilities in the drug metabolism (28,29). Zotiraciclib was reported to be predominantly metabolized by CYP1A2 and CYP3A4, but does not significantly induce these two enzymes in vitro (16). We identified a single nucleotide change in gene coding CYP1A2 (CYP1A2*5347T>C, N541N, rs2470890) that results in a synonymous amino acid substitution of unknown significance, although some have speculated this variant affects mRNA stability (30). This SNP is also associated with a significant difference in zotiraciclib pharmacokinetics in a cohort of 13 patients. rs2470890 has also been reported to be associated with increased hematologic toxicity in head and neck cancer patients with chemotherapy treatments (31,32). We plan to validate these findings in future clinical trials.
To better inform future studies, a subset analysis was performed in patients stratified by IDH mutation status of their tumors. Although it may be argued that the longer PFS in IDH-mutant glioma reflects its own favorable prognosis, all study subjects were heavily pre-treated prior to study enrollment. Despite patients with IDH-mutant glioma have a longer PFS, accelerated progression after first recurrence has been reported (33). In addition, the IDH mutation status was not found to be predictive of PFS in recurrent glioblastoma trials (34). Furthermore, studies have shown decreased oxidative phosphorylation and reduced ATP level can be induced by 2-hydroxyglutarate, an oncometabolite in IDH-mutant gliomas (35). Based on our preclinical data, zotiraciclib reduces ATP production as a single agent and more significantly when combined with TMZ, supporting our hypothesis that the therapeutic benefit in IDH-mutant gliomas may be in part related to metabolic targets (11). The combination of our preclinical data and preliminary clinical activity from the current clinical trial supports the need for a preplanned subset analysis in IDH-mutant and wildtype gliomas in future clinical trials.
In summary, zotiraciclib is an anti-cancer drug with novel mechanisms of action. To our knowledge, this is the first report of a clinical trial of zotiraciclib in the treatment of recurrent high-grade gliomas. In this early stage clinical investigation of zotiraciclib, we performed a two-stage, two-arm randomized phase 1 study to determine the optimal dosing of zotiraciclib and TMZ. In addition, the study also includes robust PK, PG, assessment of PROs and most importantly a thorough interrogation of the previously unrecognized rapid onset neutropenia. For this latter observation, without the in-depth analysis concluding that this transient event did not compromise patient safety, the study and further development of this novel CDK9 inhibitor would have been interrupted. We believe this is an important message to the oncology community as novel targets are uncovered, leading to innovative treatments and the potential for previously unrecognized “off target” effects.
Supplementary Material
Translational relevance.
Zotiraciclib, a cyclin-dependent kinase 9 (CDK9) inhibitor, was found to induce glioblastoma cell death and demonstrated synergistic anti-glioma effects when combined with temozolomide in our preclinical studies. Supported by the strong preclinical evidence, we performed the first clinical trial of a CDK9 inhibitor combined with temozolomide in recurrent high-grade astrocytoma. In addition to establishing the optimal dosing schedule for zotiraciclib and temozolomide, in-depth analyses of zotiraciclib-induced neutropenia suggest the transient nature of the drastic alteration in neutrophil biology. A close monitoring rather than discontinuation of the drug is warranted. The PK/PG analysis identified a polymorphism that potentially alters the pharmacokinetics of zotiraciclib, suggesting further investigations are warranted for a genotype-guided dosing to reduce the toxicities. Symptomatic toxicities occur but may stabilize with continued treatments, supporting the tolerability of the treatment. The comprehensive information from this multi-faceted early-stage investigation paved a way for success in further clinical trials of zotiraciclib.
Acknowledgements:
The authors thank NCI Intramural Research Program and NIH Lasker Clinical Research Scholars Program for the funding and support for the research in this manuscript. We also thank Adastra Pharmaceuticals for providing the drug supply for clinical trial. Finally, our special thanks go to all patients and their families for their participation in the clinical trial and the support for our clinical research.
Financial support:
Funding from NIH Lasker Clinical Research Scholars Program and NCI Intramural Research Program was used towards the design, implementation of the clinical trial, data collection, analysis, and generating the manuscript. This research project was supported in part from the NIH, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. Adastra Pharmaceuticals provided the drug supply for clinical trial. All authors have the full access to the data used to generate the report and agreed with the findings.
Footnotes
Conflicts of Interest:
The authors declare no potential conflicts of interest.
References
- 1.Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol 2019;16(8):509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levin VA, Abrey LE, Heffron TP, Tonge PJ, Dar AC, Weiss WA, et al. CNS Anticancer Drug Discovery and Development: 2016 conference insights. CNS Oncol 2017;6(3):167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352(10):987–96. [DOI] [PubMed] [Google Scholar]
- 4.Wong ET, Hess KR, Gleason MJ, Jaeckle KA, Kyritsis AP, Prados MD, et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol 1999;17(8):2572–8. [DOI] [PubMed] [Google Scholar]
- 5.Goh KC, Novotny-Diermayr V, Hart S, Ong LC, Loh YK, Cheong A, et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia 2012;26(2):236–43. [DOI] [PubMed] [Google Scholar]
- 6.Loyer P, Trembley JH, Katona R, Kidd VJ, Lahti JM. Role of CDK/cyclin complexes in transcription and RNA splicing. Cell Signal 2005;17(9):1033–51. [DOI] [PubMed] [Google Scholar]
- 7.Romano G, Giordano A. Role of the cyclin-dependent kinase 9-related pathway in mammalian gene expression and human diseases. Cell Cycle 2008;7(23):3664–8. [DOI] [PubMed] [Google Scholar]
- 8.Nechaev S, Adelman K. Pol II waiting in the starting gates: Regulating the transition from transcription initiation into productive elongation. Biochim Biophys Acta 2011;1809(1):34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ponder KG, Matulis SM, Hitosugi S, Gupta VA, Sharp C, Burrows F, et al. Dual inhibition of Mcl-1 by the combination of carfilzomib and TG02 in multiple myeloma. Cancer Biol Ther 2016;17(7):769–77.27246906 [Google Scholar]
- 10.Hofmeister CC, Berdeja JG, Vesole DH, Suvannasankha A, Parrott T, Abonour R. TG02, an Oral CDK9-Inhibitor, in Combination with Carfilzomib Demonstrated Objective Responses in Carfilzomib Refractory Multiple Myeloma Patients. Blood 2015;126(23):3025. [Google Scholar]
- 11.Su YT, Chen R, Wang H, Song H, Zhang Q, Chen LY, et al. Novel Targeting of Transcription and Metabolism in Glioblastoma. Clin Cancer Res 2018;24(5):1124–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu S, Yuan Y. Bayesian optimal interval designs for phase I clinical trials. Journal of the Royal Statistical Society: Series C (Applied Statistics) 2015;64(3). [Google Scholar]
- 13.Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 2010;28(11):1963–72. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong TS, Mendoza T, Gning I, Coco C, Cohen MZ, Eriksen L, et al. Validation of the M.D. Anderson Symptom Inventory Brain Tumor Module (MDASI-BT). J Neurooncol 2006;80(1):27–35. [DOI] [PubMed] [Google Scholar]
- 15.Priel DL, Kuhns DB. Assessment of Neutrophil Function. In: Rbert R Rich TAF, editor. Clinical Immunology. 5th ed: ScienceDirect; 2019. [Google Scholar]
- 16.Pasha MK, Jayaraman R, Reddy VP, Yeo P, Goh E, Williams A, et al. Preclinical metabolism and pharmacokinetics of SB1317 (TG02), a potent CDK/JAK2/FLT3 inhibitor. Drug Metab Lett 2012;6(1):33–42. [DOI] [PubMed] [Google Scholar]
- 17.Louis DN, Wesseling P, Aldape K, Brat DJ, Capper D, Cree IA, et al. cIMPACT-NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol 2020;30(4):844–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016;131(6):803–20. [DOI] [PubMed] [Google Scholar]
- 19.Wick A, Felsberg J, Steinbach JP, Herrlinger U, Platten M, Blaschke B, et al. Efficacy and tolerability of temozolomide in an alternating weekly regimen in patients with recurrent glioma. J Clin Oncol 2007;25(22):3357–61. [DOI] [PubMed] [Google Scholar]
- 20.Perry JR, Belanger K, Mason WP, Fulton D, Kavan P, Easaw J, et al. Phase II trial of continuous dose-intense temozolomide in recurrent malignant glioma: RESCUE study. J Clin Oncol 2010;28(12):2051–7. [DOI] [PubMed] [Google Scholar]
- 21.Zhou H, Murray TA, Pan H, Yuan Y. Comparative review of novel model-assisted designs for phase I clinical trials. Stat Med 2018;37(14):2208–22. [DOI] [PubMed] [Google Scholar]
- 22.Zhou H, Yuan Y, Nie L. Accuracy, Safety, and Reliability of Novel Phase I Trial Designs. Clin Cancer Res 2018;24(18):4357–64. [DOI] [PubMed] [Google Scholar]
- 23.Leitch AE, Lucas CD, Marwick JA, Duffin R, Haslett C, Rossi AG. Cyclin-dependent kinases 7 and 9 specifically regulate neutrophil transcription and their inhibition drives apoptosis to promote resolution of inflammation. Cell Death Differ 2012;19(12):1950–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang X, Wu TH, Rubin RL. Bridging of neutrophils to target cells by opsonized zymosan enhances the cytotoxicity of neutrophil-produced H2O2. J Immunol 1997;159(5):2468–75. [PubMed] [Google Scholar]
- 25.Makni-Maalej K, Chiandotto M, Hurtado-Nedelec M, Bedouhene S, Gougerot-Pocidalo MA, Dang PM, et al. Zymosan induces NADPH oxidase activation in human neutrophils by inducing the phosphorylation of p47phox and the activation of Rac2: involvement of protein tyrosine kinases, PI3Kinase, PKC, ERK1/2 and p38MAPkinase. Biochem Pharmacol 2013;85(1):92–100. [DOI] [PubMed] [Google Scholar]
- 26.Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science 1999;286(5439):487–91. [DOI] [PubMed] [Google Scholar]
- 27.McLeod HL, Evans WE. Pharmacogenomics: unlocking the human genome for better drug therapy. Annu Rev Pharmacol Toxicol 2001;41:101–21. [DOI] [PubMed] [Google Scholar]
- 28.Ulrich CM, Robien K, McLeod HL. Cancer pharmacogenetics: polymorphisms, pathways and beyond. Nat Rev Cancer 2003;3(12):912–20. [DOI] [PubMed] [Google Scholar]
- 29.Zanger UM, Turpeinen M, Klein K, Schwab M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem 2008;392(6):1093–108. [DOI] [PubMed] [Google Scholar]
- 30.Tiwari AK, Deshpande SN, Lerer B, Nimgaonkar VL, Thelma BK. Genetic susceptibility to Tardive Dyskinesia in chronic schizophrenia subjects: V. Association of CYP1A2 1545 C>T polymorphism. Pharmacogenomics J 2007;7(5):305–11. [DOI] [PubMed] [Google Scholar]
- 31.De Marchi P, Melendez ME, Laus AC, Kuhlmann PA, de Carvalho AC, Arantes L, et al. The role of single-nucleotide polymorphism (SNPs) in toxicity of induction chemotherapy based on cisplatin and paclitaxel in patients with advanced head and neck cancer. Oral Oncol 2019;98:48–52. [DOI] [PubMed] [Google Scholar]
- 32.Cusato J, Allegra S, Massano D, De Francia S, Piga A, D’Avolio A. Influence of single-nucleotide polymorphisms on deferasirox C trough levels and effectiveness. Pharmacogenomics J 2015;15(3):263–71. [DOI] [PubMed] [Google Scholar]
- 33.Miller JJ, Loebel F, Juratli TA, Tummala SS, Williams EA, Batchelor TT, et al. Accelerated progression of IDH mutant glioma after first recurrence. Neuro Oncol 2019;21(5):669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mandel JJ, Cachia D, Liu D, Wilson C, Aldape K, Fuller G, et al. Impact of IDH1 mutation status on outcome in clinical trials for recurrent glioblastoma. J Neurooncol 2016;129(1):147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karpel-Massler G, Ishida CT, Bianchetti E, Zhang Y, Shu C, Tsujiuchi T, et al. Induction of synthetic lethality in IDH1-mutated gliomas through inhibition of Bcl-xL. Nat Commun 2017;8(1):1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.