

Abstract
The RNA methyltransferase METTL3 catalyzes N6-methyladenosine (m6A) modification of messenger RNAs (mRNAs). It is overexpressed in many types of cancer, including acute myelogenous leukemia (AML), and promotes cancer cell growth and tumorigenicity. Now, a selective small molecule inhibitor of METTL3 shows significant anti-leukemic effects in preclinical AML models, highlighting the promise of pharmacological METTL3 inhibition as a new cancer therapy.
Keywords: METTL3, N6-methyladenosine, m6A, mRNA, Epitranscriptome, Cancer, Leukemia
While the importance of DNA methylation and histone modification in gene regulation is well established and chromatin-associated proteins are the subject of current drug-development efforts to treat cancer and other diseases1, RNA modifications are less well understood and appreciated. m6A is the most prevalent mRNA modification. It occurs at a particular sequence motif and is enriched at sites close to the translation stop codon of a large subset of the transcriptome2,3. A protein complex comprising METTL3 (methyltransferase-like 3) together with its essential cofactor METTL14 and accessory proteins is responsible for the deposition of almost all m6A on mRNA. The m6A ‘epitranscriptome’ plays important roles in the posttranscriptional control of gene expression including mRNA splicing, nuclear export, stability, and translation4.
METTL3 belongs to the large family of class I methyltransferases (MTases) that contain a Rossmann fold that binds the S-Adenosyl Methionine (SAM) methyl donor. Over recent years it has become increasingly clear that METTL3 functions as an oncogene in a variety of different cancer types. Studies in human cancer cell lines and mouse models have established METTL3 as a possible new anti-cancer5–8. Reporting now in Nature, Yankova, Blackaby et al., identified a selective inhibitor of METTL3 catalytic activity and demonstrate its efficacy in vitro and in vivo9.
The authors employed a RapidFire™ mass spectrometry-based assay to monitor METTL3 activity by measuring levels of S-Adenosyl homocysteine (SAH) generated by the MTase reaction A high throughput screen (HTS) of 250,000 compounds identified only two non-SAM-related hits. Chemical optimization dramatically improved the potency of one of the original hits, leading to the development of the small molecule STM2475 with an IC50 of 16.9 nM. Surface plasmon resonance (SPR) and a crystal structure of STM2475-bound METTL3 show that STM2457 adopts a co-factor competitive mode and binds within the SAM-binding pocket of METTL3 with high affinity (Kd = 1.4 nM) (Figure 1a). STM2457 exhibited high selectivity for METTL3 compared to a broad MTase panel and potently inhibited METTL3/14 catalytic activity. Using a structure-based drug discovery approach, an independent group recently also identified a selective small molecule inhibitor (UZH1a) of METTL3 activity. Like STM2457, UZH1a occupies the SAM binding site (Figure 1a) of METTL310. The strong selectivity of both of these inhibitors can be explained at least in part by the conformational reorganization of lysine 513 (K513) upon binding of the small molecule. This feature is common to STM2457 and UZH1a, but does not occur in METTL3 bound to SAM or the non-specific MTase inhibitor Sinefungin (Figure 1a). As expected, considering the SAM-competitive mechanism of action of these METTL3 inhibitors and the relatively high cellular concentration of SAM, a much higher concentration of STM2457 (IC50 = 2.2 uM), or UZH1a (IC50 = 4.6 uM) was required to inhibit METTL3 in cells as compared to biochemical assays and to reduce the overall m6A mRNA modification level.
Figure 1. METTL3 inhibition is a promising therapy against acute myelogenous leukemia (AML).
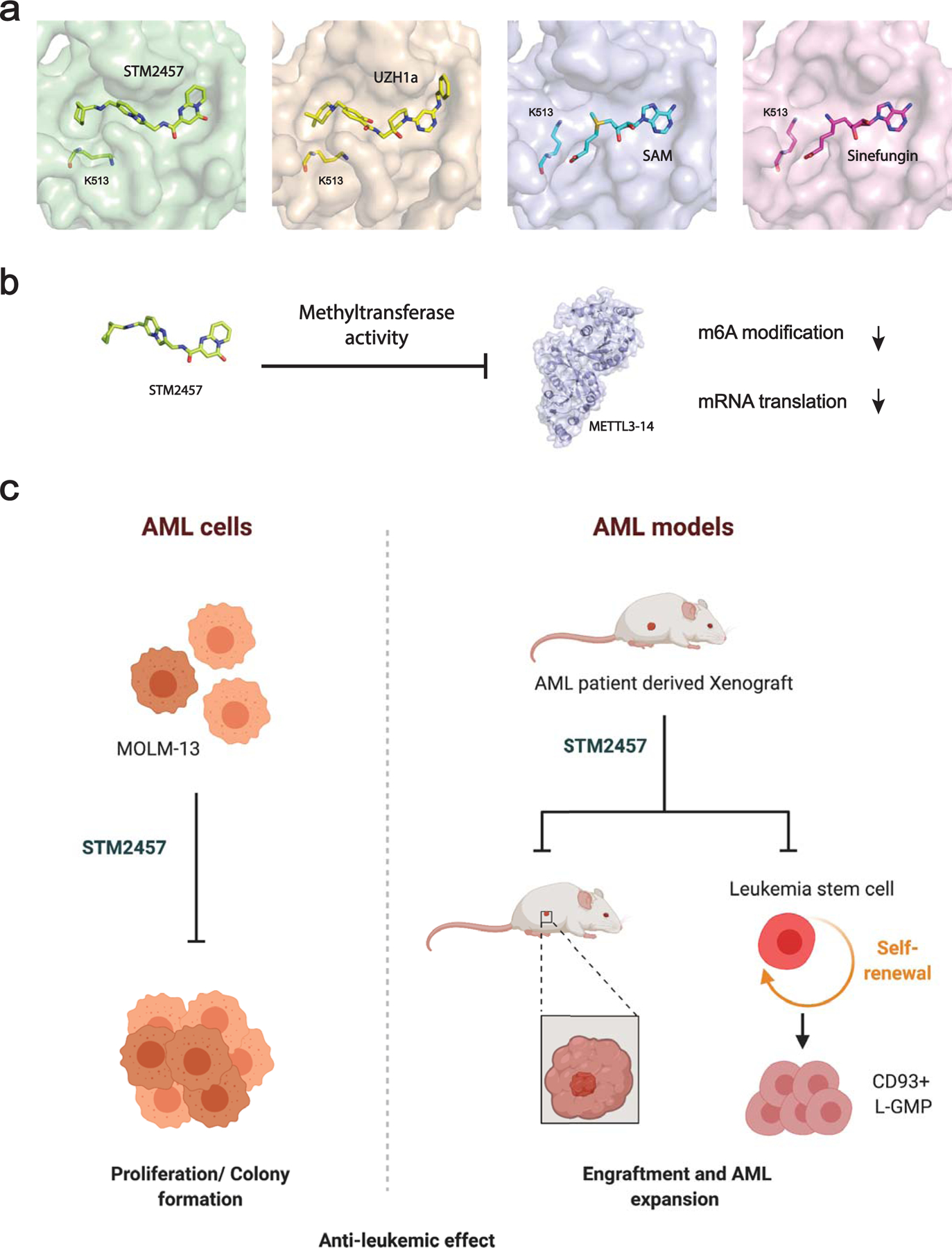
(a) The position of small molecule or ligands within the SAM-binding pocket of human METTL3. Surface representation of the crystal structure of METTL3 bound to STM2457 (PDB 7O2I), UZH1a (PDB 7ACD), SAM (PDB 5K7U), and Sinefungin (PDB 6Y4G) are displayed. Residue K513 of METTL3 is highlighted. (b) Inhibition of METTL3 methyltransferase activity affects m6A modification and translation levels of METTL3-modified mRNAs. (c) STM2457 treatment reduces proliferation and colony formation of AML cells, and impairs AML cell engraftment and expansion in human PDX models by inhibiting the self-renewal of leukemic stem cells.
Consistent with previous METTL3-depletion studies, inhibition of the catalytic activity of METTL3 with either STM2457 or UZH1a blocked proliferation of a panel of human AML cell lines including MOLM-13 (Figure 1b). Moreover, while METTL3 inhibition by STM2457 strongly reduced the clonogenic capacity and induced apoptosis of mouse primary AML cells, it had no effect on normal mouse or human hematopoietic stem and progenitor cells (HSPCs) in these in vitro assays.
These dramatic results raise the question of the underlying mechanism responsible for these phenotypes. While the biological importance and pathological relevance of METTL3 is now well-established, the molecular function of METTL3 and m6A mRNA modification remains unresolved and somewhat contentious, because effects on many different aspects of mRNA metabolism have been reported to-date. To address this question in the context of AML, Yankova and, Blackaby et al. initially performed RNA-seq on control and STM2457-treated MOLM-13 cells. Gene Ontology analysis showed that differentially expressed transcripts were enriched for genes related to myeloid differentiation and leukemia. Given the analyzed cell-type and the phenotypic changes caused by METTL3 inhibition, this result is not unexpected and at least some of the expression changes are likely due to indirect effects on the cellular state. The authors then performed m6A methylated RNA immuno-precipitation and sequencing (meRIP-Seq) to identify m6A-modified mRNAs. Identification of mRNAs with reduced m6A levels upon METTL3 inhibition is challenging, because meRIP-Seq is not very quantitative. Nevertheless, the authors perform quantitative RT-PCR to support the observation that m6A modification of several mRNAs is strongly decreased upon STM2457 treatment. These mRNAs include SP1 and BRD4, two mRNAs that are known to be regulated by METTL3 and relevant to AML.
m6A modification has been linked to various aspects of posttranscriptional mRNA control. While much focus has been on the role of m6A in targeting mRNAs for decay11,12, we previously found no major changes in steady state mRNA levels or mRNA stability upon METTL3 depletion. Instead, we observed a pronounced decrease in translation of multiple m6A-modified mRNAs5,6. Similarly, Yankova, Blackaby et al. found a substantial decrease in translation upon METTL3 inhibition . Importantly, they observe that m6A-modified target mRNAs, including SP1 and BRD4, shifted from the heavy polysome fraction to sub-polysome fractions upon METTL3 inhibition. No change in mRNA levels were detected. Overall, these findings support a major role for METTL3 and m6A in promoting translation of these (and many other) mRNAs.
Conversely, YTHDF2, one of several m6A reader proteins linked to mRNA decay, reportedly plays an important role in AML13. Even more confounding are results reporting that the putative m6A demethylases ALKBH5, and FTO promote AML and that a small molecule inhibitor of FTO effectively suppresses AML progression in mouse models14–17. The apparent discrepancy in the observation that blocking either the m6A-writer METTL3 or the m6A-erasers ALKBH5 and FTO suppress AML might be explained at least in part by recent reports that the major substrate of FTO is m6Am, a modification that occurs specifically at the 5’ CAP structure of mRNA. Moreover, FTO might primarily regulate small nuclear RNA (snRNA) rather than mRNAs18,19. More work is needed to fully reconcile these findings.
A highlight of the work by Yankova, Blackaby et al. is the striking anti-leukemic effect of STM2457 in vivo. The authors use both AML patient-derived xenografts (PDX) as well as a primary murine MLL-AF9/Flt3Itd/+ model to test the efficacy of pharmacological inhibition of METTL3. Strikingly, they found that daily injection of STM2457 suppressed AML engraftment and expansion of PDXs with different genetic cancer drivers, and extended the lifespan of recipient mice. Furthermore, these effects correlate with decreased global m6A levels in treated mice as measured by mass-spectrometry as well as reduced protein expression of several m6A-modified mRNAs. Since AML is a cancer with a well-defined cancer stem cell contribution, the authors also examined the effect of STM2457 treatment on the CD93+ cell population and found strongly diminished numbers of these leukemia stem cells upon METTL3 inhibition (Figure 1c). Prolonged survival and comparable molecular effects were also found in the mouse AML model upon treatment with STM2457. The results from secondary cell transplantations treated with STM2457 (or control) provide additional functional support that METTL3 inhibition depletes the leukemic stem cell component and results in impaired AML propagation and extended survival of mice receiving STM2457-treated cells. Gratifyingly, no overt toxicity due to systemic METTL3 inhibition was reported in the STM2457-treated mice.
Overall, these findings represent an important step towards the development of METTL3 inhibitors and highlight the potential of RNA MTases and the epitranscriptome as a largely unexplored territory for cancer drug discovery. Nevertheless, questions remain: why are cancer cells more sensitive to METTL3 inhibition than their non-transformed counterparts? What downstream gene(s) are responsible for the phenotypic effects of METTL3 inhibition? How does METTL3 influence mRNA translation and what is the relevance of altered mRNA stability to its oncogenic role? How is METTL3 expression or its activity dysregulated in AML? Are there particular AML patient subsets or cancer genotypes that are expected to be more responsive to METTL3 inhibition? Which biomarkers might help identify cancer patients for METTL3 inhibitor therapy and monitor responsiveness? Despite these open questions, this landmark study provides a proof-of-concept that pharmacological inhibition of METTL3 and manipulation the m6A epitranscriptome can suppress cancer and substantially extend survival in mouse AML models.. Given this precedent, and considering that there are well over 100 different RNA modifications present on various classes of RNAs including mRNA, rRNA, tRNA, snRNA, and other non-coding RNAs (ncRNA), it is reasonable to speculate that other RNA-modifying enzymes may represent similarly promising cancer drug targets20.
Acknowledgments
Funding: R.I.G. is supported by an Outstanding Investigator Award (R35CA232115) and R01 grant (R01CA233671) from the National Cancer Institute (NCI) of the NIH.
Footnotes
Declaration of interests: J.L. declares no competing interests. R.I.G. is a co-founder and scientific advisory board member of 28–7 Therapeutics, and Theon Therapeutics.
References
- 1.Morera L, Lubbert M & Jung M Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenetics 8, 57 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dominissini D et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–6 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Meyer KD et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149, 1635–46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer KD & Jaffrey SR The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol 15, 313–26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin S, Choe J, Du P, Triboulet R & Gregory RI The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell 62, 335–45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choe J et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 561, 556–560 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vu LP et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 23, 1369–1376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barbieri I et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yankova E et al. Small molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature (2021). [DOI] [PMC free article] [PubMed]
- 10.Moroz-Omori Elena V., Huang Danzhi, Bedi Rajiv Kumar, Cheriyamkunnel Sherry J., Bochenkova Elena, Dolbois Aymeric, Rzeczkowski Maciej D., Wiedmer Lars, Caflisch Amedeo. bioRxiv 2020.09.25.311803; doi: 10.1101/2020.09.25.311803 [DOI]
- 11.Wang X et al. N-methyladenosine-dependent regulation of messenger RNA stability. Nature (2013). [DOI] [PMC free article] [PubMed]
- 12.Zaccara S & Jaffrey SR A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell 181, 1582–1595 e18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paris J et al. Targeting the RNA m(6)A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 25, 137–148 e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 31, 127–141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang Y et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 35, 677–691 e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen C et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell 27, 64–80 e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vu LP, Cheng Y & Kharas MG The Biology of m(6)A RNA Methylation in Normal and Malignant Hematopoiesis. Cancer Discov 9, 25–33 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Mauer J et al. Reversible methylation of m(6)Am in the 5’ cap controls mRNA stability. Nature 541, 371–375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mauer J et al. FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat Chem Biol 15, 340–347 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barbieri I & Kouzarides T Role of RNA modifications in cancer. Nat Rev Cancer 20, 303–322 (2020). [DOI] [PubMed] [Google Scholar]