

Abstract
Halogens have been reported to play a major role in the inhibition of monoamine oxidase (MAO), relating to diverse cognitive functions of the central nervous system. Pyrazoline/halogenated pyrazolines were investigated for their inhibitory activities against human monoamine oxidase-A and -B. Halogen substitutions on the phenyl ring located at the fifth position of pyrazoline showed potent MAO-B inhibition. Compound 3-(4-ethoxyphenyl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazole (EH7) showed the highest potency against MAO-B with an IC50 value of 0.063 µM. The potencies against MAO-B were increased in the order of –F (in EH7) > –Cl (EH6) > –Br (EH8) > –H (EH1). The residual activities of most compounds for MAO-A were > 50% at 10 µM, except for EH7 and EH8 (IC50 = 8.38 and 4.31 µM, respectively). EH7 showed the highest selectivity index (SI) value of 133.0 for MAO-B, followed by EH6 at > 55.8. EH7 was a reversible and competitive inhibitor of MAO-B in kinetic and reversibility experiments with a Ki value of 0.034 ± 0.0067 µM. The molecular dynamics study documented that EH7 had a good binding affinity and motional movement within the active site with high stability. It was observed by MM-PBSA that the chirality had little effect on the overall binding of EH7 to MAO-B. Thus, EH7 can be employed for the development of lead molecules for the treatment of various neurodegenerative disorders.
Keywords: halogenated pyrazolines, monoamine oxidase inhibitors, kinetics, reversibility, molecular dynamics
1. Introduction
Monoamine oxidases (MAOs) are the principal metabolizing enzymes responsible for the oxidative degradation of various biogenic amines that are related to diverse cognitive functions of the central nervous system (CNS) [1]. Considering the difference in the structure, substrate specificity, biological functions, and catalytic mechanism of MAOs, two types of isoforms are available, namely, MAO-A and MAO-B [2]. Ammonia, aldehyde, and hydrogen peroxide are the major intermediate products formed during MAO-based catalyzed oxidative deamination [3]. These can produce substantial issues, such as astrocyte swelling, unbalancing of the signaling process in the neurotransmission, neuronal loss, and mitochondrial dysfunctions [4]. Eventually, these toxic by-products can lead to numerous neurodegenerative disorders, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) [5]. Selegiline is a selective/irreversible MAO-B inhibitor used as an adjuvant therapy with L-DOPA, which is considered a safe therapy for PD [6,7]. The latest research has also documented that potent, reversible dual targeted MAO-B/AChE inhibitors can decrease the production of β-amyloid peptide levels in AD-related neurons [8]. In 2017, safinamide, as the first selective and reversible MAO-B inhibitor approved by the USFDA for PD treatment, along with levodopa, has promising neuroprotective effects on MPTP-treated mice [9,10].
In the exploration of novel, selective, and reversible MAO-B inhibitors, a general blueprint of drug design has been widely accepted recently, and it consists of a molecular framework of two hydrophobic rings of phenyl/heteroaryl, which are separated by an electron-rich and flexible short spacer unit (Figure 1) [11]. Many of the molecules from this diverse class, such as pyrazolines, enamides, carboxamides, and α, β-unsaturated ketones, were identified as potent MAO-B inhibitors [12,13,14,15,16,17,18,19,20,21]. The presence of a rotatable bond and electron-rich Michael acceptor unit in the spacer can efficiently make appropriate orientations and binding affinities in the entrance and substrate cavity of MAO-B. The flexibility of the molecules provides good recognition of aromatic systems of the ligand to the hydrophobic cavity of the inhibitor binding cavity of MAO-B by Pi–Pi stacking or Pi–cation interactions. The binding requirements of the selective MAO-B inhibitors include one or more hydrophobic ring, preferably an aromatic or heteroaromatic nucleus with small lipophilic substituents, such as dimethylamino, ethoxy, methoxy, and halogens. The presence of a hydrogen bond acceptor (HBA) and hydrogen bond donor (HBD) linker with optimal rotatable bonds between these hydrophobic units had a pivotal role in the design of potent MAO-B inhibitors [22,23,24,25,26].
Figure 1.
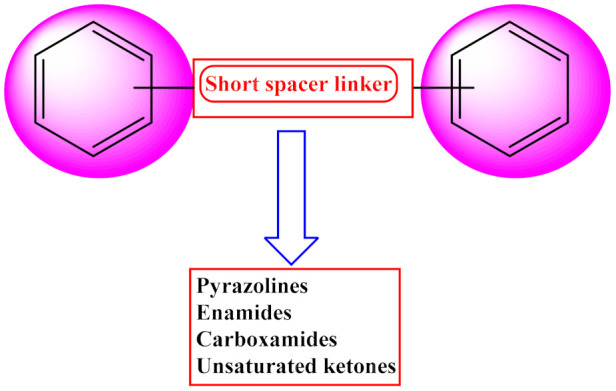
General blueprint of the design of MAO-B inhibitors.
Pyrazoline, or dihydropyrazole, is the partially reduced form of pyrazole, and 2-pyrazoline is the most stable tautomeric from in this class [27]. The presence of an asymmetric carbon atom at the C5 position of pyrazoline was revealed to be due to two epimers of the R and S forms [28]. The ring closure reaction of α, β-unsaturated ketones with hydrazine derivatives in the presence of basic medium is the commonly used synthetic methodology for 2-pyrazolines [29]. The separation of the R and S forms from the racemic mixture of 2-pyrazoline is a considerable task in the identification of biologically more active isomers [30]. Many studies of the literature have documented that the substitution of the 3rd and 5th position of pyrazolines with aryl/heteroaryl units can favor MAO-B inhibition [31]. At the same time, the length and bulkiness of the groups anchoring from the N1 position of pyrazolines can shift the selectivity from MAO-B to MAO-A. The presence of lipophilic groups, such as halogens, hydroxyl, and methoxy groups, on the phenyl ring of the third and fifth positions of the pyrazoline nucleus, can cause significant MAO-B inhibition [32,33,34,35].
Regarding the role of halogens in MAO inhibition, it is generally known that FDA-approved MAO inhibitors like clorgyline, moclobemide, safinamide, and lazabemide are chlorine-containing drugs [36]. The evidence shows that more than 40% of drugs in FDA-approved or clinical trials are in the halogenated form [37]. Recently, our group emphasized the presence and orientation of various halogens on chemical scaffolds, such as chalcones, chromones, coumarins, hydrazothiazoles, and pyrazolines, thereby providing in-depth knowledge on MAO inhibition and B isoform selectivity. It was noted that this can have a new type of impact on the drug–target interaction of these MAO inhibitors in order to fill the small lipophilic pockets in the inhibitor binding cavity (IBC) of MAOs [38].
The objective of the present study was focused on the in vitro evaluation of MAO-A and MAO-B inhibitions of pyrazoline/halogenated pyrazoline derivatives. The lead molecules were further studied for kinetics, reversibility, and blood–brain barrier (BBB) permeation. Detailed molecular dynamics established precisely the protein–ligand binding interactions of the halogenated molecules in their respective active sites of enzyme targets and the role of chirality towards MAO-B inhibition.
2. Results and Discussion
2.1. Inhibitory Activities against MAO Enzymes
At 10 µM, all derivatives in the EH sequence had potent inhibitory action against MAO-B, with residual activities of about 50% (Table 1). Fluorine-containing compound EH7 showed the greatest inhibitory activities against MAO-B with an IC50 value of 0.063 µM, followed by chlorine-containing EH6 (IC50 = 0.40 µM), and bromine-containing EH8 (IC50 = 0.69 µM). Comparing EH1 (IC50 = 5.38 µM) with EH7, the potencies for MAO-B greatly increased, i.e., by 85.4 times, based on their IC50 values, with the presence of –F instead of –H. Most of the compounds weakly inhibited MAO-A at 10 µM, with residual activities of >50%, except EH7 and EH8. Among them, EH7 showed the highest selectivity index (SI) value for MAO-B with 133.0, followed by EH6 with 55.8.
Table 1.
Inhibitions of recombinant human MAO enzymes by compounds of the EH series a.
| Compounds | Residual Activity at 10 µM (%) | IC50 (µM) | SI b | ||||
|---|---|---|---|---|---|---|---|
| MAO-A | MAO-B | AChE | MAO-A | MAO-B | AChE | ||
| EH1 | 99.1 ± 0.27 | 33.7 ± 0.96 | 81.0 ± 6.21 | >40 | 5.38 ± 0.16 | >40 | >7.43 |
| EH6 | 68.1 ± 0.18 | 19.4 ± 1.01 | 63.5 ± 4.99 | 22.3 ± 0.12 | 0.40 ± 0.051 | >40 | 55.8 |
| EH7 | 46.9 ± 1.25 | 4.03 ± 0.53 | 74.0 ± 0.71 | 8.38 ± 0.88 | 0.063 ± 0.0042 | >40 | 133.0 |
| EH8 | 33.5 ± 0.65 | 11.3 ± 0.13 | 53.7 ± 2.15 | 4.31 ± 0.013 | 0.69 ± 0.017 | 10.8 ± 0.15 | 6.3 |
| Toloxatone | 1.08 ± 0.0025 | - | - | ||||
| Lazabemide | - | 0.11 ± 0.016 | - | ||||
| Clorgyline | 0.0070 ± 0.00070 | - | - | ||||
| Pargyline | - | 0.14 ± 0.0059 | - | ||||
| Tacrine | - | - | 0.27 ± 0.019 | ||||
a Results are expressed as the means ± standard errors of duplicate or triplicate experiments. b SI values are expressed for IC50 of MAO-B/IC50 of MAO-A. Values for reference compounds were determined after preincubation for 30 min with enzymes.
The enzyme inhibition results obtained in the current study revealed that all of the halogenated pyrazoline derivatives showed good selective MAO-B inhibitory activity. The study mainly confirmed the effect of halogens on the para-position of the B ring of ethoxylated pyrazolines. Among the halogens, fluorine had the greatest impact on MAO-B inhibition with a sub-micromolar level range. It is worth noting that a highly electronegative fluorine atom-containing compound is two times more potent than marketed drugs for the inhibition of MAO-B, such as reversible-type lazabemide (IC50 = 0.11 µM) and irreversible pargyline (IC50 = 0.14 µM). Recent reports state that the anisotropy of charge circulation around the fluorine atom, which is the reason for the directional and stabilizing property of the active site, proves that halogen enhances the analogy of drugs of new molecules [38].
Recently, we evaluated MAO-B inhibitory activities for ethoxylated head of chalcone derivatives and found that chlorine-, fluorine-, or bromine-containing chalcones had good IC50 values of 0.57, 0.053, and 2.01 µM, respectively [39]. Cyclization with hydrazine hydrate of the same halogenated molecules retained higher MAO-B inhibitory activities than the unsubstituted one. This finding clearly shows the importance of halogen incorporation both in ethoxylated chalcones and their pyrazolines moieties for MAO-B inhibition. Interestingly, in both series, fluorine-containing compounds showed dominant MAO-B inhibition profiles. For the purpose of the development of compounds with good MAO-B inhibitory activity, the bonding of fluorine to the molecule was an auspicious lead.
2.2. Kinetics of MAO Inhibition
Kinetic studies on MAO-A and MAO-B inhibitions by EH6 and EH7 were performed. Lineweaver–Burk plots and secondary plots showed that EH6 was a competitive inhibitor of MAO-A and MAO-B (Figure 2A,C), with Ki values of 10.8 ± 1.03 and 0.19 ± 0.016 µM, respectively (Figure 2B,D). Lineweaver–Burk plots and secondary plots exhibited that EH7 was a competitive inhibitor of MAO-A and MAO-B (Figure 3A,C), with Ki values of 3.64 ± 0.18 and 0.034 ± 0.0067 µM, respectively (Figure 3B,D). These results suggest that EH6 and EH7 are selective and competitive inhibitors of MAO-B.
Figure 2.


Lineweaver–Burk plots for MAO-A and MAO-B inhibitions by EH6 (A,C) and their respective secondary plots (B,D) of slopes vs. inhibitor concentrations.
Figure 3.
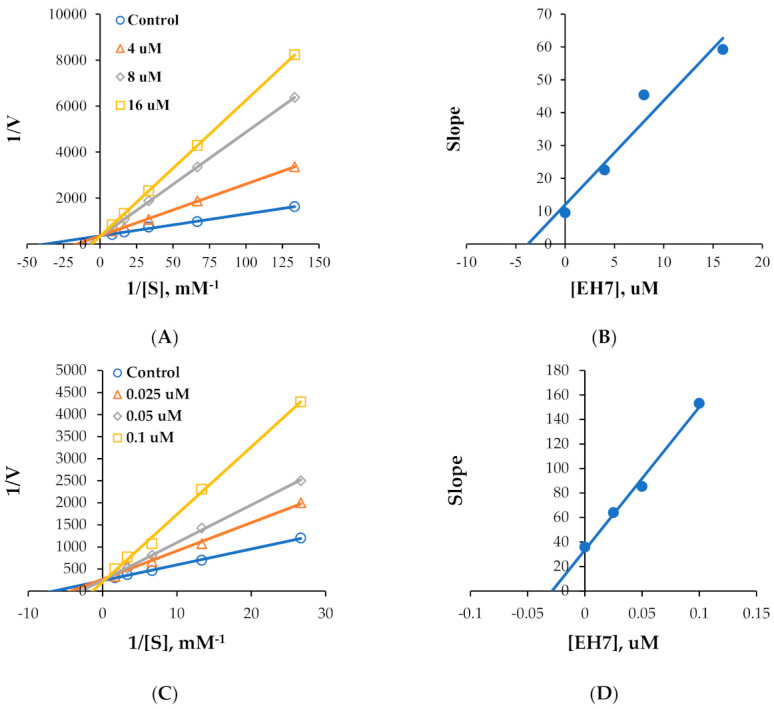
Lineweaver–Burk plots for MAO-A and MAO-B inhibitions by EH7 (A,C) and their respective secondary plots (B,D) of slopes vs. inhibitor concentrations.
2.3. Reversibility Study
Reversibility studies on MAO-B inhibition were carried out on EH7. In these experiments, inhibition of MAO-B by EH7 was recovered from 12.5% (value of AU) to 89.5% (value of AD). The recovery value was similar to that of the reversible reference lazabemide (from 35.3% to 75.7%); however, it was different from that of the irreversible inhibitor pargyline (from 41.3% to 44.9%), which was not recovered (Figure 4). Similar to the reversible reference level, inhibition of MAO-B by EH7 was reversed in these experiments, indicating that EH7 is a reversible inhibitor of MAO-B.
Figure 4.
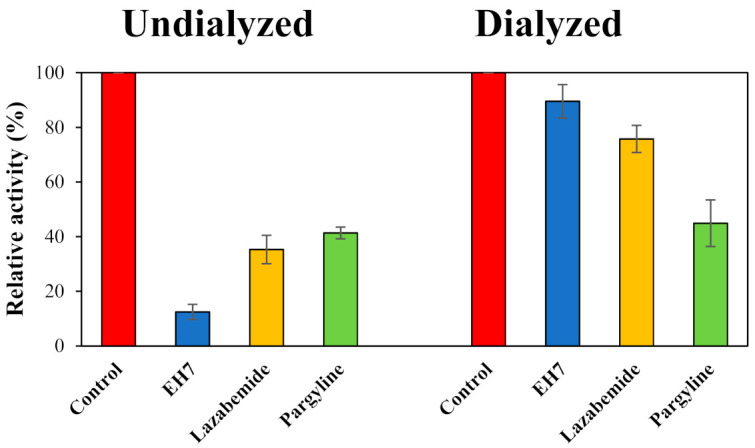
Recovery of MAO-B inhibition by EH7, using dialysis experiments. The concentrations used were EH7 at 0.15 µM, lazabemide (a reversible reference inhibitor) at 0.20 µM, and pargyline (an irreversible reference inhibitor) at 0.30 µM.
2.4. Blood–Brain Barrier (BBB) Permeation Studies
The CNS bioavailability of the molecules was confirmed by a parallel artificial membrane permeability assay (PAMPA) [40]. From the assay, highly effective permeabilities and high CNS bioavailabilities were observed for the halogenated pyrazolines with Pe ranges of 14.13 ~ 14.56 × 10−6 cm/s (Table 2). All halogenated substituted derivatives showed higher CNS permeabilities than unsubstituted ones.
Table 2.
BBB assay of standard and EH compounds.
| Compounds | Bibliography Pe (×10−6 cm/s) a |
Experimental Pe (×10−6 cm/s) |
Prediction |
|---|---|---|---|
| Progesterone | 9.3 | 9.02 ± 0.11 | CNS+ |
| Verapamil | 16.0 | 15.53 ± 0.24 | CNS+ |
| Piroxicam | 2.5 | 2.43 ± 0.30 | CNS+/− |
| Lomefloxacin | 1.1 | 1.12 ± 0.01 | CNS− |
| Dopamine | 0.2 | 0.22 ± 0.01 | CNS− |
| EH1 | 10.34 ± 0.33 | CNS+ | |
| EH6 | 14.26 ± 0.80 | CNS+ | |
| EH7 | 14.13 ± 0.71 | CNS+ | |
| EH8 | 14.56 ± 0.26 | CNS+ |
CNS+ (high): Pe (10−6 cm/s) > 4.00; CNS− (low): Pe (10−6 cm/s) < 2.00; CNS± (uncertain): Pe (10−6 cm/s) from 4.00 to 2.00. a From [40].
2.5. Molecular Dynamics Studies
2.5.1. EH1, EH6, EH7, and EH8 Exert Differential Impacts on the Stability of MAO-A and MAO-B
The root mean square deviation (RMSD) can be used to estimate the conformational stability of a system during a production run [41]. The differential structural perturbatory effect of the ligands on the structure of MAO-B relative to MAO-A was first explored. MAO-B_EH6, MAO-B_EH7, and MAO-B_EH8 attained equilibration at around 20 ns (Figure 5). However, MAO-B_EH1 achieved convergence earlier in the simulation run but eventually elicited a somewhat high structural movement at around 45 ns. In comparison, EH1, EH6, EH7, and EH8 did not elicit similar stabilizing effects on the MAO-A protein; this could be due to the IC50 values experimentally reported, suggesting their low inhibitory potential towards MAO-A. The superior stabilizing effect elicited by the ligands against MAO-B when compared to MAO-A binding points to the high selective targeting of the ligands for MAO-B. As indicated by the IC50, EH7 had the highest stabilizing impact on MAO-B with an average RMSD value of 1.49 Å, while EH1, EH6, and EH8 had 2.29, 1.61, and 1.79 Å, respectively (Table 3).
Figure 5.

Comparative root mean square deviations (RMSDs) of the Cα carbon atoms of the compounds in whole systems for MAO-B (A) and MAO-A (B) and in active sites for MAO-B (C) and for MAO-A (D). (A,C) EH1, black; EH6, green; EH7, blue; and EH8 red. (B,D) EH1, black; EH6, red; EH7, green; and EH8, blue.
Table 3.
Average RMSD, RMSD_AS, RMSF, and IC50 of the Apo and bound systems.
| Complexes | RMSD | RMSD_AS | RMSF | IC50 (µM) |
|---|---|---|---|---|
| MAO-A_EH1 | 3.35 (0.73) | 1.93 (0.18) | 1.47 (1.28) | >40 |
| MAO-A_EH6 | 2.63 (0.72) | 1.44 (0.27) | 1.15 (1.04) | 22.3 |
| MAO-A_EH7 | 2.74 (0.59) | 2.09 (0.29) | 1.07 (0.96) | 8.38 |
| MAO-A_EH8 | 3.26 (0.74) | 1.96 (0.34) | 1.40 (1.28) | 4.31 |
| MAO-B_EH1 | 2.29 (0.34) | 2.85 (0.37) | 0.88 (0.59) | 5.38 |
| MAO-B_EH6 | 1.61 (0.22) | 1.54 (0.14) | 0.83 (0.60) | 0.40 |
| MAO-B_EH7 | 1.49 (0.26) | 1.60 (0.16) | 0.68 (0.72) | 0.063 |
| MAO-B_EH8 | 1.79 (0.24) | 1.70 (0.25) | 0.75 (0.61) | 0.69 |
Zeroing on structural influence of the ligands on the mechanistic behavior of the active site could provide more clues regarding their activity. The RMSD of the MAO-A and MAO-B active sites upon EH1, EH6, EH7, and EH8 binding revealed that EH6 and EH7 had the highest active site-stabilizing effect on MAO-B, with overall average RMSD_AS values of 1.61 and 1.49 Å, respectively.
The orientation and position of a ligand are considerably indicative of how reactive the ligands can be within the active site. The characteristics of the ligands were therefore investigated. Interestingly, while EH7 elicited some motional movement within the active site, which made it flexible enough to interact with crucial amino acid residues, it also exhibited good stability—just enough to be adequately anchored for overall binding (Figure 6).
Figure 6.

Stability plot of compounds EH1, EH6, EH7, and EH8.
2.5.2. EH7 Exhibited Low Residual Motion and Trajectory Movement upon Binding to MAO-A and MAO-B
Principal component analysis (PCA) is a common molecular dynamic simulation analysis used in the estimation of the essential production run of a system when computed on a low-dimensional free energy space [42]. This estimation is associated with correlated vibrational modes; the translation of the MD trajectory towards the geometric center abolishes the initial rotational and translational movements of the atoms. The decomposition of the trajectory along PC1 and PC2 reveals that upon EH7 binding to MAO-B, the system exhibits very low diagonalization when compared with other systems (Figure 7A). This further corroborates the results discussed above in the stability section, which attest to the superior activity of EH7 over other ligands in the series. In contrast, due to the low specificity of EH1, EH6, EH7, and EH8 towards MAO-A, less high trajectory motion was observed in the ligand-bound MAO-A (Figure 7B).
Figure 7.

Principal component analysis of the compounds bound to MAO-B (A) and to MAO-A (B), and root mean square fluctuation (RMSF) of the compounds bound to MAO-B (C) and to MAO-A (D). (A,B) EH1, black; EH6, green; EH7, blue; and EH8, red. (C,D) EH1, black; EH6, green; EH7, blue; and EH8, red.
The root mean square fluctuation (RMSF) computes the residual deviation of a protein over a period of time using a reference position, such as the averaged residual position. RMSF estimates the level of fluctuation of residue from its original position [43]. As depicted in the RMSF plot, the average RMSF values of MAO-B_EH1, MAO-B_EH6, MAO-B_EH7, and MAO-B_EH8 were 0.88, 0.83, 0.68, and 0.75 Å, respectively (Figure 7C), while those of MAO-A_EH1, MAO-A_EH6, MAO-A_EH7, and MAO-A_EH8 were 1.47, 1.15, 1.07, and 1.40 Å, respectively (Figure 7D).
2.5.3. Estimation of Free Binding Energies of EH1, EH6, EH7, and EH8 on MAO-A and MAO-B
The estimation of free binding energy using an MM/PBSA protocol can be employed to gain insight into the models and further obtain the semiquantitative values of their stability. We therefore used a representative snapshot and active site residue decomposition to further explore the time-wise bond interaction occurring between major residues in the active sites of MAO-A/MAO-B and the ligands. MM/PBSA has found useful application in the drug design space used in the estimation of binding affinity between ligands and biomolecules [44].
As indicated in Table 4, EH7 had higher binding affinity (−36.29 kcal/mol) towards MAO-B than the other ligands, while the binding of EH1, EH6, and EH8 was recorded to be −4.11, −11.5, and −5.48 kcal/mol, respectively. Of note is the surprising high binding affinity (−32.55 kcal/mol) of EH7 for MAO-A. This could be due to the high electronegativity potential of fluorine, which is characteristic of compound EH7. In agreement with the IC50, the binding affinity of the compounds also followed the same trend.
Table 4.
The total free binding energy contributions of EH, EH6, EH7, and EH8.
| Complexes | Energy (kcal/mol) | ||||||
|---|---|---|---|---|---|---|---|
| ∆EvdW | ∆Eele | ∆Ggas | ∆GGB | ∆GSA | ∆Gsol | ∆Gbind | |
| MAO-B_EH1 | −6.68 | −34.66 | −35.32 | 35.91 | −1.68 | 35.74 | −4.11 |
| (±0.19) | (±4.28) | (±4.20) | (±3.99) | (±0.01) | (±3.98) | (±0.36) | |
| MAO-B_EH6 | −25.46 | −94.22 | −96.77 | 108.35 | −4.28 | 107.92 | −11.15 |
| (±0.15) | (±2.01) | (±2.06) | (±2.64) | (±0.01) | (±2.63) | (±0.88) | |
| MAO-B_EH7 | −36.79 | −32.46 | −85.96 | 55.09 | −5.12 | 49.67 | −36.29 |
| (±0.15) | (±0.54) | (±0.53) | (±0.50) | (±0.01) | (±0.50) | (±0.13) | |
| MAO-B_EH8 | −36.79 | −101.78 | −105.46 | 106.54 | − 5.35 | 106 | −5.48 |
| (±0.15) | (±1.73) | (±1.74) | (±1.56) | (±0.01) | (±1.56) | (±0.37) | |
| MAO-A_EH1 | −33.17 | −12.98 | −13.31 | −12.85 | −4.65 | 12.81 | −5.06 |
| (±0.22) | (±4.07) | (±4.20) | (±2.86) | (±0.01) | (±2.85) | (±1.49) | |
| MAO-A_EH6 | −36.75 | −14.50 | −14.88 | 13.78 | −4.86 | 13.73 | −11.33 |
| (±0.18) | (±2.36) | (±2.39) | (±1.61) | (±0.01) | (±1.61) | (±0.97) | |
| MAO-A_EH7 | −32.75 | −13.21 | −13.54 | −36.79 | −4.85 | 13.21 | −32.55 |
| (±0.15) | (±2.06) | (±2.06) | (±0.15) | (±0.01) | (±1.36) | (±1.07) | |
| MAO-A_EH8 | −7.52 | −12.07 | −12.14 | 12.92 | −2.61 | 12.89 | −7.48 |
| (±0.15) | (±2.49) | (±2.56) | (±2.30) | (±0.01) | (±0.15) | (±0.15) | |
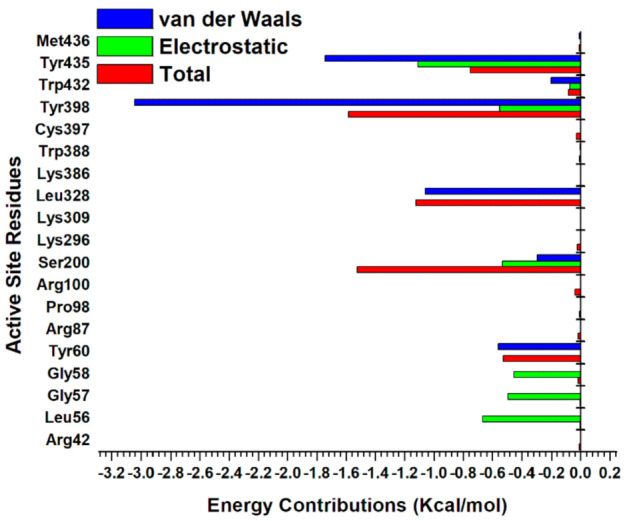
The individual energy contributions of the active sites were determined using the MM/PBSA per residue energy decomposition protocol [45]. The high binding energy exhibited by EH7 was contributed by electrostatic interactions of Arg42, Arg87, Arg98, Lys296, and Lys386, with energy values of −28.4, −12.8, −15.3, −41.9, and −20.6 kcal/mol, respectively (Figure 8C). In the interaction between compound EH6 and MAO-B, residues Arg42, Gly58, Tyr60, Arg87, Pro98, and Arg100 contributed an electrostatic energy of −2.68, −0.04, −0.12, −15.17, −0.14, and −17.27 kcal/mol, respectively (Figure 8B). Despite the electrostatic interactions and other favorable bonds elicited by EH1, EH6, and EH8, compound EH7 still had the best binding energy and highest contribution of electrostatic interactions. This behavior may not be unrelated to the impact of fluorine found on compound EH7. Furthermore, the energetically favored disposition assumed by EH7 could be attributed to the hydrogen bond formed between the fluorine of EH7 and hydrogen of Trp386 (Figure 9C). The ligand interaction profile of the interaction of EH1 (A), EH6 (B), EH7 (C), and EH8 (D) with MAO-B is shown in Figure 9.
Figure 8.

Per residue decomposition of EH1 bound to MAO-B (A), EH6 bound to MAO-B (B), EH7 bound to MAO-B (C), and EH8 bound to MAO-B (D) systems.
Figure 9.

Ligand interaction profile of the interaction of EH1 (A), EH6 (B), EH7 (C), and EH8 (D) with MAO-B.
2.5.4. Estimation of Free Binding Energies of EH1, EH6, EH7, and EH8 on MAO-A and MAO-B
Since our compounds contain a pyrazoline moiety, it is assumed that they possess an asymmetric carbon atom and, hence, exist as enantiomers. We therefore generated the R and S enantiomers of EH7 as a representative structure of the compounds, taking into consideration the fact that it has the best IC50 and total free energy values. We explored the differential binding and estimated the total free binding energy of the R isomeric form of EH7 (MAO-B_EH7R) and its S counterpart (MAO-B_EH7S) using MM/PBSA calculations. Our calculations revealed that MAO-B_EH7R and MAO-B_EH7S possess binding energies of −36.29 and −36.17 kcal/mol (Table 5). This suggests that the chirality does not change considerably in the overall binding of EH7 to MAO-B. However, MAO-B_EH7S has more van der Waals interactions than MAO-B_EH7R. Despite having total free binding energy values that are close, dissimilar residues present in the active sites are responsible for the binding interactions. Chiefly among the residues are Tyr435, Tyr398, and Leu328 (jointly shared by MAO-B_EH7R), which contribute van der Waals binding energies of −2.93, −1.58, and −1.00 kcal/mol, respectively (Figure 10).
Table 5.
The differential free binding energy of MAO-B_EH7R and MAO-B_EH7S enantiomers.
| Energy (kcal/mol) | |||||||
|---|---|---|---|---|---|---|---|
| Complexes | ∆EvdW | ∆Eele | ∆Ggas | ∆GGB | ∆GSA | ∆Gsol | ∆Gbind |
| MAO-B_EH7R | −36.79 | −32.46 | −85.96 | 55.09 | −5.12 | 49.67 | −36.2 |
| (±0.15) | (±0.54) | (±0.53) | (±0.50) | (±0.01) | (±0.50) | (±0.13) | |
| MAO-B_EH7S | −53.42 | −17.59 | −22.93 | 19.88 | −5.62 | 19.31 | −36.17 |
| (±0.08) | (±1.56) | (±0.50) | (±1.49) | (±0.01) | (±0.47) | (±0.12) | |
Figure 10.
Per residue decomposition of MAO-B_EH7S enantiomer.
2.6. Target Predicition and ADME Analysis
The biological target and ADME predictions of the lead molecule of EH7 were assessed by the online web-based program SwissTarget. The lead molecule showed good prediction results on biological targets of MAO and AChE (Figure 11A). The bioavailability radar gave saturation, size, flexibility, insolubility, and polarity of the molecule, which provided a quick judgement of the drug-likeness of the molecule. The lead molecule was recognized to be within the pink region (optimal ranges) and could be considered a compound endowed with drug-like characteristics (Figure 11B). The BOILED-Egg model also predicted the passive human BBB permeation and gastrointestinal absorption (HIA) for the compound (Figure 11C). The results revealed that EH7 had comparatively higher probability of BBB and HIA.
Figure 11.

Swiss target/ADME prediction: (A) Biological target prediction of lead molecule EH7; (B) bioavailability radar of EH7; (C) the BOILED-Egg construction of EH7.
3. Materials and Methods
3.1. Synthesis
A series of ethoxylated chalcone (0.0078 M) was refluxed with (0.0629 M) hydrazine hydrate under ethanol medium. After 14–16 h of reflux, the product was cooled, acidified with dil. HCl, washed thoroughly with water, filtered, dried, and recrystallized from ethanol. The progress of the formation of pyrazoline product was monitored by thin-layer chromatography (TLC; ethylacetate–hexane = 1:9). The synthetic route is depicted in Scheme 1.
Scheme 1.
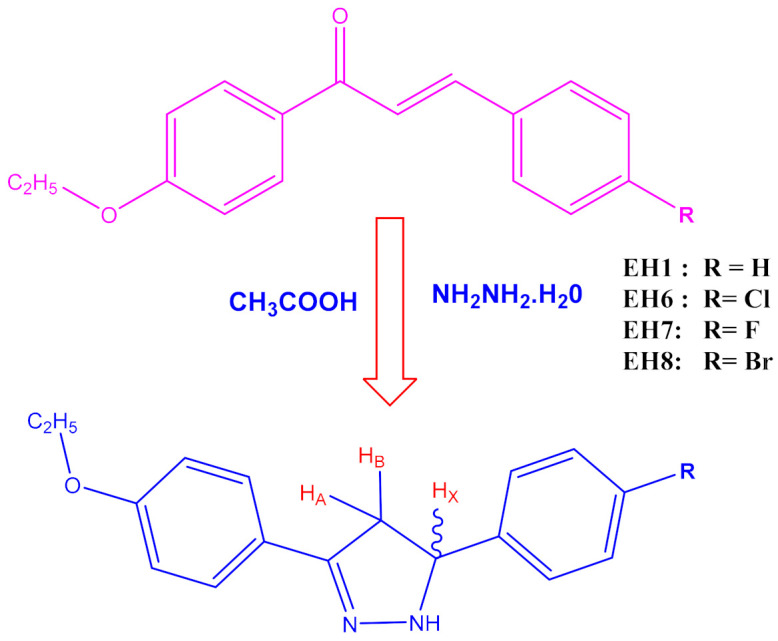
Synthetic route of the compounds under the present study.
3-(4-ethoxyphenyl)-5-phenyl-4,5-dihydro-1H-pyrazole (EH1): 1H NMR (400 MHz, CDCl3) δ: 1.47–1.45 (t, 3H, J = 8.0 Hz, CH3), 4.15–4.13 (d, 2H, J = 8.0 Hz, CH2), 3.76 (d, 1H, HA), 4.02 (d, 1H, HB), 5.51 (d, 1H, HX), 7.89–7.10 (d, 9H, ArH), 9.21(S, NH). MS (ESI) m/z calcd. for C17H18N2O: 266.3376, found 266.3345.
5-(4-chlorophenyl)-3-(4-ethoxyphenyl)-4,5-dihydro-1H-pyrazole (EH6): 1H NMR (400 MHz, CDCl3) δ: 1.47–1.45 (t, 3H, J = 8.0 Hz, CH3), 4.14–4.12 (D, 2H, J = 8.0 Hz, CH2), 3.75 (d, 1H, HA), 4.01 (d, 1H, HB), 5.51 (d, 1H, HX), 7.99–7.08 (d, 8H, ArH) 9.24 (S, NH). MS (ESI) m/z calcd. for C17H17N2OCl: 284.3280, found 284.3276.
3-(4-ethoxyphenyl)-5-(4-fluorophenyl)-4,5-dihydro-1H-pyrazole (EH7): 1H NMR (400 MHz, CDCl3) δ: 1.47–1.45 (t, 3H, J = 8.0 Hz, CH3), 4.15–4.13 (d, 2H, J = 8.0 Hz, CH2), 3.72 (d, 1H, HA), 4.04 (d, 1H, HB), 5.51 (d, 1H, HX), 7.96–7.03 (d, 8H, ArH) 9.23 (S, NH). MS (ESI) m/z calcd. for C17H17N2OF: 266.3376, found 266.3345.
5-(4-bromophenyl)-3-(4-ethoxyphenyl)-4,5-dihydro-1H-pyrazole (EH8):1H NMR (400 MHz, CDCl3) δ: 1.47–1.45 (t, 3H, J = 8.0 Hz, CH3), 4.15–4.13 (q, 2H, J = 8.0 Hz, CH2), 3.76 (d, 1H, HA), 4.02 (d, 1H, HB), 5.51 (d, 1H, HX), 7.89–7.70 (d, 8H, ArH), 9.24(S, NH). C17H17N2OBr: 345.2336, found 345.2340.
3.2. Enzyme Assays
MAO activities were assayed as described previously, using recombinant human MAO-A and MAO-B and kynuramine (0.06 mM) and benzylamine (0.3 mM) as substrates, respectively; both substrate concentrations were 1.5 × Km (Km = 0.041 and 0.20 mM, respectively). The enzymes and chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) [17].
3.3. Analysis of Enzyme Inhibitions and Kinetics
The inhibitory activities of the four compounds against MAO-A and MAO-B were first observed at a concentration of 10 µM. IC50 values for MAO-B by compounds showing residual activities of <50% were determined. Kinetic studies were performed on the most potent inhibitors, i.e., EH6 and EH7 for MAO-A and MAO-B, at five concentrations of the substrates and three inhibitor concentrations, as previously described [18].
3.4. Analysis of Inhibitor Reversibilities
Reversibilities of EH7 were analyzed using a dialysis method after preincubating with MAO-B for 30 min, as previously described. The concentrations used were EH7 at 0.15 µM, lazabemide (a reversible MAO-B reference inhibitor) at 0.20 µM, and pargyline (an irreversible MAO-B reference inhibitor) at 0.30 µM. The relative activities for undialyzed (AU) and dialyzed (AD) samples were compared to determine the reversibility patterns [19].
3.5. Molecular Dynamics Study
The 3D structures of MAO-A (PDB ID: 2Z5X and MAO-B: 2V5Z), hereafter referred to as MAO-A and MAO-B, respectively, were retrieved from the Protein Data Bank (PDB) [46,47]. Molecules that were co-crystallized with MAO-A and MAO-B were deleted with the exception of the co-enzyme FAD. Missing residues were filled with the aid of MODELLER tools found in the graphic user interface of Chimera [48,49]. Marvin Sketch was employed to draw the 2D structures of EH1, EH6, EH7, and EH8; afterwards, geometrical and structural optimizations of these ligands were carried out using B3LYP/6-311 ++G (d,p) and the UFF forcefield of Gaussian 16, respectively [50]. Active site identification was achieved by centering the grid box around a co-crystallized ligand found on the MAO-A and MAO-B proteins downloaded from PDB. Molecular docking was then carried out using AutoDock Vina [51]. The systems were then subjected to a simulation run of 50 ns using Amber18 software (San Francisco, CA, USA) [52]. Eight systems were set up for simulation (MAO-A:EH1, MAO-A:EH6, MAO-A:EH7, MAO-A:EH8, MAO-B:EH1, MAO-B:EH6, MAO-B:EH7, and MAO-B:EH8). The simulation process was carried out in accordance with an in-house protocol. The CPPTRAJ module of Amber18 software was used to analyze the trajectories emanating from the simulation process [53,54].
3.6. Target Predicition and ADME Analysis
The biological target and ADME predictions of the lead molecule of EH7 were assessed at http://www.swisstargetprediction.ch (access on 23 May 2021) [55,56].
4. Conclusions
A series of four halogenated pyrazolines with ethoxylated heads (EH1, EH6, EH7, and EH8) was synthesized from their corresponding chalcones. All four compounds were explored for their MAO-A and MAO-B inhibitory activities. On the basis of previous studies regarding the role of halogens in various scaffolds with MAO inhibition activities, we mainly focused on the effect of various halogens on the pyrazoline core with the ethoxylated head. According to the results of this study, the synthesized halogenated pyrazolines exhibited good selective MAO-B inhibition. Fluorine-containing pyrazoline (EH7) exhibited an improved selectivity profile against the MAO-B isoform. Kinetics and reversibility studies revealed that EH7 is a selective and competitive inhibitor of MAO-B, with a recovery value similar to that of the reversible reference drug lazabemide. Pyrazolines are said to be optically active compounds, and, from the MM/PBSA values obtained, it can be clearly observed that chirality has minimal influence on the binding of the compound to MAO-B. It may be suggested that the absence of bulky groups on the N1 position of pyrazoline limited the role of chirality in the binding mode of the lead molecule in the current study.
Acknowledgments
Authors acknowledge Taif University Researchers Supporting Project number (TURSP-2020/68), Taif University, Taif, Saudi Arabia.
Author Contributions
Conceptualization: L.K.P., B.M., and H.K.; biological activity: J.-M.O., A.K., and M.A.A.; kinetics: J.-M.O.; molecular dynamics: O.S. and M.E.S.; synthesis: A.S.N., V.P.K., S.K., S.T.S., and B.M.; data curation: B.M. and J.-M.O.; writing—original draft preparation: J.-M.O. and B.M.; writing—review and editing: L.K.P., B.M., and H.K.; supervision: H.K.; funding acquisition: H.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Research Foundation of Korea (NRF) grant provided by the Korean Government (NRF-2019R1A2C1088967 to H. Kim), Republic of Korea, and by a grant from Amrita Vishwa Vidyapeetham University (Seed Grant Number K-PHAR-20-628 to B. Mathew).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors have no conflict of interest to declare.
Sample Availability
Samples of the compounds are available from the authors.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Carradori S., Petzer J.P. Novel monoamine oxidase inhibitors: A patent review (2012−2014) Expert Opin. Ther. Pat. 2015;25:91–110. doi: 10.1517/13543776.2014.982535. [DOI] [PubMed] [Google Scholar]
- 2.Youdim M.B., Bakhle Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson´s disease and depressive illness. Br. J. Pharm. 2006;147:S287–S296. doi: 10.1038/sj.bjp.0706464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramsay R.R. Inhibitor design for monoamine oxidases. Curr. Pharm. Des. 2013;19:2529–2539. doi: 10.2174/1381612811319140004. [DOI] [PubMed] [Google Scholar]
- 4.Tripathi R.K.P., Ayyannan S.R. Monoamine oxidase-B inhibitors as potential neurotherapeutic agents: An overview and update. Med. Res. Rev. 2019;39:1603–1706. doi: 10.1002/med.21561. [DOI] [PubMed] [Google Scholar]
- 5.Guglielmi P., Carradori S., Ammazzalorso A., Secci D. Novel approaches to the discovery of selective human monoamine oxidase-B inhibitors: Is there room for improvement? Expert Opin. Drug Discov. 2019;14:995–1035. doi: 10.1080/17460441.2019.1637415. [DOI] [PubMed] [Google Scholar]
- 6.Kumar B., Sheetal S., Mantha A.K., Kumar V. Recent developments on the structure-activity relationship studies of MAO inhibitors and their role in different neurological disorders. RSC Adv. 2016;6:42660–42683. doi: 10.1039/C6RA00302H. [DOI] [Google Scholar]
- 7.Kumar B., Gupta V.P., Kumar V. A perspective on monoamine oxidase enzyme as drug target. Challenges and opportunities. Curr. Drug Targets. 2017;18:87–97. doi: 10.2174/1389450117666151209123402. [DOI] [PubMed] [Google Scholar]
- 8.Tipton K. 90 years of monoamine oxidase: Some progress and some confusion. J. Neural Transm. 2018;125:1519–1551. doi: 10.1007/s00702-018-1881-5. [DOI] [PubMed] [Google Scholar]
- 9.Leuratti C., Sardina M., Ventura P., Assandri A., Muller M., Brunner M. Disposition and metabolism of safinamide, a novel drug for Parkinson’s disease, in healthy male volunteers. Pharmacology. 2013;92:7–16. doi: 10.1159/000354805. [DOI] [PubMed] [Google Scholar]
- 10.Mao Q., Qin W., Zhang A., Ye N. Recent advances in dopaminergic strategies for the treatment of Parkinson’s disease. Acta Pharmacol. Sin. 2020;41:471–482. doi: 10.1038/s41401-020-0365-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hagenow J., Hagenow S., Grau K., Khanfar M., Hefke L., Proschak E., Stark H. Reversible small molecule inhibitors of MAO A and MAO B with anilide motifs. Drug Des. Dev. Ther. 2020;14:371–393. doi: 10.2147/DDDT.S236586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathew B., Suresh J., Anbazhagan S., Mathew G.E. Pyrazoline. A promising scaffold for the inhibition of monoamine oxidase. Cent. Nerv. Syst. Agents Med. Chem. 2013;13:195–206. doi: 10.2174/1871524914666140129122632. [DOI] [PubMed] [Google Scholar]
- 13.Matos M.J., Vina D., Vazquez-Rodriquez S., Uriarte E., Santana L. Focusing on new monoamine oxidase inhibitors. Differently substituted coumarins as an interesting scaffold. Curr. Top. Med. Chem. 2012;12:2210–2233. doi: 10.2174/156802612805220002. [DOI] [PubMed] [Google Scholar]
- 14.Kavully F.S., Oh J.M., Dev S., Kaipakasseri S., Palakkathondi A., Vengamthodi A., Azeez R.F.A., Tondo A.R., Nicolotti O., Kim H., et al. Design of enamides as new selective monoamine oxidase-B inhibitors. J. Pharm. Pharmacol. 2020;72:916–926. doi: 10.1111/jphp.13264. [DOI] [PubMed] [Google Scholar]
- 15.Chimenti F., Secci D., Bolasco A., Chimenti P., Bizzarri B., Granese A., Carradori S., Yáñez M., Orallo F., Ortuso F., et al. Synthesis, molecular modeling, and selective inhibitory activity against human monoamine oxidases of 3-carboxamido-7-substituted coumarins. J. Med. Chem. 2009;52:1935–1942. doi: 10.1021/jm801496u. [DOI] [PubMed] [Google Scholar]
- 16.Gaspar A., Texeira F., Uriarte E., Milhazes N., Melo A., Cordeiro M.N.D.S., Ortuso F., Alcaro S., Borges F. Towards the discovery of novel class of monoamine oxidase inhibitors: Structure property activity and docking studies on chromones amides. Chem. Med. Chem. 2011;6:628–632. doi: 10.1002/cmdc.201000452. [DOI] [PubMed] [Google Scholar]
- 17.Parambi D.G.T., Oh J.M., Baek S.C., Lee J.P., Tondo A.R., Nicolotti O., Hoon K., Mathew B. Design, synthesis and biological evaluation of oxygenated chalcones as potent and selective MAO-B inhibitors. Bioorg. Chem. 2019;93:103335. doi: 10.1016/j.bioorg.2019.103335. [DOI] [PubMed] [Google Scholar]
- 18.Reeta, Baek S.C., Lee J.P., Rangarajan T.M., Ayushee, Singh R.P., Singh M., Mangiatordi G.F., Nicolotti O., Kim H., et al. Ethyl acetohydroxamate incorporated chalcones: Unveiling a novel class of chalcones for multitarget monoamine oxidase-B inhibitors against Alzheimer’s disease. CNS Neurol. Disord. Drug Targets. 2019;8:643–654. doi: 10.2174/1871527318666190906101326. [DOI] [PubMed] [Google Scholar]
- 19.Oh J.M., Rangarajan T.M., Chaudhary R., Singh R.P., Singh M., Singh R.P., Tondo A.R., Gambacorta N., Nicolotti O., Mathew B., et al. Novel class of chalcone oxime ethers as potent monoamine oxidase-B and acetylcholinesterase inhibitors. Molecules. 2020;25:2356. doi: 10.3390/molecules25102356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathew B., Baek S.C., Parambi D.G.T., Lee J.P., Mathew G.E., Jayanthi S., Devaraji D., Raphael C., Vinod D., Kondarath S.S., et al. Potent and highly selective dual-targeting monoamine oxidase-B inhibitors: Fluorinated chalcones of morpholine versus imidazole. Arch. Pharm. 2019;352:e1800309. doi: 10.1002/ardp.201800309. [DOI] [PubMed] [Google Scholar]
- 21.Guglielmi P., Mathew B., Secci D., Carradori S. Chalcones: Unearthing their therapeutic possibility as monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2020;205:112650. doi: 10.1016/j.ejmech.2020.112650. [DOI] [PubMed] [Google Scholar]
- 22.Mathew B., Parambi D.G.T., Mathew G.E., Uddin M.S., Inasu S.T., Kim H., Marathakam A., Unnikrishnan M.K., Carradori S. Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch. Pharm. 2019;352:e1900177. doi: 10.1002/ardp.201900177. [DOI] [PubMed] [Google Scholar]
- 23.Finberg J.P.M., Rabey J.M. Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology. Front. Pharmacol. 2016;7:340. doi: 10.3389/fphar.2016.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carradori S., Silvestri R. New frontiers in selective human MAO-B inhibitors. J. Med. Chem. 2015;58:6717–6732. doi: 10.1021/jm501690r. [DOI] [PubMed] [Google Scholar]
- 25.Gaspar A., Silva T., Yáñez M., Viña D., Orallo F., Ortuso F., Uriarte E., Alcaro S., Borges F. Chromone, a privileged scaffold for the development of monoamine oxidase inhibition. J. Med. Chem. 2011;54:5165–5173. doi: 10.1021/jm2004267. [DOI] [PubMed] [Google Scholar]
- 26.Reis J., Cagide F., Chavarria D., Silva T.B., Fernandes C., Gaspar A., Uriarte E., Remiao F., Alcaro S., Ortuso F., et al. Discovery of new chemical entities of old targets. Insight on the lead optimization of chromones based monoamine oxidase B inhibitors. J. Med. Chem. 2016;59:5879–5893. doi: 10.1021/acs.jmedchem.6b00527. [DOI] [PubMed] [Google Scholar]
- 27.Shaaban M.R., Mayhoub A.S., Farag A.M. Recent advances in the therapeutic applications of pyrazolines. Expert Opin. Ther. Patents. 2012;22:253–291. doi: 10.1517/13543776.2012.667403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bansal R., Singh R. Steroidal pyrazolines as a promising scaffold in drug discovery. Future Med. Chem. 2020;12:949–959. doi: 10.4155/fmc-2019-0325. [DOI] [PubMed] [Google Scholar]
- 29.Badavath V.N., Baysal I., Ucar G., Sinha B.N., Jayaprakash V. Monoamine oxidase inhibitory activity of novel pyrazoline analogues: Curcumin based design and synthesis. ACS Med. Chem. Lett. 2016;7:56–61. doi: 10.1021/acsmedchemlett.5b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goksen U.S., Sarigul S., Bultinck P., Herrebout W., Dogan I., Yelekci K., Ucar G., Gokhan Kelekci N. Absolute configuration and biological profile of pyrazoline enantiomers as MAO inhibitory activity. Chirality. 2019;31:21–33. doi: 10.1002/chir.23027. [DOI] [PubMed] [Google Scholar]
- 31.Vishnu Nayak B., Ciftci-Yabanoglu S., Jadav S.S., Jagrat M., Sinha B.N., Ucar G., Jayaprakash V. Monoamine oxidase inhibitory activity of 3,5-biaryl-4,5-dihydro-1H-pyrazole-1-carboxylate derivatives. Eur. J. Med. Chem. 2013;69:762–767. doi: 10.1016/j.ejmech.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 32.Tong X., Chen R., Zhang T.T., Han Y., Tang W.J., Liu X.H. Design and synthesis of novel 2-pyrazoline-1-ethanone derivatives as selective MAO inhibitors. Bioorg. Med. Chem. 2015;23:515–525. doi: 10.1016/j.bmc.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 33.Evranos-Aksoz B., Ucar G., Tas S.T., Aksoz E., Yelekci K., Erikci A., Sara Y., Iskit A.B. New Human Monoamine Oxidase A Inhibitors with Potential Anti- Depressant Activity: Design, Synthesis, Biological Screening and Evaluation of Pharmacological Activity. Comb. Chem. High Throughput Screen. 2017;20:461–473. doi: 10.2174/1386207320666170504113158. [DOI] [PubMed] [Google Scholar]
- 34.Guglielmi P., Carradori S., Poli G., Secci D., Cirilli R., Rotondi G., Chimenti P., Petzer A., Petzer J.P. Design, Synthesis, Docking Studies and Monoamine Oxidase Inhibition of a Small Library of 1-acetyl- and 1-thiocarbamoyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazoles. Molecules. 2019;24:484. doi: 10.3390/molecules24030484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hitge R., Smit S., Petzer A., Petzer J.P. Evaluation of nitrocatechol chalcone and pyrazoline derivatives as inhibitors of catechol-O-methyltransferase and monoamine oxidase. Bioorg. Med. Chem. Lett. 2020;30:127188. doi: 10.1016/j.bmcl.2020.127188. [DOI] [PubMed] [Google Scholar]
- 36.Bolasco A., Carradori S., Fioravanti R. Focusing on new monoamine oxidase inhibitors. Expert Opin. Ther. Patents. 2010;20:909–939. doi: 10.1517/13543776.2010.495716. [DOI] [PubMed] [Google Scholar]
- 37.Fang W.Y., Ravindar L., Rakesh K.P., Manukumar H.M., Shantharam C.S., Alharbi N.S., Qin H.L. Synthetic approaches and pharmaceutical application of chlorine containing molecules for drug discovery: A critical review. Eur. J. Med. Chem. 2019;173:117–153. doi: 10.1016/j.ejmech.2019.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathew B., Carradori S., Guglielmi P., Uddin M.S., Kim H. New aspects of monoamine oxidase B inhibitors. The key role of halogens to open the golden door. Curr. Med. Chem. 2021;28:266–283. doi: 10.2174/0929867327666200121165931. [DOI] [PubMed] [Google Scholar]
- 39.Lakshminarayanan B., Baek S.C., Lee J.P., Kannappan N., Mangiatordi G.F., Nicolotti O., Subburaju T., Kim H., Mathew B. Ethoxylated head of chalcones as a new class of multi-targeted MAO inhibitors. ChemistrySelect. 2019;4:6614–6619. doi: 10.1002/slct.201901093. [DOI] [Google Scholar]
- 40.Di L., Kerns E.H., Fan K., McConnell O.J., Carter G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003;38:223–232. doi: 10.1016/S0223-5234(03)00012-6. [DOI] [PubMed] [Google Scholar]
- 41.Pitera J.W. Expected distributions of root-mean-square positional deviations in proteins. J. Phys. Chem. B. 2014;118:6526–6530. doi: 10.1021/jp412776d. [DOI] [PubMed] [Google Scholar]
- 42.David C.C., Jacobs D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014;1084:193–226. doi: 10.1007/978-1-62703-658-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martínez L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS ONE. 2015;10:e0119264. doi: 10.1371/journal.pone.0119264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soremekun O.S., Olotu F.A., Agoni C., Soliman M.E.S. Drug promiscuity: Exploring the polypharmacology potential of 1, 3, 6-trisubstituted 1, 4-diazepane-7-ones as an inhibitor of the ‘god father’ of immune checkpoint. Comput. Biol. Chem. 2019;80:433–440. doi: 10.1016/j.compbiolchem.2019.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Tubert-Brohman I., Sherman W., Repasky M., Beuming T. Improved docking of polypeptides with glide. J. Chem. Inf. Model. 2013;53:1689–1699. doi: 10.1021/ci400128m. [DOI] [PubMed] [Google Scholar]
- 46.Son S.Y., Ma J., Kondou Y., Yoshimura M., Yamashita E., Tsukihara T. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA. 2008;105:5739–5744. doi: 10.1073/pnas.0710626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Binda C., Wang J., Pisani L., Caccia C., Carotti A., Salvati P., Edmondson D.E. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007;50:5848–5852. doi: 10.1021/jm070677y. [DOI] [PubMed] [Google Scholar]
- 48.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 50.Weedbrook C., Pirandola S., García-Patrón R., Cerf N.J., Ralph T.C., Shapiro J.H., Lloyd S. Gaussian Quantum Information. Rev. Mod. Phys. 2012;84:621. doi: 10.1103/RevModPhys.84.621. [DOI] [Google Scholar]
- 51.Morris G.M., Goodsell D.S., Pique M.E., Lindstrom L., Heuy R., Forli S., Hart W.E., Halliday S., Belew R., Olson A.J. AutoDock Version 4.2. User Guide. [(accessed on 27 May 2021)];2009 :1–49. Available online: http://autodock.scripps.edu/faqs-help/manual/autodock-4-2-user-guide/AutoDock4.2_UserGuide.pdf.
- 52.Lee T.S., Cerutti D.S., Mermelstein D., Lin C., LeGrand S., Giese T.J., Roitberg A., Case D.A., Walker R.C., York D.M. GPU-Accelerated Molecular Dynamics and Free Energy Methods ion Amber18: Performance Enhancements and New Features. J. Chem. Inf. Model. 2018;58:2043–2050. doi: 10.1021/acs.jcim.8b00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soremekun O.S., Olotu F.A., Agoni C., Soliman M.E.S. Recruiting monomer for dimer formation: Resolving the antagonistic mechanisms of novel immune check point inhibitors against Programmed Death Ligand-1 in cancer immunotherapy. Mol. Simul. 2019;45:777–789. doi: 10.1080/08927022.2019.1593977. [DOI] [Google Scholar]
- 54.Soremekun O.S., Soliman M.E.S. From genomic variation to protein aberration: Mutational analysis of single nucleotide polymorphism present in ULBP6 gene and implication in immune response. Comput. Biol. Med. 2019;111:103354. doi: 10.1016/j.compbiomed.2019.103354. [DOI] [PubMed] [Google Scholar]
- 55.Daina A., Michielin O., Zoete V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019;47:W357–W364. doi: 10.1093/nar/gkz382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daina A., Zoete V. A boiled-egg too predict gastrointestinal absorption and brain penetration of small molecules. Chem. Med. Chem. 2016;11:1117–1121. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]