

Abstract
Blockade of the programmed cell death 1 (PD-1)/programmed cell death-ligand 1 (PD-L1) interaction is currently the focus in the field of cancer immunotherapy, and so far, several monoclonal antibodies (mAbs) have achieved encouraging outcomes in cancer treatment. Despite this achievement, mAbs-based therapies are struggling with limitations including poor tissue and tumor penetration, long half-life time, poor oral bioavailability, and expensive production costs, which prompted a shift towards the development of the small-molecule inhibitors of PD-1/PD-L1 pathways. Even though many small-molecule inhibitors targeting PD-1/PD-L1 interaction have been reported, their development lags behind the corresponding mAb, partly due to the challenges of developing drug-like small molecules. Herein, we report the discovery of a series of novel inhibitors targeting PD-1/PD-L1 interaction via structural simplification strategy by using BMS-1058 as a starting point. Among them, compound A9 stands out as the most promising candidate with excellent PD-L1 inhibitory activity (IC50 = 0.93 nM, LE = 0.43) and high binding affinity to hPD-L1 (KD = 3.64 nM, LE = 0.40). Furthermore, A9 can significantly promote the production of IFN-γ in a dose-dependent manner by rescuing PD-L1 mediated T-cell inhibition in Hep3B/OS-8/hPD-L1 and CD3-positive T cells co-culture assay. Taken together, these results suggest that A9 is a promising inhibitor of PD-1/PD-L1 interaction and is worthy for further study.
Keywords: small molecule PD-1/PD-L1 inhibitor, PD-1/PD-L1, structural simplification strategy
1. Introduction
Programmed cell-death receptor (PD-1) and programmed cell death-ligand 1 (PD-L1) inhibitors have achieved significant success in the oncology community both preclinically and clinically [1,2,3]. PD-L1, typically expressed on the surface of tumor cells, is bound to PD-1 on tumor-infiltrating lymphocytes, thereby inhibiting T-cell functions and consecutively inducing tumor immune escape [4,5,6,7]. Consequently, blocking PD-1/PD-L1 interactions has become a prospective approach in cancer treatment, which can restore immune system and elicit a favorable tumor-specific T cells response [8]. In the past few years, several monoclonal antibodies (mAbs) targeting PD-1 or PD-L1 have been approved in clinical use by the U.S. Food and Drug Administration (FDA) [9], exhibiting significant benefits with durable clinical responses and acceptable treatment-related toxicities in several types of solid tumors [10,11,12]. Although these mAbs have transformed cancer immunotherapy forever, they still exhibit several disadvantages such as very long half-life, immune-related adverse effects (irAEs), low permeability, immunogenicity, complex production process, and exorbitant treatment costs from another perspective [13]. Among the reported immunological adverse events, the overall incidences of all-grade adverse events were 66.0% and of grade 3 or higher adverse events were 14.0%. The most common side effects are endocrinological disturbances, dermatologic manifestations, and gastrointestinal disturbances. Apart from this, the most common grade 3 or higher adverse events were fatigue, anemia, and aspartate aminotransferase increase [14,15]. Compared to mAbs, small-molecule drugs have attracted tremendous attentions in immuno-oncology in recent years, due to their better pharmacokinetic properties and diffusion rates and convenient manufacture [16].
Recently, several small molecules derived from 2-methyl-3-biphenyl methanol scaffold were disclosed as effective inhibitors of PD-1/PD-L1 interaction (Figure 1) [17,18,19,20,21]. Most of the inhibitors show moderate to excellent activities of blocking PD-1/PD-L1 protein–protein interaction (PPI) with IC50 values between picomolar to nanomolar level through europium (Eu)−allophycocyanin (APC) homogeneous time-resolved fluorescence (HTRF) binding assay. The first co-crystal complex structure of small-molecule BMS-202 and hPD-L1 reveals that this compound induces and stabilize the formation of PD-L1 homodimers, leading to the blockade of PD-1/PD-L1 interaction [20,22,23].
Figure 1.

Structures of the reported compounds with binding PD-1/PD-L1 interaction. Biphenyl moiety is colored by red. IC50 values are established by the HTRF binding assay.
Since small molecules for immuno-oncology therapeutics have not been approved yet, future research should continue to focus on the discovery of novel small molecules activating the immune system with orally bioavailable compounds to combat cancer. Although some compounds bearing biphenyl moiety and their bivalent derivatives can inhibit the interaction of PD-1 with PD-L1 with IC50 values of ≤10 nM [22,23,24], most of the inhibitors have inherent disadvantages such as high molecular weight (MW), molecular polarity, and degree of dissociation, which might be responsible for the poor pharmacokinetic (PK) profile in preclinical and clinical studies [20,25,26]. Therefore, it still needs further studies to design small-molecule inhibitors of PD-1/PD-L1 interaction with better physicochemical properties. In the present study, we designed and synthesized a novel series of 2-(2-methyl-[1,1′-biphenyl]-3-yl) pyridine derivatives targeting PD-1/PD-L1 interaction via structural simplification strategy. During our design process, several structural indices, including MW, ALogP, and polar surface area (PSA), were considered to monitor their potential drug ability. The ligand efficiency (LE), a measure for the activity normalized by the number of non-H atoms, was also used as a valuable criterion for evaluating the quality of molecular design. Note that utilization of this metric during the overall drug design process to control the balance of molecular size and potency will significantly improve success rates [27]. Fortunately, our research leads to the identification of compound A9 that shows wonderful inhibitory activity at molecular level with a high LE value (HTRF IC50 = 0.93 nM, LE = 0.43; surface plasmon resonance (SPR) KD = 3.64 nM, LE = 0.40). In addition, A9 potently induces the release of IFN-γ in a Hep3B/OS-8/hPD-L1 and primary T cell co-culture system, deserving further investigation on its role in restoring immune surveillance.
2. Results and Discussion
2.1. Design and Screening
In our pursuit of novel small molecule inhibitors targeting the PD-1/PD-L1 pathway, we focused our effort on designing molecules with better drug-likeness property. BMS-1058 (IC50 = 0.48 nM) and BMS-202 (IC50 = 18 nM), which inhibited the PD-1/PD-L1 interaction in HTRF binding assay, were chosen as the template compounds for structural optimization. Structural analysis of the X-ray crystal complex of BMS-202 and hPD-L1 showed that it comprises three essential pharmacophores: (1) The biphenyl core, the minimal fragment essential for the hPD-L1 binding; (2) the polar moiety, the fragment interacting with the polar residues in solvent-oriented region; (3) the linker connecting these two regions (Figure 2A). We noticed that BMS-1058, with excellent IC50 values of 0.48 nM but possessing high MW and PSA value, has two polar parts, located at opposite ends of this molecule. Thus, it provided us a clue to design compounds by removing one side of the polar parts of BMS-1058, leading to the discovery of compound A1 (Figure 2B). It has moderate IC50 value of 113.6 nM but shows higher ligand-binding efficiency (LE = 0.38) than that of BMS-1058 (LE = 0.24) and BMS-202 (LE = 0.33) (Table 1), which gave us a valuable starting point for further optimization. In order to rationally design more potent inhibitors without reducing LE value, the in-silico docking prediction was conducted using Glide module in Schrödinger [28]. As shown in Figure 2C, the biphenyl core of A1 anchors at the bottom of the pocket, and two methyl groups form hydrophobic interactions with neighboring hydrophobic residues. In addition, one of the phenyl rings forms a T-stacking interaction with the sidechain of ATyr56. The protonated piperidin-4-ol moiety faces solvent-exposed environment and interacts with AAsp122 and BTyr56 by ionic and cation-π interactions, respectively. On the basis of this analysis, optimization of A1 would be employed by the following considerations: Substitutes on the distal phenyl ring, the linker, and the polar group in the tail.
Figure 2.


(A) Crystal structure of PD-L1 (chain A, yellow; chain B, purple)/BMS-202(white) complex (PDB code: 5J89). Receptor surfaces, covering atoms that are close to the current ligand, are colored by solvent accessibility surface (SAS). The default color spectrum used is green–white–blue, and small values (green) correspond to buried residues, whereas large values (blue) correspond to exposed residues. This picture was drawn by Discovery Studio Visualizer [29]. (B) Strctural simplification strategy of this work. (C) Putative binding modes of A1 in the binding pocket of PDL1 dimer structure. Dashed lines represent the inter-interaction between A1 and protein, specifically, green ones indicate hydrogen bond, dark green ones indicate cation–π interaction, purple ones indicate ionic interaction, and sky-blue ones indicate π–π interaction.
Table 1.
Activities of compounds A1–A4 in inhibition of the PD-1/PD-L1 interaction.
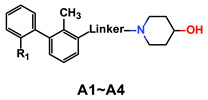
| Compd. | R1 | Linker | Inhibitory Rate (%) | IC50 (nM) a | MW | ALogP b | PSA b | LE c | |
|---|---|---|---|---|---|---|---|---|---|
| 1 μM | 0.1 μM | ||||||||
| A1 | CH3 |
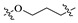
|
90 | 56 | 113.6 | 339.22 | 2.38 | 33.90 | 0.38 |
| A2 | F |
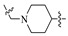
|
6.7 | 8.5 | N.D. | 382.24 | 2.09 | 27.91 | ND |
| A3 | F |
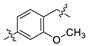
|
96.2 | 74.5 | 18.4 | 405.21 | 5.05 | 32.70 | 0.35 |
| A4 | F |
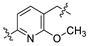
|
98.0 | 71.0 | 21.9 | 406.21 | 4.86 | 45.59 | 0.36 |
| BMS1058 | 0.48 d | 753.33 | 4.04 | 132.41 | 0.24 | ||||
| BMS202 | 18.0 | 419.22 | 4.04 | 72.48 | 0.33 | ||||
On closer inspection, however, it becomes clear that the binding conformation of aliphatic chain linker may be unfavorable for ligand binding owing to the adverse conformational entropy effects [30]. Thus, as the initial step, compounds with several cyclic linkers were prepared, where the methyl group of the distal phenyl ring were also replaced by the fluoro group in order to reduce its electron density (Table 1). Although the replacement using piperidine linker leads to a significant drop in potency, compounds with methoxy substituted benzene and pyridine rings do exhibit a significant improvement in activity as well as LE values (A3 IC50: 18.4 nM, LE: 0.35; A4 IC50: 21.9 nM, LE: 0.36). Docking studies suggest that A4 establishes an additional–π interaction with BTyr56 compared with A1, and the methoxy group fills the hydrophobic cavity defined by BIle54 and BVal68 (Figure 3), which may contribute to the ligand binding. Although A3 and A4 show a certainly decrease in activity compared with BMS-1058, they have higher LE values and proper drug-likeness properties such as ALogP and PSA, which encouraged us to make further modifications. Given the lower electron density of the central pyridine ring and solubility property, A4 was chosen to explore new molecules blocking PD-1/PD-L1 axis.
Figure 3.

(A) The optimization strategy of the linker part. (B) Putative binding modes of A4 in the binding pocket of PD-L1 dimer structure (PDB code: 5J89). Dashed lines represent the inter-interaction between A4 and protein, specifically, green ones indicate hydrogen bond, purple ones indicate ionic interaction, and sky-blue ones indicate π–π interaction.
Continuing to search for ways to improve the affinity, we turned our attention to the modification of the polar moiety in the tail, and a series of compounds were designed and synthesized. The results of biological activities are summarized in Table 2. Compounds A5, A7, and A8, decorated with 4-aminocyclohexan-1-ol, piperidine-4-carboxamide, and piperidine-4-carboxylic acid, respectively, are somewhat better tolerated than compound A6 with piperidine-3-carboxamide moiety. To our delight, the introduction of liner β-alaninamide to the polar tail (A9, IC50 = 0.93 nM) leads to an increase in the activity of more than 23-fold as compared with A4. Our CADD studies demonstrate that secondary aliphatic amine in the β-alaninamide, which is protonated at pH 7.4, forms a strong attractive charge interaction with AAsp122. This kind of strong interaction was observed in all the top-ranked docking poses. The amine group of an amide simultaneously forms two hydrogen bonds with BTyr56 and ATyr123, and its carbonyl oxygen also interacts with AArg125 by a hydrogen bonding interaction, possibly contributing to the high affinity (Figure 4A). Compared with BMS-202 (Figure 4B), A9 had similar spatial tendencies within the binding interface of dimer-PD-L1. However, more residues have participated in the formation of strong polar interactions between A9 and PD-L1. Based on the biological data results of A9, β-alaninamide group has been proved to be the most suitable R3 substituent.
Table 2.
Activities of compounds A5–A18 in inhibition of the PD-1/PD-L1 interaction.
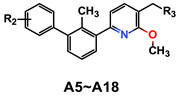
| Compd. | R2 | R3 | IC50 (nM) a | MW | ALogP b | PSA b | LE c |
|---|---|---|---|---|---|---|---|
| A5 | 2-F |
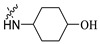
|
14.3 | 420.22 | 4.29 | 58.96 | 0.35 |
| A6 | 2-F |
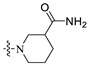
|
121.3 | 433.22 | 5.04 | 68.44 | 0.30 |
| A7 | 2-F |
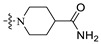
|
10.7 | 433.22 | 3.35 | 69.65 | 0.34 |
| A8 | 2-F |
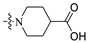
|
20.6 | 434.20 | 2.50 | 66.69 | 0.33 |
| A9 | 2-F |
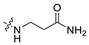
|
0.93 | 393.19 | 2.77 | 81.81 | 0.43 |
| A10 | 2-CH3 |
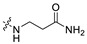
|
10.54 | 389.21 | 3.05 | 81.81 | 0.38 |
| A11 | 2-Cl |
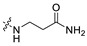
|
6.18 | 409.16 | 3.23 | 81.81 | 0.39 |
| A12 | 2-CF3 |
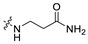
|
51.5 | 443.18 | 3.50 | 81.81 | 0.31 |
| A13 | H |
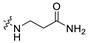
|
1.94 | 375.19 | 2.56 | 81.81 | 0.43 |
| A14 | 3-F |
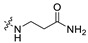
|
1.75 | 393.19 | 2.77 | 81.81 | 0.41 |
| A15 | 4-F |
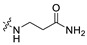
|
0.97 | 393.19 | 2.77 | 81.81 | 0.43 |
| A16 | 2,4-di-F |
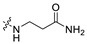
|
60.0 | 411.18 | 2.97 | 81.81 | 0.33 |
| A17 | 4-Cl-2-F |
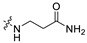
|
866.0 | 427.15 | 3.43 | 81.81 | 0.28 |
| A18 | 4-CF3-2-F |
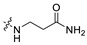
|
790.9 | 461.17 | 3.71 | 81.81 | 0.25 |
a The data were generated from two independent experiments. b Estimated physicochemical parameters by Discovery Studio [29]. c The results were calculated by the equation: LE = −1.35 logIC50/Nheavy atoms, Nheavy atoms is the number of non-H atoms.
Figure 4.

(A) Putative binding modes of A9 in the binding pocket of PD-L1 dimer structure (PDB code: 5J89); (B) binding modes of BMS-202 in the binding pocket of PD-L1 dimer structure (PDB code: 5J89). Dashed lines represent the inter-interaction between A9 or BMS-202 and protein, specifically, green ones indicate hydrogen bond, purple ones indicate ionic interaction, and sky-blue ones indicate π-π interaction.
Decorating the distal phenyl ring with various substituents provided further structure-activity relationships (SARs). The replacement of 2-fluoro by the bulkier 2-methyl (A10, IC50 = 10.54 nM) or 2-trifluoromethyl group (A12, IC50 = 51.5 nM) leads to a decrease in the activity of more than 10-fold as compared with A9. The chloro substituted compound (A11, IC50 = 6.18 nM) exhibited slightly higher inhibitory activity compared to A10. As expected, the unsubstituted phenyl ring A13 (A13, IC50 = 1.94 nM) yields an approximately 3-fold increase in potency over compound A11, which, however, does not reach the potency of 2-fluoro derivative A9. Moving the 2-fluoro to the 3-or 4-position shows comparable activity with A9, which indicates that the position of mono substituted fluoro has no significant effect on activity. This may be attributed by the more favorable π–π interaction between the electron-deficient fluorobenzene of these compounds and the residue ATyr56. However, the activity is dramatically reduced as for disubstituted derivatives A16–A18 (A16, IC50 = 60.0 nM; A17, IC50 = 866.0 nM; A18, IC50 = 790.9 nM). This is probably due to the unfavorable steric interactions between atom pairs in close contacts. Although the conformation of ATyr56 can be moved to accommodate to the size of substituent group on the distal phenyl ring, the binding affinity cannot be compensated for the energy loss by conformational change of ATyr56. In general, we have obtained the target compound A9, which has potential activity and moderate water solubility (126.37 μg/mL at pH 7.4 vs. BMS-202 89.99 μg/mL at pH 7.4), and we would conduct subsequent biological activity evaluations of A9.
2.2. Biophysical Characterization of Compounds Binding to Recombinant Human PD-L1 (hPD-L1)
To provide definitive evidence of direct binding to PD-L1 with small-molecule inhibitors, an SPR-based binding assay was performed using Biacore T200 instrument, which facilitates measurement of the kinetic parameters of ligand–protein complex formation. In this determination, hPD-L1/Fc was loaded on a Series S Sensor Chip Protein A. A9 and A15, with similar bioactivities in HTRF assay, were gradually subjected to this assay, and their kinetic parameters and the binding affinities were determined with the aid of Biacore evaluation software. Analyses of the resulting data reveals that both A9 and A15 can efficiently interact with the immobilized hPD-L1 protein, as demonstrated by the concentration-dependent responses for association and dissociation, respectively (Figure 5, Table S1). The equilibrium dissociation constant (KD) could be calculated and shows that compound A9 binds hPD-L1 with an excellent KD value of 3.64 nM, 5-fold greater than that of A15 (KD = 19.9 nM). Noteworthily, their kinetic measurements demonstrate that A9 and A15 have nearly the same association rate but show conspicuous differences in dissociation rates that may lead to selection of A9 with a slower off-rate as a more effective blocker of PD-1/PD-L1 interaction.
Figure 5.

Association and dissociation kinetic curves of compounds A9 (A) and A15 (B) at different concentrations with hPD-L1.
2.3. Non-Specific Toxicity of (2-Methyl-[1,1′-Biphenyl]-3-yl)Pyridine Derivatives
Considering that the toxicity of the compound might strongly interfere with the assay if the tested compounds could block PD-1/PD-L1 interaction to activate T cells for killing tumors, we examined their cell toxicities of MDA-MB-231 and Jurkat T cells in MTT assay. As shown in Table 3, compounds A9, A11, A13, A14, and A15 do not directly kill tumor cells and normal T cells. Furthermore, Jurkat T cells were used to simulate primary T cells for investigating the immune toxicity of the treated compounds with the regular concentrations within 48 h. This assay also shows that A9, A11, and A15 exhibit lower cell toxicity, in which their EC50 values are much higher than their respective IC50 values at the molecular level.
Table 3.
Cytotoxic of inhibitors of the PD-1/PD-L1 interaction.
| Compound | EC50 (μM)/MDA-MB-231 Toxicity |
EC50 (μM)/Jurkat Toxicity |
|---|---|---|
| A9 | 11.1 | 7.6 |
| A11 | 7.1 | 5.5 |
| A13 | >10 | 7.73 |
| A14 | >10 | >10 |
| A15 | 10.2 | 5.5 |
2.4. IFN-γ Production Effect of A9 in T-Cell Function Assay
Considering the excellent potency in biochemical assays and acceptable drug-likeness properties, we concluded that A9 is optimal, and would be chosen for further study at T cell function assay. It is well-known that the expression of interferon-γ (IFN-γ) in the tumor microenvironment is increased by a productive T-cell response against tumor-associated antigens [6,31,32,33]. However, it will be prevented once PD-L1 is overexpressed on tumor surface through PD-1/PD-L1 interaction. In the presence of PD-1/PD-L1 blockers, the interaction between PD-1 and PD-L1 will be destroyed, and the expression of IFN-γ would be restored by activated T cells.
To evaluate if compound A9 could restore the T cells-mediated immunity responses, which was previously repressed by the PD-1/PD-L1 pathway, Hep3B cells, engineered to stably express OS-8 and hPD-L1, were co-cultured with primary CD3+ T Cells in the presence of many PD-1/PD-L1 blockers (Figure 6). A PD-1 monoclonal antibody Keytruda and small molecule BMS-202 were synchronously used as positive controls. Obviously, A9 can promote the dose-dependent release of IFN-γ in this co-culture system. Impressively, the promoting effect on IFN-γ production of A9 at 5 μM was comparable to that of Keytruda at 5 μg/mL and is significantly higher than that of BMS-202. This result implies that A9 can restore T cells-mediated immune responses by blocking the PD-1/PD-L1 interaction in a tumor microenvironment.
Figure 6.

Effects of A9 on IFN-γ expression in a Hep3B/OS-8/hPD-L1 and CD3+ T-cell co-culture assay. A9 and BMS-202, and three independent experiments for blank and Keytruda. Data are shown as mean ± SD (A9 and BMS-202: n = 2; Blank and Keytruda: n = 3), * p < 0.05, ** p < 0.01, *** p < 0.001 vs. blank group.
2.5. Chemistry
The synthesis of compounds A1 and A2 is shown in Scheme 1. The key intermediate 8 was prepared through the Sandmeyer reaction of the starting material 7 and reacted with Br(CH2)3Br to obtain intermediate 9 by Williamson ether synthesis. Then, the intermediate 10 was obtained by N-alkylation reaction with piperidin-4-ol, which was converted to the desired compound A1 through Suzuki coupling reaction with 2-methylphenyl boronic acid.
Scheme 1.

Regents and conditions: (a) 1M H2SO4, 30% NaNO2, Toluene, 0–100 °C, 1 h, 89.6%; (b) Br(CH2)3Br, K2CO3, acetone, 65 °C, overnight, 81.7%; (c) piperidin-4-ol, acetonitrile, DIPEA, 80 °C, overnight, 80.2%; (d) 2-methylphenylboronic acid, Pd(PPh3)4, K2CO3, 1,4-dioxane/H2O (V:V = 10/1), 80 °C, 8 h, 82%; (e) BH3-THF, THF, 0 °C then r.t, 12 h, 97.0%; (f) SOCl2, reflux, 2 h, 98.0%; (g) 4-piperidinone, acetonitrile, DIPEA, 80 °C, overnight, 72.1%; (h) piperidin-4-ol, HOAc, CH2Cl2/MeOH, r.t, 2 h; NaBH(OAc)3, r.t., 8 h, 58.3%; (i) 2-fluorophenylboronic acid, Pd(PPh3)4, K2CO3, 1,4-dioxane/H2O(V:V = 10:1), 80 °C, 8 h, 89.3%.
Intermediate 12, which was prepared from 11 by BH3-THF mediated reductive reaction, was reacted with SOCl2 to afford 13, followed by the reaction with piperidone to provide 14. Then, 14 was converted into 15 by NaBH(OAc)3-mediated reductive amination reaction. Finally, the Suzuki-coupling reaction was conducted between 15 and 2-fluorophenylboronic acid to give the target compound A2.
The target compounds A3–19 were prepared according to the methods summarized in Scheme 2. Commercially, material 7 was coupled with various substituted phenylboronic acid to give 16a–j by Suzuki coupling reaction, which was then converted to 17a–j by the Sandmayer reaction. The resulting intermediates 17a–j were directly used to synthesize the intermediates 19a–k with 18 via Suzuki coupling reaction, which were finally reacted with appropriate amines to yield target compounds A3–19 through the NaBH(OAc)3 mediated reductive amination reaction.
Scheme 2.

Regents and conditions: (a) Substituted phenylboronic acid, Pd(PPh3)4, K2CO3, 1,4-dioxane/H2O (V:V = 10/1), 80 °C, 8 h, 80.2–99.2%; (b) 3M HCl, 30% NaNO2, B2pin2, MeOH, water, 0 °C ~rt, 40.1–68.8%; (c) 18a or 18b, Pd(PPh3)4, K2CO3, 1,4-dioxane/H2O (V:V = 10/1), 80–90 °C, 12 h, 20.8–95.3%; (d) alkyl amine, HOAc, CH2Cl2/MeOH (V:V = 3/1), r.t., 4–12 h; NaBH(OAc)3, r.t., 8 h, 19.9–64.4%.
3. Materials and Methods
3.1. Synthesis
Starting materials and solvents were purchased from commercial suppliers and used without any additional purification. 1H NMR and 13C NMR spectra were obtained on a Bruker AV-300 nuclear instrument or Bruker AVANCE NEO 400 MHz in CDCl3 or DMSO-d6 using TMS as internal standard, operating at 300 MHz or 400 MHz and 75 MHz, respectively. Chemical shifts (δ) are expressed in ppm and coupling constants J are given in Hz. Analytical thin layer chromatography (TLC) was purchased from Yantai Chemical Industry Research Institute (Cat. no. HSGF254, Yantai, China). All the reactions were monitored by thin layer chromatography in UV absorbance (254 nm). Flash chromatography was performed using silica gel (200–300 or 300–400 mesh). Melting points were measured with an RY-I melting point apparatus. The HPLC analysis was performed on a Shimadzu LC-20AT machine with a BDS Hypersil C18 column, and the column temperature was at 31 °C. Mobile phase B (100% Acetonitrile) and mobile phase A (NaH2PO4 and H3PO4 buffer solution, pH = 7.5) were used in a gradient elution program (0 min: 25% (B), 5 min: 25% (B), 12 min: 75% (B), 20 min: 75% (B), 23 min: 25% (B), 25 min: 25% (B)) with a flow rate of 1.0 mL/min at 254 nM.
3.1.1. 3-Bromo-2-Methylphenol (8)
A mixture of 3-bromo-2-methylaniline and H2SO4 (1M, 65 mL) was stirred for 30 min at room temperature, then 30% NaNO2 (2.25 g, 32.60 mmol) was added to the mixture dropwise at 0–5 °C. After 30 min, toluene (50 mL) was then added to the reaction mixture and allowed to stir at 100 °C for approximately 1 h. The mixture was extracted with ethyl acetate (50 mL × 3) and washed with brine (50 mL × 2). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography (Petroleum ether/ethyl acetate = 100/1–60/1) to afford compound 8 (white solid, 4.50 g, yield: 89.6%). m.p. 95.0–98.0 °C. 1H NMR (300 MHz, Chloroform-d) δ 7.19 (dd, J = 8.0, 1.2 Hz, 1H, ArH), 6.97 (t, J = 8.0 Hz, 1H, ArH), 6.76 (d, J = 7.9 Hz, 1H, ArH), 4.82 (s, 1H, OH), 2.38 (s, 3H, CH3).
3.1.2. 1-Bromo-3-(3-Bromopropoxy)-2-Methylbenzene (9)
A mixture of 8 (5.00 g, 26.90 mmol), K2CO3 (9.28 g, 67.25 mmol), and 1,3-dibromopropane (10.75 g, 53.80 mmol) in acetone (30 mL) was refluxed for 8 h. The mixture was diluted with water (100 mL), extracted with ethyl acetate (50 mL × 3), and washed with brine (50 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography (Petroleum ether = 100%) to afford compound 9 (colorless oil, 6.72 g, yield: 81.7%).1H NMR (300 MHz, Chloroform-d) δ 7.16 (dd, J = 8.1, 1.2 Hz, 1H, ArH), 7.00 (td, J = 8.1, 0.6 Hz, 1H, ArH), 6.79 (dd, J = 8.2, 1.2 Hz, 1H, ArH), 4.10 (t, J = 5.7 Hz, 2H, OCH2), 3.62 (t, J = 6.4 Hz, 2H, BrCH2), 2.39–2.32 (m, 2H, CH2CH2CH2), 2.31 (s, 3H, CH3).
3.1.3. 1-(3-(3-Bromo-2-Methylphenoxy)Propyl)Piperidin-4-ol (10)
A mixture of 9 (1.00 g, 3.27 mmol), DIPEA (1.10 mL, 6.54 mmol), and 4-hydroxypiperidine (0.97 g, 3.27 mmol) in MeCN (10 mL) was refluxed for 12 h. The formed precipitates were filtrated and washed with petroleum ether and less water to obtain compound 10 (yellow solid, 0.86 g, yield: 80.8%). m.p.138.0–140.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.20 (dd, J = 8.0, 1.3 Hz, 1H, ArH), 7.13 (t, J = 8.1 Hz, 1H, ArH), 6.99 (d, J = 7.9 Hz, 1H, ArH), 5.02–4.96 (m, 1H, OH), 4.09 (t, J = 5.9 Hz, 2H, OCH2), 4.01–3.92 (m, 1H, CHOH), 3.77–3.40 (m, 2H, CH2N), 3.20–2.86 (m, 4H, NCH2), 2.28 (s, 3H, CH3), 2.23–2.08 (m, 2H, CH2CH2CH2), 2.08–1.69 (m, 4H, CH2CHOH).
3.1.4. (3-Bromo-2-Methylphenyl)Methanol (12)
BH3-THF (1 M, 35 mL) was dropwise added to a solution of 3-bromo-2-methylbenzoic acid (5.00 g, 23.20 mmol) in dry THF (25 mL) at 0 °C under N2. The mixture was stirred at an ambient temperature for 12 h. The reaction quenched with 1 M HCl (30 mL), and then extracted with ethyl acetate (20 mL × 3), followed by brine (20 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum to give 12 without any further purification (white solid, 4.5 g, yield, 97.0%). m.p. 103.0–104.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.49 (d, J = 7.9 Hz, 1H, ArH), 7.39 (d, J = 7.8 Hz, 1H, ArH), 7.12 (t, J = 8.1 Hz, 1H, ArH), 5.27 (br, 1H, OH), 4.52 (s, 2H, CH2), 2.30 (s, 3H, ArCH3).
3.1.5. 1-Bromo-3-(Chloromethyl)-2-Methylbenzene (13)
A solution of 12 (4.50 g, 22.50 mmol) in SOCl2 (10 mL) was stirred at 80 °C for 2 h. The solvent was removed in vacuum to obtain compound 13 (colorless oil, 4.80 g, yield, 98.0%). 1H NMR (300 MHz, Chloroform-d) δ 7.47 (d, J = 7.0 Hz, 1H, ArH), 7.25 (d, J = 6.8 Hz, 1H, ArH), 7.04 (t, J = 8.1 Hz, 1H, ArH), 3.90 (s, 2H, CH2), 2.43 (s, 3H, CH3).
3.1.6. 1-(3-Bromo-2-Methylbenzyl)Piperidin-4-One (14)
This compound was prepared following the synthetic procedure similar to that of 10. Compound 14 (colorless oil 0.93 g, yield, 72.1%). 1H NMR (300 MHz, Chloroform-d) δ 7.52 (dd, J = 8.0, 1.6 Hz, 1H, ArH), 7.23 (d, J = 6.4 Hz, 1H, ArH), 7.03 (t, J = 7.8 Hz, 1H, ArH) 3.61 (s, 2H, CH2N), 2.77 (t, J = 6.1 Hz, 4H, 2NCH2), 2.51 (s, 3H, CH3), 2.46 (t, J = 6.2 Hz, 4H, 2CH2CO).
3.1.7. 1′-(3-Bromo-2-Methylbenzyl)-[1,4′-Bipiperidin]-4-ol (15)
A mixture of 14 (400 mg, 1.42 mmol) and 4-hydroxypiperidine (172 mg, 1.71 mmol) in DCM (10 mL) was stirred for 2 h at room temperature. NaBH(OAc)3 (2.30 mmol) was added to the mixture slowly at 0 °C and stirred for another 8 h. The reaction was adjusted to pH 7–8 with saturated NaHCO3 aqueous solution. The mixture was extracted with DCM (10 mL × 3) and washed with brine (10 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography (dichloromethane/methanol = 100/1–30/1) to afford compound 15 (yellow solid, yield, 58.3%). m.p. 102.0–105.0 °C. 1H NMR (300 MHz, Chloroform-d) δ 7.51 (d, J =7.9Hz, 1H, ArH), 7.23 (d, J = 7.4 Hz, 1H, ArH), 7.03 (t, J = 7.7 Hz, 1H, ArH), 4.34–4.27 (m, 1H, CHOH), 3.79–3.66 (m, 1H, CHN), 3.47 (s, 2H, ArCH2), 3.02–2.80 (m, 4H, NCH2), 2.46 (s, 3H, ArCH3), 2.44–2.28 (m, 4H, NCH2), 2.05–1.92 (m, 4H, 2CH2), 1.55–1.70 (m, 4H, 2CH2).
3.1.8. The Synthesis of Compounds 16a–j
Compounds 16a–j were synthesized by the following procedure. To a solution of 3-bromo-2-methylaniline (1.0 mmol), phenylboronic acid (1.2 mmol), K2CO3 (2.8 mmol) in 1,4-dioxane/water (1.0 mL/0.1 mL), Pd(PPh3)4 (0.05 mmol) was added. The mixture was stirred at 80 °C for 8 h under N2 atmosphere. The undissolved solid was filtered off, and the filtrate was concentrated under vacuum. The residue was dissolved in ethyl acetate, and washed with water, followed by brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by silica gel chromatography (petroleum ether/ethyl acetate = 100/1–80/1) to afford compounds 16a-j.
2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-amine (16a)
Brown solid. Yield, 95.6%. m.p. 61.0–62.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.48–7.35 (m, 1H, ArH), 7.34 – 7.22 (m, 3H, ArH), 6.98 (t, J = 7.7 Hz, 1H, ArH), 6.70 (dd, J = 8.0, 1.4 Hz, 1H, ArH), 6.42 (dd, J = 7.5, 1.3 Hz, 1H, ArH), 5.00 (s, 2H, NH2), 1.85 (d, J = 1.8 Hz, 3H, ArCH3).
2,2′-Dimethyl-[1,1′-biphenyl]-3-amine (16b)
Yellow oil. Yield, 90%. m.p. 52.0–55.0 °C. 1H NMR (400 MHz, Chloroform-d) δ 7.34 – 7.29 (m, 1H, ArH), 7.28–7.22 (m, 2H, ArH), 7.15 (d, J = 7.0 Hz, 1H, ArH), 7.10 (t, J = 7.7 Hz, 1H, ArH), 6.75 (dd, J = 7.9, 1.4 Hz, 1H, ArH), 6.63 (dd, J = 7.5, 1.4 Hz, 1H, ArH), 3.75 (s, 2H, ArNH2), 2.11 (s, 3H, ArCH3), 1.91 (s, 3H, ArCH3).
2′-Chloro-2-methyl-[1,1′-biphenyl]-3-amine (16c)
Yellow oil. Yield, 93.1%. m.p. 71.5–74.5 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.58–7.46 (m, 1H, ArH), 7.43–7.30 (m, 2H, ArH), 7.30–7.18 (m, 1H, ArH), 6.94 (t, J = 7.6 Hz, 1H, ArH), 6.67 (dd, J = 8.0, 1.4 Hz, 1H, ArH), 6.32 (dd, J = 7.5, 1.4 Hz, 1H, ArH), 4.96 (s, 2H, NH2), 1.76 (s, 3H, ArCH3).
2′-Chloro-2-methyl-[1,1′-biphenyl]-3-amine (16d)
Yellow solid. Yield, 82.1%. m.p. 87.5–90.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.79 (dd, J = 7.9, 1.6 Hz, 1H, ArH), 7.67 (td, J = 7.6, 1.7 Hz, 1H, ArH), 7.56 (t, J = 7.6, 1H, ArH), 7.25 (d, J = 7.5 Hz, 1H, ArH), 6.90 (t, J = 7.7 Hz, 1H, ArH), 6.67 (dd, J = 8.0, 1.4 Hz, 1H, ArH), 6.31 (d, J = 7.3 Hz, 1H, ArH), 4.98 (s, 2H, NH2), 1.68 (s, 3H, ArCH3).
2-Methyl-[1,1′-biphenyl]-3-amine (16e)
White solid. Yield, 89.6%. 1H NMR (400 MHz, Chloroform-d) δ 7.46–7.40 (m, 2H, ArH), 7.40–7.32 (m, 3H, ArH), 7.11 (t, J = 7.8 Hz, 1H, ArH), 6.75 (d, J = 7.8 Hz, 2H, ArH), 3.82 (s, 2H, NH2), 2.10 (s, 3H, ArCH3).
3′-Fluoro-2-methyl-[1,1′-biphenyl]-3-amine (16f)
Yellow oil. Yield, 88.0%.1H NMR (400 MHz, DMSO-d6) δ 7.44 (td, J = 8.1, 6.1 Hz, 1H, ArH), 7.25–7.15 (m, 1H, ArH), 7.12–7.04 (m, 2H, ArH), 6.94 (t, J = 7.8 Hz, 1H, ArH), 6.67 (dd, J = 8.0, 1.5 Hz, 1H, ArH), 6.42 (dd, J = 7.5, 1.4 Hz, 1H, ArH), 5.05 (s, 2H, NH2), 1.93 (d, J = 1.9 Hz, 3H, CH3).
4′-Fluoro-2-methyl-[1,1′-biphenyl]-3-amine (16g)
White solid. Yield, 88.0%. m.p. 76.5–78.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.32–7.23 (m, 3H, ArH), 7.20 (d, J = 8.8 Hz, 1H, ArH), 6.93 (t, J = 7.7 Hz, 1H, ArH), 6.66 (d, J = 7.9 Hz, 1H, ArH), 6.40 (d, J = 7.5 Hz, 1H, ArH), 4.94 (s, 2H, NH2), 1.92 (s, 3H, ArCH3).
2′,4′-Difluoro-2-methyl-[1,1′-biphenyl]-3-amine (16h)
Yellow solid. Yield, 88.9%. m.p. 81.5–83.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.38–7.27 (m, 2H, ArH), 7.16 (td, J = 8.5, 2.2 Hz, 1H, ArH), 6.97 (t, J = 7.8 Hz, 1H, ArH), 6.70 (dd, J = 8.1, 1.3 Hz, 1H, ArH), 6.40 (d, J = 7.4 Hz, 1H, ArH), 5.01 (s, 2H, NH2), 1.84 (d, J = 1.8 Hz, 3H, ArCH3).
4′-Chloro-2′-fluoro-2-methyl-[1,1′-biphenyl]-3-amine (16i)
Yellow oil. Yield, 99.2%. 1H NMR (300 MHz, DMSO-d6) δ 7.52 (dd, J = 9.8, 1.9 Hz, 1H, ArH), 7.37 (dd, J = 8.3, 2.0 Hz, 1H, ArH), 7.35–7.28 (m, 1H, ArH), 6.99 (t, J = 7.7 Hz, 1H, ArH), 6.72 (dd, J = 8.0, 1.4 Hz, 1H, ArH), 6.41 (dd, J = 7.5, 1.3 Hz, 1H, ArH), 5.03(s, 2H, NH2), 1.85 (d, J = 1.8 Hz, 3H, ArCH3).
2. ′-Fluoro-2-methyl-4′-(trifluoromethyl)-[1,1′-biphenyl]-3-amine (16j)
Yellow oil. Yield, 80.6%. 1H NMR (400 MHz, DMSO-d6) δ 7.74 (dd, J = 9.8, 2.1 Hz, 1H, ArH), 7.64 (dd, J = 8.0, 2.1 Hz, 1H, ArH), 7.51 (t, J = 7.6 Hz, 1H, ArH), 6.99 (t, J = 7.8 Hz, 1H, ArH), 6.73 (d, J = 8.0 Hz, 1H, ArH), 6.42 (dd, J = 7.5, 1.4 Hz, 1H, ArH), 5.04 (bs, 2H, NH2), 1.84 (d, J = 1.9 Hz, 3H, ArCH3).
3.1.9. The Synthesis of Compounds 17a–j
Compounds 17a–j were synthesized by the following procedure. HCl (24.84 mL, 3 M) was added to a solution of 16a (4.00 g, 19.9 mmol) in MeOH (40 mL), followed by the addition of H2O (20 mL) at room temperature. The mixture was stirred for 30 min, and NaNO2 (9.92 mL, 2.2 M) was added dropwise very slowly at 0–5 °C. After being stirred for another 30 min, B2pin2 (15.12 g, 59.7 mmol) in MeOH (40 mL) was added to the mixture solution and warmed to room temperature for 2 h. Then the mixture was extracted with DCM (50 mL × 3), washed with brine (50 mL × 2), and dried over anhydrous Na2SO4. The organic layer was concentrated in vacuum and purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 250/1–150/1) to the target compound.
2-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17a)
Yellow solid. Yield, 60.4%. m.p. 94.0–95.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.70 (dd, J = 5.4, 3.7 Hz, 1H, ArH), 7.53–7.36 (m, 1H, ArH), 7.34–7.25 (m, 5H, ArH), 2.27 (d, J = 1.4 Hz, 3H, ArCH3), 1.31 (s, 12H, CH3).
2-(2,2′-Dimethyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17b)
Colorless oil. Yield, 40.1%. 1H NMR (300 MHz, DMSO-d6) δ 7.67 (dd, J = 7.4, 1.8 Hz, 1H, ArH), 7.34–7.21 (m, 4H, ArH), 7.15 (dd, J = 7.5, 1.8 Hz, 1H, ArH), 7.10–6.95 (m, 1H, ArH), 2.17 (s, 3H, ArCH3), 1.99 (s, 3H, ArCH3), 1.33 (s, 12H, CCH3).
2-(2′-Chloro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17c)
Light yellow solid. Yield, 62.9%. m.p. 101.0–103.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.71 (dd, J = 7.3, 1.8 Hz, 1H, ArH), 7.60 (dd, J = 5.8, 3.5 Hz, 1H, ArH), 7.48–7.41 (m, 2H, ArH), 7.33–7.25 (m, 2H, ArH), 7.21 (dd, J = 7.6, 1.8 Hz, 1H, ArH), 2.22 (s, 3H, ArCH3), 1.34 (s, 12H, CH3).
4,4,5,5-Tetramethyl-2-(2-methyl-2′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)-1,3,2-dioxaborolane (17d)
Colorless oil. Yield, 68.8%. 1H NMR (300 MHz, DMSO-d6) δ 7.83 (dd, J = 7.9, 1.6 Hz, 1H, ArH), 7.79–7.70 (m, 2H, ArH), 7.68–7.61 (m, 1H, ArH), 7.29 (d, J = 7.5 Hz, 1H), 7.25–7.13 (m, 2H, ArH), 2.12 (s, 3H, ArH), 1.31 (s, 12H, CH3).
4,4,5,5-Tetramethyl-2-(2-methyl-[1,1′-biphenyl]-3-yl)-1,3,2-dioxaborolane (17e)
Colorless oil. Yield, 55.3%. 1H NMR (400 MHz, DMSO-d6) δ 7.69 (d, J = 7.8 Hz, 1H, ArH), 7.57–7.40 (m, 1H, ArH), 7.39–7.20 (m, 4H, ArH), 7.03 (s, 1H, ArH), 6.99 (d, J = 7.9 Hz, 1H, ArH), 2.26 (d, J = 1.3 Hz, 3H, ArCH3), 1.32 (s, 12H, CH3).
2-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17f)
White solid. Yield, 56.8%. 1H NMR (300 MHz, DMSO-d6) δ 7.69 (dd, J = 6.4, 2.5 Hz, 1H, ArH), 7.50 (td, J = 8.1, 6.2 Hz, 1H, ArH), 7.34–7.26 (m, 2H, ArH), 7.26–7.21 (m, 1H, ArH), 7.19–7.16 (m, 2H, ArH), 2.37 (s, 3H, ArCH3), 1.34 (s, 12H, CH3).
2-(4′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17g)
Yellow solid. Yield, 60.0%. 1H NMR (300 MHz, DMSO-d6) δ 7.71–7.63 (m, 1H, ArH), 7.38–7.34 (m, 1H, ArH), 7.34–7.29 (m, 2H, ArH), 7.29–7.23 (m, 3H, ArH), 2.35 (s, 3H, ArCH3), 1.33 (s, 12H, CH3).
2-(2′,4′-Difluoro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17h)
Light yellow solid. Yield, 67.5%. m.p. 59.5–61.5 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.79–7.68 (m, 1H, ArH), 7.43–7.32 (m, 2H, ArH), 7.28 (d, J = 4.4 Hz, 2H, ArH), 7.19 (td, J = 8.5, 2.7 Hz, 1H, ArH), 2.28 (d, J = 1.3 Hz, 3H, ArCH3), 1.33 (s, 12H, CH3).
2-(2′,4′-Difluoro-2-methyl-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17i)
Light yellow solid. Yield, 63.5%. m.p. 91.5–94.5 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.72 (q, J = 4.0 Hz, 1H, ArH), 7.57 (dd, J = 9.8, 2.0 Hz, 1H, ArH), 7.41 (dd, J = 8.2, 2.0 Hz, 1H, ArH), 7.37 (d, J = 7.7 Hz, 1H, ArH), 7.30 (d, J = 4.5 Hz, 2H, ArH), 2.28 (s 3H, ArCH3), 1.34 (s, 12H, CH3).
2-(2′-Fluoro-2-methyl-4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17j)
Brown solid. Yield, 63.5%. m.p. 76.5–78.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.80 (d, J = 9.8Hz, 1H, ArH), 7.74 (d, J = 6.3Hz, 1H, ArH), 7.70–7.66 (m, 1H, ArH), 7.59–7.52 (m, 1H, ArH), 7.33–7.28 (m, 2H, ArH), 2.27 (s, 3H, CH3), 1.31 (s, 12H, CH3).
3.1.10. 2″-Fluoro-3-Methoxy-2′-Methyl-[1,1′:3′,1″-Terphenyl]-4-Carbaldehyde (19a)
To a solution of 17 (2.50 g, 8.01 mmol), 18a (1.51 g, 8.81 mmol), and K2CO3 (3.10 g, 22.4 mmol) in 1,4-dioxane/water (25 mL/2.5 mL), Pd(PPh3)4 (0.78 g, 0.67 mmol) was added, and the mixture was allowed to stir at 90 °C for 8 h under N2 atmosphere. The undissolved solid was filtered off, and the filtrate was concentrated under vacuum. The residue was dissolved in ethyl acetate (25 mL × 3) and washed with water (25 mL × 2), followed by brine (25 mL × 2). The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography (Petroleum ether/ ethyl acetate = 150/1–80/1) to afford compound 19a (white solid 2.45 g, yield: 95.3%). m.p. 82.0–83.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H, CHO), 7.77 (d, J = 7.9 Hz, 1H, ArH), 7.50–7.43 (m, 1H, ArH), 7.41–7.36 (m, 2H, ArH), 7.36–7.30 (m, 3H, ArH), 7.30–7.24 (m, 1H, ArH), 7.20 (s, 1H, ArH), 7.08 (dt, J = 7.9, 1.0 Hz, 1H, ArH), 3.96 (s, 3H, OCH3), 2.21 (s, 3H, ArCH3).
3.1.11. The Synthesis of Compounds 19b–k
Compounds 19b–k were prepared analogously to compound 16a.
6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19b)
White solid powder. Yield, 72.1%. m.p. 78.5–81.5 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.32 (s, 1H, CHO), 8.22 (d, J = 7.7 Hz, 1H, ArH), 7.59–7.54 (m, 1H, ArH), 7.51 (d, J = 7.8 Hz, 1H, ArH), 7.46 (d, J = 7.3 Hz, 1H, ArH), 7.42 (d, J = 5.4 Hz, 1H, ArH), 7.40–7.31 (m, 4H, ArH), 4.07 (s, 3H, OCH3), 2.18 (s, 3H, ArCH3).
6-(2,2′-Dimethyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19c)
Colorless oil. Yield, 74.5%. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, CHO), 8.19 (d, J = 7.7 Hz, 1H, ArH), 7.50 (dd, J = 7.8, 1.5 Hz, 1H, ArH), 7.42–7.36 (m, 2H, ArH), 7.35–7.24 (m, 3H, ArH), 7.20 (dd, J = 7.5, 1.5 Hz, 1H, ArH), 7.14 (dd, J = 6.9, 2.0 Hz, 1H, ArH), 4.04 (s, 3H, OCH3), 2.05 (s, 3H, ArCH3), 2.04 (s, 3H, ArCH3).
6-(2′-Chloro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19d)
Colorless oil, Yield, 61.7%. 1H NMR (300 MHz, DMSO-d6) δ 10.32 (s, 1H, CHO), 8.22 (d, J = 7.7 Hz, 1H, ArH), 7.65–7.61 (m, 1H, ArH), 7.57 (dd, J = 7.8, 1.7 Hz, 1H, ArH), 7.54–7.46 (m, 2H, ArH), 7.46–7.37 (m, 3H, ArH), 7.28 (dd, J = 7.6, 1.5 Hz, 1H, ArH), 4.07 (s, 3H, OCH3), 2.12 (s, 3H, ArCH3).
2-Methoxy-6-(2-methyl-2′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)nicotinaldehyde (19e)
White solid. Yield, 48.4%. 1H NMR (300 MHz, DMSO-d6) δ 10.31 (s, 1H, CHO), 8.20 (d, J = 7.7 Hz, 1H, ArH), 7.89 (d, J = 6.6 Hz, 1H, ArH), 7.77 (t, J = 6.7 Hz, 1H, ArH), 7.66 (t, J = 7.7 Hz, 1H, ArH), 7.56 (dd, J = 7.8, 1.5 Hz, 1H, ArH), 7.52–7.43 (m, 3H, ArH), 7.25 (d, J = 7.8 Hz, 1H, ArH), 4.05 (s, 3H, OCH3), 2.03 (s, 3H, ArCH3).
2-Methoxy-6-(2-methyl-[1,1′-biphenyl]-3-yl)nicotinaldehyde (19f)
White solid. Yield, 56.8%. 1H NMR (300 MHz, DMSO-d6) δ 10.42 (s, 1H, CHO), 7.79 (d, J = 7.8 Hz, 1H, ArH), 7.54–7.45 (m, 1H, ArH), 7.44–7.25 (m, 6H, ArH), 7.23 (s, 1H, ArH), 7.10 (d, J = 7.9 Hz, 1H, ArH), 3.99 (s, 3H, OCH3), 2.04 (s, 3H, ArCH3).
6-(3′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19g)
White solid. Yield, 68.8%. m.p. 131.0–132.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.31 (s, 1H, CHO), 8.22 (dd, J = 8.2, 5.4 Hz, 1H, ArH), 7.52 (dq, J = 8.9, 3.7, 3.1 Hz, 2H, ArH), 7.46–7.31 (m, 3H, ArH), 7.26 (t, J = 7.5 Hz, 3H, ArH), 4.06 (s, 3H, OCH3), 2.24 (s, 3H, ArCH3).
6-(3′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19h)
White solid. Yield, 72.3%. m.p. 46.0–48.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.18 (s, 1H, ArH), 8.12 (d, J = 7.9 Hz, 1H, ArH), 7.64 (dd, J = 5.7, 3.3 Hz, 1H, ArH), 7.42–7.10 (m, 7H, ArH), 4.01 (s, 3H, OCH3), 2.33 (s, 3H, ArCH3).
6-(2′,4′-Difluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19i)
White solid. Yield, 20.8%. m.p. 119.5–222.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, CHO), 8.20 (d, J = 7.6 Hz, 1H, ArH), 7.54 (dd, J = 7.8, 1.6 Hz, 1H, ArH), 7.48–7.41(m, 2H, ArH), 7.40–7.35 (m, 2H, ArH), 7.33 (dd, J = 7.6, 1.6 Hz, 1H, ArH), 7.22 (td, J = 8.5, 2.3 Hz, 1H, ArH), 4.04 (s, 3H, OCH3), 2.14 (s, 3H, ArCH3).
6-(4′-Chloro-2′-fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxynicotinaldehyde (19j)
White solid. Yield, 29.8%. m.p. 132.0–134.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, CHO), 8.20 (d, J = 7.8 Hz, 1H, ArH), 7.61–7.53 (m, 2H, ArH), 7.47–7.40 (m, 3H, ArH), 7.37 (d, J = 7.6 Hz, 1H, ArH), 7.34 (dd, J = 7.6, 1.6 Hz, 1H, ArH), 4.04 (s, 3H, OCH3), 2.14 (s, 3H, ArCH3).
6-(2′-Fluoro-2-methyl-4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)-2-methoxy nicotinaldehyde (19k)
White solid. Yield, 47.5%. m.p. 62.0–64.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H, CHO), 8.20 (d, J = 7.6 Hz, 1H, ArH), 7.84 (dd, J = 9.9, 1.9 Hz, 1H, ArH), 7.74–7.69 (m, 1H, ArH), 7.65 (t, J = 7.6 Hz, 1H, ArH), 7.58 (dd, J = 7.6, 1.6 Hz, 1H, ArH), 7.46 (t, J = 7.5 Hz, 1H, ArH), 7.41-7.32 (m, 2H, ArH), 4.04 (s, 3H, OCH3), 2.15 (s, 3H, ArCH3).
3.1.12. The Synthesis of Compounds A1 and A2
Compounds A1 and A2 were prepared analogous to compounds 16a–j.
1-(3-((2,2′-Dimethyl-[1,1′-biphenyl]-3-yl)oxy)propyl)piperidin-4-ol hydrochloride (A1).
White solid. Yield, 82.0%. m.p. 118.0–201.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.27 (dd, J = 6.6, 1.5 Hz, 2H, ArH), 7.26–7.22 (m, 1H, ArH), 7.19 (t, J = 7.8 Hz, 1H, ArH), 7.09–7.00 (m, 1H, ArH), 6.94 (d, J = 8.1 Hz, 1H, ArH), 6.66 (d, J = 7.4 Hz, 1H, ArH), 4.60 (s, 1H, OH), 4.09–3.99 (m, 3H, OCH3), 3.54–3.42 (m, 2H, NCH2), 2.87–2.72 (m, 2H, NCH2-piperidinol), 2.14 (m, 2H, CH2CH2CH2), 1.99 (s, 3H, ArCH3), 1.94 (s, 2H, NCH2-piperidinol), 1.83 (s, 3H, ArCH3), 1.73 (s, 2H, CH2-piperidinol), 1.44 (s, 2H, CH2-piperidinol). 13C NMR (75 MHz, DMSO-d6) δ 156.82, 142.68, 141.40, 135.61, 130.22, 129.45, 127.72, 126.74, 126.13, 124.03, 121.91, 110.52, 65.62, 60.03, 50.72, 47.37, 31.89, 29.75, 19.94(2C), 13.21(2C). ESI-HRMS: m/z [M+H]+calculated for C22H31NO2, 340.2277; Found: 340.2268. LCMS: tR 9.97 min, purity, 95.1%.
1′-((2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)methyl)-[1,4′-bipiperidin]-4-ol (A2).
Light yellow powder. Yield, 89.3%. m.p. 110.0–113.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.52–7.41 (m, 1H, ArH), 7.36–7.25 (m, 4H, ArH), 7.23 (d, J = 7.5 Hz, 1H, ArH), 7.11 (dd, J = 7.5, 1.6 Hz, 1H, ArH), 4.54 (s, 1H, CHOH), 3.46 (s, 3H, ArCH2N/ CHOH), 2.89 (d, J = 11.0 Hz, 2H, NCH2-piperidine), 2.83–2.69 (m, 2H, NCH2-piperidine), 2.36–2.15 (m, 3H, CHN, NCH2-piperidine), 2.12 (s, 3H, CH3), 1.99 (t, J = 10.7 Hz, 2H, NCH2-piperidine), 1.82–1.61 (m, 4H, CH2-piperidine), 1.57–1.18 (m, 4H, CH2-piperidine).13C NMR (75 MHz, DMSO-d6) δ 161.07, 157.84, 137.64, 136.18, 135.79, 132.11, 129.81, 129.14, 125.51, 124.96, 116.01, 115.72, 67.13, 61.95, 61.08, 53.49(2C), 47.13(2C), 35.25(2C), 28.32(2C), 16.09. ESI-HRMS: m/z [M+H]+calculated for C24H32FN2O, 383.2499 ; Found: 383.2495. LCMS: tR 10.69 min, purity, 96.5%.
3.1.13. The Synthesis of Compounds A3–A8
Compounds A3–A8 were synthesized by the following procedure. A mixture of 19a or 19b (0.92 mmol), amide (1.11 mmol), and HAc (0.11 mL, 1.84 mmol) in DCM/MeOH (5 mL/1 mL) was stirred for 4 h at room temperature. NaBH(OAc)3 (2.30 mmol) was added to the mixture slowly at 0 °C and stirred for another 8 h. The reaction was adjusted to pH 7–8 with saturated NaHCO3 aqueous solution. The mixture was extracted with DCM (10 mL × 3) and washed with brine (10 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography to afford the product.
1-((2″-Fluoro-3-methoxy-2′-methyl-[1,1′:3′,1″-terphenyl]-4-yl)methyl)piperidin-4-ol (A3).
White solid. Yield, 66.4%. m.p. 61.5–63.5 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.52–7.43 (m, 1H, ArH), 7.41–7.33 (m, 3H, ArH), 7.32–7.26 (m, 3H, ArH), 7.21 (dd, J = 7.3, 1.9 Hz, 1H, ArH), 6.92 (d, J = 8.4 Hz, 2H, ArH), 4.57 (s, 1H, OH), 3.81 (s, 3H, OCH3), 3.47 (br, 3H, ArCH2/piperidine-CH), 2.75 (b r, 2H, piperidine-CH2), 2.11 (b r, 2H, piperidine-CH2), 2.01 (s, 3H, ArCH3), 1.79–1.63 (m, 2H, piperidine-CH2), 1.50–1.36 (m, 2H, piperidine-CH2). 13C NMR (75 MHz, DMSO-d6) δ 161.08, 157.85(F-C), 157.45(F-C), 142.66, 141.71, 136.56, 132.13(d), 132.08, 130.12(d), 129.93, 129.51, 129.29, 126.04, 125.25, 125.02(d), 121.37, 116.12, 115.83, 112.12, 66.71, 55.85 (2C), 55.73, 51.59, 34.82, 18.35 (2C). ESI-HRMS: m/z [M+H]+ calculated for C26H28FNO2, 406.2104; Found: 406.2177. LCMS: tR 10.48 min, purity, 98.4%.
1-((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)piperidin-4-ol (A4).
White solid powder. Yield, 56.4%. m.p. 84.0–86.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.76 (d, J = 7.5 Hz, 1H, ArH), 7.52 (dd, J = 7.6, 1.7 Hz, 1H, ArH), 7.42–7.28 (m, 5H, ArH), 7.24–7.16 (m, 1H, ArH), 7.09 (d, J = 7.3 Hz, 1H, ArH), 4.02 (s, 3H, OCH3), 3.77 (dt, J = 8.8, 4.5 Hz, 1H, CHOH), 3.61 (s, 2H, ArCH2N), 2.90 (dt, J = 10.8, 4.7 Hz, 2H, CH2-piperidine), 2.37–2.28 (m, 2H, CH2-piperidine), 2.27 (s, 3H, ArCH3), 2.05–1.96 (m, 2H, CH2-piperidine), 1.84 (br, 1H, OH) 1.78–1.68 (m, 2H, CH2-piperidine), 1.47–1.36 (m, 2H, CH2-piperidine). 13C NMR (75 MHz, Chloroform-d) δ 161.16, 155.99, 140.94, 138.50, 136.90, 134.92, 131.75, 130.08, 129.69, 129.58, 129.17, 129.06, 125.43, 124.00, 118.64, 117.24, 115.70, 115.40, 67.97, 55.53, 53.49 (2C), 51.19, 34.51 (2C),18.05. ESI-HRMS: m/z [M+H]+ calculated for C26H29FNO2, 407.2182; Found: 407.2137. LCMS: tR 11.76 min, purity, 96.1%.
4-(((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)cyclohexan-1-ol (A5).
White solid. Yield, 51.3%. m.p. 80.0–82.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.82 (d, J = 7.5 Hz, 1H, ArH), 7.54–7.44 (m, 2H, ArH), 7.43–7.33 (m, 4H, ArH), 7.32–7.25 (m, 1H, ArH), 7.14 (d, J = 7.3 Hz, 1H, ArH), 4.52 (d, J = 4.5 Hz, 1H, OH), 3.90 (s, 3H, OCH3), 3.72 (s, 2H, CH2NH), 3.45–3.40 (m,1H, CHOH), 2.43–2.32 (m, 1H, NHCH), 2.13 (s, 3H, ArCH3), 1.94–1.78 (m, 4H, CH2-cyclohexane), 1.21–1.01 (m, 4H, CH2-cyclohexane). 13C NMR (75 MHz, DMSO-d6) δ 160.55, 157.88(F-C), 155.05(F-C), 141.03, 138.01, 136.75, 134.40, 132.12,130.12, 130.23, 129.95, 129.3(d), 126.05, 125.04, 122.11, 117.48, 116.11, 115.82, 69.29, 55.91, 53.59, 44.53, 34.32(2C), 31.26(2C), 18.17. ESI-HRMS: m/z [M+H]+ calculated for C26H30FN2O2, 421.2291; Found: 421.2284. LCMS: tR 9.46 min, purity, 95.1%.
1-((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)piperidine-3-carboxamide (A6).
White solid powder. Yield, 41.0%. m.p. 88.0–91.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.78 (d, J = 7.5 Hz, 1H, ArH), 7.55–7.44 (m, 2H, ArH), 7.42–7.33 (m, 4H, ArH, 1/2CONH2), 7.32–7.26 (m, 2H, ArH), 7.16 (d, J = 7.5 Hz, 1H), 6.82 (br, 1H, 1/2CONH2), 3.91 (s, 3H, OCH3), 3.50 (s, 2H, ArCH2), 2.89–2.71 (m, 2H, piperidine-CH2), 2.45–2.33 (m, 1H, piperidine-CHCO), 2.23–2.00 (m, 5H, ArCH3, piperidine-CH2), 1.84–1.62 (m, 2H, piperidine-CH2), 1.56–1.36 (m, 2H, piperidine-CH2). 13C NMR (75 MHz, DMSO-d6) δ 176.04, 161.00, 157.8(C-F), 155.51(C-F), 140.95, 139.15, 136.76, 134.41, 132.14, 130.28, 129.97, 129.39, 129.16, 126.07, 125.09, 119.21, 117.53, 115.9(d), 56.57, 56.05, 53.86, 53.65, 42.77, 27.54, 24.76, 18.19. ESI-HRMS: m/z [M+H]+ calculated for C26H29FN3O2, 434.2244; Found: 434.2238. LCMS: tR 12.04 min, purity, 95.6%.
1-((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)piperidine-4-carboxamide (A7).
White solid powder. Yield, 44.7%. m.p. 194.0–196.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.80 (d, J = 7.5 Hz, 1H, ArH), 7.53–7.45 (m, 2H, ArH), 7.43–7.34 (m, 3H, ArH), 7.33–7.26 (m, 2H, ArH), 7.25 (s, 1H, 1/2CONH2), 7.17 (d, J = 7.5 Hz, 1H, ArH), 6.75 (s, 1H, 1/2CONH2), 3.91 (s, 3H, OCH3), 3.54 (s, 2H, ArCH2), 2.91 (m, 2H, piperidine-CH2), 2.18–2.12 (s, 3H, ArCH3), 2.11–1.99 (m, 2H, piperidine-CH2), 1.78–1.54 (m, 4H, piperidine-CH2). 13C NMR (75 MHz, Chloroform-d) δ 177.63, 161.37, 161.16,158.16(C-F) 156.27(C-F), 140.83, 138.79, 136.90, 134.90, 131.76, 130.12, 129.74, 129.66, 129.52, 129.17(d), 125.43, 124.01, 117.29, 115.53(d), 55.64, 53.51(2C), 53.03, 42.38, 28.70(2C), 18.04. ESI-HRMS: m/z [M+H]+ calculated for C26H29FN3O2, 434.2244; Found: 434.2243. LCMS: tR 10.05 min, purity, 96.3%.
1-((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)piperidine-4-carboxylic acid (A8).
White solid powder. Yield, 19.9%. m.p. 218.0–220.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 12.27 (Br, 1H, COOH), 7.78 (d, J = 7.3 Hz, 1H, ArH), 7.53–7.44 (m, 2H, ArH), 7.43–7.32 (m, 4H, ArH), 7.32–7.24 (m, 1H, ArH), 7.15 (d, J = 7.3 Hz, 1H, ArH), 3.90 (s, 3H, OCH3), 3.49 (s, 2H, ArCH2N), 2.83 (d, J = 12.0 Hz, 2H, piperidine-CH2), 2.32–2.16 (m, 1H, CHCOOH), 2.17–2.03 (m, 5H, ArCH3/ piperidine-CH2), 1.83 (d, J = 13.0 Hz, 2H, piperidine-CH2), 1.68–1.54 (m, 2H, piperidine-CH2). 13C NMR (75 MHz, DMSO-d6) δ 176.78, 160.90, 157.85(C-F), 155.41(C-F), 140.94, 138.98, 136.75, 134.40, 132.16, 130.28, 130.16, 129.96, 129.35, 129.14, 126.08, 125.07(d), 119.20, 117.59, 115.97(d), 55.66, 53.65(2C), 53.16, 40.76, 28.62(2C),18.19. ESI-HRMS: m/z [M+H]+ calculated for C26H28FN2O3, 435.2084; Found: 435.2806. LCMS: tR 6.52 min, purity, 99.1%.
3.1.14. The Synthesis of Compounds A9–A18
Compounds A9–A18 were synthesized by the following procedure. A mixture of aldehyde (0.92 mmol), β-alanine amide hydrochloride (1.11 mmol), and Et3N (1.84 mmol) in DCM/MeOH (5 mL/1 mL) was stirred for 12 h at room temperature. NaBH(OAc)3 (2.30 mmol) was added to the mixture slowly at 0 °C and stirred for another 8 h. The reaction was adjusted to pH 7–8 with saturated NaHCO3 aqueous solution. The mixture was extracted with DCM (10 mL × 3) and washed with brine (10 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The residue was purified by silica gel chromatography to afford the product.
3-(((6-(2′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A9)
White solid powder. Yield, 54.1%. m.p. 218.0–222.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.81 (d, J = 7.5 Hz, 1H, ArH), 7.52–7.45 (m, 2H, ArH), 7.45–7.33 (m, 5H, ArH, 1/2CONH2), 7.33–7.26 (m, 1H, ArH), 7.15 (d, J = 7.3 Hz, 1H, ArH), 6.82 (s, 1H, 1/2CONH2), 3.92 (s, 3H, OCH3), 3.72 (s, 2H, ArCH2), 2.77 (t, J = 6.8 Hz, 2H, NHCH2), 2.29 (t, J = 6.8 Hz, 2H, CH2CO), 2.14 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 172.08, 161.02, 158.13(F-C), 157.85(F-C), 141.56, 140.48, 136.87, 134.42, 132.12, 130.69,130.34, 130.24, 129.97, 129.01(d), 126.21, 125.11, 117.70, 115.85, 113.26, 54.12, 44.95, 43.43, 30.87, 18.16. ESI-HRMS: m/z [M+H]+ calculated for C23H25FN3O2, 394.1931; Found: 394.1929. LCMS: tR 8.19 min, purity, 96.7%.
3-(((6-(2,2′-Dimethyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino) propenamide (A10).
White solid powder. Yield, 55.5%. m.p. 70.0–73.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.77 (d, J = 7.5 Hz, 1H, ArH), 7.42 (dd, J = 7.6, 1.6 Hz, 2H, ArH), 7.36–7.25 (m, 4H, ArH, 1/2CONH2), 7.17–7.10 (m, 3H, ArH), 6.82 (s, 1H, 1/2CONH2), 3.89 (s, 3H, OCH3), 3.88 (s, 1H, NH), 3.70 (s, 2H, ArCH2), 2.74 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.26 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.05 (s, 3H, ArCH3), 2.00 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 174.03, 160.68, 155.56, 142.51, 141.86, 140.88, 138.20, 135.65, 133.61, 130.29, 129.58, 129.37, 129.18, 127.78, 126.25, 125.92, 120.84, 117.49, 53.58, 47.04, 45.50, 35.68, 20.10, 18.03. ESI-HRMS: m/z [M+H]+ calculated for C24H28N3O2, 390.2182; Found: 390.2183. LCMS: tR 9.04 min, purity, 95.1%.
3-(((6-(2′-Chloro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A11).
White solid powder. Yield, 52.9%. m.p. 49.0–51.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.81 (d, J = 7.5 Hz, 1H, ArH), 7.58–7.54 (m, 1H, ArH), 7.50 (s, 1H, 1/2CONH2), 7.46–7.40 (m, 3H, ArH), 7.40–7.31 (m, 2H, ArH), 7.24–7.02 (m, 2H, ArH), 6.89 (s, 1H, 1/2CONH2), 3.91 (s, 3H, OCH3), 3.74 (s, 2H, ArCH2), 2.80 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.33 (t, J = 6.7 Hz, 2H, NHCH2CH2), 2.07 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 174.15, 160.70, 155.26, 140.83, 140.69, 140.38, 138.15, 134.20, 133.99, 132.82, 131.76, 129.87, 129.71, 129.59, 127.74, 125.94, 121.13, 117.52, 53.57, 47.11, 45.57, 35.80, 17.99. ESI-HRMS: m/z [M+H]+ calculated for C23H25ClN3O2, 410.1635; Found: 410.1633. LCMS: tR 9.02 min, purity, 98.7%.
3-(((2-Methoxy-6-(2-methyl-2′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)pyridin-3-yl)methyl)amino)propenamide (A12).
White solid powder. Yield, 57.5%. m.p. 50.5–53.5 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.85 (t, J = 7.5 Hz, 2H, ArH), 7.74 (t, J = 7.2 Hz, 1H, ArH), 7.63 (t, J = 7.7 Hz, 1H, ArH), 7.50 (s, 1H, 1/2CONH2), 7.46 (d, J = 7.7Hz, 1H, ArH), 7.40 (d, J = 7.3 Hz, 1H, ArH), 7.33 (t, J = 7.7 Hz, 1H, ArH), 7.15 (dd, J = 7.6, 5.2 Hz, 2H, ArH), 6.90 (s, 1H, 1/2CONH2), 3.90 (s, 3H, OCH3), 3.82 (s, 2H, ArCH2), 2.85 (t, J = 6.9 Hz, 2H, NHCH2CH2), 2.36 (t, J = 6.8 Hz, 2H, NHCH2CH2), 1.98 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 173.58, 160.77, 155.86, 140.71, 140.43, 140.03, 138.98, 133.93, 132.84, 132.31, 129.91, 129.65, 128.58, 128.16(β-CF3), 127.77(β-CF3), 127.39(β-CF3), 127.00 (β-CF3), 126.47(CF3), 126.39, 125.26, 122.75(CF3), 120.61(CF3), 119.24, 117.59, 53.68, 46.60, 45.06, 34.75, 18.53. ESI-HRMS: m/z [M+H]+ calculated for C24H25F3N3O2, 444.1899; Found: 444.1894. LCMS: tR 6.68 min, purity, 95.9%.
3-(((2-Methoxy-6-(2-methyl-[1,1′-biphenyl]-3-yl)pyridin-3-yl)methyl)amino)propenamide (A13).
White solid powder. Yield, 50.3%. m.p. 127.0–128.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.78 (d, J = 7.4 Hz, 1H, ArH), 7.50–7.43 (m, 2H, ArH), 7.43–7.31 (m, 6H, ArH, 1/2CONH2), 7.24 (dd, J = 7.4, 1.7 Hz, 1H, ArH), 7.12 (d, J = 7.4 Hz, 1H, ArH), 6.81 (s, 1H, 1/2CONH2), 3.90 (s, 3H, OCH3), 3.71 (s, 2H, ArCH2), 2.74 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.26 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.18 (s, 3H, ArCH3). 13C NMR (100 MHz, Chloroform-d) δ 175.15, 161.09, 156.99, 143.21, 142.21, 140.93, 138.21, 133.65, 129.94, 129.42(2C), 128.91, 128.08(2C), 126.84, 125.44, 119.35, 117.15, 53.52, 48.30, 44.85, 35.16, 18.64. ESI-HRMS: m/z [M+H]+ calculated for C23H26N3O2, 376.2025; Found: 376.2022. LCMS: tR 8.51 min, purity, 95.6%.
3-(((6-(3′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A14)
White solid powder. Yield, 48.6%. m.p. 48.0–51.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.80 (d, J = 7.5 Hz, 1H, ArH), 7.58–7.48 (m, 1H, ArH), 7.48–7.41 (m, 3H, ArH, 1/2CONH2), 7.33–7.18 (m, 4H, ArH), 7.14 (d, J = 7.3 Hz, 1H, ArH), 6.81 (s, 1H, 1/2CONH2), 3.92 (s, 3H, OCH3), 3.71 (s, 2H, ArCH2), 2.76 (t, J = 6.9 Hz, 2H, NHCH2), 2.28 (t, J = 6.8 Hz, 2H, CH2CONH2), 2.21 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 172.31, 164.00, 161.02, 160.77, 158.03, 144.37, 141.78, 141.33, 140.85, 133.33, 130.78, 130.19, 129.67, 126.24, 117.71, 116.33, 114.26, 113.69, 54.11, 45.81, 43.61, 31.17, 18.77. ESI-HRMS: m/z [M+H]+ calculated for C23H25FN3O2, 394.1931; Found: 394.1926. LCMS: tR 4.95 min, purity, 96.1%.
3-(((6-(4′-Fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A15)
White solid powder. Yield, 49.3%. m.p. 120.0–122.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.82 (d, J = 7.5 Hz, 1H, ArH), 7.47–7.42 (m, 4H, ArH, 1/2CONH2), 7.40–7.25 (m, 4H, ArH), 7.15 (d, J = 7.5 Hz, 1H, ArH), 6.85 (s, 1H, 1/2CONH2), 3.93 (s, 3H, OCH3), 3.76 (s, 2H, ArCH2), 2.80 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.31 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.19 (s, 3H, ArCH3). 13C NMR (100 MHz, Chloroform-d) δ 175.34, 163.17(F-C), 161.08(F-C), 160.17, 156.78, 142.14, 141.07, 138.18(2C), 133.72, 130.96(2C), 129.93, 129.07, 125.50, 119.67, 117.11, 115.09, 114.85, 53.50, 48.27, 44.93, 35.32, 18.61. ESI-HRMS: m/z [M+H]+ calculated for C23H25FN3O2, 394.1931; Found: 394.1931. LCMS: tR 8.47 min, purity, 95.6%.
3-(((6-(2′,4′-Difluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A16)
White solid powder. Yield, 43.9%. m.p. 113.0–115.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.78 (d, J = 7.4 Hz, 1H, ArH), 7.49–7.43 (m, 1H, ArH), 7.43–7.39 (m, 2H, ArH), 7.39–7.33 (m, 2H, ArH), 7.27–7.16 (m, 2H, ArH, 1/2CONH2), 7.12 (d, J = 7.3 Hz, 1H, ArH), 6.80 (s, 1H, 1/2CONH2), 3.89 (s, 3H, OCH3), 3.64 (s, 2H, ArCH2), 2.74 (t, J = 6.8 Hz, 2H, NHCH2CH2CO), 2.26 (t, J = 6.8 Hz, 2H, NHCH2CH2CO), 2.10 (s, 3H, ArCH3). 13C NMR (100 MHz, DMSO-d6) δ 174.01, 160.71, 155.38, 141.04, 138.31, 135.85, 134.58, 133.03(d, 2C), 130.38, 130.10, 126.07, 125.59(d), 120.88, 117.46, 112.21(d), 104.38(d, 2C), 53.58, 47.00, 45.49, 35.60, 18.09. ESI-HRMS: m/z [M+H]+ calculated for C23H24F2N3O2, 412.1837; Found: 412.1831. LCMS: tR 8.92 min, purity, 98.7%.
3-(((6-(4′-Chloro-2′-fluoro-2-methyl-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A17)
White solid powder. Yield, 46.9%. m.p. 122.0–124.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.80 (d, J = 7.5 Hz, 1H, ArH), 7.56 (d, J = 9.4 Hz, 1H, ArH), 7.50–7.37 (m, 5H, ArH,1/2CONH2), 7.25 (d, J = 7.5 Hz, 1H, ArH), 7.13 (d, J = 7.3 Hz, 1H, ArH), 6.83 (s, 1H, 1/2CONH2), 3.89 (s, 3H, OCH3), 3.73 (s, 2H, ArCH2), 2.77 (t, J = 5.9 Hz, 2H, NHCH2CH2), 2.28 (t, J = 6.2 Hz, 2H, NHCH2CH2), 2.11 (s, 3H, ArCH3). 13C NMR (75 MHz, DMSO-d6) δ 173.95, 161.05, 160.70, 157.77(F-C), 155.20(F-C), 141.08, 138.23, 135.61, 134.43, 133.69, 133.55, 133.37(d), 130.27, 128.33(d), 126.19, 125.35(d), 121.18, 117.51, 116.66(d), 53.63, 47.04, 45.55, 35.76, 18.15. ESI-HRMS: m/z [M+H]+ calculated for C23H24ClFN3O2, 428.1541; Found: 428.1537. LCMS: tR 9.62 min, purity, 95.2%.
3-(((6-(2′-Fluoro-2-methyl-4′-(trifluoromethyl)-[1,1′-biphenyl]-3-yl)-2-methoxypyridin-3-yl)methyl)amino)propenamide (A18)
White solid powder. Yield, 50.9%. m.p. 137.0–139.0 °C. 1H NMR (300 MHz, DMSO-d6) δ 7.87–7.76 (m, 2H, ArH), 7.71 (dd, J = 8.5, 1.8 Hz, 1H, ArH), 7.64 (t, J = 7.6 Hz, 1H, ArH), 7.50 (dd, J = 7.7, 1.6 Hz, 1H, ArH), 7.45–7.37 (m, 2H, ArH, 1/2CONH2), 7.30 (dd, J = 7.6, 1.6 Hz, 1H, ArH), 7.13 (d, J = 7.3 Hz, 1H, ArH), 6.80 (s, 1H, 1/2CONH2), 3.89 (s, 3H, OCH3), 3.69(s, 2H, ArCH2), 2.74 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.26 (t, J = 6.8 Hz, 2H, NHCH2CH2), 2.12 (s, 3H, ArCH3). 13C NMR (100 MHz, Chloroform-d) δ 175.29, 161.15, 160.63(F-C),158.16(F-C), 156.22, 140.95, 138.15, 135.48, 134.68, 133.49(d), 132.47(d), 131.51(dd), 130.23, 127.44(CF3), 125.69, 124.72(CF3), 122.04(CF3), 121.03, 119.95,119.33(CF3), 117.17, 113.13(d), 53.52, 48.33, 44.90, 35.35, 18.00. ESI-HRMS: m/z [M+H]+ calculated for C24H24F4N3O2, 462.1805; Found: 462.1799. LCMS: tR 9.72 min, purity, 96.8%.
3.2. Pharmacology
3.2.1. HTRF-Based PD-1/PD-L1 Binding Assay
The PD-1/ PD-L1 binding assay kits (Cisbio, Codolet, France, Cat. no. 64ICP01PEG) were purchased from Cisbio. The experiments were performed according to the manufacturer’s guidelines. The HTRF assay used Perkin Elmer EnVision to read the fluorescence intensity at 665 nm and 615 nm and calculate the HTRF ratio (665 nm emission/615 nm emission). The IC50 values were calculated by four-parameter logistic curve in Graphpad prism.
3.2.2. In Vitro Non-Specific Cytotoxicity Assay
The MDA-MB-231 and Jurkat cell line was provided by the Chinese Academy of Sciences Cell Bank. The cytotoxicity of the test compounds was determined using the MTT assay. Briefly, MDA-MB-231 and Jurkat cells were incubated at 37 °C in a humidified 5% CO2 incubator for 24 h in 96-microwell plates. Then, 100 μL of culture medium containing the test compounds at different concentrations was added to each well and incubated at 37 °C for another 48 h. The MTT was added and incubated for another 4 h, and the optical density was detected with a microplate reader at 570 nm. The IC50 values were calculated according to the dose-dependent curves.
3.2.3. Binding Affinity Assay
SPR analysis was carried out according to the method described by a previous article [23]. The kinetic analysis for the interaction of small molecules with hPD-L1 was performed on a Biacore T200 (GE Healthcare Bio-sciences, Uppsala, Sweden) using Series S Sensor Chip Protein A (GE Healthcare Bio-sciences, Uppsala, Sweden, catalog # 29139131-AA). Briefly, Fc-hPD-L1 (Acro Biosystems, Cambridge, MA, USA, catalog #PD1-H5258) was diluted using 1 × PBS-P+ buffer (phosphate buffer 1×, GE Healthcare Bio-sciences, catalog # 28-9950-84, enriched with 0.05% surfactant P20) containing 5% DMSO (Sigma-Aldrich, St. Louis, MO, America, catalog # D4540) and captured as the ligand (i.e., the target of small molecules) on protein A chip surface following Biacore T200 build-in protocol for kinetic/affinity analysis at a concentration of 10 μg/mL. The contact time and flow rates were set as 120 s and 10 μL/min for the ligand capture. Each experiment was performed using a pair of flow cells, one with ligand captured and the other kept as reference. A concentration gradient of analytes was freshly prepared in 1× PBS-P+ buffer with 5% DMSO with at least five concentrations, and the concentration range was optimized according to different analytes. Analytes could flow through both ligand-captured flow cells and reference flow cells at the same rate (30 μL/min) and contact time (90 s). Solvent correction was included to avoid the impact of DMSO on surface plasmon effect during binding analysis. Extra washing of the flow system using 50% DMSO in 1×PBS-P+ buffer was allowed to control carry over effects. For the regeneration of surface after each cycle (one-time ligand capture and binding of analyte at a single concentration) was carried out using 10 mM glycine pH 1.5 (GE Healthcare Bio-sciences, Uppsala, Sweden, catalog #BR-1003-54) at a flow rate of 30 μL/min for 30 s. Kinetic analysis was performed at 25 °C. Maximum binding response (Rmax) and equilibrium dissociation rate constant (KD) values were calculated using Biacore T200 Evaluation software following the 1:1 binding model.
3.2.4. IFN-γ Production Effect in T Cells Function Assay
The tumor/T cells coculture assay was conducted by ChemPartner [19]. Briefly, Hep3B cells were engineered to stably express OS-8 (anti-CD3 single chain variable fragment) and human PD-L1 (hPD-L1). Fresh PBMCs were isolated from a healthy donor by density gradient centrifugation. CD3+ T cells were isolated from fresh PBMCs by EasySep Human T Cells Isolation Kit (negative selection, STEMCELL Technologies, Vancouver, Canada). Hep3B-OS8- hPD-L1 cells were harvested and treated with 10 μg/mL mitomycin C at 37 °C for 1.5 h and washed 4 times with PBS. Hep3B-OS8-hPD-L1 and T cells (2.5 × 104 in 50 μL and 5 × 104 in 100 μL complete media, respectively) were added to the 96-well plates, followed by the addition of 4× final concentration of test compound in 50 μL complete media. The supernatants (150 μL) were harvested after 72 h of coculture to determine IFN-γ levels by ELISA.
3.3. Molecular Modelling
Molecular docking simulation was performed in the Schrödinger suite [28]. The molecules were washed and processed, the 3D conformation of small molecules were prepared by LigPrep module. The X-ray crystallographic structure of PD-L1 was obtained from the RCSB Protein Data Bank (PDB ID: 5J89). The protein was prepared to remove the water molecules and add hydrogens and minimized using Protein Preparation Wizard module. Then, the binding site was defined using the original ligand in the complex by Receptor Grid Generation module. The Glide Docking module in standard precision (SP) was used to investigate the interactions between compounds and PD-L1. Other parameters were set as the default. All the figures illustrating the molecular docking results were visualized and generated using the Maestro or Pymol software.
3.4. Water Solubility Studies
The aqueous solubility was estimated by HPLC. The HPLC analysis was performed on a Shimadzu LC-20AT machine with a BDS Hypersil C18 column, and the column temperature was at 28 °C. Mobile phase B (100% MeOH) and mobile phase A (100% water) were used in a gradient elution program (0 min: 30% (B), 2 min: 50% (B), 8 min: 95% (B), 14 min: 95% (B), 18 min: 70% (B), 20 min: 30% (B)) with a flow rate of 1.0 mL/min at 254 nM. Methanol was used to configure an accurate compounds concentration to calculate the standard curve. The excess test compound was dissolved in PBS (KH2PO4 0.24 g + KCl 0.2 g + NaCl 8 g + Na2HPO4·12H2O 3.58 g + 1 L water) and then analyzed by HPLC.
4. Conclusions
The blockade of the PD-1/PD-L1 interaction by small molecules has been highly anticipated as a promising alternative or complementary therapeutic to mAbs in the field of cancer immunotherapy. Currently, the field of developing small-molecule inhibitors of PD-1/PD-L1 interaction is intensively explored and makes great progress. Moreover, several small molecules, derived from the skeleton of BMS-202, have entered clinical trials to treat advanced solid tumors. Although several co-crystal complexes of inhibitors binding to PD-L1 dimer have been reported, which provided a definite guide in the rational design of PD-L1 small molecular inhibitors, the highly hydrophobic PD-1/PD-L1 binding interface remain a challenge for designing anti-PD-1/PD-L1 small-molecule inhibitors.
In this research, a novel series of (2-methyl-[1,1′-biphenyl]-3-yl) pyridine derivatives were designed and synthesized as inhibitors targeting PD-1/PD-L1 pathways based on structural simplification strategy. The representative compound A9 presents significantly improved potency in biochemical assay compared with BMS-202. Notably, the bioactivity of A9 is equivalent to that of BMS-1058, but possesses lower MW, PSA, and higher LE value. Subsequently, we utilized SPR assay to measure the binding affinity and kinetics of A9 to hPD-L1 (KD: 3.64 nM), and the results show that A9 possesses the feature of fast association and slow dissociation rate. Furthermore, small-molecule inhibitor A9 (5 μM) and mAbs Keytruda (5 μg/mL) show comparable promoting effect on production of INF-γ in a dose-dependent manner in Hep3B/OS-8/hPD-L1 and CD3-positive T cells co-culture assay, implying that A9 may reverse hPD-1/hPD-L1 pathway-mediated immunosuppression. Taken together, these results suggest that A9 is a promising inhibitor of PD-1/PD-L1 interaction and is worthy for further study.
Acknowledgments
Thank you for support of the public platform of the State Key Laboratory of Natural Medicines, China Pharmaceutical University.
Supplementary Materials
The following are available online, Table S1: The Kinetic Parameters and Binding Affinities of compounds A7, A9, A11 and A15 with hPD-L1.
Author Contributions
Designed and synthesized the target compounds, H.Z., Y.X. (Yu Xia) and H.L.; in vitro HTRF assay, H.Z., S.H. and H.D.; in vitro non-specific cytotoxicity assay, C.Y.; software, Y.Z.; writing—original draft preparation, H.Z. and J.L.; writing—review and editing, H.Z. and Y.Z.; funding acquisition, Q.Z. and Y.X. (Yungen Xu). All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Outstanding Scientific and Technological Innovation Team Projects of Jiangsu Province, China (2015).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The article contains complete data used to support the findings of this study.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Sample Availability
Samples of the compounds described in this paper are available from the authors.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Algazi A.P., Tsai K.K., Shoushtari A.N., Munhoz R.R., Eroglu Z., Piulats J.M., Ott P.A., Johnson D.B., Hwang J., Daud A.I., et al. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer. 2016;122:3344–3353. doi: 10.1002/cncr.30258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reck M., Rodríguez-Abreu D., Robinson A.G., Hui R., Csőszi T., Fülöp A., Gottfried M., Peled N., Tafreshi A., Cuffe S., et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 3.Swami U., Monga V., Bossler A.D., Zakharia Y., Milhem M. Durable clinical benefit in patients with advanced cutaneous melanoma after discontinuation of anti-PD-1 therapies due to immune-related adverse events. J. Oncol. 2019;2019:1856594. doi: 10.1155/2019/1856594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou W., Wolchok J.D., Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boussiotis V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016;375:1767–1778. doi: 10.1056/NEJMra1514296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keir M.E., Butte M.J., Freeman G.J., Sharpe A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boxberg M., Steiger K., Lenze U., Rechl H., Von Eisenhart-Rothe R., Wörtler K., Weichert W., Langer R., Specht K. PD-L1 and PD-1 and characterization of tumor-infiltrating lymphocytes in high grade sarcomas of soft tissue—Prognostic implications and rationale for immunotherapy. Oncoimmunology. 2018;7:e1389366. doi: 10.1080/2162402X.2017.1389366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwai Y., Ishida M., Tanaka Y., Okazaki T., Honjo T., Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu J.X., Hodge J.P., Oliva C., Neftelinov S.T., Hubbard-Lucey V.M., Tang J. Trends in clinical development for PD-1/PD-L1 inhibitors. Nat. Rev. Drug Discov. 2020;19:163–164. doi: 10.1038/d41573-019-00182-w. [DOI] [PubMed] [Google Scholar]
- 10.Cha E., Wallin J., Kowanetz M. PD-L1 Inhibition with MPDL3280A for Solid Tumors. Semin. Oncol. 2015;42:484–487. doi: 10.1053/j.seminoncol.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Dirix L.Y., Takacs I., Jerusalem G., Nikolinakos P., Arkenau H.T., Forero-Torres A., Boccia R., Lippman M.E., Somer R., Smakal M., et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast Cancer Res. Treat. 2018;167:671–686. doi: 10.1007/s10549-017-4537-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vandeveer A.J., Fallon J.K., Tighe R., Sabzevari H., Schlom J., Greiner J.W. Systemic Immunotherapy of Non-Muscle Invasive Mouse Bladder Cancer with Avelumab, an Anti–PD-L1 Immune Checkpoint Inhibitor. Cancer Immunol. Res. 2016;4:452–462. doi: 10.1158/2326-6066.CIR-15-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang J., Shalabi A., Hubbard-Lucey V. Comprehensive analysis of the clinical immuno-oncology landscape. Ann. Oncol. 2018;29:84–91. doi: 10.1093/annonc/mdx755. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y., Zhou S., Yang F., Qi X., Wang X., Guan X., Shen C., Duma N., Aguilera J.V., Chintakuntlawar A., et al. Treatment-Related Adverse Events of PD-1 and PD-L1 Inhibitors in Clinical Trials. JAMA Oncol. 2019;5:1008–1019. doi: 10.1001/jamaoncol.2019.0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baraibar I., Melero I., Ponz-Sarvise M., Castanon E. Safety and Tolerability of Immune Checkpoint Inhibitors (PD-1 and PD-L1) in Cancer. Drug Saf. 2019;42:281–294. doi: 10.1007/s40264-018-0774-8. [DOI] [PubMed] [Google Scholar]
- 16.Adams J.L., Smothers J., Srinivasan R., Hoos A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discov. 2015;14:603–622. doi: 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- 17.Chupak L.S., Ding M., Marin S.W., Zheng X., Hewawasam P., Connolly T.P., Xu N., Yeung K.-S., Zhu J., Langley D.R., et al. Compounds Useful as Immunomodulators. WO2,015,160,641. WO Patent. 2015 Oct 22;
- 18.Yeung K.S., Grant-Young K.A., Zhu J., Saulnier M.G., Frennesson D.B., Langley D.R., Hewawasam P., Wang T., Zhang Z., Meng Z. Compounds Useful as Immunomodulators. WO2,017,066,227. WO Patent. 2017 Apr 20;
- 19.Qin M., Cao Q., Zheng S., Tian Y., Zhang H., Xie J., Xie H., Liu Y., Zhao Y., Gong P. Discovery of [1,2,4]Triazolo[4,3-a]pyridines as Potent Inhibitors Targeting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction. J. Med. Chem. 2019;62:4703–4715. doi: 10.1021/acs.jmedchem.9b00312. [DOI] [PubMed] [Google Scholar]
- 20.Basu S., Yang J., Xu B., Magiera-Mularz K., Skalniak L., Musielak B., Kholodovych V., Holak T.A., Hu L. Design, Synthesis, Evaluation, and Structural Studies of C2-Symmetric Small Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein–Protein Interaction. J. Med. Chem. 2019;62:7250–7263. doi: 10.1021/acs.jmedchem.9b00795. [DOI] [PubMed] [Google Scholar]
- 21.Chen T., Li Q., Liu Z., Chen Y., Feng F., Sun H. Peptide-based and small synthetic molecule inhibitors on PD-1/PD-L1 pathway: A new choice for immunotherapy? Eur. J. Med. Chem. 2019;161:378–398. doi: 10.1016/j.ejmech.2018.10.044. [DOI] [PubMed] [Google Scholar]
- 22.Guzik K., Zak K.M., Grudnik P., Magiera K., Musielak B., Törner R., Skalniak L., Dömling A., Dubin G., Holak T.A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017;60:5857–5867. doi: 10.1021/acs.jmedchem.7b00293. [DOI] [PubMed] [Google Scholar]
- 23.Zak K.M., Grudnik P., Guzik K., Zieba B.J., Musielak B., Dömling A., Dubin G., Holak T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1) Oncotarget. 2016;7:30323–30335. doi: 10.18632/oncotarget.8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ganesan A., Ahmed M., Okoye I., Arutyunova E., Babu D., Turnbull W.L., Kundu J.K., Shields J., Agopsowicz K.C., Xu L., et al. Comprehensive in vitro characterization of PD-L1 small molecule inhibitors. Sci. Rep. 2019;9:12392. doi: 10.1038/s41598-019-48826-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skalniak L., Zak K.M., Guzik K., Magiera K., Musielak B., Pachota M., Szelazek B., Kocik J., Grudnik P., Tomala M., et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget. 2017;8:72167–72181. doi: 10.18632/oncotarget.20050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin X., Lu X., Luo G., Xiang H. Progress in PD-1/PD-L1 pathway inhibitors: From biomacromolecules to small molecules. Eur. J. Med. Chem. 2020;186:111876. doi: 10.1016/j.ejmech.2019.111876. [DOI] [PubMed] [Google Scholar]
- 27.Hopkins A.L., Groom C.R., Alex A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 28.Glide. Schrödinger, LLC; New York, NY, USA: 2009. [(accessed on 25 May 2021)]. Available online: https://www.schrodinger.com/products/glide. [Google Scholar]
- 29.Dassault Systèmes. BIOVIA Discovery Studio; San Diego, CA, USA: 2019. [Google Scholar]
- 30.AlMahmoud S., Zhong H.A. Molecular Modeling Studies on the Binding Mode of the PD-1/PD-L1 Complex Inhibitors. Int. J. Mol. Sci. 2019;20:4654. doi: 10.3390/ijms20184654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J., Jiang C.C., Jin L., Zhang X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016;27:409–416. doi: 10.1093/annonc/mdv615. [DOI] [PubMed] [Google Scholar]
- 32.Akinleye A., Rasool Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019;12:92. doi: 10.1186/s13045-019-0779-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalbasi A., Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020;20:25–39. doi: 10.1038/s41577-019-0218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The article contains complete data used to support the findings of this study.