

Abstract
The direct 4-alkoxylation of 4-iodo-1H-pyrazoles with alcohols was achieved by a CuI-catalyzed coupling protocol. The optimal reaction conditions employed excess alcohol and potassium t-butoxide (2 equiv) in the presence of CuI (20 mol%) and 3,4,7,8-tetramethyl-1,10-phenanthroline (20 mol%) at 130 °C for 1 h under microwave irradiation. The present method was efficiently applied to the synthesis of withasomnine and its six- and seven-membered cyclic homologs.
Keywords: synthesis, 4-alkoxypyrazole, CuI, coupling reaction, microwave, withasomnine, homologue
1. Introduction
Owing to their diverse bioactivities, both natural and synthetic pyrazoles and pyrazole-fused heterocycles have been widely exploited as pharmaceutical or pesticide active ingredients [1,2,3,4]. Therefore, the efficient synthesis of substituted pyrazoles possessing characteristic functionalities at specific positions is an important objective in organic and medicinal chemistry, as well as in drug discovery. In this context, we recently reported palladium- or copper-catalyzed C–N coupling reactions at the C-4 positions of pyrazoles [5]. Although metal-catalyzed C–O coupling reactions have been widely reported, owing to their wide-ranging potentials [6,7,8,9,10,11,12], the direct C4-O-functionalization of pyrazoles has not yet been studied satisfactorily [13,14] despite the important bioactivities that have been demonstrated for several 4-alkoxypyrazoles, as presented in Figure 1.
Figure 1.

Bioactive 4-alkoxypyrazoles.
4-Hyroxypyrazole, a metabolite of pyrazole, exhibits various biological activities such as anti-inflammatory, antipyretic, antitumor, antifungal effects [15]. 4-Methoxy-, 4-ethoxy-, 4-n-propoxy-, and 4-isopropoxypyrazoles have been reported to inhibit liver alcohol dehydrogenase (LAD) in humans, rats, and horses [16]. In particular, 4-ethoxypyrazole and 4-propyloxypyrazole have been recognized as a cytochrome P-450 inducer [17] and cytochrome P450 2E1 inhibitor, respectively [18]. Two C4-O-functionalized pyrazoles have been patented: 1-methyl-3,5-diphenyl-4-propoxypyrazole, as a fungicide [19], and the alkyl (4-alkoxy-1-phenyl)pyrazolylcarboxylates, which possess antipyretic, sedative, anti-inflammatory, and analgesic activities [20]. A compound bearing a 4-(2,4-difluorophenyl)oxy group was revealed as a human dihydroorotate dehydrogenase (DHODH) inhibitor [21].
4-Allyloxy-1H-1-tritylpyrazole 4a (Scheme 1), derived from 4-iodopyrazole (1), played a key role as a versatile intermediate in our previous studies for the total synthesis of the pyrazole alkaloid, withasomnine (7) [22,23], and its six-membered homolog 11 [23,24,25,26,27], which were reported to exhibit COX-2 inhibitory activities [23,27,28]. Compound 4a was also extensively utilized as an important intermediate for the construction of new pyrazole-fused heterobicyclic molecules 9 via ring-closing metathesis (RCM) (Scheme 1) [29,30,31]. However, the synthesis of compound 4a requires six steps from commercially available pyrazole, through 4-iodopyrazole (1), 4-iodo-1H-1-tritylpyrazole (2a), and aldehyde 3. If direct O-allylation from 1 could be realized, the synthesis of several synthetic targets would be remarkably shortened. Thus, we focused our attention on the direct C–O coupling reactions of 1, based on our prior report of the C–N couplings of 4-halopyrazoles [5]. Herein, we disclose CuI-catalyzed coupling reactions of 4-iodo-1H-pyrazoles and alcohols. Furthermore, the developed method was applied to improve the synthesis of withasomnine and its homologs containing six- or seven-membered ring systems.
Scheme 1.

Versatile intermediate 4-allyloxy-1H-tritylpyrazole (4a), developed in our previous works (adapt from references [22,23,24,29,30,31]).
2. Results and Discussions
2.1. Investigation of 4-O-Allylation of 4-Iodopyrazole
Initially, we attempted the Pd(dba)2-catalyzed reaction between 2a and allyl alcohol (2 equivalent (equiv)) in the presence of tBuDavePhos as a ligand and potassium tert-butoxide (tBuOK) as a base under the reaction conditions in our previous report [5]; however, none of the desired coupling product was obtained (Table 1, entry 1). The corresponding 4-bromo-1H-1-tritylpyrazole was not effective in the palladium-catalyzed coupling reaction. Then, the CuI-catalyzed reaction between 4-iodo-1H-1-tritylpyrazole 2a and allyl alcohol was examined; the results are summarized in Table 1. All reactions were performed using 2a (50 mg) in a solvent (2.0 mL). In the presence of ligand 2-isobutyroylcyclo-hexanone (L2) or 1,10-phenanthroline (L3) in N,N-dimethylformamide (DMF) [5], reactions of 2a and allyl alcohol (2 equiv) did not afford 4a (entries 2 and 3, respectively). However, when allyl alcohol was used as a solvent for this reaction with L3 at 100 °C overnight, the desired C4-O-allylation product 4a was obtained in 51% yield (entry 4). Next, microwave (MW) assistance was applied to reduce the reaction time (entries 5–9). In these experiments, the reaction time was fixed at 1 h and the ligand was changed to 3,4,7,8-tetramethyl-1,10-phenanthroline (L4). From entry 6, the optimum reaction temperature was determined to be 130 °C, giving 4a in 66% yield. At 160 °C, the reaction mixture turned black with a poor yield of 4a (16%, entry 8). In addition, shortening the reaction time (30 min) or reducing the amount of CuI to 10 mol% afforded 4a in lower yields (entry 7:24%; entry 9:37%, respectively). Based on these results, the optimum conditions obtained in entry 6 were applied in the following coupling reactions of 4-iodopyrazoles with various alcohols.
Table 1.
Optimization of CuI-catalyzed reaction between 4-iodo-1H-1-tritylpyrazole (2a) and allyl alcohol.

| Entry | Catalyst | Ligand a | Solvent | Temperature (°C) | Time | 4a Yield, % |
|---|---|---|---|---|---|---|
| 1 b,c | Pd(dba)2 | L1 | xylene | 160 (MW) | 30 min | 0 |
| 2 c | CuI | L2 | DMF | 100 | overnight | 0 |
| 3 c | CuI | L3 | DMF | 100 | overnight | 0 |
| 4 | CuI | L3 | allyl alcohol | 100 | overnight | 51 |
| 5 | CuI | L4 | allyl alcohol | 100 (MW) | 1 h | 31 |
| 6 | CuI | L4 | allyl alcohol | 130 (MW) | 1 h | 66 |
| 7 | CuI | L4 | allyl alcohol | 130 (MW) | 30 min | 24 |
| 8 | CuI | L4 | allyl alcohol | 160 (MW) | 1 h | 16 |
| 9 d | CuI | L4 | allyl alcohol | 130 (MW) | 1 h | 37 |
a. 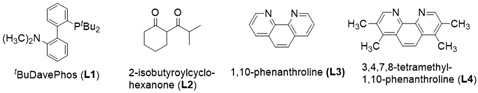 . b. 10 mol% Pd(dba)2 was used, c. 2 equiv of allyl alcohol was added, d. 10 mol% CuI was used.
. b. 10 mol% Pd(dba)2 was used, c. 2 equiv of allyl alcohol was added, d. 10 mol% CuI was used.
2.2. C4-Alkoxylation of 4-iodopyrazole with Alcohols Using CuI-Catalyzed Coupling
To study the scope and limitations of this transformation, the CuI-catalyzed reactions of iodopyrazoles 2 (50 mg) with various alcohols (2.0 mL, excess amount) were carried out under the optimal conditions (Table 1, entry 6). The results are summarized in Table 2. The reactions of 2a with linear short-chain primary alcohols (methanol, ethanol, and n-propanol) afforded the corresponding products 4c, 4d, and 4e in moderate yields (61–76%, entries 1–3), while the reaction with a longer-chain primary alcohol (n-butanol) resulted in a lower yield (33%, entry 4). The reactions of 2a with branched primary alcohols (isobutyl and isoamyl alcohols) provided 4i (45%) and 4k (37%), respectively (entries 7 and 9), but with secondary isopropanol, gave 4g in only 9% yield (entry 5). The presence of sec- or tert-butyl groups in the alcohol was not compatible with the present reaction conditions (entries 6 and 8), probably due to steric hindrance. In contrast, the reactions with cyclic secondary alcohols did proceed (entries 10, 11, and 12), but the respective isolated yields of the coupled products 4l, 4m, and 4n were 59%, 18%, and 25%, respectively. In these reactions, 1.0 mL of cyclic alcohol was used with respect to substrate 2a (50 mg); the high boiling points (cyclobutanol: 123 °C/733 mmHg; cyclopentanol: 139–140 °C; cyclohexanol: 160–161 °C) of these materials complicated product isolation by chromatography. Furthermore, when 2 equivalents of the cyclic alcohols and acetonitrile (2.0 mL) as a co-solvent were used, no coupled products could be detected. Although the reaction with benzyl alcohol (bp: 205 °C) was also difficult, the use of benzyl alcohol (1.0 mL) and toluene as a co-solvent (1.0 mL) afforded the corresponding product (4o) in poor yield (12%, entry 13). With phenols, no desired coupling products were obtained under various reaction conditions (entries 14 and 15). In the case of p-methoxyphenol (entry 15), a detailed analysis of the reaction mixture revealed a trace amount of 5,5′-dimethoxy-2,2′-biphenyldiol, which has been reported to have radical scavenging or antibacterial activities [32,33]. The initially formed dihydroxybiphenyls [34] might inhibit the attempted C–O coupling reaction.
Table 2.
CuI-catalyzed coupling reaction between iodopyrazoles and various alcohols.

| Entry | Substrate | R2OH | Product | Yield (%) |
|---|---|---|---|---|
| 1 | 2a: R1 = Tr | R2 = Me | 4c: R1 = Tr, R2 = Me | 61 |
| 2 | 2a | R2 = Et | 4d: R1 = Tr, R2 = Et | 76 |
| 3 | 2a | R2 = n-Pr | 4e: R1 = Tr, R2 = n-Pr | 64 |
| 4 | 2a | R2 = n-Bu | 4f: R1 = Tr, R2 = n-Bu | 33 |
| 5 | 2a | R2 = iPr | 4g: R1 = Tr, R2 = iPr | 9 |
| 6 | 2a | R2 = sec-Bu | 4h: R1 = Tr, R2 = sec-Bu | 0 |
| 7 | 2a | R2 = iBu | 4i: R1 = Tr, R2 = iBu | 45 |
| 8 | 2a | R2 = tert-Bu | 4j: R1 = Tr, R2 = tert-Bu | 0 |
| 9 | 2a | R2 = isoamyl | 4k: R1 = Tr, R2 = isoamyl | 37 |
| 10 a | 2a | R2 = cyclobutyl | 4l: R1 = Tr, R2 = cyclobutyl | 59 |
| 11 a | 2a | R2 = cyclopentyl | 4m: R1 = Tr, R2 = cyclopentyl | 18 |
| 12 a | 2a | R2 = cyclohexyl | 4n: R1 = Tr, R2 = cyclohexyl | 25 |
| 13 b | 2a | R2 = Bn | 4o: R1 = Tr, R2 = Bn | 12 |
| 14 | 2a | R2 = Ph | 4p: R1 = Tr, R2 = Ph | 0 |
| 15 c | 2a | R2 = p-MeOPh | 4q: R1 = Tr, R2 = p-MeOPh | 0 |
| 16 d | 2a | R2 = allyl | 4a: R1 = Tr, R2 = allyl | 66 |
| 17 | 2b: R1 = 1-propenyl | R2 = 1-propenyl | 4r: R1 = 1-propenyl, R2 = allyl | 65 |
| 18 | 2c: R1 = allyl | R2 = allyl | 4s: R1 = R2 = allyl | 58 |
| 19 | 2d: R1 = 3-butenyl | R2 = allyl | 4t: R1 = 3-butenyl, R2 = allyl | 64 |
| 20 e | 2a | R2 = OH | 4u: R1 = Tr, R2 = H | 0 |
| 21 | 1 | R2 = allyl | 4v: R1 = H, R2 = allyl | 0 |
a. 1.0 mL of cyclic alcohol was used, b. 1.0 mL of BnOH was used with toluene (1.0 mL) as co-solvent, c. Trace amount of 5,5’-dimethoxy-2,2’-biphenyldiol was isolated, d. Table 1, entry 6, e. 2a was recovered (89%).
The direct introduction of the allyloxy group at the C4 position of N-alkenyl-4-iodo-1H-pyrazoles (2b, 2c, 2d) by CuI-mediated reaction afforded the expected products 4r, 4s, and 4t in moderate yields (entries 17–19). These products were subsequently applied in the synthesis of withasomnine and its analogs (Scheme 2). Neither the C–O coupling reaction of 2a with water nor of N-nonprotected iodopyrazole 1 with allyl alcohol was successful (entries 20 and 21).
Scheme 2.

Application to improved synthesis of withasomnine (7).
In preliminary experiments, the Pd(dba)2-catalyzed coupling of 2a with four types of alcohols (methanol, ethanol, n-propanol, and tert-butyl alcohol) under the same conditions as mentioned above was examined; however, these trials did not give the desired coupling products, yielding only hydrogenated 1H-1-tritylpyrazole in 52, 63, 64, and 8% yields, respectively.
2.3. Application to Improved Synthesis of Withasomnine and Six- and Seven-Membered Cyclic Homologs
A modified synthesis of withasomnine and its homologs using the products described above was performed to demonstrate the usefulness of the present method. The improved synthesis of withasomnine (7) is summarized in Scheme 2. 4-Iodo-1H-pyrazole (1) was treated with allyl bromide under basic conditions to give N-allylated compound 2c in 97% yield. The double bond in the N-allyl group in 2c was migrated by treatment with a ruthenium hydride catalyst (RuClH(CO)(PPh3)3) to give an E/Z mixture of 2b in 96% yield, which was transformed to 4r by CuI-catalyzed coupling, as described above (Table 2, entry 17). The Claisen rearrangement of 4r gave (E/Z)-12 (87%), which was subsequently O-triflated by treatment with trifluoromethanesulfonic anhydride (Tf2O) in the presence of triethylamine at −20 °C to yield ring-closing metathesis (RCM) substrate 13 in 90% yield. Treatment of 13 with Grubbs2nd catalyst in toluene at 100 °C under MW irradiation gave the desired RCM product 14 in 0–58% yields with unsatisfied reproducibility.
Alternatively, CuI-assisted RCM [24,35] of 13 in CH2Cl2 under milder conditions using MW-aided heating at 80 °C for 1 h successfully afforded pyrrole-[1,2-b] pyrazole 14 (63%), which was immediately hydrogenated under a hydrogen gas atmosphere with Pd/C in MeOH to give penultimate product 6 in 90% yield. As the transformation from 6 to 7 via a Suzuki-Miyaura coupling reaction has already been reported [22,23], the present approach constitutes a formal total synthesis of withasomnine (7). The overall yield of 7 in this case was 24% over nine steps from commercially available pyrazole, whereas that of our previous method was 8% in 13 steps. Therefore, the current synthesis realizes a four-step reduction and nearly threefold improvement in overall yield [22,23].
The syntheses of the six-and seven-membered cyclic homologs 11 and 15 are summarized in Scheme 3. The total yield of 11 was improved by ~1.6-fold over our former synthesis based on the yields of transformations from 1 to 2c (seen in Scheme 2) and 2c to 4s (Table 2, entry 18) [24]. Our synthesis of another withasomnine homolog, 15, previously achieved by Allin via radical cyclization [25,26], began with the transformation of 1 to 2d in 88% yield. Compound 2d was O-allylated using the present method to 4t, as described previously (Table 2, entry 19). Then, 4t was rearranged into 16 (81% yield) under MW-assisted heating, and subsequent O-triflation afforded 17 (83% yield). RCM substrate 17 was similarly cyclized to seven-membered intermediate 18 in 72% yield, which was then subjected to Suzuki-Miyaura coupling with phenylboronic acid to afford 19 in 87% yield. The synthesis of 15 was completed in 92% yield by the Pd-C-catalyzed hydrogenation of 19. An alternative route to 15 comprised the transformation of 1 to 4b in 52% yield via a five-step process, and subsequent N-butenylation to give the common intermediate 4t in 69% yield. Therefore, the present route to 15 using the CuI-catalyzed coupling achieved a 1.6-fold increase in overall yield compared to the prior procedure.
Scheme 3.

Synthesis of withasomnine six- and seven-membered cyclic homologs 11 and 15.
3. Conclusions
In this study, a range of 4-alkoxy-1H-pyrazoles was synthesized using the CuI-catalyzed coupling reaction of 4-iodopyrazoles with an excess amount of alcohol. Improved syntheses of withasomnine and its homologs were achieved using the products obtained with the present method. The current withasomnine synthetic route was reduced by four steps with a threefold-improvement in the overall yield compared to our previous report [22,23]. However, reducing the amounts of catalysts, ligands, and alcohols will be required to increase the practicality of this reaction in the future.
4. Materials and Methods
4.1. General Information
NMR spectra were recorded at 27 °C on Agilent 400- and 600-MR-DD2 spectrometers (Agilent Tech., Inc., Santa Clara, CA, USA) in CDCl3 with tetramethylsilane (TMS) as an internal standard. HRMS was performed using a JEOL JMS-700 (2) mass spectrometer (JEOL, Tokyo, Japan). Melting points were determined using a Yanagimoto micromelting point apparatus (Yamagimoto, Kyoto, Japan) and were uncorrected. Liquid column chromatography was conducted using silica gel (Fuji Silysia FL-60D). Analytical TLC was performed on precoated Merck glass plates (silica gel 60 F254), and compounds were detected by dipping the plates in an ethanol solution of phosphomolybdic acid, followed by heating. All microwave-aided reactions were performed using a Biotage Initiator® (Biotage, Uppsala, Sweden). Allyl alcohol, n-propanol, isobutyl alcohol, cyclobutanol, cyclopentanol, cyclohexanol, Phenol, p-methoxyphenol, allylbromide, and Pd(dba)2, were purchased from Tokyo Chemical Industry (TCI) Co. (Tokyo, Japan). t-BuOK, CuI, 1,10-phenanthroline (L3), 3,4,7,8-tetramethyl-1,10-phenanthroline (L4), isopropanol, tert-butanol, isoamyl alcohol, toluene, cesium acetate, trifluoromethanesulfonic anhydride, and triethylamine were purchased from Nacalai Tesque, Inc. (Kyoto, Japan). Dry xylene, dry DMF, n-butanol, sec-butanol, and 1,2-dimethoxyethane, were purchased from FUJIFILM Wako Pure Chemical Co. (Osaka, Japan). tBuDavePhos (L1), 2-isobutyrylcyclohexanone (L2), RuClH(CO)(PPh3)3, Grubbs2nd, and XPhos were purchased from Sigma-Aldrich Co. LLC (St. Louis, MO, USA).
1H- and 13C-NMR spectra of all new compounds with compound 15 are provided in Supplementary Materials as Figures S1–S48.
4.2. CuI-Catalyzed Coupling Reactions of 4-iodo-1H-1-tritylpyrazole with Alcohols (Table 1 and Table 2)
General procedure (Table 1, entry 6): To a solution of 2a (50.0 mg, 0.12 mmol) in allyl alcohol (2.0 mL, 29 mmol) in a microwave vial (0.5–2.0 mL) were added 3,4,7,8-tetramethyl-1,10-phenanthroline (5.8 mg, 0.026 mmol, 20 mol%), CuI (4.4 mg, 0.023 mmol, 20 mol%), and tBuOK (28.8 mg, 0.26 mmol, 2.0 equiv). The mixture was stirred to make a solution, sealed, and heated at 130 °C for 1 h under MW irradiation. The cooled mixture was checked by TLC (hexane/AcOEt = 8:1), quenched by the addition of saturated (sat.) aqueous (aq.) NH4Cl (1 mL), and extracted with dichloromethane (CH2Cl2; 1.0 mL × 3). The combined organic layers were dried over MgSO4, filtered, and evaporated to give a crude residue that was purified by silica gel column chromatography (eluent:hexane/AcOEt = 20:1) to afford previously reported 4a (27.9 mg, 66%).
4-Methoxy-1H-1-tritylpyrazole (4c): Colorless needles (CH2Cl2); mp 149–152 °C; 1H-NMR (400 MHz, CDCl3): δ 7.40 (1H, s, pyrazole-H), 7.32–7.28 (9H, m, Tr-H), 7.18–7.14 (6H, m, Tr-H), 7.01 (1H, s, pyrazole-H), 3.68 (3H, s, 4-OMe); 13C-NMR (100 MHz, CDCl3): δ 145.7, 143.2, 130.1, 127.6, 127.4, 127.2, 117.2, 78.6, 58.7; HREIMS m/z calculated (calcd) for C23H20N2O (M+) 340.1575, found 340.1577.
4-Ethoxy-1H-1-tritylpyrazole (4d): Colorless needles (CH2Cl2); mp 141–144 °C; 1H-NMR (400 MHz, CDCl3): δ 7.40 (1H, s, pyrazole-H), 7.01 (1H, s, pyrazole-H), 7.31–7.28 (9H, m, Tr-H), 7.18–7.14 (6H, m, Tr-H), 3.87 (2H, q J = 6.6 Hz, -OCH2CH3), 1.32 (3H, t, J = 6.6 Hz, CH2CH3); 13C-NMR (100 MHz, CDCl3): δ 144.4, 143.2, 130.1, 127.8, 127.6, 127.6, 117.9, 78.6, 67.1, 14.9; HREIMS m/z calcd for C24H22N2O (M+) 354.1732, found 354.1735.
4-n-Propyloxy-1H-1-tritylpyrazole (4e): Colorless needles (CH2Cl2); mp 118–120 °C; 1H-NMR (400 MHz, CDCl3): δ, 7.40 (1H, s, pyrazole-H), 7.30–7.27 (9H, m, Tr-H), 7.18–7.14 (6H, m, Tr-H), 7.01 (1H, s, pyrazole-H), 3.76 (2H, t, J = 6.6 Hz, -OCH2CH2-), 1.72 (2H, qt, J = 7.4, 6.6 Hz, -CH2CH2CH3), 0.97 (3H, t, J = 7.4 Hz, -CH2CH3); 13C-NMR (100 MHz, CDCl3): δ 144.6, 143.2, 130.1, 127.8, 127.61, 127.59, 117.7, 78.5, 73.1, 22.7, 10.4; HREIMS m/z calcd for C25H24N2O (M+) 368.1889, found 368.1889.
4-n-Butoxy-1H-1-tritylpyrazole (4f): Colorless needles (CH2Cl2); mp 127–130 °C; 1H-NMR (400 MHz, CDCl3): δ 7.40 (1H, s, pyrazole-H), 7.31–7.28 (9H, m, Tr-H), 7.18–7.14 (6H, m, Tr-H), 7.01 (1H, s, pyrazole-H), 3.80 (2H, q J = 6.6 Hz, -OCH2CH2-), 1.68 (2H, quint, J = 6.7 Hz, -CH2CH2CH2-), 1.42 (2H, sext, J = 6.6 Hz, -CH2CH2CH3), 0.93 (3H, t, J = 6.7 Hz, -CH2CH3); 13C-NMR (100 MHz, CDCl3): δ 144.7, 143.2, 130.1, 127.8, 127.6, 127.6, 117.7, 78.5, 71.3, 31.4, 19.1, 13.9; HREIMS m/z calcd for C26H26N2O (M+) 382.2035, found 382.2040.
4-Isopropyloxy-1H-1-tritylpyrazole (4g): White powder; mp 91–93 °C; 1H-NMR (400 MHz, CDCl3): δ 7.39 (1H, d, J = 1.0 Hz, pyrazole-H), 7.31–7.28 (9H, m, Tr-H), 7.18–7.14 (6H, m, Tr-H), 7.02 (1H, d, J = 0.7 Hz, pyrazole-H), 4.12 (1H, sept, J = 6.1 Hz, -OCH(CH3)2), 1.26 (6H, d, J = 6.1 Hz, -CH(CH3)2); 13C-NMR (100 MHz, CDCl3): δ 143.2, 142.7, 130.1, 129.2, 127.6, 119.8, 78.6, 74.0, 21.9; HREIMS m/z calcd for C25H24N2O (M+) 368.1889, found 368.1885.
4-Isobutoxy-1H-1-tritylpyrazole (4i): White powder; mp 95–98 °C; 1H-NMR (400 MHz, CDCl3): δ 7.40 (1H, s, pyrazole-H), 7.30–7.28 (9H, m, Tr-H), 7.18–7.14 (6H, m, Tr-H), 7.02 (1H, s, pyrazole-H), 3.56 (2H, d, J = 6.7 Hz, -OCH2CH-), 1.99 (1H, nonet, J = 6.7 Hz, -CH2CH(CH3)2), 0.96 (6H, d, J = 6.7 Hz, -CH(CH3)2); 13C-NMR (100 MHz, CDCl3): δ 144.8, 143.2, 130.1, 127.8, 127.65, 127.61, 127.58, 117.6, 78.5, 78.0, 28.4, 19.1; HREIMS m/z calcd for C26H26N2O (M+) 382.2035, found 382.2040.
4-Isoamyloxy-1H-1-tritylpyrazole (4k): White powder; mp 77–75 °C; 1H-NMR (400 MHz, CDCl3): δ 7.40 (1H, s, pyrazole-H), 7.31–7.29 (9H, m, Tr-H), 7.18–7.14 (6H, m, Tr-H), 7.01 (1H, s, pyrazole-H), 3.83 (2H, q, J = 6.6 Hz, -OCH2CH2-), 1.76 (1H, nonet, J = 6.7 Hz, -CH2CH(CH3)2), 1.58 (2H, q, J = 6.7 Hz, -CH2CH2CH-), 0.92 (6H, d, J = 6.7 Hz, -CH (CH3)2); 13C-NMR (100 MHz, CDCl3): δ 22.6, 24.8, 38.1, 70.0, 78.6, 117.8, 127.6, 127.7, 127.8, 130.1, 143.2, 144.6; HREIMS m/z calcd for C26H26N2O (M+) 396.2201, found 396.2201.
4-Cyclobutyloxy-1H-1-tritylpyrazole (4l): White powder; mp 119–121 °C; 1H-NMR (400 MHz, CDCl3): δ 7.34 (1H, br s, pyrazole-H), 7.31–7.28 (9H, m, Tr-H), 7.17–7.12 (6H, m, Tr-H), 6.95 (1H, br s, pyrazole-H), 4.40–4.33 (1H, m, -OCH(CH2)2-), 2.31–2.33 (2H, m), 2.13–2.03 (2H, m), 1.81–1.73 (1H, m), 1.62–1.52 (1H, m); 13C-NMR (100 MHz, CDCl3): δ 143.2, 142.4, 130.1, 128.3, 127.6, 118.6, 78.6, 74.4, 30.3, 12.6; HREIMS m/z calcd for C26H24N2O (M+) 380.1889, found 380.1890.
4-Cyclopentyloxy-1H-1-tritylpyrazole (4m): White powder; mp 104–106 °C; 1H-NMR (400 MHz, CDCl3): δ 7.37 (1H, d, J = 0.6 Hz, pyrazole-H), 7.37–7.28 (9H, m, Tr-H), 7.17–7.12 (6H, m, Tr-H), 6.98 (1H, d, J = 0.8 Hz, pyrazole-H), 4.44–4.42 (1H, m, -OCH(CH2)2-), 1.82–1.74 (4H, m), 1.72–1.56 (2H, m); 13C-NMR (100 MHz, CDCl3): δ 143.2, 130.1, 128.7, 127.8, 127.7, 127.6, 119.0, 83.1, 78.5, 32.6, 23.8; HREIMS m/z calcd for C27H26N2O (M+) 394.2045, found 394.2043.
4-Cyclohexyloxy-1H-1-tritylpyrazole (4n): White powder; mp 132–135 °C; 1H-NMR (400 MHz, CDCl3): δ 7.39 (1H, s, pyrazole-H), 7.31–7.28 (9H, m, Tr-H), 7.17–7.12 (6H, m, Tr-H), 7.03 (1H, s, pyrazole-H), 3.78–3.83 (1H, m, -OCH(CH2)2-), 1.95–1.92 (2H, m), 1.57–1.51 (2H, m), 1.48–1.38 (2H, m) 1.32–1.23 (2H, m); 13C-NMR (100 MHz, CDCl3): δ 143.2, 142.5, 130.1, 129.5, 127.6, 120.1, 79.6, 78.6, 31.8, 25.6, 23.6; HREIMS m/z calcd for C28H28N2O (M+) 408.2202, found 408.2201.
4-Benzyloxy-1H-1-tritylpyrazole (4o): White powder; mp 154–156 °C; 1H-NMR (400 MHz, CDCl3): δ 7.44 (1H, s, pyrazole-H), 7.35–7.28 (14H, m, Tr-H, Ph-H), 7.15–7.12 (6H, m, Tr-H, Ph-H), 7.02 (1H, s, pyrazole-H), 4.86 (2H, br s, -OCH2Ph); 13C-NMR (100 MHz, CDCl3): δ 144.1, 143.1, 136.7, 130.1, 128.5, 128.0, 127.8, 127.6, 118.7, 78.6, 73.7; HREIMS m/z calcd for C29H24N2O (M+) 416.1889, found 416.1889.
(E/Z)-4-(Allyloxy)-1-(prop-1-en-1-yl)-1H-pyrazole (4r): Colorless oil; 1H-NMR (400 MHz, CDCl3): δ 7.38 (0.1H, s, pyrazole-H), 7.32 (0.7H, br s, pyrazole-H), 7.28 (0.3H, d, J = 0.6 Hz, pyrazole-H), 7.23 (0.7H, d, J = 0.8 Hz, pyrazole-H), 6.74 (0.7H, dq, J = 14.2, 0.7 Hz, (E)-NCH=CHCH3), 6.68 (0.3H, dq, J = 9.4, 0.7 Hz, (Z)-NCH=CHCH3), 6.08–5.97 (1H, m, -OCH2CH=CH2), 5.85 (0.7H, dq, J = 14.2, 7.0 Hz, (E)-NCH=CHCH3), 5.40 (1H, br d, J = 17.2 Hz, -CH=CHH), 5.29 (1H, br d, J = 9.4 Hz, -CH=CHH), 5.26 (0.3H, dq, J = 9.4, 7.9, (Z)-NCH=CHCH3), 4.44 (0.6H, dt, J = 5.5, 1.2Hz, -OCH2CH=CH2), 4.42 (1.4H, dt, J = 5.5, 1.5 Hz, -OCH2CH=CH2), 1.95 (0.9H, dd, J = 7.4, 1.8 Hz, (Z)-NCH=CHCH3), 1.81 (2.1H, dd, J = 7.1, 0.6 Hz, (E) -NCH=CHCH3); 13C-NMR (100 MHz, CDCl3): δ 146.0, 133.1, 133.0, 128.8, 128.72, 127.67, 127.65, 118.1, 115.0, 111.8, 111.1, 72.55, 72.47,, 14.7, 12.8 (3 carbons are overlapped); HREIMS m/z calcd for C9H12N2O (M+) 164.0950, found 164.0949.
4s: known [24]
4-Allyloxy-1-(3-buten-1-yl)pyrazole (4t): Colorless oil; 1H-NMR (400 MHz, CDCl3): δ 7.24 (1H, d, J =1.2 Hz, pyrazole-H), 7.08 (1H, d, J =1.0 Hz, pyrazole-H), 6.01 (1H, ddt, J = 17.2, 10.5, 5.5 Hz, -OCH2CH=CH2), 5.74 (1H, ddt, J = 17.2, 10.4, 6.8 Hz, -CH2CH=CH2), 5.38 (1H, dq, J = 17.2, 1.6 Hz, -CH2CH=CHH), 5.28 (1H, dq, J = 10.4, 1.4 Hz,-CH2CH=CHH), 5.04–5.10 (2H, overlapped, 2 × -CH=CHH), 4.40 (2H, dt, J = 5.4, 1.5 Hz, -OCH2CH=), 4.07 (2H, t, J = 7.1 Hz, NCH2CH2-), 2.57 (2H, qt, J = 7.0, 1.2 Hz, -CH2CH2CH=CH2); 13C-NMR (100 MHz, CDCl3): δ144.9, 134.1, 133.3, 127.0, 117.8, 117.4, 115.0, 72.5, 52.1, 34.5; HREIMS m/z calcd for C10H14N2O (M+) 178.1106, Found 178.1105.
4.3. Modified Synthesis of Withasomnine, (Scheme 2)
4.3.1. Synthesis of 1-allyl-4-iodo-1H-pyrazole (2c)
To a solution of 4-iodopyrazole 1 (500.0 mg, 2.6 mmol) in acetone (5 mL), 20% NaOH aq. (0.5 mL, 1.5 equiv) was added with stirring followed by allyl bromide (0.2 mL, 3.9 mmol, 1.5 equiv). The reaction mixture was stirred at room temperature for 1 h. After checking by TLC (hexane/AcOEt = 2:1), sat. aq. NH4Cl (5 mL) was added to the reaction mixture to quench the reaction. The mixture was extracted with CH2Cl2 (10 mL × 3) and the combined organic layers were washed with brine (5 mL × 2), dried over MgSO4, and filtered. The solvent was removed under reduced pressure to give a crude residue that was purified by silica gel column chromatography (eluent:hexane/AcOEt = 2:1) to afford 2c (566.2 mg, 96%): colorless oil; 1H-NMR (400 MHz, CDCl3): δ 7.53 (1H, s, pyrazole-H), 7.45 (1H, s, pyrazole-H), 6.00 (1H, ddt, J = 17.1, 10.2, 6.1 Hz, -NHCH2CH=CH2), 5.30 (1H, dq, J = 10.2, 1.2 Hz, -CH2CH=CHH), 5.26 (1H, dq, J = 17.1, 1.4 Hz, -CH2CH=CHH), 4.75 (2H, dt, J = 6.3, 1.4 Hz, -NCH2CH=CH2); 13C-NMR (100 MHz, CDCl3) δ 144.4, 133.4, 132.2, 119.3, 56.2, 55.1; HREIMS m/z calcd for C6H7N2I (M+) 233.9654, found 233.9653.
4.3.2. Synthesis of (E/Z)-4-iodo-1-(prop-1-en-1-yl)-1H-pyrazole (2b)
To a MW vial containing a solution of 2c (571.5 mg, 2.4 mmol) in toluene (2 mL) was added the ruthenium hydride catalyst, RuClH(CO)(PPh3)3 (116.3 mg, 0.12 mmol, 5 mol%). The reaction mixture in the sealed vial was heated at 160 °C for 10 min under MW irradiation. After removal of the solvent from the mixture, the residue was purified by silica gel column chromatography (eluent:hexane/AcOEt = 10:1) to afford 2b (546.6 mg, 96%) as a colorless oil: 1H-NMR (400 MHz, CDCl3): δ 7.62 (0.2H, s, pyrazole-H), 7.61 (0.2H, s, pyrazole-H), 7.58 (0.8H, s, pyrazole-H), 7.55 (0.8H, s, pyrazole-H), 6.80 (0.8H, dq, J = 14.0, 1.6 Hz, -CHE=CHCH3), 6.76 (0.2H, dq, J = 9.2, 1.8 Hz, -CHZ=CHCH3), 6.04 (0.8H, dq, J = 13.8, 7.0 Hz, -CH=CHECH3), 5,45 (0.2H, br quint, J = 7.3 Hz, -CH=CHZCH3), 1.94 (0.4H, dd, J = 7.4, 1.8 Hz, -CH=CHCH3), 1.82 (2.6H, dd, J = 6.9, 1.8 Hz, -CH=CHCH3); 13C-NMR (100 MHz, CDCl3): δ 145.0, 144.8, 134.0, 133.8, 133.6, 131.3, 128.7, 128.5, 128.4, 127.4, 126.7, 116.7, 114.1, 57.6, 57.4, 14.7, 12.9; HREIMS m/z calcd for C6H7N2I (M+) 233.9654, found 233.9652.
4.3.3. Synthesis of (E/Z)-5-allyl-1-(prop-1-en-1-yl)-1H-pyrazol-4-yl trifluoromethane-sulfonate (12)
A solution of 4r (418.9 mg, 2.6 mmol) in 1,2-dimethoxethane (DME, 2 mL) in a sealed vial was heated at 180 °C for 30 min under MW irradiation. The reaction mixture was concentrated directly under reduced pressure to give a crude residue that was purified by silica gel column chromatography (eluent:hexane/AcOEt = 2:1) to give (E/Z)-12 (364.3 mg, 87%) as a colorless oil. 1H-NMR (400 MHz, CDCl3): δ 7.30 (0.3H, s, pyrazole-H), 7.24 (0.7H, s, pyrazole-H), 6.62 (0.7H, dd, J = 13.7, 1.6 Hz, (E)-NCH=CHCH3), 6.50 (0.3H, dd, J = 8.7, 1.6 Hz, (Z)-NCH=CHCH3), 6.15 (0.7H, dq, J = 13.9, 6.9 Hz, (E)-CH=CHCH3), 5.83–5.94 (1H, m, -CH2CH=CH2), 5.54 (0.3H, dq, J = 8.7, 7.4 Hz, (Z)-CH=CHCH3), 5.12–5.17 (1H, m, -CH2CH=CHH), 5.00 (1H, br, -OH), 5.04 (1H, ddd, J = 17.0, 7.2, 1.1 Hz, -CH2CH=CHH), 3.43 (1.4H, d, J = 5.7 Hz, -CH2CH=CH2), 3.38 (0.6H, d, J = 6.1 Hz, -CH2CH=CH2), 1.87 (0.9H, dd, J = 7.2, 1.4 Hz, -CH=CHCH3), 1.80 (2.1H, dd, J = 6.9, 1.2 Hz, -CH=CHCH3); 13C-NMR (100 MHz, CDCl3): δ 139.2, 138.8, 133.5, 133.3, 129. 4, 129.1, 126.9, 125.8, 124.8, 124.6, 123.4, 116.7, 116.5, 114.8, 27.0, 26.5, 15.1, 12.9; HREIMS m/z calcd for C9H12N2O (M+) 164.0950, found 164.0949.
4.3.4. Synthesis of (E/Z)-5-allyl-1-(prop-1-en-1-yl)-1H-pyrazol-4-yl trifluoromethanesulfonate (13)
To a solution of 12 (264.7 mg, 1.6 mmol) in CH2Cl2 (4 mL) was added triethylamine (0.3 mL, 2.4 mmol, 1.5 equiv) at −20 °C with stirring. After stirring for 10 min, Tf2O (0.4 mL, 2.4 mmol, 1.5 equiv) was added dropwise to the reaction mixture. After stirring at room temperature for another 1 h, the reaction was quenched by the addition of sat. aq. NH4Cl (5 mL) and extracted with CH2Cl2 (5 mL × 3). The combined organic layers were washed with brine (5 mL × 2), dried over MgSO4, filtered, and evaporated. The obtained residue was purified by silica gel column chromatography (eluent:hexane/AcOEt = 2:1) to give 13 (430.4 mg, 90%) as an (E/Z) mixture. (Z)-13: Colorless oil; 1H-NMR (600 MHz, CDCl3): δ 7.58 (1H, s, pyrazole-H), 6.54 (1H, dq, J = 8.8, 1.8 Hz, -CH=CHCH3), 5.79 (1H, ddt, J = 17.0, 10.0, 5.9 Hz, -CH2CH=CH2), 5.71 (1H, dq, J = 8.8, 7.3 Hz, -CH=CHCH3), 5.17 (1H, br dq, J = 10.3, 1.8 Hz, -CH2CH=CHH), 5.06 (1H, br dq, J = 17.0, 1.8 Hz, -CH2CH=CHH), 3.42 (2H, dt, J = 6.1, 1.6 Hz, -CH2CH=CH2), 1.87 (3H, dd, J = 7.4, 1.8 Hz, -CH=CHCH3); 13C-NMR (100 MHz, CDCl3): δ 132.4, 131.4, 131.2, 130.9, 124.4, 123.9, 118.7 (q, J = 321.43 Hz, -CF3), 118.2, 27.2, 12.9; HREIMS m/z calcd for C10H11F3N2O3S (M+) 296.0442, found 296.0443. (E)-13: colorless oil; 1H-NMR (600 MHz, CDCl3): δ 7.58 (1H, s, pyrazole-H), 6.54 (1H, dq, J = 8.8, 1.8 Hz, -CH=CHCH3), 5.79 (1H, ddt, J = 17.0, 10.0, 5.9 Hz, -CH2CH=CH2), 5.83 (1H, dq, J = 8.8, 7.3 Hz, -CH=CHCH3), 5.21 (1H, br dq, J = 10.3, 1.7 Hz, -CH2CH=CHH) 5.06 (1H, br dq, J = 17.0, 1.8 Hz, -CH2CH=CHH), 3.47 (2H, dt, J = 5.9, 1.8 Hz, -CH2CH=CH2), 1.84 (3H, dd, J = 7.0, 1.9 Hz, -CH=CHCH3); 13C-NMR (100 MHz, CDCl3): δ 131.6 131.4, 131.0, 130.9, 124.3, 118.7 (q, J = 321.3Hz, -CF3), 118.3, 117.9, 26.7, 15.0; HREIMS m/z calcd for C10H11F3N2O3S (M+) 296.0442, found 296.0443.
4.3.5. Synthesis of 4H-pyrrolo [1,2-b]pyrazol-3-yl trifluoromethanesulfonate (14)
To a solution of 13 (50.0 mg, 0.17 mmol) in CH2Cl2 (2 mL) were added Grubbs2nd catalyst (7.1 mg, 0.0085 mmol, 5 mol%) and CuI (0.8 mg, 0.0042 mmol, 2.5 mol%). The sealed reaction mixture was heated at 80 °C for 1 h under MW irradiation. The solvent was removed from the mixture under reduced pressure to give a crude material that was purified by silica gel column chromatography (eluent:hexane/AcOEt = 4:1) to afford 14 (27.0 mg, 63%). 14: Colorless oil; 1H-NMR (400 MHz, CDCl3): δ 7.55 (1H, s, pyrazole-H), 7.21 (1H, dt, 4.1, 2.2 Hz, -CH2CH=CH-), 6.08 (1H, m, -CH2CH=CH-), 3.55 (2H, br t, J = 2.3 Hz, ArCH2CH=CH-); 13C-NMR (100 MHz, CDCl3): δ 135.0, 134.0, 130.0, 129.0, 118.6 (q, J = 322.0 Hz, -CF3), 118.4, 30.3; HREIMS m/z calcd for C7H5F3N2O3S (M+) 253.9973, found 253.9977.
4.3.6. Synthesis of 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl trifluoromethanesulfonate (6)
To a solution 14 (50.0 mg, 0.20 mmol) in MeOH (5 mL) was added Pd/C (5.00 mg, 10 mol%). The mixture was stirred for 24 h at room temperature under hydrogen gas at 1 atm. After removal of Pd/C by filtration, the solvent was evaporated to give a crude mixture that was purified by silica gel column chromatography (eluent:hexane/AcOEt = 8:1) to afford 6 (45.6 mg, 90%) [22,23].
4.4. Synthesis of Withasomnine Homolog 15 (Scheme 3)
4.4.1. Synthesis of 1-allyl-4-iodo-1H-pyrazole (2d)
To a solution of 1 (300.0 mg, 1.5 mmol) in acetone (9 mL) was added 20% NaOH aq. (6 mL, excess) with stirring, followed by allyl bromide (0.3 mL, 3.0 mmol), and the reaction mixture was stirred at rt for 1 h. After quenching with sat. aq. NH4Cl (10 mL), the mixture was extracted with CH2Cl2 (20 mL × 3), and the combined organic layers were washed with brine (5 mL × 2), dried over MgSO4, and filtered. The solvent was removed under reduced pressure to give a crude residue that was purified by silica gel column chromatography (eluent:hexane/AcOEt = 3:1) to afford 2d (373.9 mg, 88%). 2d: Colorless oil;1 H-NMR (400 MHz, CDCl3): δ 7.50 (1H, s, pyrazole-H), 7.42 (1H, s, pyrazole-H), 5.72 (1H, ddt, J = 17.4, 9.9, 7.0 Hz, -CH2CH=CH2), 5.08 (1H, br d, J = 17. 4 Hz, -CH=CHH), 5.08 (1H, br d, J = 9.9 Hz, -CH=CHH), 4.18 (2H, t, J = 7.0 Hz, NHCH2CH2-), 2.60 (2H, br q, J = 7.0 Hz, -CH2CH2CH=); 13C-NMR (100 MHz, CDCl3) δ 144.2, 133.7, 133.5, 117.9, 55.6, 52.0, 34.5 (one aromatic or olefinic carbon signal is overlapped); HREIMS m/z calcd for C7H9N2I (M+) 247.9810, found 247.9810.
4.4.2. Synthesis of 5-allyl-1-(but-3-en-1-yl)-4-hydroxy-1H-pyrazole (16)
A solution of 4t (81.8 mg, 0.46 mmol) in DME (2 mL) in a sealed vial was heated at 200 oC for 1 h under MW irradiation. The reaction mixture was concentrated directly under reduced pressure to give a crude residue that was purified by silica gel column chromatography (eluent:hexane/AcOEt = 2:1) to give 16 (70.0 mg, 81%). 16: Colorless oil; 1H-NMR (400 MHz, CDCl3): δ 7.12 (1H, s, pyrazole-H), 6.91 (1H, br s, -OH), 5.87 (1H, ddt, J = 17.0, 10.0, 5. 8 Hz, -OCH2CH=CH2), 5.71 (1H, ddt, J = 17.0, 10.4, 7.0, -CH2CH=CH2), 5.11 (1H, dq, J = 10.2, 1.4 Hz, -CH=CHH), 5.01–5.07 (3H, overlapped. 3 × -CH=CHH), 3.98 (2H, t, J = 7.4 Hz, NCH2CH2-), 3.39 (2H, dt, J = 5.8, 1.5 Hz, ArCH2CH=), 2.49 (2H, br q, J = 7. 2 Hz, -CH2CH2CH=); 13C-NMR (100 MHz, CDCl3): δ 138.3, 134.1, 133.7, 127.6, 126.0, 116.6, 116.4, 48.9, 34.5, 27.0; HREIMS m/z calcd for C10H14N2O (M+) 178.1106, Found 178.1104.
4.4.3. Synthesis of 5-allyl-1-(but-3-en-1-yl)-1H-pyrazol-4-yl trifluoromethanesulfonate (17)
To a solution of 16 (70.0 mg, 0.39 mmol) in CH2Cl2 (10 mL) was added triethylamine (0.06 mL, 0.43 mmol, 1.1 equiv) at −20 °C with stirring. After stirring for 10 min, Tf2O (0.1 mL, 0.6 mmol, 1.5 equiv) was added dropwise to the reaction mixture. After stirring at room temperature for another 1 h, the reaction was quenched by the addition of sat. aq. NH4Cl (5 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were washed with brine (5 mL × 2), dried over MgSO4, filtered, and concentrated. The obtained residue was purified by silica gel column chromatography (eluent:hexane/AcOEt = 2:1) to give 17 (100.6 mg, 83%). 17: Colorless oil; 1H-NMR (400 MHz, CDCl3): δ 7.48 (1H, s, pyrazole-H), 5.88–5.67 (2H, overlapped, 2 × -CH2CH=CH2), 5.20 (1H, dq, J = 10.2, 1.2 Hz, -CH=CHH), 5.12–5.03 (3H, overlapped. 3 × -CH=CHH), 4.06 (2H, t, J = 7.3 Hz, NCH2CH2-), 3.44 (2H, dt, J = 5.8, 1.6 Hz, ArCH2CH=), 2.57 (2H, br q, J = 7. 4 Hz, -CH2CH2CH=); 13C-NMR (100 MHz, CDCl3): δ 133.2, 131.5, 130.6, 129.7, 118.4 (q, JC-F = 321.2 Hz), 117.8, 117.7, 100.5, 49.4, 33.8, 26.9; HREIMS m/z calcd for C11H13F3N2O3S (M+) 310.0599, Found 310.0598.
4.4.4. Synthesis of 7,8-dihydro-4H-pyrazolo[1,5-a]azepin-3-yl trifluoromethanesulfonate (18)
To a solution of 17 (99.1 mg, 0.32 mmol) in CH2Cl2 (2 mL) were added Grubbs2nd catalyst (13 mg, 0.016 mmol, 5 mol%) and CuI (1.0 mg, 0.005 mmol, 2.5 mol%). The sealed reaction mixture was heated at 80 °C for 1 h under MW irradiation. Solvent was removed under reduced pressure to give a crude mixture that was purified by silica gel column chromatography (eluent:hexane/AcOEt = 4:1) to afford 18 (65.1 mg, 72%). 18: Amorphous solid; mp 72–75 °C; 1H-NMR (400 MHz, CDCl3): δ 7.38 (1H, s, pyrazole-H), 5.77–5.70 (2H, m, 2 × -CH=CHCH2-), 4.46 (2H, dd, J = 5.6, 4.5 Hz, NCH2CH2-), 3.47–3.45 (2H, m, ArCH2CH=), 2.49–2.45 (2H, m, -CH2CH2CH=); 13C-NMR (100 MHz, CDCl3): δ 134.0, 129.8, 129.16, 129.12, 122.4, 118.6 (q, JC-F = 321.2 Hz), 50.9, 27.8, 21.7; HREIMS m/z calcd for C9H9F3N2O3S (M+) 282.0286, found 282.0282.
4.4.5. Synthesis of 3-phenyl-7,8-dihydro-4H-pyrazolo[1,5-a]azepine (19)
To a solution of 18 (54.7 mg, 0.19 mmol) in DME/H2O = 9:1 (5 mL) in a MW vial were added XPhos (9.2 mg, 0.019 mmol, 10 mol%), Pd(dba)2 (11.0 mg, 0.019 mmol, 10 mol%), cesium carbonate (126.4 mg, 0.38 mmol, 2.0 eq.), and phenylboronic acid (50.0 mg, 0.41 mmol, 2.0 eq.). The sealed vial was heated under MW irradiation at 130 °C for 1 h. The cooled reaction mixture was quenched with sat. aq. NH4Cl solution (40 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were dried over MgSO4, filtered, and the solvent was removed under reduced pressure to give a crude residue that was purified by silica gel column chromatography (eluent:hexane/AcOEt = 2:1) to afford 19 (35.4 mg, 87%). 19: Oil; 1H-NMR (400 MHz, CDCl3): δ 7.49 (1H, s, pyrazole-H), 7.42–7.34 (2H, m, Ph-H), 7.34–7.26 (3H, m, Ph-H), 5.73–5.56 (2H, m, -NCH2CH=CH-, -CH=CHCH2-), 4.54–4.51 (m, 2H), 3.61–3.59 (2H, m), 2.50–2.46 (2H, m); 13C-NMR (100 MHz, CDCl3): δ 138.9, 136.8, 133.6, 129.1, 128.9, 128.6, 128.2, 126.3, 123.8, 120.7, 49.5, 28.4, 23.1; HREIMS m/z calcd for C14H14N2 (M+) 210.1157, found 210.1156.
4.4.6. Synthesis of 3-phenyl-5,6,7,8-tetrahydro-4H-pyrazolo[1,5-a]azepine (15)
To a solution of 19 (3.0 mg, 0.014 mmol) in MeOH (10 mL) was added Pd/C (0.2 mg, 10 mol%). The mixture was stirred overnight at room temperature under hydrogen gas at 1 atm. After removal of Pd/C by filtration, the solvent was evaporated to give a crude residue that was purified by column chromatography (eluent:hexane/AcOEt = 4:1) to afford 15 (2.8 mg, 92%). 15: 1H-NMR (400 MHz, CDCl3): δ 7.44 (1H, s, pyrazole-H), 7.37–7.42 (2H, m, Ph-H), 7.33–7.25 (3H, m, Ph-H), 4.35–4.30 (2H, m, -NCH2CH2-), 2.91–2.86 (2H, m, ArCH2CH2-), 1.93–1.86 (2H, m), 1.86–1.80 (2H, m), 1.74–1.67 (2H, m); lit. 1H-NMR (400 MHz, CDCl3): δ 7.43 (1H, s, pyrazole-H), 7.40–7.26 (5H, m, Ph-H), 4.33–4.31 (2H, m, -NCH2CH2-), 2.89–2.86 (2H, m, ArCH2CH2-) 1.89–1.79 (4H, m), 1.75–1.67 (2H, m) [26]; 13C-NMR (100 MHz, CDCl3): δ 140.7, 136.6, 134.1, 128.6, 128.3, 126.1, 121.0, 53.4, 31.0, 28.0, 26.8, 24.4; lit. 13C-NMR (100 MHz, CDCl3): δ 140.7, 136.5, 134.1, 128.5, 128.3, 126.1, 121.0, 53.3, 30.9, 28.0, 26.8, 24.4; HREIMS m/z calcd for C14H16N2 (M+) 212.1313, found 212.1312 [26].
4.4.7. Synthesis of 4t from 4b
To a solution of 4b (48.6 mg, 0.39 mmol) in acetone (1 mL) was added 20% NaOH aq. (0.12 mg, 1.5 eq.) with stirring, followed by 1-bromo-3-butene (0.08 mL, 0.08 mmol, 2 eq.). The reaction mixture was heated at 80 °C for 10 min under MW irradiation. After addition of sat. aq. NH4Cl (1 mL) to the reaction mixture, it was extracted with CH2Cl2 (10 mL × 3) and the combined organic layers were washed with brine (5 mL × 2), dried over MgSO4, and filtered. The solvent was removed under reduced pressure to give a crude residue that was purified by silica gel column chromatography (eluent:hexane/AcOEt = 3:1) to afford 4t (48.1 mg, 69%).
Acknowledgments
We thank K. Minoura and Fujitake for the NMR and MS measurements, respectively. K. Sumimoto of our laboratory is also appreciated for providing inspiration and suggestions on the improved synthesis of withasomnine at an early stage.
Supplementary Materials
Figures S1–S48: 1H- and 13C-NMR spectra of compounds 2c, 2d, 4c, 4d, 4e, 4f, 4g, 4i, 4k, 4l, 4m, 4n, 4o, 4r, 4s, 4t, and 12–19.
Author Contributions
Y.U. conceived and designed experiments. Y.K., N.K., T.T., J.O., K.N., A.N. and Y.T. performed experimental work. N.H., S.H. and H.Y. made discussions and suggestions on this work and wrote the manuscript with Y.U. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fustero S., Sánchez-Roselló M., Barrio P., Simón-Fuentes A. From 2000 to Mid-2010: A fruitful decade for the synthesis of pyrazoles. Chem. Rev. 2011;111:6984–7034. doi: 10.1021/cr2000459. [DOI] [PubMed] [Google Scholar]
- 2.Ansari A., Ali A., Asif M., Shamsuzzaman S. Review: Biologically active pyrazole derivatives. New J. Chem. 2017;41:16–41. doi: 10.1039/C6NJ03181A. [DOI] [Google Scholar]
- 3.Karrouchi K., Radi S., Ramli Y., Taoufik J., Mabkhot Y.N., Al-Aizari F.A., Ansar M. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules. 2018;23:134. doi: 10.3390/molecules23010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown A.W. Recent developments in the chemistry of pyrazoles. Adv. Heterocycl. Chem. 2018;126:55–107. [Google Scholar]
- 5.Usami Y., Tatsui Y., Yoneyama H., Harusawa S. C4-Alkylamination of C4-halo-1H-1-tritylpyrazoles using Pd(dba)2 or CuI. Molecules. 2020;25:4634. doi: 10.3390/molecules25204634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ray R., Hartwig J.F. Oxalohydrazide ligands for copper-catalyzed C−O coupling reactions with high turnover numbers. Angew. Chem. Int. Ed. 2020;133:8284–8292. doi: 10.1002/ange.202015654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vijeta A., Casadevall C., Roy S., Reisner E. Visible-light promoted C-O bond formation with an integrated carbon nitride–nickel heterogeneous photocatalyst. Angew. Chem. Int. Ed. 2021;60:8494–8499. doi: 10.1002/anie.202016511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vorogushin A.V., Huang X., Buchwald S.L. Use of tunable ligands allows for intermolecular Pd-catalyzed C−O Bond formation. J. Am. Chem. Soc. 2005;127:8146–8149. doi: 10.1021/ja050471r. [DOI] [PubMed] [Google Scholar]
- 9.Maiti D., Buchwald S.L. Orthogonal Cu- and Pd-based catalyst system for the O- and N-arylation of aminophenols. J. Am. Chem. Soc. 2009;131:17423–17429. doi: 10.1021/ja9081815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H., Ruiz-Castillo P., Buchwald S.L. Palladium-catalyzed C-O cross-coupling of primary alcohols. Org. Lett. 2018;20:1580–1583. doi: 10.1021/acs.orglett.8b00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gowrisankar S., Sergeev A.G., Anbarasan P., Spannenberg A., Neumann H., Beller M. A general and efficient catalyst for palladium-catalyzed C−O coupling reactions of aryl halides with primary alcohols. J. Am. Chem. Soc. 2010;132:11592–11598. doi: 10.1021/ja103248d. [DOI] [PubMed] [Google Scholar]
- 12.Maiti D., Buchwald S.L. Cu-catalyzed arylation of phenols: Synthesis of sterically hindered and heteroaryl diaryl ethers. J. Org. Chem. 2010;75:1791–1794. doi: 10.1021/jo9026935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nayak M., Batra S. Copper-Catalyzed Cascade Reactions of Substituted 4-Iodopyrazolecarbaldehydes with 1,2-Phenylenediamines and 2-Aminophenols. Adv. Synth. Catal. 2020;352:3431–3437. doi: 10.1002/adsc.201000662. [DOI] [Google Scholar]
- 14.Wong D.M., Li J., Chen Q.H., Han Q., Mutunga J.M., Wysinski A., Anderson T.D. Select small core structure carbamates exhibit high contact toxicity to “carbamate-resistant” strain malaria mosquitoes, Anopheles gambiae (Akron) PLoS ONE. 2012;7:e46712. doi: 10.1371/journal.pone.0046712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacDonald E., Ihalainen E., Pispa J.P. Pharmacological and toxicological properties of by 4-hydroxypyrazole, a metabolite of pyrazole. Acta Pharmacol. Toxicol. 1981;48:418–423. doi: 10.1111/j.1600-0773.1981.tb01641.x. [DOI] [PubMed] [Google Scholar]
- 16.Cornell N.W., Hansch C., Kim K.H., Henegar K. The inhibition of alcohol dehydrogenase in vitro and in isolated hepatocytes by 4-substituted pyrazoles. Arch. Biochem. Biophys. 1983;227:81–90. doi: 10.1016/0003-9861(83)90349-1. [DOI] [PubMed] [Google Scholar]
- 17.Sinclair J., Cornell N.W., Zaitlin L., Hansch C. Induction of cytochrome P-450 by alcohols and 4-substituted pyrazoles: Comparison of structure-activity relationships. Biochem. Pharmacol. 1986;35:707–710. doi: 10.1016/0006-2952(86)90370-9. [DOI] [PubMed] [Google Scholar]
- 18.Jones J.P., Joswig-Jones C.A., Hebner M., Chu Y., Koop D.R. The effects of nitrogen-heme-iron coordination on substrate affinities for cytochrome P450 2E1. Chem. Biol. Interact. 2011;193:50–56. doi: 10.1016/j.cbi.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walworth B.L. Fungicidal Use of 4-alkoxypyrazoles. No. US 4000301 A 19761228. U.S. (1976) Patent. 1975 Nov 14;
- 20.Matsuo J., Takaya M., Maki Y. 1-Phenyl-4-alkoxypyrazole-5-carboxylic Acid Esters. No. JP 47042665 A. Jpn. Kokai Tokkyo Koho (1972) Patent. 1972 Dec 16;
- 21.Munier-Lehmann H., Lucas-Hourani M., Guillou S., Helynck O., Zanghi G., Noel A., Tangy F., Vidalain P.O., Janin Y.L. Original 2-(3-Alkoxy-1H-pyrazol-1-yl)pyrimidine derivatives as inhibitors of human dihydroorotate dehydrogenase (DHODH) J. Med. Chem. 2015;58:860–877. doi: 10.1021/jm501446r. [DOI] [PubMed] [Google Scholar]
- 22.Ichikawa H., Watanabe R., Fujino Y., Usami Y. Divergent synthesis of withasomnines via synthesis of 4-hydroxy-1H-pyrazoles and Claisen rearrangement of their 4-O-allylethers. Tetrahedron Lett. 2011;52:4448–4451. doi: 10.1016/j.tetlet.2011.06.061. [DOI] [Google Scholar]
- 23.Usami Y., Watanabe R., Fujino Y., Shibano M., Ishida C., Yoneyama H., Harusawa S., Ichikawa H. Divergent synthesis and evaluation of inhibitory activities against Cyclooxygenases-1 and −2 of natural withasomnines and analogues. Chem. Pharm. Bull. 2012;60:1550–1560. doi: 10.1248/cpb.c12-00725. [DOI] [PubMed] [Google Scholar]
- 24.Usami Y., Tatsui Y., Sumimoto K., Miyamoto A., Koito N., Yoneyama H., Harusawa S. 3-Trifluoromethansulfonyloxy-4,7-dihidropyrazolopyridine via ring-closing metathesis: Synthesis and transformation to withasomnine homologs. Heterocycles. 2021;103:284–299. doi: 10.3987/COM-20-S(K)13. [DOI] [Google Scholar]
- 25.Allin S.M., Barton W.R.S., Bowman W.R., McInally T. Radical cyclisation onto pyrazoles: Synthesis of withasomnine. Tetrahedron Lett. 2002;43:4191–4193. doi: 10.1016/S0040-4039(02)00763-3. [DOI] [Google Scholar]
- 26.Allin S.M., Barton W.R.S., Russell Bowman W., Bridge (née Mann) E., Elsegood M.R.J., McInally T., McKee V. Bu3SnH-mediated radical cyclisation onto azoles. Tetrahedron. 2008;64:7745–7758. doi: 10.1016/j.tet.2008.06.014. [DOI] [Google Scholar]
- 27.Xia T., Hu Z., Ji W., Zhang S., Shi H., Liu C., Pang B., Liu G., Liao X. Synthesis of withasomnine and pyrazole derivatives via intramolecular dehydrogenative cyclization, as well as biological evaluation of withasomnine-based scaffolds. Org. Chem. Front. 2018;5:850–854. doi: 10.1039/C7QO00847C. [DOI] [Google Scholar]
- 28.Wube A.A., Wenzig E.M., Gibbons S., Asres K., Bauer R., Bucar F. Constituents of the stem bark of Discopodium penninervium and their LTB4 and COX-1 and -2 inhibitory activities. PhytoChemistry. 2008;69:982–987. doi: 10.1016/j.phytochem.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 29.Usami Y., Kohno A., Yoneyama H., Harusawa S. Synthesis of dihydrooxepino[3,2-c]pyrazoles via Claisen rearrangement and ring-closing metathesis from 4-allyloxy-1H-pyrazoles. Molecules. 2018;23:592. doi: 10.3390/molecules23030592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usami Y., Sumimoto K., Kishima A., Tatsui Y., Yoneyama H., Harusawa S. Synthesis of dihydropyrano[3,2-c]pyrazoles via double bond migration and ring-closing metathesis. Molecules. 2019;24:296. doi: 10.3390/molecules24020296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Usami Y., Tsujiuchi Y., Machiya Y., Chiba A., Ikawa T., Yoneyama H., Harusawa S. Synthetic challenges in the construction of 8- to 10-Membered pyrazole-fused rings via ring-closing metathesis. Heterocycles. 2020;101:496–511. doi: 10.3987/COM-19-S(F)32. [DOI] [Google Scholar]
- 32.Kadoma Y., Murakami Y., Ogiwara T., Machino M., Yokoe I., Fujisawa S. Radical-scavenging activity and cytotoxicity of p-methoxyphenol and p-cresol dimers. Molecules. 2010;15:1103–1112. doi: 10.3390/molecules15031103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X.B., Chen G.Y., Liu R.J., Zheng C.J., Song X.M., Han C.R. A new biphenyl derivative from the mangrove endophytic fungus Phomopsis longicolla HL-2232. Nat. Prod. Res. 2017;31:2264–2267. doi: 10.1080/14786419.2017.1300799. [DOI] [PubMed] [Google Scholar]
- 34.Allen S.E., Walvoord R.R., Padilla-Salinas R., Kozlowski M.C. Aerobic copper-catalyzed organic reactions. Chem. Rev. 2013;113:6234–6458. doi: 10.1021/cr300527g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamal F., Colombel-Rouen S., Dumas A., Guégan J.P., Roisnel T., Dorcet V., Baslé O., Rouen M., Mauduit M. Activation of olefin metathesis complexes containing unsymmetrical unsaturated N-heterocyclic carbenes by copper and gold transmetalation. Chem. Commun. 2019;55:11583–11586. doi: 10.1039/C9CC05776E. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.