

Abstract
SARS-CoV-2, the causative agent for COVID-19, infect human mainly via respiratory tract, which is heavily inhabited by local microbiota. However, the interaction between SARS-CoV-2 and nasopharyngeal microbiota, and the association with metabolome has not been well characterized. Here, metabolomic analysis of blood, urine, and nasopharyngeal swabs from a group of COVID-19 and non-COVID-19 patients, and metagenomic analysis of pharyngeal samples were used to identify the key features of COVID-19. Results showed lactic acid, l-proline, and chlorogenic acid methyl ester (CME) were significantly reduced in the sera of COVID-19 patients compared with non-COVID-19 ones. Nasopharyngeal commensal bacteria including Gemella morbillorum, Gemella haemolysans and Leptotrichia hofstadii were notably depleted in the pharynges of COVID-19 patients, while Prevotella histicola, Streptococcus sanguinis, and Veillonella dispar were relatively increased. The abundance of G. haemolysans and L. hofstadii were significantly positively associated with serum CME, which might be an anti-SARS-CoV-2 bacterial metabolite. This study provides important information to explore the linkage between nasopharyngeal microbiota and disease susceptibility. The findings were based on a very limited number of patients enrolled in this study; a larger size of cohort will be appreciated for further investigation.
Keywords: Metabolome, Nasopharyngeal microbiome, COVID-19, SARS-CoV-2, Susceptibility
1. Introduction
COVID-19 is a respiratory illness caused by SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2), and over 95 million people worldwide have been infected as of Jan 17, 2021. Through investigating the extensive features of the COVID-19 patients, those individuals who are elderly, having other clinical comorbidities [1], blood group A [2,3], or genetic variation [4,5] might have increased risk for the infection with SARS-CoV-2 or develop into severe COVID-19 cases.
SARS-CoV-2 primarily causes lung infection through binding of ACE2 receptors present on the alveolar epithelial cells [6], but it also infects intestinal epithelial cells [7], oral and nasal epithelial cells [8]. Gut microbiota may play a role in the susceptibility and as a diagnostic biomarker for COVID-19 [[9], [10], [11]]. Nasopharyngeal microbiome comprises an abundance of microorganism species that interact with the local epithelial and immune cells, and together, they form a unique micro-ecological system. Respiratory tract microbiota is associated with resistance against the development of respiratory tract infections [12]. A single-cell analysis showed airway epithelium-immune cell interaction is associated with COVID-19 severity [13]. SARS-CoV-2 is associated to the significant change in the respiratory microbiota [14], but whether nasopharyngeal microbiome could affect the susceptibility to COVID-19 and regulate olfactory and gustatory disorders remain to be elucidated. Some opportunistic pathogens including Streptococcus, Prevotella and Campylobacter were enriched in pharyngeal microbiota of COVID-19 patients, and moreover, two Streptococcus strains could stimulate the expression of ACE2 of Vero cells in vitro [15]. The Prevotella proteins are found to be involved in multiple interactions with NF-κB and can promote SARS-CoV-2 infection [16]. Therefore, some pathogens in the pharynxes of COVID-19 patients may change the expression of the ACE2 or modulating the host's immune system involved in virus-host interactions. In addition, several metabolomic studies have shown metabolic alteration in COVID-19 patients [[17], [18], [19]], which may shed light on diagnostic biomarkers and therapy development for COVID-19.
In this study, we integrated non-targeted metabolomic analysis of blood, urine, and nasopharyngeal swabs from 9 COVID-19 patients and 6 non-COVID-19 patients from a Chinese population, and found several metabolites were significantly reduced in the serum of COVID patients. Then we analyzed the nasopharyngeal microbiome and association of COVID-19-related clinical phenotypes with microbial species, which revealed potential biological mechanisms linking nasopharyngeal microbiota to the COVID-19 susceptibility. Due to the emergency of COVID-19 pandemic and the enrollment policy changed, only a very limited number of patients eventually participated in this study, a larger size of cohort will be appreciated for further investigation.
2. Materials and methods
2.1. Study design and enrolled patients
The cohort consisted of 9 COVID-19 and 6 non-COVID-19 patients with matched age and gender. All patients were admitted to department of infectious diseases, the Third Affiliated Hospital of Sun Yat-sen University (TAH-SYSU), Guangzhou, China, during January and February 2020. This study complied with all relevant ethical regulations and was approved by ethics committee of TAH-SYSU with the ethical number [2020]-02-018-01. SARS-CoV-2 infections were confirmed using nucleic acid test (NAT) by Guangzhou Center for Disease Control and Prevention. 6 non-COVID-19 patients had similar clinical characteristics including fever, cough, and ground-glass opacity in the lung as COVID-19 patients however negative in the NAT. Non-COVID-19 controls experienced NAT for at least two sequential NAT (four out of six underwent three sequential NAT), and all showed negative results. As for the severity, one of the COVID-19 patients was severe, while two of the controls were severe. None of them were admitted to a critical care unit or on the ventilator.
2.2. Subjects and sample collection
Nasopharyngeal swabs and anal swabs from all study subjects were collected on admission. The viral load of SARS-CoV-2 RNA in the nasopharynx was relatively higher than in the oropharynx according to our experience and literature report [20]. The anal swabs were also used for NAT, as an auxiliary means of examination. All blood and urine samples used were also collected on admission. Throughout the hospitalization period, patients were provided with standard meals per day and received standard treatment. The examination and medication of COVID-19 group and non-COVID-19 group remained the same, until the nucleic acid results confirmed. The standard meals included steamed fish, scrambled egg with sweet pepper, lettuces, corn and carrot soup, etc. Blood specimens were taken for analyzing hematological indexes, using automated equipment and routine clinical laboratory methods at TAH-SYSU.
2.3. DNA extraction and metagenome sequencing
DNA from nasopharyngeal samples of 15 patients were extracted with the DNA Mini Kit (Qiagen). DNA concentrations were measured using the Qubit quantification system (Thermo Scientific, Wilmington, US), and then metagenomic libraries were constructed. Metagenomic shotgun sequencing was performed using the Illumina NovaSeq platform (Illumina, San Diego, California). Raw 2 × 150 bp paired-end Illumina reads were quality-filtered using fastp (v0.20.0) with the default parameters (-W 4 -M 20). Human reads were removed by mapping the reads to the human reference genome (hg38) with Bowtie2 (v2.3.4.3). After removing human reads, the average and range of read lengths were 148bp and 90–150bp, respectively.
2.4. Metagenomics analysis of taxonomic profiling and functional profiling
HUMAnN2 v0.11.2 [21] was used to for taxonomic classification and estimation of microbial species abundances. HUMAnN2 efficiently identifies known microbial species in a sample by screening DNA reads with MetaPhlAn2 [22]. Functional capabilities of the microbial community were described by the MetaCyc metabolic pathways, and assessed using the UniRef90 proteomic database annotations. This analysis grouped all gene families to 298 pathways. HUMAnN2 was also used to evaluate the percentage of species contributing to the abundance of each microbial metabolic pathway.
2.5. Non-targeted metabolomic profiling and metabolites identification
We used UPLC-MS/MS untargeted metabolomics approach to analyze the serum, urine, and nasopharyngeal samples. Each type of samples thawed on ice and quality-control (QC) samples were prepared by mixing each aliquot with a pooled sample, and then analyzing them in parallel using the same method. Agilent 1290 infinity II (Agilent Technologies) equipped with ACQUITY UPLC HSS T3 (1.8 μm 2.1 × 100 mm, Waters) was used for chromatographic separation. Metabolomic profiling was performed by Agilent 6545A QTOF (Agilent Technologies). All samples were analyzed in positive and negative mode. The raw MS files were converted to ABF file format using the freely available Reifycs file converter. Peak picking and alignment were performed using MS-DIAL version 4.10 [23], and metabolites were identified using database METLIN (https://metlin.scripps.edu/).
2.6. Bioinformatic and statistical analyses
For clinical data, continuous variables were expressed as median and compared with the two-tailed unpaired Student's t-test; categorical variables were expressed as number (%) and compared by Fisher's exact test between COVID-19 and non-COVID-19 groups. For metabolomic data, partial-least-squares discrimination analysis (PLS-DA) was applied to eliminate the effect of inter-subject variability among the participants and identify metabolites that significantly contributed to the classification. Metabolites were ranked according to their variable importance in the projection (VIP) scores from PLS-DA model and metabolites with VIP scores >1.0 are considered as the significant contributors. To identify the most relevant metabolic pathways involved in COVID-19, the pathway analysis was employed by MetaboAnalyst 3.0 [24].
For metagenome data, the alpha diversity (Shannon index) of each sample was calculated with R package VEGAN (v2.5.3) on the relative abundance of all species. principal coordinate analysis (PCoA) was performed on the family level using VEGAN. To test the difference in the microbial composition between two groups, PERMANOVA was employed based on the Bray-Curtis dissimilarity. Species and functional pathways were tested for enrichment or depletion in individuals with COVID-19 or non-COVID-19 according to the non-parametric method, Wilcoxon rank-sum test. Linear discriminant analysis (LDA) Effect Size (LEfSe) was also used to identify differentially abundant taxa within the different groups. The threshold of the LDA score (log10) is 3. Moreover, we analyzed the Spearman's association between serum metabolites and clinical phenotypes, and between nasopharyngeal metagenome and clinical phenotypes, with permutational P-value< 0.05. Heat maps were hierarchically clustered to represent the associated patterns based on the correlation distance, and the analyses and visualizations were implemented in R (v3.6.3).
3. Results
3.1. Clinical characteristics of participants
Characteristics of the study population are presented in Table 1. There were no significant differences in age (p = 0.40) and gender (p = 0.75) between the COVID-19 group and non-COVID-19 group. Non-COVID-19 group had higher rates of smoking than COVID-19 group (50% VS. 11%), but there is no statistically significant difference (p = 0.28). One out of nine (11.1%) in COVID-19 patients and one out of six (16.7%) in non-COVID-19 had co-existing medical condition, hypertension. Fever was present in 100% of the COVID-19 patients and 83.3% of the non-COVID-19 patients on admission. Moreover, the percentage of sputum production, fatigue or myalgia, sore throat, chest tightness and ground-glass opacity in the lung were also calculated, all of which showed no significant difference between the two groups. Results of anal swabs NAT indicated two of the nine COVID-19 patients were positive, and one of the two had intestinal symptoms, diarrhea (Table 1). Most of hematological indexes including lymphocyte (LYM) count, haemoglobin (HGB) level, and platelet (PLT) count also showed no remarkable difference between COVID-19 and non-COVID-19 patients. Notably, the white blood cell (WBC) count was significantly reduced in COVID-19 (p < 0.01, Table 1). Regarding inflammatory markers, there was no significant difference in procalcitonin (PCT), as well as in C-reactive protein (CRP) between two groups of patients. All patients were cured at last, no death, and discharged from hospital.
Table 1.
Clinical characteristics of the study population.
| non-COVID-19 (N = 6) | COVID-19 (N = 9) | p-value | |
|---|---|---|---|
| Age, median (IQR), year | 45.3 (31–76) | 38.9 (27–62) | 0.40 |
| Female sex – no. (%) | 2/6 (33.3%) | 5/9 (55.6%) | 0.75 |
| Smoke – no. (%) | 3/6 (50%) | 1/9 (11.1%) | 0.28 |
| Coexisting disorder – no. (%) | |||
| Hypertension | 1/6 (16.7%) | 1/9 (11.1%) | 1 |
| Symptoms – no. (%) | |||
| fever on admission | 5/6 (83.3%) | 9/9 (100%) | 0.83 |
| temperature on admission (°C) | 38.5 (36.4–40) | 38.1(37.3–38.6) | 0.30 |
| cough | 5/6 (83.3%) | 6/9 (66.7%) | 0.91 |
| Sputum production | 3/6 (50%) | 3/9 (33.3%) | 0.91 |
| fatigue or myalgia | 1/6 (16.7%) | 3/9 (33.3%) | 0.91 |
| sore throat | 0/6 (0%) | 2/9 (22.2%) | 0.64 |
| chest tightness | 1/6 (16.7%) | 3/9 (33.3%) | 0.91 |
| ground-glass opacity in the lung | 4/6 (66.7%) | 8/9 (88.9%) | 0.69 |
| Diarrhea | 0/6 (0%) | 1/9 (11.1%) | 1 |
| Red blood cell (RBC) count, × 1012/L | 4.58 (3.34–5.25) | 4.67 (3.88–5.73) | 0.81 |
| White blood cell (WBC) count, × 10⁹/L | 8.11 (5.42–9.95) | 5.19 (3.8–7.3) | <0.01 |
| Lymphocyte (LYM) count, × 10⁹/L | 1.03 (0.63–1.67) | 1.25 (0.65–2.98) | 0.50 |
| Haemoglobin (HGB) level, g/L | 134.7 (108–151) | 138.1 (108–164) | 0.72 |
| Platelet count (PLT), × 10⁹/L | 225.5 (174–286) | 202.8 (115–298) | 0.44 |
| Aspartate aminotransferase (AST), U/L | 26.7 (15–40) | 19.7 (16–24) | 0.09 |
| Alanine aminotransferase (ALT), U/L | 27.7 (11–63) | 16.4 (7–37) | 0.14 |
| albumin (ALB), g/L | 44.8 (30.9–53.8) | 47.9 (39.2–56.7) | 0.39 |
| total bilirubin (TBIL), μmol/L | 11.4 (4.8–17.5) | 7.5 (4.3–16.9) | 0.20 |
| blood urea nitrogen (BUN), mmol/L | 3.81 (2.97–4.54) | 4.06 (2.79–6.33) | 0.66 |
| Creatinine (CREAT), μmol/L | 73.3 (58–103) | 66.3 (55–94) | 0.37 |
| creatine kinase (CK), U/L | 75.5 (41–130) | 83 (46–186) | 0.73 |
| Lactate dehydrogenase (LDH), U/L | 241.5 (139–553) | 183.7 (152–198) | 0.28 |
| myoglobin (MGB), ng/mL | 26.8 (13.9–55.6) | 27.8 (6.6–45.7) | 0.90 |
| pro B type natriuretic peptide (Pro-BNP) ≤100 ng/L – no. (%) | 4/6 (66.7%) | 8/9 (88.9%) | 0.69 |
| Cardiac troponin I (CTnI), μg/L | 0.027 (0.002–0.111) | 0.007 (0.001–0.035) | 0.24 |
| prothrombin time (PT), S | 13.5 (12.5–15.3) | 12.8 (12.4–13.2) | 0.07 |
| international normalized ratio (INR) | 1.03 (0.94–1.2) | 0.96 (0.92–1.0) | 0.06 |
| Procalcitonin (PCT) level, μg/L | 0.195 (0.02–0.76) | 0.027 (0.02–0.06) | 0.09 |
| C-reactive protein (CRP) level ≥5 mg/L – no. (%) | 4/6 (66.7%) | 7/9 (77.8%) | 1 |
| erythrocyte sedimentation rate (ESR), mm/h | 33.5 (7–85) | 17.9 (7–37) | 0.22 |
3.2. Metabolomics data analysis showed the altered metabolites in serum, urine, and nasopharyngeal swabs of COVID-19 patients
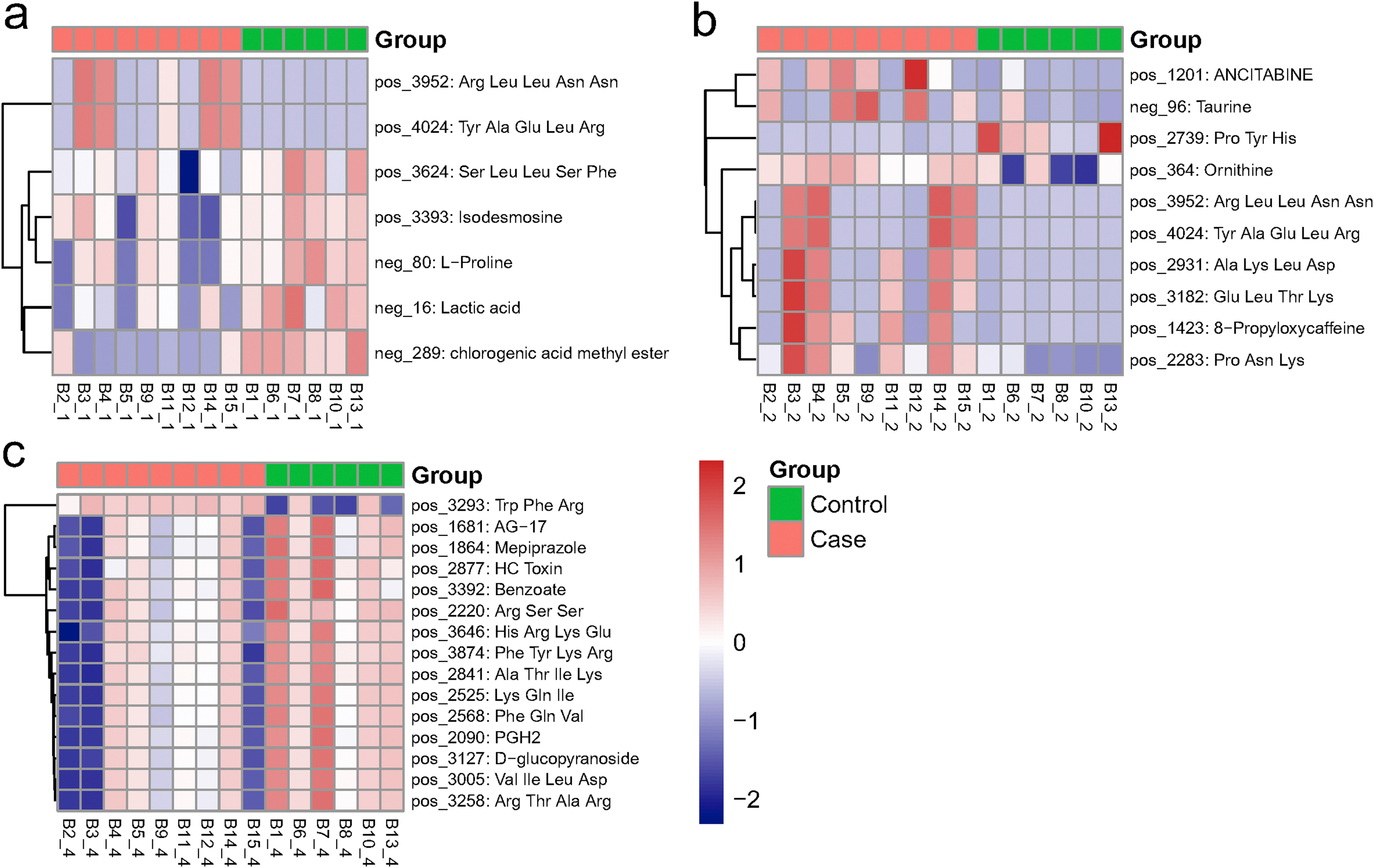
The metabolomes of 15 serum, urine and nasopharyngeal samples from COVID-19 patients and non-COVID-19 patients were identified and quantified. Using principal component analysis (PCA), we observed a clear differentiation of the serum metabolomes from COVID-19 and non-COVID-19 groups in both negative and positive ion modes (Fig. 1. a-b). PERMANOVA indicated the difference between the two groups was not significant overall (p = 0.22), but one of the principal components, PC3, was significantly different for both negative ion modes (p = 0.011) and positive ion modes (p = 0.005). For negative ion modes, PC1 and PC3 account for 39.1% and 9.2% of the total variation, respectively; while for positive ion modes, they account for 12.5% and 7.3%, respectively. For urine, there was also some difference between two groups of samples (Fig. 1. c-d). The samples from distinct groups were largely separated according to the results of PLS-DA (Fig. 1. e-f). In contrast to non-COVID-19, COVID-19 samples had exhibited reduced metabolites including isodesmosine, lactic acid, l-proline, and chlorogenic acid methyl ester (CME) in the serum (Fig. 1. g, Table S1). On the other hand, metabolites including ornithine, 8-propyloxycaffeine, and taurine were up-regulated in the urine of COVID-19 patients (Fig. 1. h, Table S2). In addition, we found several metabolites, including benzoate and PGH2 (prostaglandin H2), were significantly down-regulated in the nasopharyngeal metabolome from COVID-19 patients (Fig S1, Table S3).
Fig. 1.

The serum and urine metabolome. (a–b) The negative (a) and positive (b) ion modes of the serum metabolome between COVID-19 and non-COVID-19 patients. (c–d) The negative (c) and positive (d) ion modes of the urine metabolome between COVID-19 and non-COVID-19 pneumonia. Red indicates the COVID-19 samples, and green indicates the non-COVID-19 samples, while blue refers to quality-control (QC) samples. (e–f) PLS-DA score plot of all the 632 metabolites of serum (e) and 972 metabolites of urine (f). Case (red color) and control (green color) indicate the samples from COVID-19 and non-COVID-19, respectively. (g–h) Volcano plots (p-value ≤ 0.05 and fold change (FC) >1.2) identify statistically significant metabolites in serum (g) and urine (h) between COVID-19 and non-COVID-19 pneumonia. Red indicates up-regulated in COVID-19, while green indicates down-regulated in COVID-19.
According to the results of differentially abundant metabolic pathways, lactate- or l-proline-enriched 17 pathways such as pyruvate metabolism, HIF-1 signaling pathway, prodigiosin biosynthesis, central carbon metabolism in cancer, styrene degradation, glucagon signaling pathway, mineral absorption, and carbapenem biosynthesis were relatively depleted in the serum of COVID-19 patients (Fig. 2. a, Table S4). As for urine, the 5 significantly up-regulated pathways in COVID-19 patients were taurine and hypotaurine metabolism, sulfur metabolism, primary bile acid biosynthesis, ABC transporters, and neuroactive ligand-receptor interaction, all of which were taurine-related (Fig. 2. b, Table S5). In the metabolome of nasopharyngeal swabs, 5 PGH2-related pathways including platelet activation, retrograde endocannabinoid signaling, oxytocin signaling pathway, serotonergic synapse, and arachidonic acid metabolism were down-regulated in COVID-19 patients (Table S6).
Fig. 2.

Bubble plot of KEGG pathway enrichment for differential metabolites in serum (a), and differential metabolites in urine (b) of COVID-19 patients. 17 pathways were all down-regulated in serum (a) and they were contributed by lactate and l-Proline, while 5 pathways were all up-regulated in urine (b) that contributed by taurine. There were no up-regulated pathways in serum and down-regulated pathways in urine in COVID-19 patients compared to controls. The point size indicates the count of differential metabolites that involve in the corresponding pathways, and the color indicates the adjusted p value.
3.3. Metagenome analysis showed the altered nasopharyngeal microbiota between COVID-19 and non-COVID-19 patients
To explore whether the nasopharyngeal microbiota would exhibit differentially upon SARS-CoV-2 infection or not, we analyzed the microbial community and function within the COVID-19 and non-COVID-19 patients by metagenome sequencing. After DNA extraction, 6 of 15 libraries failed library construction, even with the addition of carrier DNA and adaptor concentration adjustment to increase ligation efficiencies. As a result, 9 samples (6 from COVID-19 and 3 from non-COVID-19 patients) showed successful construction of libraries, then to perform the shot-gun metagenomic sequencing.
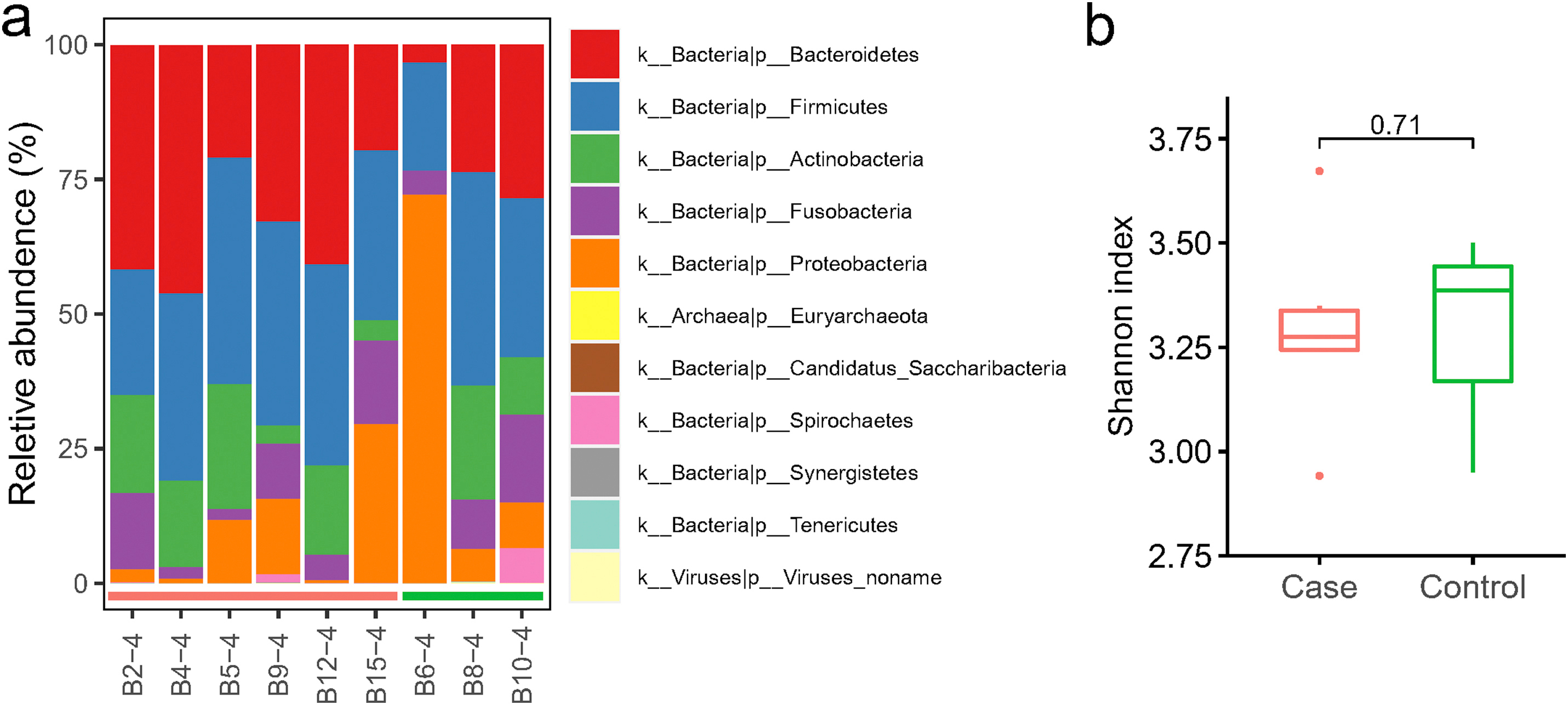
Metagenome analysis revealed the microbial composition consists of a variety of bacteria and a small proportion of archaea and viruses (Fig S2. a). Firmicutes and Bacteroidetes were the two most abundant phyla in the 9 nasopharyngeal samples, comprising on average 32.9% and 28.6%, respectively. The relative abundance of Proteobacteria, Actinobacteria, and Fusobacteria were 16.2%, 12.6% and 8.8%, respectively. Spirochaetes was the next group of bacteria with the proportion less than 1% (0.91%). Viruses were present in 3 out of 9 samples, and on average, making up 0.03%. Through comparing the alpha-diversity of nasopharyngeal community between COVID-19 and non-COVID-19 groups, we found there was no significant difference (p = 0.71, Fig S2. b). PCoA of Bray-Curtis distances indicated variance in the nasopharyngeal microbiota between two groups of patients (Fig. 3 a). At the genus level, communities were dominated by Prevotella (21.1%), Veillonella (11.7%), Neisseria (8.9%), and Actinomyces (7.5%) (Table S7). More specifically, the abundance of five species including Prevotella histicola, Megasphaera micronuciformis, Lautropia mirabilis, Streptococcus sanguinis, and Veillonella dispar were higher in COVID-19 patients, although not to a statistically significant level (Fig. 3 b). LEfSe analysis identified Gemella morbillorum, Gemella haemolysans, Campylobacter gracilis, Stenotrophomonas maltophilia, and Leptotrichia hofstadii were significantly more abundant in non-COVID-19 patients (Fig. 3 c), which is consistent with the results of wilcoxon rank-sum test (Fig. 3 d).
Fig. 3.

The metagenome analysis of the nasopharyngeal microbiome in COVID-19 and non- COVID-19 patients. (a) PCoA plot shows the composition of nasopharyngeal microbiota at the family level in COVID-19 (Case, n = 6) and non-COVID-19 (Control, n = 3) patients. (b) The species that were more abundant in the nasopharyngeal samples of COVID-19 patients with p value less than 0.1 (but not less than 0.05). The mean values and error bars were shown in the boxplots. P values, Wilcoxon rank-sum test, of each species were labelled above the boxplots. (c) The LDA Effect Size (LEfSe) analysis of the species in relative abundance between COVID-19 (Case) and non-COVID-19 (Control) patients. Five species were enriched in non-COVID-19 (Control) patients according to the LDA scores. (d) The significantly different species in relative abundance of nasopharyngeal microbiome between COVID-19 (Case) and non-COVID-19 (Control) patients. *, p value < 0.05.
Additionally, gene families and functional pathways enriched in the nasopharyngeal microbiota of COVID-19 patients relative to non-COVID-19 were analyzed. 29 gene families were significantly different, including TonB-dependent siderophore receptor, addiction module toxin, cytosine-specific methyltransferase, tetratricopeptide repeat protein, iron-sulfur cluster biosynthesis protein, response regulator receiver domain-containing protein, and signal peptidase I, which were all lower in COVID-19 group (p < 0.01). The gene family TonB-dependent siderophore receptor can be contributed by Campylobacter showae, while cytosine-specific methyltransferase and tetratricopeptide repeat protein could be contributed by L. hofstadii (Fig. 4 a). For iron-sulfur cluster biosynthesis protein, response regulator receiver domain-containing protein, and signal peptidase I, G. morbillorum is the important contributor.
Fig. 4.

The significantly different gene families and KEGG pathways between COVID-19 (Case) and non-COVID-19 (Control) patients. (a) Heat map of differential gene families (Wilcoxon rank-sum test, p < 0.01) in the nasopharyngeal samples. Specific species contributing to the abundance of these gene families were labelled in the figure. The horizontal color bar indicates the group of each sample. (b) Boxplots of significantly different KEGG pathways in two groups and the pathway names were colored according to direction of enrichment. Red indicated to be enriched in COVID-19 (Case), while green indicated to be enriched in non- COVID-19 (Control). *, p < 0.05.
Moreover, the relative enrichment of 13 KEGG pathways varied significantly between the two groups. Pathways related to pyruvate fermentation to isobutanol, superpathway of purine nucleotides de novo biosynthesis, seleno-amino acid biosynthesis, superpathway of L-methionine biosynthesis, lactose and galactose degradation I, and guanosine nucleotides degradation II were enriched in non-COVID-19 group (Fig. 4 b). On the contrary, pathways involved in superpathway of L-serine and glycine biosynthesis I, NAD biosynthesis I (from aspartate), superpathway of menaquinol-8 biosynthesis I, superpathway of demethylmenaquinol-8 biosynthesis, and superpathway of menaquinol-8 biosynthesis II were significantly increased in COVID-19 samples (Fig. 4 b).
3.4. Nasopharyngeal microbiome associated with serum metabolomics in COVID-19 and non-COVID-19 patients
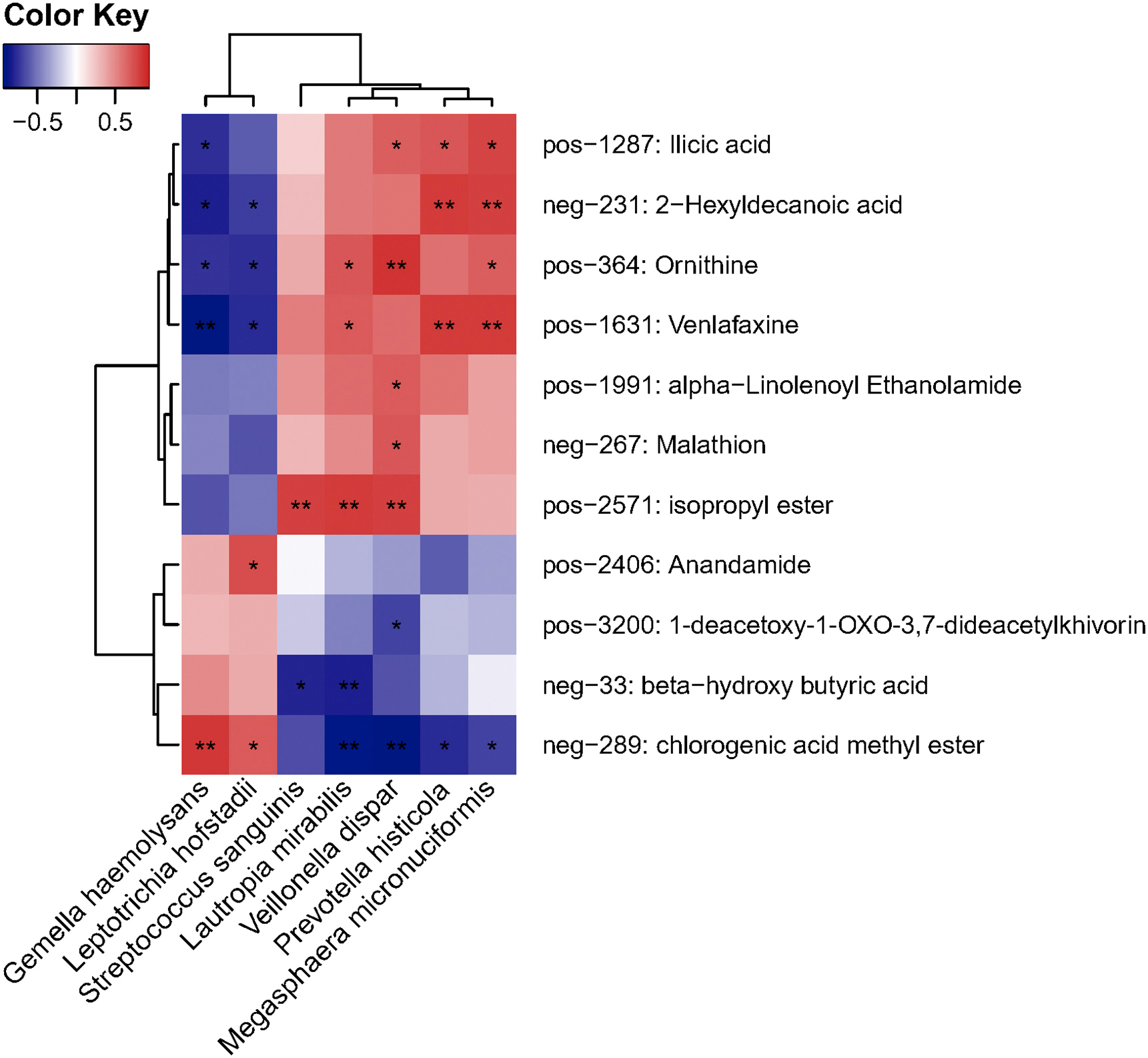
To understand the extent to which the altered nasopharyngeal microbiota in COVID-19 patients was linked with circulating metabolites in our patients, we performed the association analysis of the microbial abundance and serum metabolome. G. haemolysans and L. hofstadii, relatively depleted in COVID-19 patients, were significantly positively associated with CME in serum (p < 0.05, Fig. 5 d). Besides, these two species were also correlated with beta-hydroxy butyric acid.
Fig. 5.

The association of the serum metabolome and clinical phenotypes. (a) Boxplot of serum WBC between COVID-19 (Case) and non-COVID-19 (Control) patients. (b) The level of serum metabolite neg-289 in COVID-19 (Case) and non-COVID-19 (Control) patients. The metabolite's name of neg-289 is chlorogenic acid methyl ester (CME). P values were labelled above the boxplots (a, b). (c) Heat map showed the association of serum metabolome and clinical phenotypes in 15 patients. RBC, red blood cell; WBC, white blood cell; LYM, lymphocyte; HGB, haemoglobin; PLT, platelet count; AST, aspartate aminotransferase; ALT, alanine aminotransferase; ALB, albumin; TBIL, total bilirubin; BUN, blood urea nitrogen; CREAT, creatinine; CK, creatine kinase; LDH, Lactate dehydrogenase; MGB, myoglobin; Pro-BNP, pro B type natriuretic peptide; CTnI, cardiac troponin I; PT, prothrombin time; INR, international normalized ratio; PCT, procalcitonin level; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate. (d) The significant association of several specific nasopharyngeal bacteria and serum metabolome. Red indicates the positive correlation and blue indicates the negative correlation (c–d). *, P < 0.05; **, P < 0.01.
We also performed the association analysis of the microbial component and hematological indexes from COVID-19 patients and controls. The abundance of Haemophilus parainfluenzae, Neisseria flavescens, Neisseria subflava, Gemella sanguinis, Gemella morbillorum, Gemella haemolysans, and Streptococcus australis were positively correlated with WBC, while were negatively correlated with ALB (Fig S3). Rothia dentocariosa, Eubacterium brachy, Corynebacterium durum, Bulleidia extructa, and Bacteroidetes oral taxon 274 were consistently negatively correlated with AST, MGB, and CK (p < 0.05). AST and CK reflect the degree of myocardial and skeletal muscle damage. Actinomyces graevenitzii, Actinomyces odontolyticus, Prevotella loescheii, Rothia aeria, Selenomonas noxia, Selenomonas flueggei, Streptococcus tigurinus, and Streptococcus salivarius were negatively correlated with LDH. Moreover, four species from Streptococcus (S. parasanguinis, S. constellatus, S. anginosus, and S. intermedius) were uniformly negatively associated with AST (p < 0.05).
In addition, serum metabolome may reveal the alteration of COVID-19 patients after SARS-CoV-2 infection, so we explored the serum-related metabolites linked to clinical phenotypes. Results indicated that serum level of L-norvaline, L-isoleucine, L-norleucine, niacinamide, and camptothecin were positively associated with RBC and HGB (Fig. 5. c, Table S8). Spermidine was found to be positively associated with CK and ALB, but negatively associated with Pro-BNP, ESR and CTnI. CME was positively correlated with both CK and MGB (p < 0.05). WBC is the only factor that had significant difference between COVID-19 and non-COVID-19 patients (Fig. 5 a), and it was positively correlated with CME (neg-289) that is notably lower in COVID-19 group (Fig. 5 b). CME could be a potential beneficial metabolite for the prevention or treatment of COVID-19.
4. Discussion
COVID-19 has become a pandemic, several studies has been reported regarding the characteristics of metabolome, but there is lack of metagenome analysis of nasopharyngeal microbiome for COVID-19 infection. This study presents a comprehensive exploration of the relationship of the nasopharyngeal microbiome with serum, urine, and nasopharyngeal metabolome in 9 COVID-19 and 6 non-COVID-19 patients, which provides novel insights into the characterization of this illness. COVD-negative cases in our study also have ground glass opacity in the lung that may be viral pneumonia caused by other kinds of viruses. Several 16S rRNA gene sequencing studies had showed the alterations in the human oral and upper respiratory microbiomes in COVID-19 [25,26]. Recently, a Chinese team from Wuhan institute of virology published a metagenomic sequencing study that reveals the potential linkages between pharyngeal microbiota and COVID-19 [15]. Our study is a novel metagenomic study to reveal the nasopharyngeal microbiome features of COVID-19 patients in China, which also have been reported by other studies in USA [14] and Italy [27].
The serum metabolome between COVID-19 and non-COVID-19 patients showed distinct profiles. COVID-19 patients had down-regulated metabolites including isodesmosine, lactic acid, l-proline, and CME in the serum. The tetrasubstituted pyridinium amino acids isodesmosine and desmosine are cross-linkers of elastin and are biomarkers for various pathological conditions [28], such as chronic obstructive pulmonary disease [29]. Lactate contributes to enhanced immune responses by inducing GPR31-mediated dendrite protrusion of intestinal CX3CR1+ cells and high resistance to invasive Salmonella infection [30]. Additionally, pathways for the synthesis of arginine, proline, and ornithine were increased in the fecal microbiome in a pulmonary arterial hypertension (PAH) cohort compared with reference cohort [31]. Chlorogenic acid can ameliorate experimental colitis in mice by suppressing signaling pathways involved in inflammatory response and apoptosis [32]. CME significantly alleviated the pathological damage of the lung tissue, reduced the levels of PGE2 and IL-1beta in the serum and the protein expression levels of related-inflammatory factors in the lung tissue of LPS-induced mice with acute lung injury [33]. Thus, lactate and CME may be anti-inflammatory metabolites in COVID-19.
Pyruvate metabolism, HIF-1 signaling pathway, prodigiosin biosynthesis, and central carbon metabolism in cancer were Lactate- or l-Proline-enriched pathways that down-regulated in serum of COVID-19 patients. The lactate-to-pyruvate ratio is increasing in serum samples from patients with essential hypertension [34]. Prodigiosin is normally secreted by some microorganisms as a secondary metabolite, having many beneficial properties that make it a promising drug candidate [35]. Proteomics analysis of SARS-CoV-2-infected host cells revealed that inhibition of central carbon metabolism prevents viral replication [36]. Therefore, metabolites of lactate and l-proline might be helpful for reducing the risk of SARS-CoV-2 infection.
Nasopharyngeal microbiota underlying the susceptibility of healthy individuals to COVID-19 needs to be elucidated, since nasopharynx may be the frontline for the virus to land and invade human. Metagenome analysis indicated that the relative abundance of Prevotella histicola, Megasphaera micronuciformis, Lautropia mirabilis, Streptococcus sanguinis, and Veillonella dispar were higher in COVID-19 patients. P. histicola is a human gut-derived commensal bacterium that boosts anti-inflammatory immune responses and suppresses inflammatory arthritis [37], and it can also be found in the human oral cavity [38] or tongue microbiota [39]. Although Streptococcus are not typical respiratory pathogens, some species were identified as etiologic agents of pulmonary infections such as aspiration pneumonia [40]. S. sanguinis is a pioneer species of oral biofilm and promote oral health, but is well known as a cause of infective endocarditis [41]. Streptococcus suis and S. agalactiae stimulated the expression of ACE2 of Vero cells in vitro, which may promote SARS-CoV-2 infection [15]. Further research will be needed to validate the interaction between Streptococcus strains and SARS-CoV-2. In addition, V. dispar is strongly associated with autoimmune hepatitis, positively correlated with serum level of aspartate aminotransferase and liver inflammation [42]. They may involve in the host's immune regulation and then confer COVID-19 susceptibility or severity.
Gemella morbillorum, Gemella haemolysans, Campylobacter gracilis, Stenotrophomonas maltophilia, and Leptotrichia hofstadii were significantly more abundant in non-COVID-19 patients. In the oropharynx, microbial communities in both young children and adults had a significantly higher proportion of Streptococcus, Gemella, Haemophilus and Neisseria [43]. Overgrowth of lactic acid-producing bacteria like Megasphaera and Leptotrichia species in oropharyngeal samples was related to susceptibility to H7N9 infection [44]. However, different strains from one same species might carry distinct genome, which means that they could perform varied function among diverse strains. Therefore, the correlation of Leptotrichia species and respiratory virus infection needs more validation. G. morbillorum is a natural inhabitant of the human oropharyngeal and gastrointestinal flora, tending to cause endocarditis among patients with valvular diseases [45]. Both of G. morbillorum and G. haemolysans carry the genes required for incorporating phosphorylcholine into their cell walls and encoded some choline-binding proteins [46]. Gene-level analysis of nasopharyngeal metagenome indicated G. morbillorum is involved in iron-sulfur cluster biosynthesis protein, response regulator receiver domain-containing protein, and signal peptidase I.
The abundance of family Propionibacteriaceae and species Corynebacterium accolens were notably increased and decreased, respectively, in a group of USA SARS-CoV-2-positive nasopharyngeal samples [14], but they showed no significant difference in our study. Recently, a meta-transcriptomic study of a Chinese cohort showed the predominant respiratory microbial taxa of severely ill COVID-19 patients were Burkholderia cepacia complex, Staphylococcus epidermidis, and Mycoplasma spp. (including M. hominis and M. orale) [47], which was not observed in our cohort. The results above suggest that individual genetics, medical conditions, and regional difference might play important roles in influencing one's respiratory microbiome composition and function; moreover, methods for sampling and analyzing could also affect the sequencing results. Therefore, more studies need to be conducted in order to elucidate the effect of SARS-CoV-2 infection on respiratory microbiota, especially the studies including multi-regional COVID-19 patients.
Several functional pathways such as pyruvate fermentation to isobutanol, and superpathway of purine nucleotides de novo biosynthesis, were significantly decreased in COVID-19 group. Isobutanol is a promising candidate as second-generation biofuel [48], while purine nucleotides de novo biosynthesis could be modulated as a therapeutic strategy in mitochondrial myopathy that is characterized by muscle weakness [49]. Clinical symptoms at the onset of COVID-19 pneumonia were myalgia or fatigue [50], which might be regulated by microbial metabolism of purine nucleotides de novo biosynthesis. Association analysis showed G. haemolysans and L. hofstadii from nasopharynx were significantly positively associated with CME in serum, suggesting the species may have some beneficial properties for COVID-19.
However, our study has some notable limitations. First, the cohort size of participating patients is small. Due to the revised enrollment policy and rearrangement of medical resource among the city, new suspected and confirmed COVID-19 patients were asked to be sent to a different municipal designated hospital and we did not have access to those new patients, which lead to a very limited number of patients enrolled in this study. Secondly, we only obtained nasopharyngeal metagenomic sequencing results from 9 patients, since the amount of DNA extracted from the pharyngeal swabs were of low concentration in the other 6 samples and failed in the library construction. The composition of nasopharyngeal microbiota might be better determined by sequencing the 16S rRNA gene [[51], [52], [53]]. However, metagenomic sequencing has advantages for its accurate identification of species and metabolic pathways. Third, the metabolome of serum and urine, and metagenome of nasopharyngeal swabs were not collected at multiple time points and profiled, because of the priority for clinical treatment. A more comprehensive study, which including a large cohort of patients and each patient with diverse specimen at multi-time-point, would be greatly appreciated for further investigation.
5. Conclusion
We identified that the specific biomarkers, Lactate, l-Proline and CME, in non-COVID-19 patient, which may be anti-SARS-CoV-2 bacterial metabolites. Moreover, G. morbillorum, G. haemolysans and L. hofstadii were nasopharyngeal commensal bacteria that significantly depleted in COVID-19 patients, could possibly regulate host inflammatory and immune processes through their genes and encoded proteins. Continued research efforts on identification of signature microbial and metabolic biomarkers and developing appropriate procedures are warranted to both prevent and treat respiratory virus infections.
Ethics approval
This study complied with all relevant ethical regulations and was approved by ethics committee of Third Affiliated Hospital of Sun Yat-sen University.
Data availability statement
The datasets presented in this study can be found in NCBI Sequence Read Archive (SRA) database, and the accession number is PRJNA656660.
CRediT authorship contribution statement
Jing Liu: Investigation, Funding acquisition, Writing – review & editing. Sheng Liu: Data curation, Visualization, Writing – original draft. Zhao Zhang: Methodology, Software, Validation. Xuejun Lee: Methodology, Investigation. Wenxuan Wu: Investigation. Zhanlian Huang: Resources. Ziying Lei: Resources. Wenxiong Xu: Resources. Dabiao Chen: Resources. Xing Wu: Data curation, Software, Validation. Yang Guo: Investigation. Liang Peng: Resources. Bingliang Lin: Resources. Yutian Chong: Resources. Xiangyu Mou: Methodology. Mang Shi: Methodology. Ping Lan: Methodology. Tao Chen: Supervision. Wenjing Zhao: Supervision, Funding acquisition, Writing – review & editing. Zhiliang Gao: Conceptualization, Supervision, Funding acquisition.
Declaration of competing interest
The authors declare that they have no conflicts of interest.
Acknowledgements
This work has been supported by National Science and Technology Major Project (2018ZX10302204), Shenzhen Science and Technology Program (Grant No. KQTD20200820145822023), Emergency special program for 2019-nCov of Guangdong province science and technology project (2020B111105001), Guangzhou science and technology project (202008040003), Clinical Research Foundation of the third Affiliated Hospital of Sun Yat-sen University (YHJH201904), and National Natural Science Foundation of China (Grant No. 31900056).
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.synbio.2021.06.002.
Contributor Information
Wenjing Zhao, Email: Zhaowj29@mail.sysu.edu.cn.
Zhiliang Gao, Email: gaozhl@mail.sysu.edu.cn.
Appendix A. Supplementary data
The following is/are the supplementary data to this article:
figs1The significant different metabolites between COVID-19 (Case) and non-COVID-19 (Control) patients. Heat map shows the differentiated metabolites (p value<0.05, VIP score>1) in serum (a), urine (b), and nasopharyngeal swabs (c). Samples from Case or Control group were annotated on the plots.
{kind=link}
The composition and alpha-diversity of pharyngeal microbiota in COVID-19 and non-COVID-19 patients by metagenome sequencing. (a) The phylum-level composition in the nasopharyngeal microbiome. Each phylum was shown in a different color. (b) The comparison of Shannon index in COVID-19 (Case) and non-COVID-19 (Control). Case, n=6; Control, n=3.
{kind=link}
Heat map of the nasopharyngeal microbiota and clinical characteristics of patients. Red indicates the positive correlation and blue indicates the negative correlation. *, P<0.05; **, P<0.01.
{kind=link}
References
- 1.Zhou F. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Y. Relationship between ABO blood group distribution and clinical characteristics in patients with COVID-19. Clin Chim Acta. 2020;509:220–223. doi: 10.1016/j.cca.2020.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goker H. The effects of blood group types on the risk of COVID-19 infection and its clinical outcome. Turk J Med Sci. 2020;50(4):679–683. doi: 10.3906/sag-2005-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen A. Human leukocyte antigen susceptibility map for severe acute respiratory Syndrome coronavirus 2. J Virol. 2020;94(13) doi: 10.1128/JVI.00510-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellinghaus D. Genomewide association study of severe covid-19 with respiratory failure. N Engl J Med. 2020;383(16):1522–1534. doi: 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zou X. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med. 2020;14(2):185–192. doi: 10.1007/s11684-020-0754-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao F. Evidence for gastrointestinal infection of SARS-CoV-2. Gastroenterology. 2020;158(6):1831–1833 e3. doi: 10.1053/j.gastro.2020.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sungnak W. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 2020;26(5):681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu S. Alterations of the gut microbiota in patients with COVID-19 or H1N1 influenza. Clin Infect Dis. 2020;71(10):2669–2678. doi: 10.1093/cid/ciaa709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zuo T. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology. 2020;159(3):944–955 e8. doi: 10.1053/j.gastro.2020.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeoh Y.K. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut. 2021;70(4):698–706. doi: 10.1136/gutjnl-2020-323020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosch A. Maturation of the infant respiratory microbiota, environmental drivers, and health consequences. A prospective cohort study. Am J Respir Crit Care Med. 2017;196(12):1582–1590. doi: 10.1164/rccm.201703-0554OC. [DOI] [PubMed] [Google Scholar]
- 13.Chua R.L. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat Biotechnol. 2020;38(8):970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- 14.Mostafa H.H. Metagenomic next-generation sequencing of nasopharyngeal specimens collected from confirmed and suspect COVID-19 patients. mBio. 2020;11(6) doi: 10.1128/mBio.01969-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong D. Enriched opportunistic pathogens revealed by metagenomic sequencing hint potential linkages between pharyngeal microbiota and COVID-19. Virol Sin. 2021 doi: 10.1007/s12250-021-00391-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan A.A., Khan Z. COVID-2019-associated overexpressed Prevotella proteins mediated host-pathogen interactions and their role in coronavirus outbreak. Bioinformatics. 2020;36(13):4065–4069. doi: 10.1093/bioinformatics/btaa285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song J.W. Omics-driven systems interrogation of metabolic dysregulation in COVID-19 pathogenesis. Cell Metabol. 2020;32(2):188–202. doi: 10.1016/j.cmet.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas T. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight. 2020;5(14):e140327. doi: 10.1172/jci.insight.140327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen B. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. 2020;182(1):59–72. doi: 10.1016/j.cell.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou L. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N Engl J Med. 2020;382(12):1177–1179. doi: 10.1056/NEJMc2001737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franzosa E.A. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018;15(11):962–968. doi: 10.1038/s41592-018-0176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Truong D.T. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12(10):902–903. doi: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- 23.Tsugawa H. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat Methods. 2015;12(6):523–526. doi: 10.1038/nmeth.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia J. MetaboAnalyst 3.0--making metabolomics more meaningful. Nucleic Acids Res. 2015;43(W1):W251–W257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren Z. Alterations in the human oral and gut microbiomes and lipidomics in COVID-19. Gut. 2021;70(7):1253–1265. doi: 10.1136/gutjnl-2020-323826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu R. Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults. Commun Biol. 2021;4(1):240. doi: 10.1038/s42003-021-01796-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nardelli C. Nasopharyngeal microbiome signature in COVID-19 positive patients: can we definitively get a role to fusobacterium periodonticum? Front Cell Infect Microbiol. 2021;11:625581. doi: 10.3389/fcimb.2021.625581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rathod P. Quantification of desmosine and isodesmosine using MALDI-ion trap tandem mass spectrometry. Anal Bioanal Chem. 2018;410(26):6881–6889. doi: 10.1007/s00216-018-1288-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Usuki T. Biomimetic Chichibabin pyridine synthesis of the COPD biomarkers and elastin cross-linkers isodesmosine and desmosine. Org Lett. 2014;16(6):1672–1675. doi: 10.1021/ol500333t. [DOI] [PubMed] [Google Scholar]
- 30.Morita N. GPR31-dependent dendrite protrusion of intestinal CX3CR1(+) cells by bacterial metabolites. Nature. 2019;566(7742):110–114. doi: 10.1038/s41586-019-0884-1. [DOI] [PubMed] [Google Scholar]
- 31.Kim S. Altered gut microbiome profile in patients with pulmonary arterial hypertension. Hypertension. 2020;75(4):1063–1071. doi: 10.1161/HYPERTENSIONAHA.119.14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vukelic I. Chlorogenic acid ameliorates experimental colitis in mice by suppressing signaling pathways involved in inflammatory response and apoptosis. Food Chem Toxicol. 2018;121:140–150. doi: 10.1016/j.fct.2018.08.061. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L. Chlorogenic acid methyl ester exerts strong anti-inflammatory effects via inhibiting the COX-2/NLRP3/NF-kappaB pathway. Food Funct. 2018;9(12):6155–6164. doi: 10.1039/c8fo01281d. [DOI] [PubMed] [Google Scholar]
- 34.Ameta K. Essential hypertension: a filtered serum based metabolomics study. Sci Rep. 2017;7(1):2153. doi: 10.1038/s41598-017-02289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yip C.H. Recent advancements in high-level synthesis of the promising clinical drug, prodigiosin. Appl Microbiol Biotechnol. 2019;103(4):1667–1680. doi: 10.1007/s00253-018-09611-z. [DOI] [PubMed] [Google Scholar]
- 36.Bojkova D. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature. 2020;583(7816):469–472. doi: 10.1038/s41586-020-2332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marietta E.V. Suppression of inflammatory arthritis by human gut-derived Prevotella histicola in humanized mice. Arthritis Rheum. 2016;68(12):2878–2888. doi: 10.1002/art.39785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Downes J. Prevotella histicola sp. nov., isolated from the human oral cavity. Int J Syst Evol Microbiol. 2008;58(Pt 8):1788–1791. doi: 10.1099/ijs.0.65656-0. [DOI] [PubMed] [Google Scholar]
- 39.Asakawa M. Tongue microbiota and oral health status in community-dwelling elderly adults. mSphere. 2018;3(4):e00332–18. doi: 10.1128/mSphere.00332-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akata K. The significance of oral streptococci in patients with pneumonia with risk factors for aspiration: the bacterial floral analysis of 16S ribosomal RNA gene using bronchoalveolar lavage fluid. BMC Pulm Med. 2016;16(1):79. doi: 10.1186/s12890-016-0235-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu B. Streptococcus sanguinis biofilm formation & interaction with oral pathogens. Future Microbiol. 2018;13:915–932. doi: 10.2217/fmb-2018-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei Y. Alterations of gut microbiome in autoimmune hepatitis. Gut. 2020;69(3):569–577. doi: 10.1136/gutjnl-2018-317836. [DOI] [PubMed] [Google Scholar]
- 43.Stearns J.C. Culture and molecular-based profiles show shifts in bacterial communities of the upper respiratory tract that occur with age. ISME J. 2015;9(5):1268. doi: 10.1038/ismej.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu H.F. Disordered oropharyngeal microbial communities in H7N9 patients with or without secondary bacterial lung infection. Emerg Microb Infect. 2017;6(12):e112. doi: 10.1038/emi.2017.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shinha T. Endocarditis due to Gemella morbillorum. Intern Med. 2017;56(13):1751. doi: 10.2169/internalmedicine.56.8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia Lopez E., Martin-Galiano A.J. The versatility of opportunistic infections caused by Gemella isolates is supported by the carriage of virulence factors from multiple origins. Front Microbiol. 2020;11:524. doi: 10.3389/fmicb.2020.00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong H. Characterization of respiratory microbial dysbiosis in hospitalized COVID-19 patients. Cell Discov. 2021;7(1):23. doi: 10.1038/s41421-021-00257-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wess J., Brinek M., Boles E. Improving isobutanol production with the yeast Saccharomyces cerevisiae by successively blocking competing metabolic pathways as well as ethanol and glycerol formation. Biotechnol Biofuels. 2019;12:173. doi: 10.1186/s13068-019-1486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kimoloi S. Modulation of the de novo purine nucleotide pathway as a therapeutic strategy in mitochondrial myopathy. Pharmacol Res. 2018;138:37–42. doi: 10.1016/j.phrs.2018.09.027. [DOI] [PubMed] [Google Scholar]
- 50.Huang C. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boelsen L.K. The association between pneumococcal vaccination, ethnicity, and the nasopharyngeal microbiota of children in Fiji. Microbiome. 2019;7(1):106. doi: 10.1186/s40168-019-0716-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Camelo-Castillo A. Nasopharyngeal microbiota in children with invasive pneumococcal disease: identification of bacteria with potential disease-promoting and protective effects. Front Microbiol. 2019;10:11. doi: 10.3389/fmicb.2019.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verhagen L.M. Nasopharyngeal microbiota profiles in rural Venezuelan children are associated with respiratory and gastrointestinal infections. Clin Infect Dis. 2021;72(2):212–221. doi: 10.1093/cid/ciaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
figs1The significant different metabolites between COVID-19 (Case) and non-COVID-19 (Control) patients. Heat map shows the differentiated metabolites (p value<0.05, VIP score>1) in serum (a), urine (b), and nasopharyngeal swabs (c). Samples from Case or Control group were annotated on the plots.
The composition and alpha-diversity of pharyngeal microbiota in COVID-19 and non-COVID-19 patients by metagenome sequencing. (a) The phylum-level composition in the nasopharyngeal microbiome. Each phylum was shown in a different color. (b) The comparison of Shannon index in COVID-19 (Case) and non-COVID-19 (Control). Case, n=6; Control, n=3.
Heat map of the nasopharyngeal microbiota and clinical characteristics of patients. Red indicates the positive correlation and blue indicates the negative correlation. *, P<0.05; **, P<0.01.
Data Availability Statement
The datasets presented in this study can be found in NCBI Sequence Read Archive (SRA) database, and the accession number is PRJNA656660.