

Abstract
As a participant in the joint CASP13-CAPRI46 assessment, the ClusPro server debuted its new template-based modeling functionality. The addition of this feature, called ClusPro TBM, was motivated by the previous CASP-CAPRI assessments and by the proven ability of template-based methods to produce higher quality models, provided templates are available. In prior assessments, ClusPro submissions consisted of models that were produced via free docking of pre-generated homology models. This method was successful in terms of the number of acceptable predictions across targets, however, analysis of results showed that purely template-based methods produced a substantially higher number of medium quality models for targets for which there were good templates available. The addition of template-based modeling has expanded ClusPro’s ability to produce higher accuracy predictions, primarily for homomeric but also for some heteromeric targets. Here we review the newest additions to the ClusPro web server and discuss examples of CASP-CAPRI targets that continue to drive further development. We also describe ongoing work not yet implemented in the server. This includes the development of methods to improve template-based models and the use of co-evolutionary information for data-assisted free docking.
Keywords: Protein-protein interaction, modeling of protein complexes, method development, template-based, homology modeling
1. INTRODUCTION
The prediction of protein complexes remains an active and challenging field. A relatively small number of heteromeric complexes are available in the Protein Data Bank (PDB) compared to their individually crystallized components. Due to low complex availability, docking servers and modeling tools are often employed to predict such interactions. While the number of structures deposited in the PDB continues to grow, reportedly at a yearly rate of ~10%1, there is a continued need for docking and modeling tools that have the capability to handle larger structures and the ability to account for more complicated experimental data.2
Strategies for predicting protein complexes typically fall into two categories: free docking and template-based modeling. Free docking techniques take structural inputs, sample potential orientations and rotations of the two structures, and often filter or rank resulting poses using a scoring function. Template-based modeling uses the protein sequence to search available databases for related proteins to use as structural templates. Template availability of complexes is often considered a limiting factor in this approach. However, it has been shown that nearly all known protein-protein complexes can be modeled, provided there are strong homologs deposited in the PDB for each of their components.3
Community-wide assessments such as CASP (Critical Assessment of protein Structure Prediction) and CAPRI (Critical Assessment of Predicted Interactions) serve as important platforms to not only evaluate the performance of current structural prediction servers, but to also challenge participants with unique targets and encourage advances in server methodologies. ClusPro v2, a participant in CAPRI since Round 13, including all joint CASP-CAPRI rounds, has repeatedly ranked among the top servers.4–6 The ClusPro server performs three main steps: (1) Fast Fourier Transform (FFT)-based rigid-body sampling, (2) ranking via cluster population, and (3) energy minimization to remove steric clashes. This algorithm has proven itself to be an effective method for a variety of targets. Features added to ClusPro, including the ability to account for Small Angle X-ray Scattering (SAXS) profiles7,8 and pairwise distance restraints,9 have been motivated by specific CAPRI targets, where this information was made available to predictors. More recently, CASP-CAPRI targets inspired the addition of a tool for the discrimination between biological and crystallographic dimers.10
While early CAPRI targets presented participants with crystal structures for one or even both complex subunits, later rounds, including those combined with CASP rounds, have required participants to use homology models as representative subunit structures. ClusPro, as a free docking server, has not previously incorporated homology modeling into its automated protocol, instead either relying on structural predictions from CASP participants or on homology models generated by the HHPred web server.11 These models were then submitted for ClusPro docking in hopes of producing a near-native interface. This method was employed in CASP12, and produced an acceptable or better solution within the top 10 submitted models in 7 of 10 targets, 3 of which were of medium quality.6 However, compared to other servers employing template-based modeling approaches, ClusPro generally produced fewer high accuracy results. In a retrospective study12 on 15 validated homodimers from CASP11-CAPRI and CASP12-CAPRI assessments, it was shown that template-based modeling greatly increased the reliability of predictions for the 12 designated easy targets. When templates were available, higher quality predictions were produced via template-based modeling alone. Interestingly, for one of the three difficult targets (T72/T0770, T86/T0815, and T116/T0893) that did not have suitable templates, global docking yielded an acceptable model, whereas the template-based method produced none. These findings further support the need for ClusPro to incorporate both free docking and template-based searches into its predictive strategy.
Here we present the template-based modeling feature of ClusPro; this protocol will be discussed in reference to T152/T1003, a homodimer, as an example of a straightforward case where many good templates are available, followed by the discussion of targets T142/H0974 and T141/T0976 that required more complicated modeling steps. We will discuss plans to further expand ClusPro TBM by incorporating new template selection techniques, modeling/docking decision making, and inputs for experimental data such as SAXS profiles and Electron Microscopy (EM) density maps to help guide the modeling process. Finally, we will highlight a promising lead for future ClusPro server submissions based on the use of co-evolutionary information for two targets (T146/H0993, T157/H1019). Inferred residue-residue contacts submitted as restraints with free docking proved successful in the human prediction round. While this method is not yet implemented in ClusPro, the results presented here suggest that the use of co-evolutionary information in docking will substantially increase the number of complexes that can be predicted with acceptable or higher accuracy, thus providing an important but challenging direction for future development.
2. METHODS
2.1. Protocol selection
For each target, we first attempted to perform template search/homology modeling using the novel ClusPro template-based modeling functionality. Whenever templates were available, the resulting models were submitted as target predictions. When no templates were identified, we used ClusPro free docking capabilities to generate the models.
2.2. Template-based modeling
As inputs, the ClusPro template-based modeling module requires a set of sequences in FASTA format and the stoichiometry of the assembly to be modeled (Fig. 1A). Potential structural templates for each query sequence are identified using a local installation of HHpred,13 which runs HHblits and HHsearch using default settings and searches through the latest versions of uniprot20 and pdb70 databases, respectively. HHpred results are filtered by HHpred probability (>90%) and query sequence coverage (>50%), after which PDB structures that have at least one HHpred hit for each of the unique query sequences are identified (Fig. 1B). We term such a “shared” PDB file and a set of HHpred results pointing to it a “common template”. It should be noted that a single PDB template can accommodate several hits of the same query sequence in different positions and chains, and these multiple hits are included in the “common template”.
Figure 1.
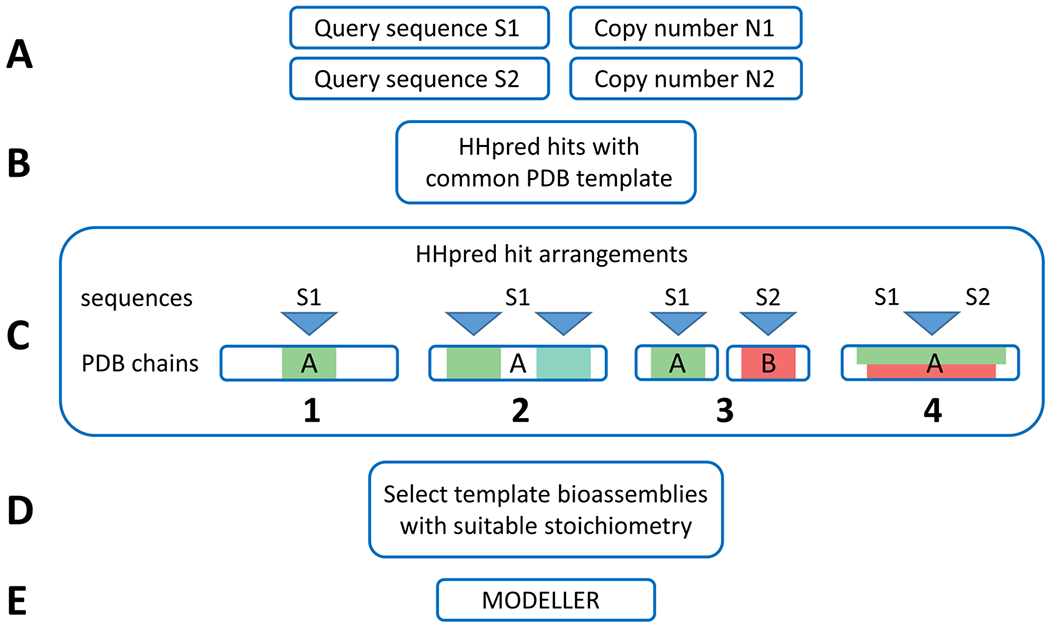
General outline of the ClusPro template-based modeling (TBM) protocol. (A) ClusPro TBM takes component sequences and their corresponding copy numbers in the modeled assembly as inputs. (B) HHpred is used to find potential templates for each query sequence, and HHpred hits sharing a common structural template are identified. (C) The HHpred hits are combined to obtain potential assembly-generating arrangements for each template structure (arrangements in the figure are examples, and are not necessarily generated as a part of a single server job). (D) The arrangements are evaluated on their ability to produce a model with user-specified stoichiometry based on biological assemblies specified in the template PDB file. (E) Arrangements passing the stoichiometry filter are used to construct the assembly models using MODELLER.
Since a single “common template” can have multiple HHpred hits from a single query sequence, various combinations of hits can be used to construct different “hit arrangements”, potentially leading to different assembly models. We combinatorially generate all such possible “hit arrangements”, with the requirement that at least one hit for each query sequence is present in the arrangement. The resultant arrangements can represent a variety of query-template relationships (see Fig. 1C).
For each generated arrangement, we iterate through all biological assemblies specified in the shared template PDB structure and check whether this template assembly can be used to produce a model of user-specified stoichiometry given a particular “hit arrangement” (Figure 1C,D). Figure 1C provides examples of some representative HHpred hit arrangements for homo and heterooligomeric multimers that were present as targets in CASP13. The leftmost in Fig. 1C.1 depicts the most straightforward homooligomeric case, where a single query sequence is aligned to a separate chain in the template PDB. If the target in this case is an A2 complex, the required template stoichiometry for this arrangement is also A2. Fig. 1C.2 shows a more complicated case, in which a single query sequence aligns to multiple regions within the same chain of a template PDB. This relationship is likely to occur when the template protein is a result of gene duplication and fusion as seen in T0976 (see Section 3.2.3). If the target is a dimer in this case, the template should be a simple monomer. Fig. 1C.3, represents the simplest heterooligomeric case, where two query sequences align to two different template chains. Similar to the simplest homooligomeric case, the template stoichiometry here should match the stoichiometry of the modeled assembly. For example, a simple heterodimer target needs a heterodimer template to be modeled correctly. Finally, Fig. 1C.4 shows a case where different query sequences align to the same region within the same template chain. Such an outcome is likely to happen if query sequences are related to each other (like in H0974). The template in such a case needs to be a homomultimer with the number of subunits equal to the combined number of S1 and S2 subunits of the target assembly. For instance, the template needs to be a homodimer if the target is a heterodimer.
If the assembly template matches the stoichiometry requirements, it is used to construct an assembly model. For each query sequence, the top-ranking HHpred hit from the arrangement is used to construct the model of the monomer. The target sequence is modeled onto a single chain of the homomultimeric template using MODELLER.14 Regions of the target which are not aligned to a template sequence are removed to avoid the addition of unstructured loops into the model, while aligned portions of the target are built with the same backbone. Once produced, the monomer model is copied and aligned to other locations of the multimer template based on HHpred hits for the given query sequence present in the “hit arrangement”, followed by interface minimization. These models are ranked based on the averaged ranks of HHpred hits used to build the models of the monomers. The server also provides an advanced option for manually curated homology modeling, allowing the users to upload their own templates and alignments.
2.3. Free docking
When assembly templates were not available, we used ClusPro free docking capabilities to generate the predictions. Monomer models were constructed using the HHpred server.11 When the HHpred server did not produce any models, we used monomer models as predicted by the CASP servers. The free docking pipeline was as described previously.15 Briefly, the FFT-based PIPER protein docking program16 is used to generate 1000 low-energy poses which are then clustered together using a 9 Å clustering radius. Clusters are ranked by their populations and cluster centers are treated as complex models. These models are subjected to local energy minimization by CHARMM17 and returned as final server predictions.
2.4. Co-minimization via CHARMM
Both for template-based and free docking models, CHARMM was used to co-minimize the modeled interface using the PARAM19 force field with polar hydrogens only. ClusPro TBM complexes were first minimized using 1000 steps of Adapted Basic Newton-Raphson (ABNR) minimization, with harmonic restraints set on the alpha carbons, to remove larger clashes that would otherwise occur in the interface. The harmonic restraints were then removed, followed by 1000 steps of unconstrained ABNR minimization. A constant dielectric was used during the energy calculations, and a distance cutoff of 15 Å when considering non-bonding interactions.
3. RESULTS AND DISCUSSION
3.1. Introduction
Similarly to previous CASP-CAPRI rounds, the majority of targets in CASP13-CAPRI were homomeric complexes. As the majority of biological assemblies in the PDB are also homomeric, the targets of this type, compared to heteromeric targets, are much more likely to have structural templates readily available. Additionally, our experience with previous CASP-CAPRI rounds suggests that structural templates, when present, enable the construction of higher accuracy models than those generated using free docking approaches.12
Motivated by these observations, we utilized a template-first pipeline to predict the structures of target complexes, in which we performed template-based modeling whenever assembly templates were available, and used free docking otherwise. Here we describe several representative cases from the last CASP-CAPRI round that highlight the new server functionality.
3.2. Selected server predictions
3.2.1. T152/T1003 - simple homodimer case
Target T152/T1003, 5′-Aminolevulinate Synthase 2, serves as an ideal template-based modeling case, having A2 stoichiometry and an abundance of available templates with high sequence similarity (up to ~48% sequence identity). Within the first 11 structures suggested by an HHpred template search, 10 were available as dimerized biological assemblies. All were listed with reported HHpred probabilities of 100, which exceeds the value (0.95) considered high enough to indicate certain homology between query and template sequences.11 As our protocol describes, for each of these templates, MODELLER produced monomer models, which were then copied onto each unit in the corresponding template assembly. The first five models submitted by ClusPro TBM were based on templates 2W8T, 2X8Y, 5TXR, 3TQX, and 2BWN, and were all evaluated as medium quality models. Figure 2A shows a representative model produced by ClusPro TBM.
Figure 2.
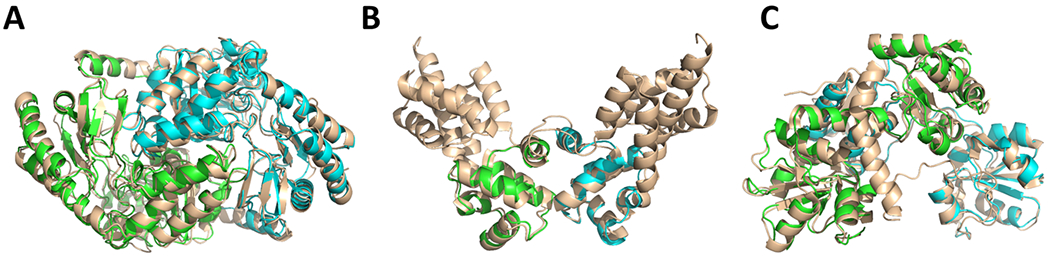
Examples of successful ClusPro TBM predictions based on different HHpred hit arrangements. (A) Model of T152/T1003 (green and cyan) overlapped with its homodimeric template (wheat, PDB 2W8T). (B) A model of T142/H0974 (green and cyan) overlapped with its homodimeric template (wheat, PDB 4RYK). (C) Modeled subunits (green and cyan) of T141/T0976 aligned to different locations on the same chain of the template protein (wheat, PDB 1YT8).
3.2.2. T142/H0974 - heterodimer based on homodimer
Target H0974 with A1B1 stoichiometry is an example of successful modeling of a heterodimer using homodimeric complexes of remote homologs as templates. The target represented a heterocomplex of DNA binding proteins, and the sequences of the target subunits were homologous to each other. Predictably, the templates identified by HHpred were predominantly homodimeric complexes, and HHpred hits for target subunits were usually mapped to the same chain of the template structure. While handling of such templates is trivial when done manually, it is less straightforward in the automatic regime. The arrangement procedure implemented in ClusPro TBM was successful in automatically determining the homomeric templates as having suitable stoichiometry and correctly aligning the monomer models onto different chains of the template assembly, producing three medium quality and one acceptable models. Figure 2B shows an example homomeric template (PDB 4RYK) together with the predicted complex model based on it.
3.2.3. T141/T0976 - homodimer based on monomer
Another notable docking target was a homodimer formed by the Rhodanese-like family protein SCHU S4. The only productive template identified by HHpred was, in fact, a monomeric fusion protein that had appeared in 3 different HHpred hits. Since the hit arrangement procedure of ClusPro TBM allows for model construction from multiple HHpred hits, 2 of these HHpred hits using non-overlapping regions of the template chain were used by the server as monomer alignment sites to construct an acceptable quality model (see Figure 2C).
4. PROSPECTIVE DEVELOPMENT
4.1. Introduction
Our template-based modeling demonstrated promising results in CASP13, however, it can be further improved by implementing more sophisticated template searches, adding follow-up free docking steps, and incorporating experimental data. In the following sections, we discuss existing limitations in the methodology and propose potential enhancements to the server.
4.2. Template-based modeling
4.2.1. Refinement of template-based solutions with focused docking
As demonstrated by target T137/T0965, routine template-based modeling may sometimes lead to low-quality models. Following an HHpred search of the provided sequence, numerous high-probability (>0.95) homodimer templates are given. The target appears an easy one, with a noticeable agreement between the top ten template interfaces. However, none of the models produced by ClusPro were evaluated as acceptable or better compared to the crystal complex (PDB 6D2V). The template complex for this target has correct contact location, however, one of the subunits is 130 degrees rotated with respect to the target structure (see Figure 3).
Figure 3.
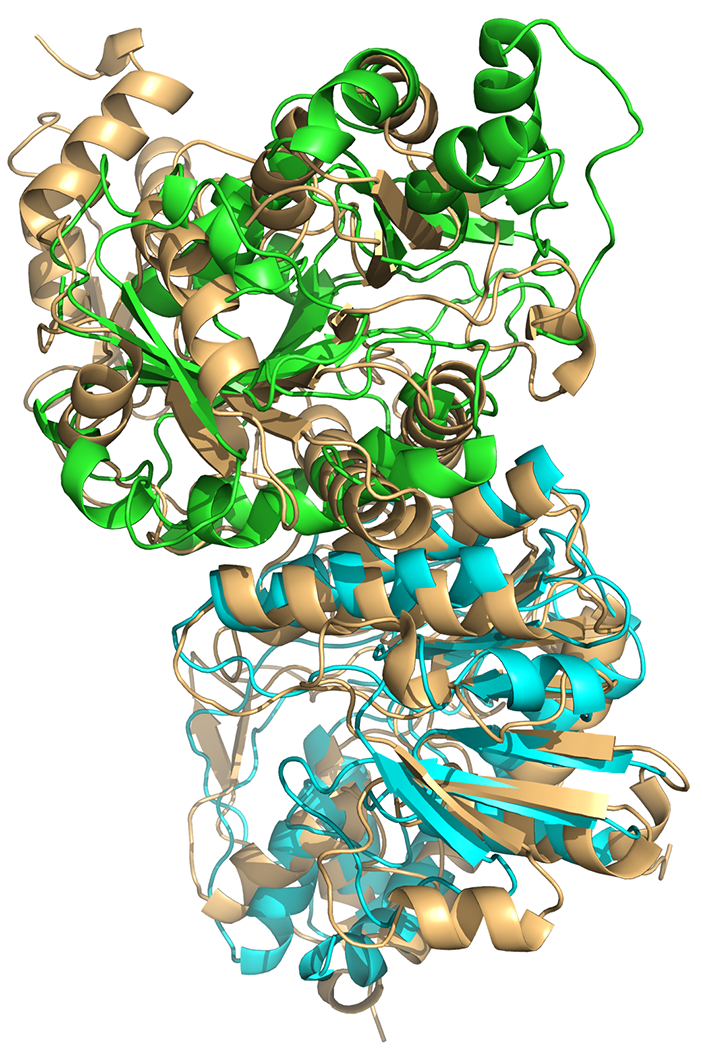
A model (green, cyan) of T137/T0965 superimposed with the native structure (wheat, PDB 6D2V).
This case demonstrates the need for a merger between template-based and free docking methodologies. While our template-based method alone would have failed, it was later shown that the complex could be solved by applying free docking with restricted sampling (i.e. “focused” docking) about the modeled interface.12 Thus a criterion should be developed which can effectively distinguish deceptive templates and switch the protocol from template-based mode to free docking.
4.2.2. Template discrimination and ranking
Ranking of the different templates is another issue of the template-based method which could be resolved by free docking. We tested an approach based on re-docking the separated subunits of the models to be evaluated. The expectation was that the more correct models would be more frequently reconstructed. This approach was inspired by the problem of discriminating between biological and crystal contacts in X-ray structures.18 For targets with several different template models, this strategy might be applied to rank and prioritize them by the number of low energy docking solutions discovered in the neighborhood, which can be a good indicator of a low free energy state. The successful example of this approach was target T75/T0776 (PDB 4Q9A) from the previous CASP11-CAPRI challenge, for which 2 different templates were available (see Figure 4). The number of docking poses near the correct template model was about twice the number of poses near the incorrect one.
Figure 4.
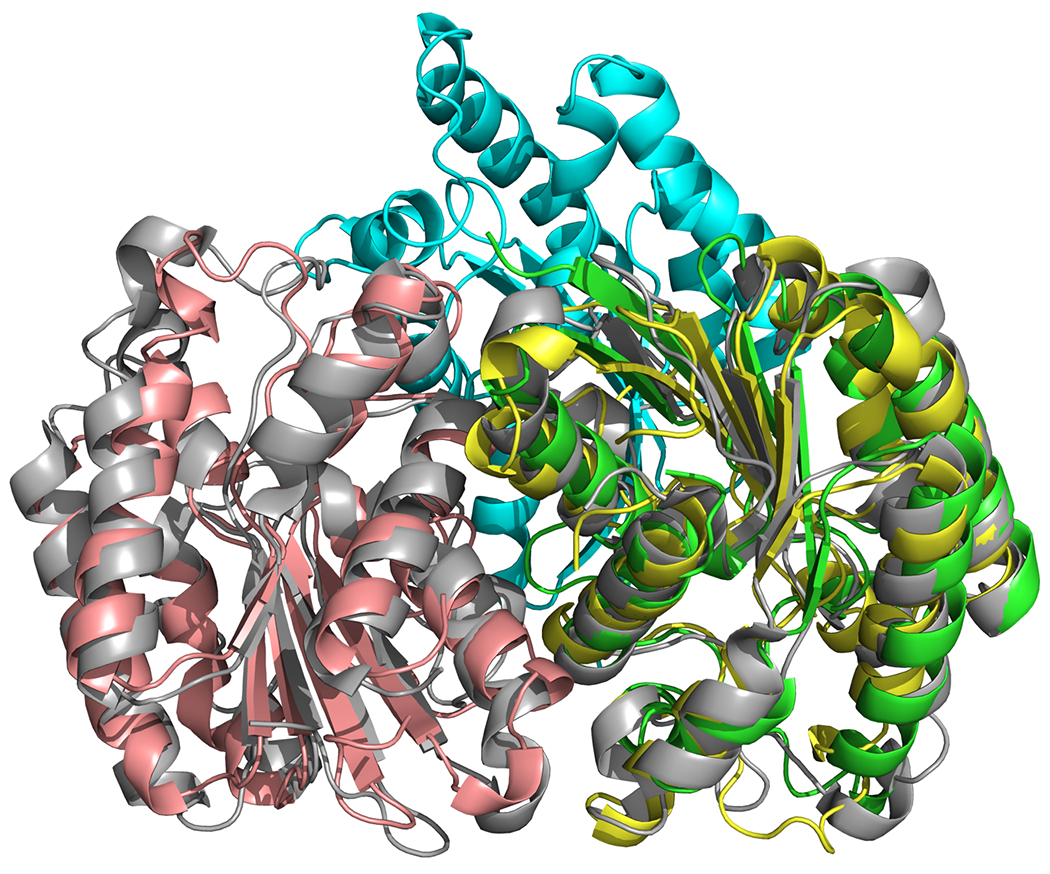
Two different T75/T0776 models (green-cyan and yellow-pink) aligned to one of the subunits of the target structure (gray, PDB 4Q9A).
4.2.3. Data-assisted template-based modeling
Over the years the ClusPro free docking procedure has been enhanced with a variety of tools for incorporating experimental data, including options for using SAXS and arbitrary restraints. At this point, however, these tools are not available as a part of the template-based modeling pipeline, which is a definite flaw of the current version of the server.
In addition to the need for making the existing tools available through the TBM interface, the latest CASP-CAPRI round has demonstrated that ClusPro needs to be enhanced with tools for handling EM data, which is currently rapidly growing in availability. One particular example where EM was used in human submission was target T159/H1021, representing a portion of a contractile insertion system and possessing an A6B6C6 stoichiometry. For this target, a low-resolution EM map (EMDB-2419) was available at the time the target was made open. Also, while there was no template for the assembly as a whole, partial templates were available (for instance, 1J9Q for the A6B6 portion and 1J2M for the B6C6 portion). Thus, for our submission as a human group, we individually fitted these partial templates into the EM map using a new version of the fast manifold Fourier transform (FMFT) software.19 During the fitting procedure, the EM density grid was correlated with the steric density grid of the template being fitted, and 350 best-scoring conformations were clustered to produce the final fitting poses. These poses were combined to build the global assembly template (see Figure 5), which we then used for homology modeling with MODELLER. The resulting models recapitulated the global geometry of the assembly, and had several interfaces evaluated as acceptable or medium quality. However, working with EM data is not yet implemented in ClusPro.
Figure 5.
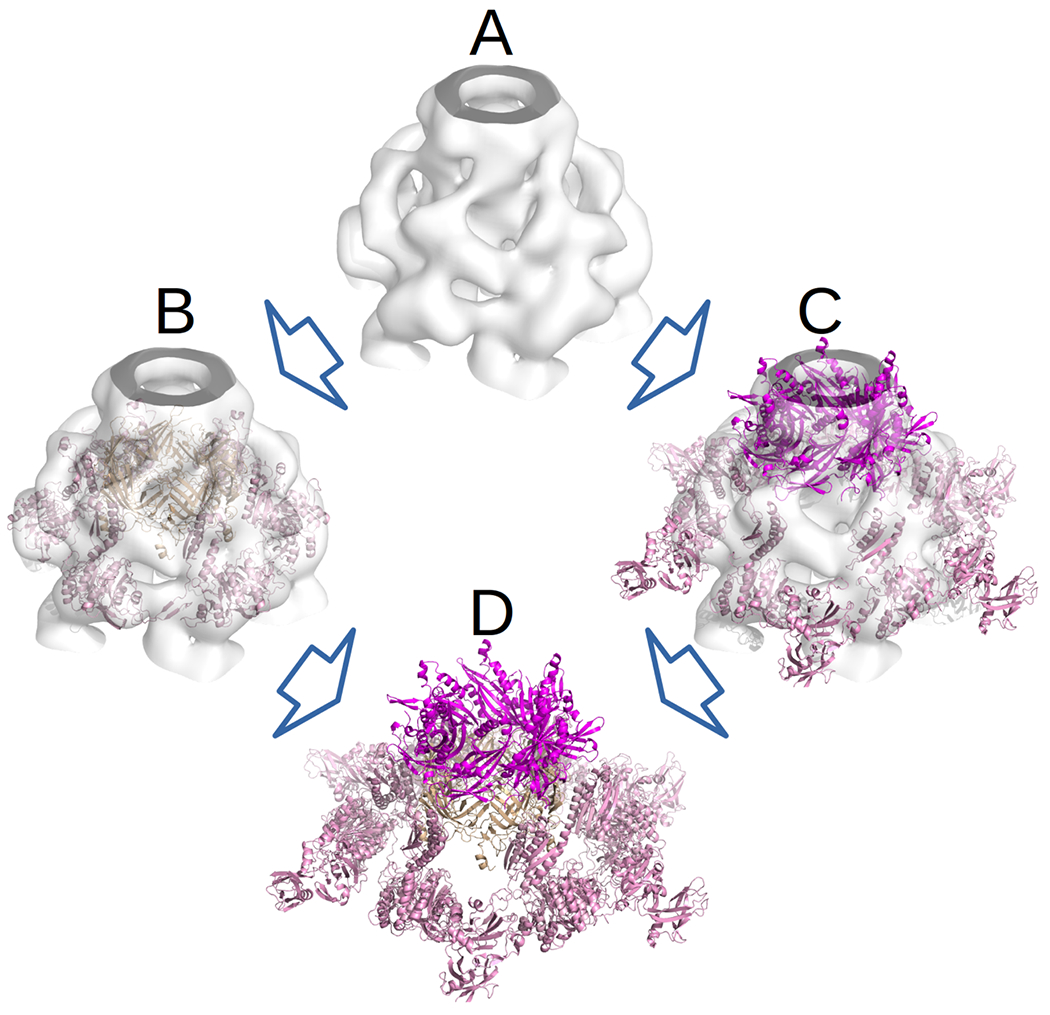
Template based modeling of target T159/H1021 assisted by low-resolution Electron Microscopy data. A) EM density map (EMDB-2419). B) Partial template for subunits A and B aligned to the EM map C) Partial template for subunits B and C aligned to the EM map. D) Mutual arrangement of the templates induced by the EM map.
4.3. Free docking
4.3.1. Data-assisted free docking
Free docking becomes a very challenging problem when models are used as structural inputs for subunits, and our attempts to use unbiased free docking in CASP13-CAPRI server predictions were largely unsuccessful. Nevertheless, the quality of free docking of homology models can be improved if docking predictions are guided by additional experimental data, and development of computational pipelines robustly incorporating these various sources of data is a promising avenue for the improvement of docking methods.
During CASP13-CAPRI we selected three targets (T146/H0993, T155/H1015, and T157/H1019) that did not have assembly templates and for which the interfacial residue-residue contacts could be inferred from co-evolution between the residues of the protein pairs in the multiple sequence alignments of homologous proteins. We then used these contacts to focus the docking predictions and submitted the resulting models for evaluation as a human group. In two cases (T146 and T157) an acceptable quality model was found among the top 5 predictions. We believe this result makes a strong point for the incorporation of evolutionary data into the standard ClusPro toolset. Below we briefly discuss the details of the methodology we employed for these targets.
To generate the contacts, we used GREMLIN.20 The GREMLIN protocol is based on the premise that if the amino acids from different proteins are correlated in multiple organisms they are likely to interact. It starts with finding the homologs of two target proteins that are present in the same organism and are located close in the genome. From this, a paired multiple sequence alignment (MSA) is built and filtered to provide adequate coverage, and then the regularized pseudo-likelihood maximization procedure21,22 is used to find coupled residues.
Target T146/H0993 was an A2B2 heterotetramer consisting of two copies of the ATP-binding protein MlaF and two copies of the cytoplasmic solute-binding protein MlaB. We prepared homology models of the MlaF homodimer and the MlaB monomer based on templates identified by HHpred (for MlaF: 3RLF and 4YER, and for MlaB: 1VC1, 3F43, and 4HYL), and focused our efforts on predicting the MlaF-MlaB interface. For this interface, seven high-confidence inter-protein contacts were identified by GREMLIN: S123:D68, P119:H65, Q115:L31, E130:N94, L120:W34, S123:L64, and L120:L62. We used these contacts to impose restraints on the distances between the atoms of the corresponding residues, requiring them to be in the range of 1-8 Å or 1-5 Å. During the docking runs, 5 out of 7 restraints had to be satisfied for the pose to be considered. Docking was carried out with the ClusPro restrained docking option9 using various combinations of component models as inputs, and manually selected representative docking models were submitted for evaluation. A model ranked fifth had the corresponding interface graded as acceptable quality.
Target T157/H1019 posed a challenge in terms of homology modeling of its subunits, so we used manually selected CASP server models instead. The six selected receptor and ligand protein models were docked to each other in an exhaustive manner in 36 ClusPro restrained docking runs. During the docking procedure, restraints were applied to the following set of residue pairs identified by GREMLIN: K14:V32, F76:Y19, K14:E28, K80:P31, and E79:S45. The distances between the atoms of coupled residues were required to be in the 0-8 Å range, and 4 out of 5 restraints had to be satisfied for the pose to be considered for further processing. Unlike the case of T146, we tried to limit manual intervention in final model selection, so we used a clustering procedure where up to 10 top-ranking models from each docking run were clustered, the clusters were ranked based on their population, and the cluster centers were submitted for CASP-CAPRI evaluation as final models. A model ranked third was graded as acceptable quality. Paired alignment for target T155/H1015 contained substantially fewer sequences than for the other two targets (199 vs 5316 and 1340 for T146/H0993 and T157/H1019 respectively), and therefore the co-evolutionary signal was likely not strong enough to result in reliable docking restraints.
4.4. Current limitations
There are several unresolved issues currently restricting both template based and free docking capabilities of ClusPro.
The main weakness of ClusPro TBM is the fact that it currently relies on the pre-filtered pdb70 HHpred database for homology search and template selection. In this database, the sequences of protein chains present in the PDB are clustered with a 70% sequence identity cutoff, and each sequence cluster is represented by a single “central” representative.13 Using this database greatly speeds up the search and reduces the redundancy of the output. Unfortunately, many potential higher-quality templates may be discarded as a result, which potentially reduces the quality of the resulting models. This issue is aggravated in the case of heteromeric targets, where each of the unique template sequences needs to be the “center” of the corresponding cluster for the template to be considered by ClusPro TBM, which increases the probability that the template is neglected even further. To address this problem, we are planning to switch to the unfiltered pdb100 database in the future.
Another issue with the current ClusPro TBM algorithm is insufficient flexibility of the stoichiometry filtering step in the modeling pipeline. While the current implementation has the ability to deal with various non-obvious cases, including fuzzy stoichiometry matching (see Section 3.2.2 regarding T142/T0974), or modeling of higher order assemblies based on lower order templates (Section 3.2.3), it is quite limited in its ability to use higher-order template assemblies to model lower-order targets. For example, it is currently unable to use a portion of a homotetrameric template to model a homodimer. Additionally, it only considers biological assemblies as potential templates, and does not take advantage of potentially useful crystallographic contacts present in the X-ray structures. Further improving the stoichiometry filtering capabilities should address these issues.
More generally, modeling performance of ClusPro TBM is limited by the quality of the templates available for the desired target sequence. When templates of high sequence identity (>30%)23,24 are not available, it becomes more difficult to select and predict templates which will produce high ranking models. We have suggested continued development of template selection methodologies, as detailed in Section 4.2.2, which may be used to filter out templates and resulting models.
The ClusPro free docking option is significantly constrained in its ability to model protein complexes in which large conformational changes play a key role. Complexes which fit this description, categorized as ‘difficult’ in the ZLab protein docking benchmark25,26, require the inclusion of flexible sampling, which is not currently implemented as part of the ClusPro server.
5. CONCLUSIONS
With the latest template modeling addition to ClusPro, the server is now able to submit models produced by template-based modeling or free docking. The protocol has been successful for a variety of cases; for example, we have shown that ClusPro TBM is well suited for modeling straightforward homooligomer targets (such as T152/T1003), as well as cases requiring less-conventional models based on a combination of HHpred hits. For T142/H0974, successful ClusPro models were produced by modeling the target, which had A1B1 stoichiometry, on homodimer templates. We also describe a case, T141/T0976, where another modeling mode was explored, in which the predicted structures of the A2 target are modeled on different regions of monomer templates.
This test of the ClusPro TBM module has been very promising as predictions were successful across the majority of the assessment targets. However, an important caveat is that most CAPRI targets were homomers, and hence the new module needs substantial further testing on heterodimers. Nevertheless, the targets of the current CAPRI rounds already inspired several new avenues to server improvement. Experimental data, like the EM maps used in H1021 prior to docking, may prove useful for future complex prediction challenges, either as a modeling guidance tool or perhaps even as a scoring method for template-based models. Template discrimination is another important aspect of our modeling approach which will require future work, but whose success would improve the efficiency of the ClusPro TBM protocol. An even more challenging problem arose in T137. Despite a strong agreement between template structures, the target complex can only be reproduced when focused docking is applied. Integration of free docking and template-based modeling into one pipeline may help to expand the number of difficult targets that can be tackled by ClusPro.
Not all targets in the latest assessment were well suited for Cluspro TBM. If no good templates were available, free docking of subunit models was used to generate predictions. Unfortunately for the few targets where this was the case, there were no acceptable or better predictions, which may be attributed to the low quality of the templates used. In fact, the side chain positions and loop conformations are usually less accurate in the homology models than in the X-ray structures of the separately crystallized constituent proteins of a complex. It appears that the methods and parameters developed for docking X-ray structures are less than optimal for docking such homology models, and there is a well-defined need for adjusting the methodology. Despite this difficulty, an important finding was made regarding the use of co-evolutionary analysis, which resulted in the addition of docking restraints that helped to recover an acceptable prediction for two targets in a later manual round. Evolutionary constraints will be pursued as a likely input in future ClusPro docking options, and may also be investigated as a means to filter template-based results. A beta version of ClusPro TBM is available at https://tbm.cluspro.org/.
ACKNOWLEDGEMENTS
This investigation was supported by grants DBI 1759277 and AF 1645512 from the National Science Foundation, and R35GM118078 and R21GM127952 from the National Institute of General Medical Sciences. Acpharis Inc. offers commercial licenses to PIPER. Dima Kozakov and Sandor Vajda own stock in the company. However, the PIPER program and the use of the ClusPro server are free for academic and governmental use.
REFERENCES
- 1.Rose PW, Prlic A, Altunkaya A, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017;45(D1):D271–D281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carroni M, Saibil HR. Cryo electron microscopy to determine the structure of macromolecular complexes. Methods. 2016;95:78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kundrotas PJ, Zhu Z, Janin J, Vakser IA. Templates are available to model nearly all complexes of structurally characterized proteins. Proc Natl Acad Sci U S A. 2012;109(24):9438–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lensink MF, Wodak SJ. Docking, scoring, and affinity prediction in CAPRI. Proteins. 2013;81(12):2082–2095. [DOI] [PubMed] [Google Scholar]
- 5.Lensink MF, Velankar S, Wodak SJ. Modeling protein-protein and protein-peptide complexes: CAPRI 6th edition. Proteins. 2017;85(3):359–377. [DOI] [PubMed] [Google Scholar]
- 6.Lensink MF, Velankar S, Baek M, Heo L, Seok C, Wodak SJ. The challenge of modeling protein assemblies: the CASP12-CAPRI experiment. Proteins. 2018;86 Suppl 1:257–273. [DOI] [PubMed] [Google Scholar]
- 7.Xia B, Mamonov A, Leysen S, et al. Accounting for observed small angle X-ray scattering profile in the protein-protein docking server ClusPro. J Comput Chem. 2015;36(20):1568–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ignatov M, Kazennov A, Kozakov D. ClusPro FMFT-SAXS: Ultra-fast filtering using small-angle X-ray scattering data in protein docking. J Mol Biol. 2018;430(15):2249–2255. [DOI] [PubMed] [Google Scholar]
- 9.Xia B, Vajda S, Kozakov D. Accounting for pairwise distance restraints in FFT-based protein-protein docking. Bioinformatics. 2016;32(21):3342–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vajda S, Yueh C, Beglov D, et al. New additions to the ClusPro server motivated by CAPRI. Proteins. 2017;85(3):435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimmermann L, Stephens A, Nam SZ, et al. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol. 2018;430(15):2237–2243. [DOI] [PubMed] [Google Scholar]
- 12.Porter KA, Desta I, Kozakov D, Vajda S. What method to use for protein-protein docking? Curr Opin Struct Biol. 2019;55:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33(Web Server issue):W244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics. 2016;54:5 6 1–5 6 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozakov D, Brenke R, Comeau SR, Vajda S. PIPER: an FFT-based protein docking program with pairwise potentials. Proteins. 2006;65(2):392–406. [DOI] [PubMed] [Google Scholar]
- 17.Brooks BR, Brooks CL 3rd, Mackerell AD Jr., et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30(10):1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yueh C, Hall DR, Xia B, Padhorny D, Kozakov D, Vajda S. ClusPro-DC: Dimer classification by the ClusPro server for protein-protein docking. J Mol Biol. 2017;429(3):372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Padhorny D, Kazennov A, Zerbe BS, et al. Protein-protein docking by fast generalized Fourier transforms on 5D rotational manifolds. Proc Natl Acad Sci U S A. 2016;113(30):E4286–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ovchinnikov S, Kamisetty H, Baker D. Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. Elife. 2014;3:e02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balakrishnan S, Kamisetty H, Carbonell JG, Lee SI, Langmead CJ. Learning generative models for protein fold families. Proteins. 2011;79(4):1061–1078. [DOI] [PubMed] [Google Scholar]
- 22.Correction for Kamisetty et al. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proceedings of the National Academy of Sciences. 2013;110(46):18734–18734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiang Z Advances in homology protein structure modeling. Current Protein & Peptide Science. 2006;7(3):217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalev I, Habeck M. HHfrag: HMM-based fragment detection using HHpred. Bioinformatics. 2011;27(22):3110–3116. [DOI] [PubMed] [Google Scholar]
- 25.Hwang H, Vreven T, Janin J, Weng Z. Protein-protein docking benchmark version 4.0. Proteins. 2010;78(15):3111–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vreven T, Moal IH, Vangone A, et al. Updates to the integrated protein-protein interaction benchmarks: Docking benchmark version 5 and affinity benchmark version 2. J Mol Biol. 2015;427(19):3031–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]