

Abstract
Background
Cancer cachexia is a multifactorial debilitating syndrome that directly accounts for more than 20% of cancer deaths while there is no effective therapeutic approach for treatment of cancer cachexia. Carnosol (CS) is a bioactive diterpene compound present in Lamiaceae spp., which has been demonstrated to have antioxidant, anti‐inflammatory, and anticancer properties. But its effects on cancer cachexia and the possible mechanism remain a mystery.
Methods
The in vitro cell models of C2C12 myotube atrophy and 3T3‐L1 mature adipocyte lipolysis were used to check the activities of CS and its synthesized analogues. C26 tumour‐bearing BALB/c mice were applied as the animal model to examine their therapeutic effects on cancer cachexia in vivo. Levels of related signal proteins in both in vitro and in vivo experiments were examined using western blotting to study the possible mechanisms.
Results
Carnosol and its analogues [dimethyl‐carnosol (DCS) and dimethyl‐carnosol‐D6 (DCSD)] alleviated myotube atrophy of C2C12 myotubes and lipolysis of 3T3‐L1 adipocytes in vitro. Interestingly, CS and its analogues exhibited stronger inhibitive effects on muscle atrophy induced by tumour necrosis factor‐α (TNF‐α) (CS, P < 0.001; DCS, P < 0.001; DCSD, P < 0.001) in C2C12 myoblasts than on muscle atrophy induced by IL‐6 (CS, P < 0.05; DCS, P = 0.08; DCSD, P < 0.05). In a C26 tumour‐bearing mice model, administration of CS or its analogue DCSD significantly prevented body weight loss without affecting tumour size. At the end of the experiment, the body weight of mice treated with CS and DCSD was significantly increased by 11.09% (P < 0.01) and 11.38% (P < 0.01) compared with that of the C26 model group. CS and DCSD also improved the weight loss of epididymal adipose tissue in C26 model mice by 176.6% (P < 0.01) and 48.2% (P < 0.05) increase, respectively. CS and DCSD treatment partly preserved gastrocnemius myofibres cross‐sectional area. CS treatment decreased the serum level of TNF‐α (−95.02%, P < 0.01) but not IL‐6 in C26 tumour‐bearing mice. Inhibition on NF‐κB and activation of Akt signalling pathway were involved in the ameliorating effects of CS and its analogues on muscle wasting both in vitro and in vivo. CS and its analogues also alleviated adipose tissue loss by inhibiting NF‐κB and AMPK signalling pathways both in vitro and in vivo.
Conclusions
CS and its analogues exhibited anticachexia effects mainly by inhibiting TNF‐α/NF‐κB pathway and decreasing muscle and adipose tissue loss. CS and its analogues might be promising drug candidates for the treatment of cancer cachexia.
Keywords: Carnosol, Cancer cachexia, Muscle atrophy, Lipolysis, NF‐κB
Introduction
Cachexia is a multifactorial wasting syndrome characterized by uncontrolled body weight loss, which is mainly reflected by the atrophy of skeletal muscles and the wasting of adipose tissue, leading to inevitable functional impairment. Such pathologic condition could be caused by various factors, including cancer. 1 Cancer cachexia is also associated with diminishing sensitivity to chemotherapy and reducing quality of life, thereby contributing to shortened survival times. 2 In advanced cancer patients, more than 50% of cancer patients display cachectic symptoms and leads to at least 20% of cancer deaths.
Currently, there is no approved medicine for treating cancer cachexia. Multimodal therapeutic approaches such as nutritional support and appetite stimulant (megestrol and medroxyprogesterone) 3 only slightly help manage the symptoms. Based on the important involvement of inflammatory factors in the development of cancer cachexia, specific antibody therapies for inflammatory factors such as the anti‐tumour necrosis factor‐α (TNF‐α) monoclonal antibody Infliximab and the anti‐interleukin (IL)‐6 monoclonal antibody Clazakizumab have been developed. Unfortunately, most of these antibody therapies in phases I and II clinical studies produced mixed results, and few drugs have demonstrated significant positive effects in larger scale phase III trials. 4 In all, over the past few years, it has been found that a single strategy, such as nutritional supply, or pharmacological intervention with a single agent aimed at inflammation, anorexia, and catabolic pathways was ineffective to treat cancer cachexia. 5 Therefore, there is an urgent need to develop effective therapeutic drugs for preventing and treating cancer cachexia, and possibly, a combinatorial approach might be beneficial because of targeting several characteristics of cancer cachexia at the same time.
Herbal medicine in treatment of cancer cachexia is a fast‐emerging area. 6 Previous studies have found that Rosemary extract has antioxidant and anti‐inflammatory activities, 7 , 8 so we wondered if it might have a therapeutic effect on cancer cachexia. In the present study, we conducted study to check the effects of the three representative components of Rosemary extract, that is, rosmarinic acid, carnosic acid, and carnosol (CS). 9 CS and structurally related diterpenes are major secondary plant metabolites in Lamiaceae spp. such as Salvia officinalis L (Sage) and Rosmarinus officinalis L (Rosemary), which exhibit numerous biological features. CS is found in considerable quantities in Rosemary such as approximately 0.2–1% in dried Rosemary and 10.3% in commercially available Rosemary extracts. 10 The molecular formula of CS is C20H26O4, and its molecular weight is 330.42. It is an ortho‐diphenolic of abietane‐type diterpene lactone, consists of an abietane carbon skeleton with hydroxyl groups at positions C‐11 and C‐12 and a lactone moiety across the B ring. 11 CS has been demonstrated to have antioxidation, 12 anti‐inflammation, 13 and anticancer properties. 14 For example, CS could partially reduce UVB‐induced reactive oxygen species elevation and thus reduce DNA damage in keratinocytes. 15 CS was reported to exert anti‐inflammatory effects by reducing cytokine release [e.g. interleukin (IL)‐1 and IL‐6] and iNOS formation. 13 Moreover, it has been reported that CS could inhibit tumour growth in C57BL/6J/Min/+ (Min/+) mice, 14 as well as growth of prostate cancer, breast cancer, skin cancer, and leukaemia. 16
However, the effects of CS on cancer cachexia have not been reported before. In order to confirm the anticachexia effects of CS and try to get new compounds with better characteristics, we modified the structure of CS and obtained two analogues, dimethyl‐carnosol (DCS) and dimethyl‐carnosol‐D6 (DCSD). In the present study, we conduct both in vitro experiments and in vivo experiments using cancer cachexia mice model to systematically and thoroughly study the anticachexia effects and mechanisms of CS and its analogues. In combining the in vitro and in vivo study results, we showed the exciting effects and the possible mechanisms of CS and its analogues on cancer cachexia.
Materials and methods
Reagents
The ethanolic extract of Rosemary was kindly provided by Dr Yiming Li from Shanghai University of Traditional Chinese Medicine. CS, with a purity of >98%, was purchased from Shanghai NatureStandard Biotechnology Co., LTD (Shanghai, P.R. China). DCS and DCSD were synthesized from the structural modification of CS. Dulbecco's modified Eagle's minimal essential medium (DMEM) (high glucose), RMPI‐1640, penicillin/streptomycin, and trypsin/EDTA were purchased from Hyclone (Los Angeles, CA, United States). Horse serum was purchased from Gibco (New York, NY, United States). Foetal bovine serum (FBS) was derived from Biological Industries (Kibbutz Beit Haemek, Israel). RIPA Lysis, Halt Protease, and Phosphatase Inhibitor Cocktail (100×) were purchased from Thermo Scientific (Rockford, IL, United States). BCA protein assay kit used to quantify protein concentration was purchased from Beyotime (Shanghai, P.R. China). TNF‐α and IL‐6 were purchased from PeproTech (Rocky Hill, CT, United States). Other chemicals, except where specially noted, were purchased from Sigma‐Aldrich Chemical Co. (St. Louis, MO, United States).
Cell culture
C26 cell line was obtained from Shanghai Institute of Materia Medica, Chinese Academy of Sciences. Cells were cultured in RMPI‐1640 medium containing 10% FBS at 37°C with 5% CO2. C2C12 murine myoblast cell line, obtained from ATCC, was cultured in high‐glucose DMEM with 10% FBS at 37°C with 5% CO2. For differentiation, cells were plated on culture plates coated with 0.1% gelatine, and the medium was switched into differentiation medium (high‐glucose DMEM containing 2% horse serum) when cell confluence reached 70%. After 5 days, multinuclear myotubes were formed. 3T3‐L1 pre‐adipocytes cell line, obtained from Shanghai Institute of Materia Medica, Chinese Academy of Sciences, was cultured in adipocytes medium (AM, high‐glucose DMEM with 10% FBS) at 37°C with 5% CO2. The 3T3‐L1 pre‐adipocytes were plated on culture plates coated with 0.1% gelatine until the confluence reached about 100% for 48 h in AM. Then, the 3T3‐L1 pre‐adipocytes were induced to differentiate by treated with differentiation media (DM I and DM II) for 48 h, respectively. DM I medium is AM containing 10 μg/mL of insulin (Solarbio, Beijing, China), 1 μM of dexamethasone, and 0.5 mM of 3‐isobutyl‐1‐methylxanthine (IBMX) while DM II medium is dexamethasone‐free and 3‐isobutyl‐1‐methylxanthine‐free DM I. Thereafter, the differentiated cells were maintained in AM changed every 2 days until used for induction of lipolysis. All cells were negative for mycoplasma contamination before use.
Animals
Male BALB/c mice (6–8 weeks old) were purchased from Shanghai SLAC Laboratory Animal Co., LTD (Shanghai, China). The care and experimental protocols for this study complied with the Chinese regulations and the Guidelines for the Care and Use of Laboratory Animals drawn up by the National Institutes of Health (United States) and were approved by the Institutional Animal Care and Use Committee of the East China Normal University. Mice were maintained on a 12:12 light–dark cycle in a temperature‐controlled (21°C–23°C) and specific pathogen free conditional room and were provided standard rodent chow and water ad libitum. All animals were acclimatized for a week before beginning the study.
C26 tumour medium collection
C26 tumour‐conditioned medium was collected as reported in our previous papers. 17 Briefly, when the confluence of C26 tumour cells reached 70%, the medium was changed into new high‐glucose DMEM medium and continue to culture for 48 h. Thereafter, conditioned medium was collected, and then, the cells and debris were removed by centrifugation (5000×g, 10 min at 4°C). The final supernatant was filtered and stored at −80°C or used immediately at a 1:1 dilution with fresh normal medium. Medium from non‐tumour cells (C2C12 cell or 3T3‐L1 cell) was used as control medium.
Cancer cachexia models in vitro
In vitro cancer cachexia models were established as reported in our previous paper. 17 Briefly, C2C12 myoblasts were differentiated into myotubes for 5 days using differentiation medium. The C2C12 myotube atrophy was induced by C26 tumour‐conditioned medium (1:1 dilution with fresh normal medium), TNF‐α (100 ng/mL), or IL‐6 (100 ng/mL), respectively, for 48 h in the presence of different concentrations of CS, DCS, DCSD, or 0.1% DMSO (solvent control). Then, cells were harvested for western blotting assay or used for morphological analysis.
3T3‐L1 pre‐adipocytes cells were differentiated into 3T3‐L1 mature adipocytes for 6 days using differentiation medium. The lipolysis of 3T3‐L1 mature adipocytes was induced by C26 tumour‐conditioned medium (1:1 dilution with fresh normal medium) and treated for 48 h in the presence of different concentrations of CS, DCS, DCSD, or 0.1% DMSO (solvent control). Then, cells were harvested for western blotting or used for morphological analysis or analysis of triglyceride (TG) and glycerol content.
MTT assay
C2C12 cells (3 × 103/mL) or 3T3‐L1 cells (3 × 103/mL) were seeded into 96‐well plates and, after cell differentiation, was treated with 100 μL of fresh medium containing various concentrations of CS, DCS, or DCSD for 48 h. Then, MTT was added into the medium and incubated at 37°C for 4 h. After discarding the medium, the formazan crystals were dissolved with 150 μL DMSO very well, and then, the absorbance at 570 nm was measured with a microplate reader.
Cancer cachexia model in vivo
Two experiments using C26 tumour‐bearing cancer cachexia mice were conducted in the present study. In the first experiment, male BALB/c mice with the same initial body weight were randomly divided into three groups (eight mice per group): healthy group without tumour (healthy group), C26 tumour‐bearing mice group (C26 model group), and C26 tumour‐bearing mice treated with 10 mg/kg CS group (C26 + CS group). In the second experiment, male BALB/c mice with the same initial body weight were randomly divided into three groups (eight mice per group): healthy group without tumour (healthy group), C26 tumour‐bearing mice group (C26 model group), and C26 tumour‐bearing mice treated with 20 mg/kg DCSD group (C26 + DCSD group). Cancer cachexia in mice was induced as reported in our previous paper. 18 Briefly, on Day 0, mice were implanted subcutaneously in the right flank with 100 μL (1.0 × 106) C26 tumour cells. Starting from the next day, C26 model group mice received daily intraperitoneal injections of sterile saline; CS‐treated mice received daily intraperitoneal injections of CS (10 mg/kg), while DCSD‐treated mice received daily intraperitoneal injections of DCSD (20 mg/kg). Body weight, body temperature, and food intake were measured daily from C26 inoculation to the end of the experiment. On Day 6 or Day 4, tumours were first detected. The shortest diameter (x) and longest diameter (y) of each tumour were recorded using callipers. The tumour volume was calculated following the formula: V = x*x*y*0.5. When the mice lost more than 10% of their body weight or when their tumour volumes reached 2000 mm3, the experiment ended. All animals were euthanized by CO2 inhalation, gastrocnemius (GA) muscles and epididymal adipose tissue (eWAT) were rapidly dissected, weighed, and frozen in liquid nitrogen, then stored at −80°C until ready for further analyses, or fixed in 4% paraformaldehyde overnight and embedded in paraffin.
Haematoxylin–eosin staining
Cells were washed three times with phosphate‐buffered saline (PBS), fixed in 4% paraformaldehyde for 1 h, and then washed three times with cold PBS. Cells were stained with haematoxylin and eosin (H&E) by standard procedures and washed three times with water. Staining was visualized by bright‐field microscopy. GA samples and eWAT samples embedded in paraffin were cut into 10 μm sections and then stained with H&E by standard procedures. Staining was visualized by bright‐field microscopy.
Assessment of haematoxylin–eosin staining
Myotube diameter of H&E stained C2C12 myotubes was quantified on images using Image J software as described in our previous report. 17 Briefly, 10 fields were randomly selected to determine the average diameters of at least 100 myotubules for each condition. The average diameter per myotube was calculated as the mean of three measurements taken along the long axis of the myotube. Results of three independent experiments were used for statistical analysis. Cross‐sectional area of myofibres and adipocytes were quantified on H&E stained tissue sections by using Image J software. Briefly, 10 images were randomly taken for each individual muscle and adipose tissue sample, and the cross‐sectional area of all myofibres and adipocytes on the image were measured. Results of the mice in the treatment groups were used for statistical analysis.
Oil Red O staining
Cells were washed three times with PBS, fixed in 4% formalin for 30 min, and then washed three times with cold PBS. Cells were stained in the Oil Red O (Sigma–Aldrich, St. Louis, MO, United States) working solution (3:2, 0.5% Oil Red O dye in isopropanol: water) for 30 min at room temperature (25°C) and washed three times with water. Staining was visualized by bright‐field microscopy.
Western blotting
C2C12 myotubes and 3T3‐L1 mature adipocytes were homogenized in RIPA buffer plus a phosphatase protease inhibitor. GA and eWAT tissues were homogenized in RIPA buffer with a tissue lyser (QIAGEN). The lysates were centrifuged at 13 000 rpm for 30 min at 4°C. The supernatant was quantified for protein concentration using the BCA Protein Assay Kit (Beyotime, Shanghai, China). Equal amounts of protein samples were separated by 10% SDS‐PAGE gel electrophoresis and transferred to a polyvinylidene fluoride membrane. The polyvinylidene fluoride membranes were blocked in 5% non‐fat milk in PBS containing 0.1% Tween 20 (TPBS) for 1 h at room temperature and then incubated with primary antibodies diluted in 5% BSA‐TPBS at 4°C overnight. The primary antibodies used were as follows: mouse anti‐MHC monoclonal antibody (1:1000, DSHB, Iowa City, IA, United States), rabbit anti‐MyoD polyclonal antibody (1:1000, Santa Cruz Biotechnology, Dallas, TX, United States), rabbit anti‐AKT monoclonal antibody (1:1000, Cell Signaling Technology), rabbit anti‐p‐AKT monoclonal antibody (1:1000, Cell Signaling Technology), rabbit anti‐NF‐κB p65 monoclonal antibody (1:1000, Cell Signaling Technology), rabbit anti‐p‐NF‐κB p65 monoclonal antibody (1:1000, Cell Signaling Technology), rabbit anti‐MuRF‐1 monoclonal antibody (1:1000, Abcam), rabbit anti‐Atrogin‐1 monoclonal antibody (1:1000, Abcam), rabbit anti‐AMPKα monoclonal antibody (1:1000, Cell Signaling Technology), rabbit anti‐p‐AMPKα monoclonal antibody (1:1000, Cell Signaling Technology), rabbit anti‐HSL polyclonal antibody (1:1000, Cell Signaling Technology), rabbit anti‐p‐HSL polyclonal antibody (1:1000, Cell Signaling Technology), rabbit anti‐UCP1 monoclonal antibody (1:1000, Cell Signaling Technology), Actin‐HRP (1: 5000, Santa Cruz Biotechnology), and GAPDH‐HRP (1: 5000, Santa Cruz Biotechnology). HRP‐conjugated goat anti‐mouse secondary antibody (1: 5000, Multi Sciences, Hangzhou, China) and goat anti‐rabbit secondary antibody (1: 5000, Multi Sciences) were incubated with membranes for 1 h in 5% non‐fat milk in TPBS. ECL Chemiluminescent Kit (Thermo Fisher, Waltham, MA, United States) was used to visualize the antibody–antigen interaction, and chemical luminescence of membranes was detected by an Amersham Imager 600 (GE).
Measurement of glycerol and triglyceride
Triglycerides in lysate of 3T3‐L1 mature adipocytes or in serum of mice were determined using a Triglyceride Quantification Kit (Applygen, Beijing, China) following the manufacturer instructions. For lipolysis experiments, glycerol accumulation in the media from 3T3‐L1 mature adipocytes and serum of mice was measured using a Lipolysis Assay Kit (Applygen, Beijing, P.R. China) following the manufacturer instructions.
Measurement of inflammatory cytokines in serum
The inflammatory cytokines of TNF‐α and IL‐6 in the mouse serum were determined by using enzyme‐linked immunosorbent assay kits (mice TNF‐α kit and mice IL‐6 kit; MultiSciences Biotech, P.R. China).
Statistical analysis
Data are expressed as mean ± SEM. Two‐tailed Student's t‐test was used for comparisons between two groups. One‐way analysis of variance test was performed to compare multiple groups followed by Bonferroni's post hoc test. A P value of 0.05 or lower was considered significant in all experiments. All analyses were performed using GraphPad Prism 8.0. P values of less than 0.05 were considered to be statistically significant and were presented as *P < 0.05, **P < 0.01, and ***P < 0.001 or #P < 0.05, ##P < 0.01, and ###P < 0.001.
Results
Activity screening and structural modification of natural compounds
In this study, we examined the effects of Rosemary extract at concentrations of 25, 50, and 100 μM on C2C12 myotube atrophy induced by C26 medium. As shown in Figure 1A, Rosemary extract could relieve the C2C12 myotube atrophy induced by C26 medium in a concentration‐dependent manner. Previous studies have shown that the three main monomer components in Rosemary extract are rosmarinic acid, carnosic acid, and CS. 9 The effects of the three natural compounds on C2C12 myotube atrophy induced by C26 medium were then compared. As shown in Figure 1B, only CS could alleviate the myotube atrophy in a concentration‐dependent way. Thus, CS was selected for further study. The chemical structure of CS was shown in Figure 1C. To confirm the effects and mechanisms of CS, structural modification of CS was conducted to obtain DCS (Figure 1C) and DCSD (Figure 1C).
Figure 1.
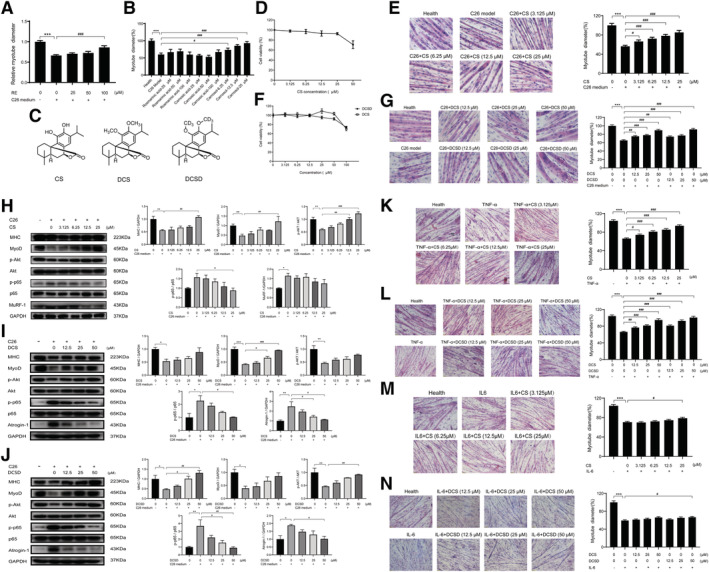
Effects of Rosemary extract, CS, DCS, and DCSD on C2C12 myotube atrophy in cancer cachexia model in vitro. C2C12 myoblasts were differentiated into myotubes for 5 days using differentiation medium. The C2C12 myotube atrophy was induced by C26‐tumour medium (1:1 dilution with fresh normal medium) and treating at the same time with different concentrations of the Rosemary extract (RE) or the compounds for 48 h, or induced by C26‐tumour medium (1:1 dilution with fresh normal medium), TNF‐α (100 ng/mL) or IL‐6 (100 ng/mL) and treated at the same time with different concentrations of CS, DCS, or DCSD respectively for 48 h. (A) Effects of RE on the cytomorphology of C2C12 myotube atrophy induced by C26‐tumour medium. (B) Effects of three representative natural compounds of RE on the cytomorphology of C2C12 myotube atrophy induced by C26‐tumour medium. (C) Structural formula of CS, DCS, and DCSD. (D) Effects of CS on the survival rate of C2C12 myotubes. (E) Effects of CS on the cytomorphology of C2C12 myotube atrophy induced by C26‐tumour medium. Scale bar, 20 μm. (F) Effects of DCS and DCSD on the survival rate of C2C12 myotubes. (G) Effects of DCS and DCSD on the cytomorphology of C2C12 myotube atrophy induced by C26‐tumour medium. Scale bar, 20 μm. (H, I, and J) Representative western blotting and quantification of MHC, MyoD, phosphorylated AKT, total AKT, phosphorylated p65, total p65, MURF‐1, or Atrogin‐1 in C2C12 myotube atrophy induced by C26‐tumour medium with or without treatment of CS, DCS, or DCSD. (K) Effects of CS on the cytomorphology of C2C12 myotube atrophy induced by TNF‐α. Scale bar, 50 μm. (L) Effects of DCS and DCSD on the cytomorphology of C2C12 myotube atrophy induced by TNF‐α. Scale bar, 50 μm. (M) Effects of CS on the cytomorphology of C2C12 myotube atrophy induced by IL‐6. Scale bar, 50 μm. (N) Effects of DCS and DCSD on the cytomorphology of C2C12 myotube atrophy induced by IL‐6. Scale bar, 50 μm. Data presented are the mean ± SEM of three independent experiments. *vs. control group; #vs. C26‐tumour medium, TNF‐α single treatment group, or IL‐6 single treatment group. ***P < 0.001, #P < 0.05, ##P < 0.01, ###P < 0.001.
CS, DCS, and DCSD alleviated myotube atrophy in cancer cachexia model in vitro and affected NF‐κB and Akt signalling pathway in C2C12 myotubes
The cytotoxicity of CS, DCS, and DCSD on C2C12 myotubes was checked. As shown in Figure 1D and 1F, CS at doses lower than 25 μM and DCS or DCSD at doses lower than 50 μM did not exhibit cytotoxicity on C2C12 myotubes. The dose‐dependent protective effects of CS, DCS, or DCSD on muscle atrophy of C2C12 myotubes induced by C26 medium were shown in Figure 1E and 1G, respectively. The three compounds all could dose‐dependently ameliorate the atrophy of C2C12 myotubes induced by C26 medium. Furthermore, to study the roles of inflammatory cytokine‐activated pathways in the effects of these compounds, the effects of the three compounds on muscle atrophy of C2C12 myotubes induced by TNF‐α or IL‐6 were also checked. As shown in Figure 1K and 1L, CS (at 3.125 to 25 μM) and DCS or DCSD (at 3.125 to 25 μM) dose‐dependently ameliorated the atrophy of C2C12 myotubes induced by TNF‐α. While, effective protection on the atrophy of C2C12 myotubes induced by IL‐6 was only observed with 25 μM CS or 50 μM DCS or DCSD (Figure 1M and 1N). These results suggested that the protective effects of these compounds on C2C12 myotubes might be mainly based on the inhibition on TNF‐α‐activated pathways. Results of western blotting assay checking the signal proteins related to muscle atrophy in C2C12 myotubes treated with C26 medium were shown in Figure 1H, 1I, and 1J. Interestingly, as shown in Figure 1H, 1I, and 1J, C26 medium induced activation of NF‐κB pathway (increase of phosphorylated‐p65), inhibition of AKT pathway (decrease in phosphorylated AKT), increase in protein degradation (increase of MuRF‐1 and Atrogin‐1), and decrease in the expression of MyoD, a myogenic transcriptional factor, and myosin heavy chain (MHC). Treatments of CS, DCS, or DCSD could all significantly inhibit the activation of NF‐κB pathway and the decrease in the expression of MyoD and MHC, and partly ameliorate inhibition of AKT pathway as well as increase in protein degradation induced by C26 medium (Figure 1H, 1I, and 1J). These results suggested that these compounds might ameliorate muscle atrophy by inhibiting the activation of NF‐κB pathway in C2C12 myotubes induced by inflammatory cytokines, keeping the balance between protein synthesis and degradation and thus protecting the myotubes from atrophy.
CS, DCS, and DCSD alleviated lipolysis in cancer cachexia model in vitro and affected NF‐κB and AMPK signalling pathway in 3T3‐L1 adipocytes
The cytotoxicity of CS, DCS, and DCSD on 3T3‐L1 adipocytes was checked. As shown in Figure 2A and 2F, CS at doses lower than 50 μM and DCS or DCSD at doses lower than 100 μM did not exhibit cytotoxicity on 3T3‐L1 adipocytes. The dose‐dependent protective effects of CS, DCS, or DCSD on lipolysis of 3T3‐L1 adipocytes induced by C26 medium were shown in Figure 2B and 2G, respectively. Results of Oil Red O staining for lipid droplets in mature 3T3‐L1 adipocytes indicated that adipocytes treated with C26 medium have fewer lipid droplets than control cells, while CS, DCS, and DCSD increased the amount of lipid droplets in a dose‐dependent manner (Figure 2B and 2G). Consistent with this finding, adipocytes treated with C26 medium released higher levels of glycerol than control cells, while CS, DCS, and DCSD significantly inhibited the release of glycerol from adipocytes (Figure 2C and 2H). Furthermore, CS, DCS, and DCSD also ameliorate the decrease of TG content in 3T3‐L1 adipocytes induced by C26 medium (Figure 2D and 2I). Interestingly, western blotting results showed that CS, DCS, and DCSD significantly inhibited the activation of NF‐κB pathway (increased phosphorylation of p65) in adipocytes induced by C26 medium (Figure 2E, 2J, and 2K). Hormone‐sensitive lipase (HSL) is a major TG lipase expressed predominantly in adipose tissue and play essential roles in energy homeostasis. The phosphorylation of HSL was remarkably up‐regulated in mature 3T3‐L1 adipocytes under C26 medium treatment, while this up‐regulation was effectively suppressed by CS, DCS, and DCSD (Figure 2E, 2J, and 2K). In order to discover the underlying mechanism, we evaluated whether AMPK signalling, one of the upstream pathways regulating HSL phosphorylation and energy balance, took part in this lipolysis. The phosphorylation of AMPKα could be significantly inhibited by CS, DCS, and DCSD treatment (Figure 2E, 2J, and 2K). Furthermore, CS, DCS, and DCSD also significantly decreased the expression of UCP1 (Figure 2E, 2J, and 2K), which suggested that the ineffective thermogenesis of adipose tissue might be blocked. Overall, these results demonstrated that CS, DCS, and DCSD were able to inhibit lipolysis by suppressing NF‐κB and AMPK signalling and keys actors in the lipolytic and lipid utilization pathways.
Figure 2.
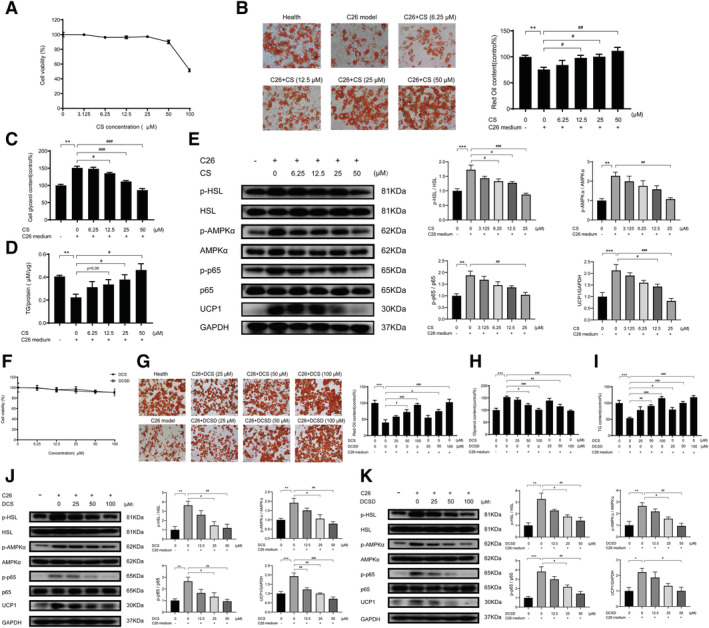
Effects of CS, DCS, and DCSD on lipolysis of 3T3‐L1 adipocyte in vitro. 3T3‐L1 pre‐adipocytes cells were differentiated into 3T3‐L1 mature adipocyte for 6 days using differentiation medium. The lipolysis of 3T3‐L1 mature adipocyte was induced by C26‐tumour medium (1:1 dilution with fresh normal medium) and treated at the same time with different concentrations of CS, DCS, or DCSD, respectively for 48 h. (A) Effects of CS on the survival rate of 3T3‐L1 mature adipocytes. (B) Oil Red O stained lipid droplets to detect the effect of CS on the lipolysis of 3T3‐L1 mature adipocyte induced by C26‐tumour medium. Scale bar, 50 μm. (C) Levels of free glycerol released in the 3T3‐L1 mature adipocyte induced by C26‐tumour medium with or without treatment of CS. (D) Concentrations of TG in the 3T3‐L1 mature adipocyte induced by C26‐tumour medium with or without treatment of CS. (E) Representative western blotting and quantification of phosphorylated HSL, total HSL, phosphorylated AMPK, total AMPK, phosphorylated p38 MAPK, total p38 MAPK, and UCP1 in the lipolysis of 3T3‐L1 mature adipocyte induced by C26‐tumour medium with or without treatment of CS. (F) Effects of DCS and DCSD on the survival rate of 3T3‐L1 adipocytes. (G) Oil Red O stained lipid droplets to detect the effect of DCS or DCSD on the lipolysis of 3T3‐L1 mature adipocyte induced by C26‐tumour medium. Scale bar, 50 μm. (H) Levels of glycerol released in the 3T3‐L1 mature adipocyte induced by C26‐tumour medium with or without treatment of DCS or DCSD. (I) Concentration of TG in the 3T3‐L1 mature adipocyte induced by C26‐tumour medium with or without treatment of DCS or DCSD. (J, K) Representative western blotting and quantification of phosphorylated HSL, total HSL, phosphorylated AMPK, total AMPK, phosphorylated p38 MAPK, total p38 MAPK, and UCP1 in the lipolysis of 3T3‐L1 mature adipocyte induced by C26‐tumour medium with or without treatment of DCS or DCSD. Data presented are the mean ± SEM of three independent experiments. *vs. control group; #vs. C26‐tumour medium. ***P < 0.01, ***P < 0.001, #P < 0.05, ##P < 0.01, ###P < 0.001.
CS and DCSD attenuated cachexia symptoms of C26 tumour‐bearing mice
The effects of CS and DCSD on physiological parameters of C26 tumour‐bearing mice were systematically evaluated, and the results are shown in Figures 3 and 4, respectively. As shown in Figure 3A, the body weight of healthy control mice continued to increase in the period of experiment (17 days). On the contrary, the body weight of mice in the C26 model group decreased quickly while CS treatment could effectively ameliorate the decrease in body weight; thus, the body weight of CS‐treated mice was significantly higher by 11.09% than that of the C26 model group at the end of the experiment (Day 17). Results of tumour‐free body weight also showed similar tendency (Figure 3B). What is more, as shown in Figure 3C and 3D, CS could partly increase the food intake of mice bearing C26 tumour. The body temperature of mice in the CS group was slightly higher than that in the C26 model group, but the difference was not significant (Figure 3E). Importantly, CS did not exhibit influence on tumour growth. As shown in Figure 3F and 3G, no difference was observed between the tumour growth in C26 model group and C26 + CS group. As shown in Figure 4, DCSD exhibited results similar to that of CS in C26 tumour‐bearing mice. DCSD significantly increased body weight by 11.38% (Figure 4A), tumour‐free body weight (Figure 4B) in C26 tumour‐bearing mice, partly improved the food intake of mice (Figure 4C and 4D) while showed almost no influence on the body temperature (Figure 4E) and tumour growth of the mice (Figure 4F). Together, these results demonstrated that CS as well as DCSD could effectively attenuate C26 tumour‐induced body weight loss, and their anti‐cachexia effects were not based on inhibition on C26 tumour growth.
Figure 3.
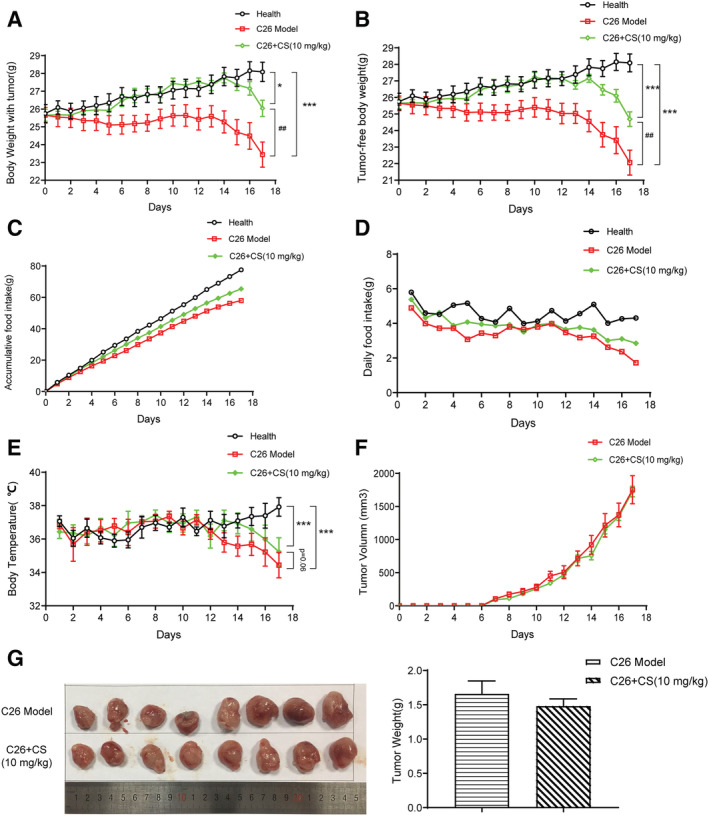
CS attenuated cachexia symptoms of C26 tumour‐bearing mice. C26 tumour‐bearing mice were administrated CS (10 mg/kg) by intraperitoneal injection every day for 17 days, and healthy BALB/c mice and C26 model group mice received equal volume solvent. Body weight, food intake, body temperature, and tumour volume were recorded every day. (A) Body weight of mice. (B) Tumour‐free body weight of mice. (C) Accumulative food intake of mice. (D) Daily food intake of mice. (E) Body temperature of mice. (F) Tumour volume of mice. (G) Photo and weight of the isolated tumour tissues. Values are expressed as mean ± SEM (n = 8). *vs. health group mice; #vs. C26 model group mice. One‐way ANOVA test was performed followed by Bonferroni's post hoc test. *P < 0.05, ***P < 0.001, ##P < 0.01.
Figure 4.
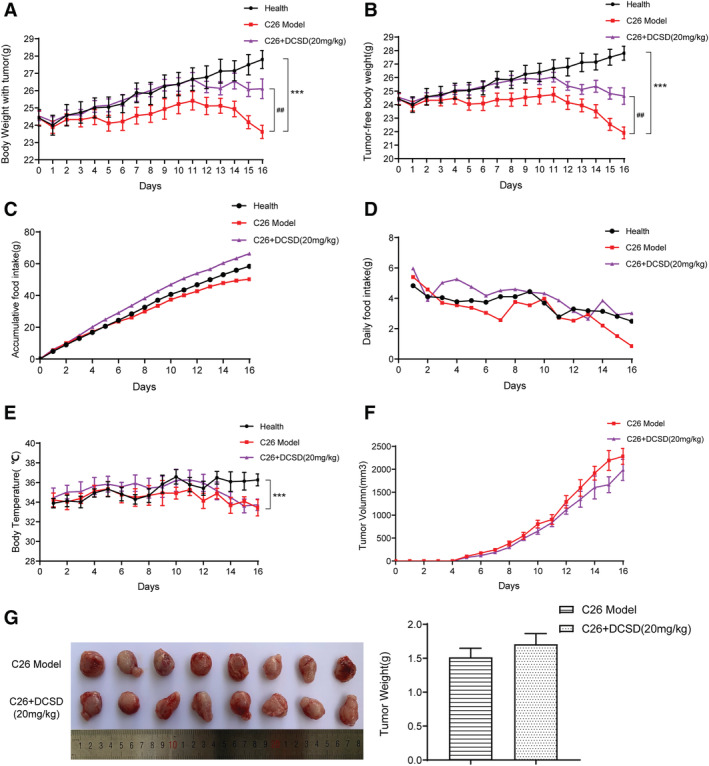
DCSD attenuated cachexia symptoms of C26 tumour‐bearing mice. C26 tumour‐bearing mice were administrated DCSD (20 mg/kg) by intraperitoneal injection every day for 16 days, and healthy BALB/c mice and C26 model group mice received equal volume solvent. Body weight, food intake, body temperature, and tumour volume were recorded every day. (A) Body weight of mice. (B) Tumour‐free body weight of mice. (C) Accumulative food intake of mice. (D) Daily food intake of mice. (E) Body temperature of mice. (F) Tumour volume of mice. (G) Photo and weight of the isolated tumour tissues. Values are expressed as mean ± SEM (n = 8). *vs. health group mice; #vs. C26 model group mice. One‐way ANOVA test was performed followed by Bonferroni's post hoc test. ***P < 0.001, ##P < 0.01.
CS and DCSD ameliorated myofibre atrophy in C26 tumour‐bearing mice
As shown in Figure 5A and 5B, C26 tumour led to a significant decrease of GA mass, which indicated muscle atrophy. CS treatment could not significantly ameliorate the decrease in the weight of GA in C26 tumour‐bearing mice (Figure 5A and 5B). Results of H&E staining of GA muscle showed that the GA myofibres cross‐sectional area remarkably decreased in C26 model mice while CS treatment could ameliorate the decrease in GA myofibres cross‐sectional area (Figure 5C and 5D). Similarly, as shown in Figure 6, DCSD treatment could partly ameliorate the decrease by an 8.29% increase in the weight of GA in C26 tumour‐bearing mice and increased GA myofibres cross‐sectional area. Overall, these data demonstrated that CS as well as DCSD could partly ameliorate muscle atrophy in C26 tumour‐bearing mice.
Figure 5.
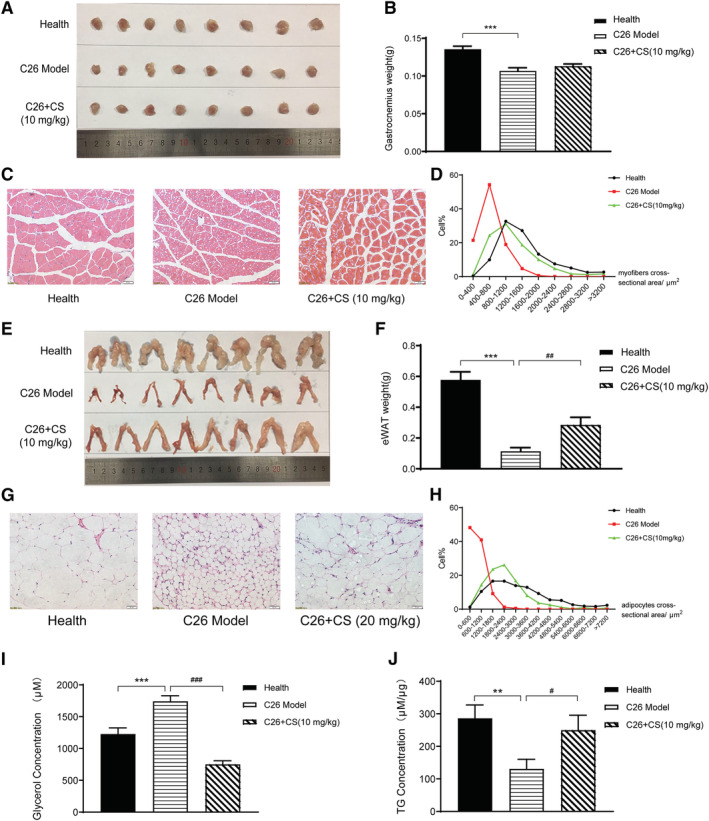
CS ameliorated muscle wasting and fat degradation in C26 tumour‐bearing mice. C26 tumour‐bearing mice were administrated CS (10 mg/kg) by intraperitoneal injection every day for 17 days, and healthy BALB/c mice and C26 model group mice received equal volume solvent. The gastrocnemius (GA) muscle tissues and eWAT were dissected, weighed, and then fixed in 4% PFA at the end of the experiment. (A, B) GA weight of each group mice with or without treatment of CS. (C, D) H&E‐stained sections of mice GA and quantify the myofibres cross‐sectional area of GA tissues with or without treatment of CS. Scale bar, 50 μm. (E, F) eWAT weight of each group mice with or without treatment of CS. (G, H) H&E‐stained sections of mice eWAT and quantify the adipocytes cross‐sectional area of eWAT with or without treatment of CS. Scale bar, 50 μm. (I, J) content of glycerol and TG in serum with or without treatment of CS. Values are expressed as mean ± SEM (n = 8). *vs. health group mice; #vs. C26 model group mice. One‐way ANOVA test was performed followed by Bonferroni's post hoc test. ***P < 0.001.
Figure 6.
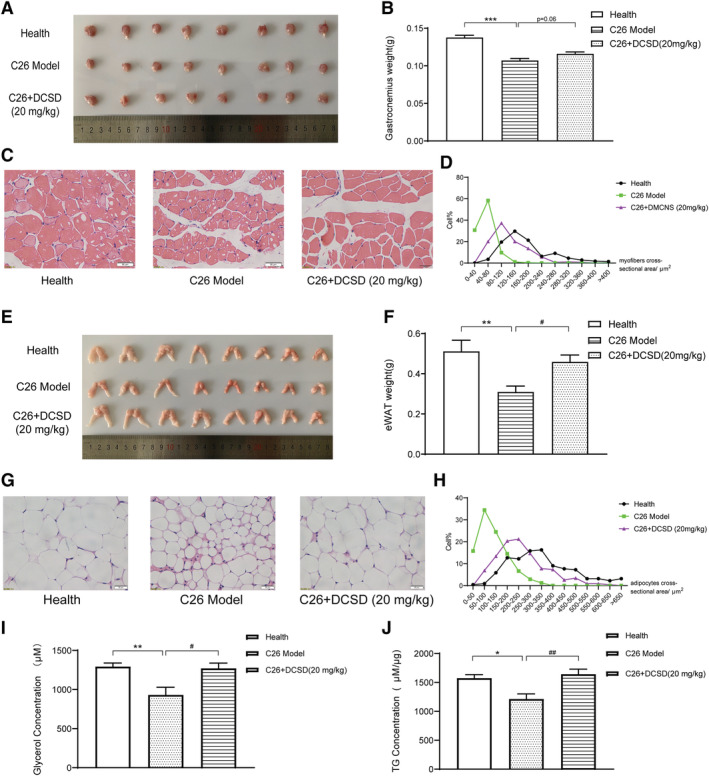
DCSD ameliorated muscle wasting and fat degradation in C26 tumour‐bearing mice. C26 tumour‐bearing mice were administrated DCSD (20 mg/kg) by intraperitoneal injection every day for 16 days, and healthy BALB/c mice and C26 model group mice received equal volume solvent. The GA muscle tissues and eWAT were dissected, weighed, and then fixed in 4% PFA at the end of the experiment. (A, B) GA weight of each group mice with or without treatment of DCSD. (C, D) H&E‐stained sections of mice GA and quantify the myofibres cross‐sectional area of GA tissues with or without treatment of DCSD. Scale bar, 50 μm. (E, F) eWAT weight of each group mice with or without treatment of DCSD. (G, H) H&E‐stained sections of mice eWAT and quantify the adipocytes cross‐sectional area of eWAT with or without treatment of DCSD. Scale bar, 50 μm. (I, J) Content of glycerol and TG in serum with or without treatment of DCSD. Values are expressed as mean ± SEM (n = 8). *vs. healthy control group mice; #vs. C26 model group mice. One‐way ANOVA test was performed followed by Bonferroni's post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, #P < 0.05, ##P < 0.01, ###P < 0.001.
CS and DCSD ameliorated fat degradation in C26 tumour‐bearing mice
As shown in Figure 5, significant decrease in eWAT weight (Figure 5E and 5F) and decrease in adipocytes cross‐sectional area of eWAT tissues (Figure 5G and 5H) were observed in C26 tumour model group. CS treatment could ameliorate the decrease in eWAT weight by a 176.6% increase and decrease in adipocytes cross‐sectional area of eWAT tissues induced by C26 tumour burden. Furthermore, the levels of glycerol and TG in mice serum were also determined. As shown in Figure 5I and 5J, CS treatment could significantly ameliorate the increase of glycerol and the decrease of TG in serum of C26 tumour‐bearing mice. These results suggested that CS treatment ameliorated fat degradation in C26 tumour‐bearing mice.
As shown in Figure 6, in the experiment checking the effects of DCSD on C26 tumour‐bearing mice, DCSD also could partly ameliorate the decrease by a 48.2% increase in eWAT weight, decrease in adipocytes cross‐sectional area of eWAT tissues, and significantly ameliorate the decrease in TG content in mice serum. While, interestingly, the serum glycerol level of the C26 model group was lower than that of the healthy control mice group in the experiment. The serum glycerol level could be affected by both the fat degradation, which would cause increase in glycerol level, and the metabolism of glycerol to produce energy, which could cause decrease in glycerol. Therefore, it is possible that the decrease in serum glycerol level might be resulted from enhancement of glycerol metabolism in C26 tumour‐bearing mice in this in vivo experiment. Importantly, DCSD treatment also could ameliorate the decrease in serum glycerol; thus, the level of serum glycerol in the C26 + DCSD group was similar to that of the healthy control group. These results suggested that DCSD might ameliorate abnormal fat metabolism in C26 tumour‐bearing mice.
CS and DCSD affected NF‐κB and related pathways in GA and eWAT tissues
Results of western blotting assay checking levels of signal proteins in GA muscle tissues were shown in Figure 7. As shown in Figure 7A and 7B, the level of MHC, a major indicator of skeletal muscle fibre type and functional property, and the level of MyoD, a myogenic factor regulating myoblast differentiation, were decreased in GA muscle tissues of C26 model mice. CS and DCSD could significantly ameliorate the decrease in MHC and MyoD thus ameliorate muscle atrophy in GA muscle tissues. Importantly, the activation of NF‐κB pathway (increased phosphorylation of p65) in GA muscle tissues of C26 model mice was partly inhibited by CS and DCSD. Furthermore, DCSD treatment could significantly ameliorated the inhibition of AKT pathway, which controls protein synthesis in GA muscle tissues of C26 model mice, and CS also partly ameliorate the inhibition of AKT pathway. MuRF1 and Atrogin‐1 are muscle‐specific E3 ubiquitin ligases participated in protein degradation. In GA muscle tissues of C26 model mice, MuRF1 was not significantly increased, whereas Atrogin‐1 was up‐regulated. DCSD treatments partly ameliorated the increased of Atrogin‐1 in cancer cachexia mice. Taken together, these results suggest that CS and DCSD treatment might influence myoblast protein synthesis and protein degradation in muscle tissues of cancer cachexia mice via regulating NF‐κB and Akt signalling pathway.
Figure 7.
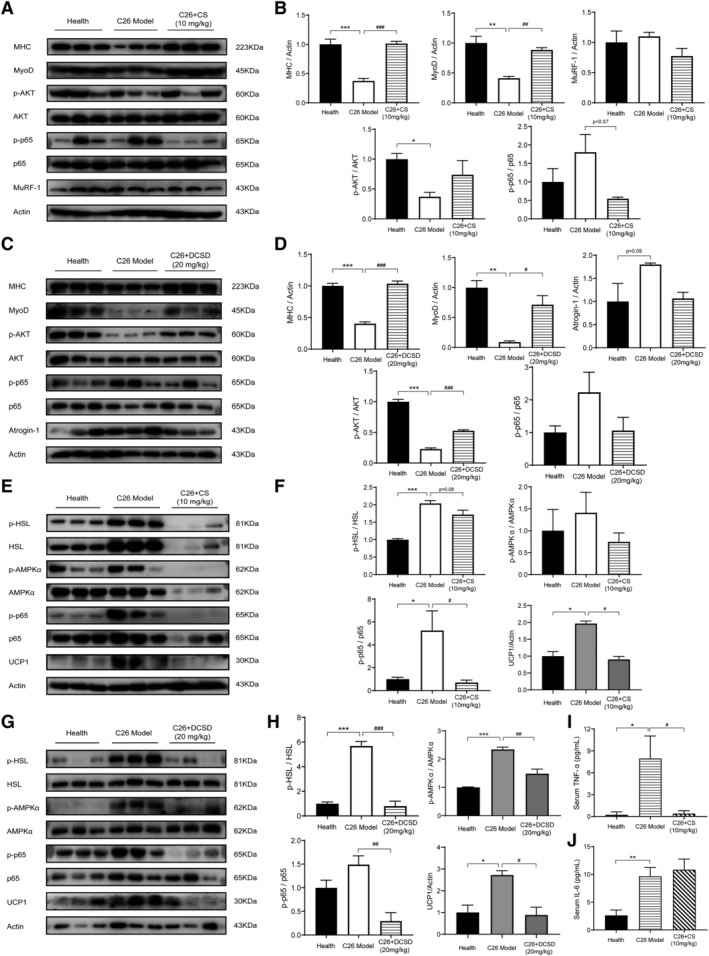
CS and DCSD affected NF‐κB and related pathways in the GA and eWAT and serum levels of inflammatory cytokines. GA and eWAT were freshly isolated and homogenized by a tissue lyser in RIPA buffer at the end of the animal experiment. The protein expression levels in GA and eWAT were measured with western blotting analysis. (A) Representative Western blotting of MHC, MyoD, MuRF‐1, phosphorylated AKT, total AKT, phosphorylated p65, and total p65 in the GA tissues with or without treatment of CS. (B) The quantification of panel (A). (C) Representative western blotting of MHC, MyoD, MuRF‐1, phosphorylated AKT, total AKT, phosphorylated p65, and total p65 in the GA tissues with or without treatment of DCSD. (D) The quantification of panel (C). (E) Representative western blotting of phosphorylated HSL, total HSL, phosphorylated AMPKa, total AMPKa, phosphorylated p65, total p65, and UCP1 in the eWAT tissue of mice with or without treatment of CS. (F) The quantification of panel (E). (G) Representative western blotting of phosphorylated HSL, total HSL, phosphorylated AMPKa, total AMPKa, phosphorylated p65, total p65, and UCP1 in the eWAT tissue of mice with or without treatment of DCSD. (H) The quantification of panel (G). (I, J) Serum level of TNF‐α and IL‐6 with or without treatment of CS. Values are expressed as mean ± SEM (n = 3). *vs. health group mice; #vs. C26 tumour‐bearing group mice. One‐way ANOVA test was performed followed by Bonferroni's post hoc test. *P < 0.05, **P < 0.01, ***P < 0.001, #P < 0.05, ##P < 0.01, ###P < 0.001.
Results of western blotting assay checking levels of signal proteins in eWAT tissues were shown in Figure 7 including Figure 7E and 7G and Figure 7F and 7H. Notably, in C26 model mice, phosphorylation of p65 was elevated in eWAT, which suggested the activation of NF‐κB pathway in fat tissues of C26 tumour‐bearing mice, while CS and DCSD could both significantly inhibit the activation of NF‐κB pathway. In eWAT fat tissues of C26 model mice, increased phosphorylation of AMPK was also observed while DCSD significantly restrained the phosphorylation of AMPK in C26 tumour‐bearing mice and CS could partly decrease the phosphorylation of AMPK. Furthermore, elevated phosphorylation of HSL and increase in the level of UCP1, a key component of the browning response, were observed in eWAT tissues of C26 model mice. CS and DCSD treatment could both significantly inhibit the elevated phosphorylation of HSL as well as the increase in UCP1 in C26 tumour‐bearing mice. These results suggested that CS and DCSD treatment might ameliorate eWAT loss in cancer cachexia through the modulation of NF‐κB and AMPK pathways.
Cumulated studies have shown that serum cytokines such as TNF‐α and IL‐6 are elevated in cachexia mice and elevated serum cytokines convey biological effects on tissue cells via the NF‐κB pathway and STAT3 pathway, respectively. 19 , 20 As shown in Figure 7I and 7J, significant elevation of IL‐6 and TNF‐α level was observed in serum of C26 tumour‐bearing mice. Notably, CS treatment could significantly decrease the TNF‐α serum level but not the IL‐6 level. The results suggested that inhibition of TNF‐α/NF‐κB signalling pathway might play a critical role in the effects of CS in ameliorating cancer cachexia.
Discussion
A total of about 800 300 patients in the EU (15.8 subjects per 10 000 people of the total EU population) and about 527 100 patients in the United States (16.5 per 10 000 people of the total US population) are recently estimated to suffer from cancer cachexia. 21 In the present study, CS, a kind of natural polyphenolic antioxidant, and its analogues exhibited considerable ameliorating effects on cancer cachexia both in vitro and in vivo. In in vitro experiments, CS and its analogues could relieve the atrophy of C2C12 myotubes induced by C26 medium or TNF‐α in a dose‐dependent manner, and its mechanism is mainly to inhibit protein degradation and promote protein synthesis. CS and its analogues also ameliorated the lipid degradation of 3T3‐L1 adipocytes induced by C26 medium in a dose‐dependent manner. Specifically, the accumulation of TG increased, and the decomposed free glycerol decreased in CS‐treated adipocytes. In in vivo experiments, CS and its analogue DCSD could attenuate cancer cachexia symptoms in C26 tumour‐bearing mice. CS and DCSD significantly prevented the loss of body weight without influencing the tumour growth and significantly alleviated the wasting of skeletal muscle and adipose tissue of the tumour‐bearing mice. These results suggested that CS and its analogues might be promising candidates for developing into new agents for therapy of cancer cachexia.
It is well established that cancer cachexia is an inflammation‐related disease and pro‐inflammatory cytokines such as TNF‐α and activation of signal pathways such as NF‐κB pathway is of great importance. 20 , 22 In cancer patients with cachexia, higher serum levels of pro‐inflammatory cytokines and increased NF‐κB activation in both muscle tissues and adipose tissues have been observed. 23 , 24 It is a vicious cycle that NF‐κB can modulate immunity and inflammation partly by inducing production of pro‐inflammatory cytokines while these cytokines can reversely activate NF‐κB, which could exacerbate cachexia symptoms. 25 Reduced protein synthesis and abnormal activation of ubiquitin‐proteasome system have all been shown to mediate muscle wasting in cancer cachexia. 26 TNF‐κB is required to up‐regulate the expressions of MuRF1 thus inhibition of NF‐κB pathway is adequate to decrease tumour‐induced muscle atrophy, in some degree, by hampering the up‐regulation of MuRF1. 25 In the present study, increase in phosphorylation of p65, that is, activation of NF‐κB pathway, was observed in both in vitro and in vivo muscle wasting models induced by C26. Notably, CS and its analogues could inhibit the activation of NF‐κB pathway and ameliorate the increase in the level of MURF1 in C2C12 myoblasts as well as muscle tissues of C26 tumour‐bearing mice. At the same time, CS and its analogues also could ameliorate the inhibition of AKT pathway (decrease in the level of phosphorylated AKT) in C2C12 myoblasts as well as muscle tissues of C26 tumour‐bearing mice. AKT controls protein synthesis and cell differentiation by regulating the mTOR signal pathway, while, in muscle atrophy, the phosphorylation of AKT was inhibited thus resulting in down‐regulation of MyoD and MHC. 27 Moreover, activation of NF‐κB also could regulate MyoD mRNA levels through the PAX pathway, inhibiting skeletal muscle differentiation and injury repair. 28 And there is crosstalk between AKT/mTOR signalling and NF‐κB in hypoxic injury of muscle cells. 29 Notably, STAT3 pathway is also a key regulator of AKT signalling and expression of MuRF‐1; thus, IL‐6 could induce cancer cachexia phenotype by STAT3 activation in vitro and in vivo. 2 , 30 In the present study, we compared the effects of CS and its analogues on muscle atrophy of C2C12 cells induced by TNF‐α or IL‐6. Interestingly, the results suggested that CS and its analogues exhibited much stronger ameliorating effects on muscle atrophy induced by TNF‐α than that by IL‐6. These results suggested that CS and its analogues might prevent muscle atrophy mainly by inhibiting NF‐κB pathway and activating AKT pathway.
Recent studies revealed that adipose tissue browning and its associated increases in energy expenditure precedes skeletal muscle atrophy and is important for the development and progression of cachexia. 31 In the present study, activation of NF‐κB pathway and increase in phosphorylation of AMPK‐α and HSL were observed in 3T3‐L1 adipocytes treated with C26 medium. HSL, the rate‐limiting enzyme in regulating adipocyte lipolysis, is a substrate for AMPK‐α, thus activation of AMPK‐α could increase phosphorylation of HSL to mobilize lipids and induce lipolysis. 32 What is more, the activation of AMPK signalling was also reported to increase the expression level of UCP1 leading to increased lipid mobilization and energy expenditure in cachectic mice. 33 Our results also showed high UCP1 level induced by C26 medium in 3T3‐L1 cells. Notably, CS and its analogues could inhibit the activation of AMPK pathway and NF‐κB signalling pathway and thus prevent the increase in the levels of phosphorylated HSL and UCP1. In addition, adipose tissue of cancer cachexia patients has been highlighted as a potential source of pro‐inflammatory cytokine production during cancer cachexia progression. 34 The inhibition of NF‐κB pathway in adipocytes by CS and its analogues might be beneficial to control the inflammation in cancer cachexia.
In our in vivo experiment using C26 tumour‐bearing mice, increase in the levels of TNF‐α and IL‐6 in mouse serum was observed. TNF‐α is a direct inducer of NF‐κB 19 while activated NF‐κB signalling pathway could induce proteasome degradation in muscle tissues 35 and promote HSL phosphorylation in adipose tissues. 36 IL‐6 could induce cancer cachexia phenotype by STAT3 activation in vitro and in vivo. While, notably, CS significantly ameliorate the increase of TNF‐α in serum of C26 tumour‐bearing mice but exhibit not significant influence on the serum level of IL‐6. The results suggested that CS might mainly affect the TNF‐α/NF‐κB signalling pathway but not the IL‐6/STAT3 signalling pathway. These in vivo study results were consistent with the in vitro study results which showed that CS could significantly and dose‐dependently ameliorate muscle atrophy of C2C12 myoblasts induced by TNF‐α but not IL‐6. The inhibitive effects of CS on NF‐κB signalling pathway in other kinds of cells have been reported before. Previous study indicated that CS inhibited TNFα‐induced IKKβ phosphorylation in endothelial cells and inhibited NF‐κB nuclear translocation and activation. 37 The inhibition on NF‐κB signalling pathway was involved in the effects of CS in regulating expression of cell adhesion molecules and chemokines. 38 CS could inhibit NF‐κB activation in keratinocytes, 15 macrophages and chondrocytes, 39 and Th17 cells. 11 In accordance with previous reports, results of the present study showed the inhibitive effects of CS on NF‐κB signalling pathway in C2C12 myoblasts, 3T3‐L1 adipocytes, as well as muscle and fat tissues. The results also suggested the important roles of NF‐κB inhibition in the anticachexia activities of CS.
Notably, CS is not only a natural NF‐κB inhibitor and anti‐inflammatory agent. Firstly, it is a potent antioxidant. The antioxidant activity of CS might be the basis of its inhibition on NF‐κB signalling pathway. 37 The antioxidant activity of CS also supports its potency as a cytoprotective agent to protect cells against various injuries. 10 Secondly, CS also could exhibit antitumour activities. 14 CS has been demonstrated to inhibit cell proliferation and survival, reduce migration and invasion, and significantly enhance apoptosis of cancer cells. Although CS, at the dose used in the present study, did not exhibit inhibiting effects on tumour growth, CS at higher dose might be able to inhibit tumour growth and thus further contribute to its ameliorating effects on cancer cachexia. Interestingly, the various activities of CS, such as antioxidant effects, anti‐inflammatory effects, and anticancer effects, are all beneficial to the treatment of cancer cachexia. The possible mechanisms of CS, as well as its analogues, in ameliorating cancer cachexia were illustrated in Figure 8.
Figure 8.
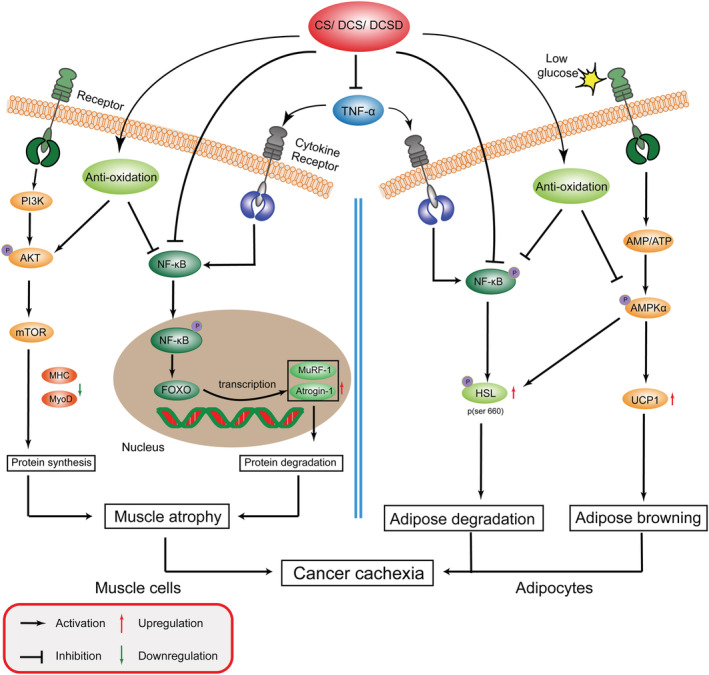
Carnosol and its analogues attenuate cancer cachexia muscle atrophy and fat loss. CS, DCS, and DCSD inhibit the TNF‐α production and inhibit the activation of NF‐κB signalling in both muscle cells and adipocytes. In muscle cells, CS and its analogues inhibit ubiquitin‐proteasome system (UPS) mediated by MURF‐1, and Atrogin‐1; activate the AKT signalling; promote the expression of MHC and MyoD; and thus ameliorate muscle atrophy. In adipocytes, CS and its analogues inhibit NF‐κB signalling mediated inflammatory response and AMPK phosphorylation activation, decrease HSL phosphorylation and expression of UCP1, and thus ameliorate fat loss.
In summary, results of the present study provided evidence for the first time that extract of Rosemary was sufficient to attenuate cancer cachexia and CS was the active component for the anticachexia activities. Synthesized CS analogues including DCS and DCSD also exhibit similar or even better effects in alleviating cancer cachexia. DCS and DCSD showed less cytotoxicity on C2C12 myoblasts and 3T3‐L1 adipocytes. These results suggested that CS might be a promising leading compound for developing new agents for the treatment of cancer cachexia. National patents of China have been applied by our lab for the use of CS (202010089693.1), DCS (202010088733.0), and DCSD (202010088747.2) in the treatment of cancer cachexia. We are still trying to conduct further structural modification of CS in order to obtain new agents with better therapeutic effects. The development and utilization of the series of compounds of carnosol family might have important reference value.
Funding
This work was supported by National Natural Science Foundation of China (81872496 and 81873056), and the Science and Technology Commission of Shanghai Municipality (20S11902200 and 16DZ2280100).
Conflicts of interest
The authors declare no conflict of interests.
Acknowledgements
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle. 40
Lu S., Li Y., Shen Q., Zhang W., Gu X., Ma M., Li Y., Zhang L., Liu X., and Zhang X. (2021) Carnosol and its analogues attenuate muscle atrophy and fat lipolysis induced by cancer cachexia, Journal of Cachexia, Sarcopenia and Muscle, 12, 779–795, 10.1002/jcsm.12710
Shanshan Lu and Yiwei Li contributed equally to this work and are co‐first authors.
Contributor Information
Xuan Liu, Email: xuanliu@shutcm.edu.cn.
Xiongwen Zhang, Email: xwzhang@sat.ecnu.edu.cn.
References
- 1. Scherbakov N, Doehner W. Cachexia as a common characteristic in multiple chronic disease. J Cachexia Sarcopenia Muscle 2018;9:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sun L, Quan XQ, Yu S. An epidemiological survey of cachexia in advanced cancer patients and analysis on its diagnostic and treatment status. Nutr Cancer 2015;67:1056–1062. [DOI] [PubMed] [Google Scholar]
- 3. Prado CM, Purcell SA, Laviano A. Nutrition interventions to treat low muscle mass in cancer. J Cachexia Sarcopenia Muscle 2020;11:366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prado BL, Qian Y. Anti‐cytokines in the treatment of cancer cachexia. Ann Palliat Med 2019;8:67–79. [DOI] [PubMed] [Google Scholar]
- 5. Aversa Z, Costelli P, Muscaritoli M. Cancer‐induced muscle wasting: latest findings in prevention and treatment. Ther Adv Med Oncol 2017;9:369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen X, Wu Y, Yang T, Wei M, Wang Y, Deng X, et al. Salidroside alleviates cachexia symptoms in mouse models of cancer cachexia via activating mTOR signalling. J Cachexia Sarcopenia Muscle 2016;7:225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alavi MS, Fanoudi S, Ghasemzadeh Rahbardar M, Mehri S, Hosseinzadeh H. An updated review of protective effects of rosemary and its active constituents against natural and chemical toxicities. Phytother Res 2021;35:1313–1328. [DOI] [PubMed] [Google Scholar]
- 8. de Macedo LM, Santos EMD, Militao L, Tundisi LL, Ataide JA, Souto EB, et al. Rosemary (Rosmarinus officinalis L., syn Salvia rosmarinus Spenn.) and its topical applications: a review. Plants (Basel) 2020;9:651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hassani FV, Shirani K, Hosseinzadeh H. Rosemary (Rosmarinus officinalis) as a potential therapeutic plant in metabolic syndrome: a review. Naunyn Schmiedebergs Arch Pharmacol 2016;389:931–949. [DOI] [PubMed] [Google Scholar]
- 10. Chen CC, Chen HL, Hsieh CW, Yang YL, Wung BS. Upregulation of NF‐E2‐related factor‐2‐dependent glutathione by carnosol provokes a cytoprotective response and enhances cell survival. Acta Pharmacol Sin 2011;32:62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li X, Zhao L, Han JJ, Zhang F, Liu S, Zhu L, et al. Carnosol modulates Th17 cell differentiation and microglial switch in experimental autoimmune encephalomyelitis. Front Immunol 2018;9:1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lo AH, Liang YC, Lin‐Shiau SY, Ho CT, Lin JK. Carnosol, an antioxidant in rosemary, suppresses inducible nitric oxide synthase through down‐regulating nuclear factor‐kappaB in mouse macrophages. Carcinogenesis 2002;23:983–991. [DOI] [PubMed] [Google Scholar]
- 13. Kashyap D, Kumar G, Sharma A, Sak K, Tuli HS, Mukherjee TK. Mechanistic insight into carnosol‐mediated pharmacological effects: recent trends and advancements. Life Sci 2017;169:27–36. [DOI] [PubMed] [Google Scholar]
- 14. Moran AE, Carothers AM, Weyant MJ, Redston M, Bertagnolli MM. Carnosol inhibits beta‐catenin tyrosine phosphorylation and prevents adenoma formation in the C57BL/6J/Min/+ (Min/+) mouse. Cancer Res 2005;65:1097–1104. [PubMed] [Google Scholar]
- 15. Tong L, Wu S. The mechanisms of carnosol in chemoprevention of ultraviolet B‐light‐induced non‐melanoma skin cancer formation. Sci Rep 2018;8:3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson JJ. Carnosol: a promising anti‐cancer and anti‐inflammatory agent. Cancer Lett 2011;305:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miao C, Lv Y, Zhang W, Chai X, Feng L, Fang Y, et al. Pyrrolidine dithiocarbamate (PDTC) attenuates cancer cachexia by affecting muscle atrophy and fat lipolysis. Front Pharmacol 2017;8:915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shen Q, Miao CX, Zhang WL, Li YW, Chen QQ, Li XX, et al. SiBaoChongCao exhibited anti‐fatigue activities and ameliorated cancer cachexia in mice. RSC Adv 2019;9:17440–17456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hayden MS, Ghosh S. Regulation of NF‐kappaB by TNF family cytokines. Semin Immunol 2014;26:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou W, Jiang ZW, Tian J, Jiang J, Li N, Li JS. Role of NF‐kappaB and cytokine in experimental cancer cachexia. World J Gastroenterol 2003;9:1567–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anker MS, Holcomb R, Muscaritoli M, von Haehling S, Haverkamp W, Jatoi A, et al. Orphan disease status of cancer cachexia in the USA and in the European Union: a systematic review. J Cachexia Sarcopenia Muscle 2019;10:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS Jr. NF‐kappaB‐induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 2000;289:2363–2366. [DOI] [PubMed] [Google Scholar]
- 23. Rhoads MG, Kandarian SC, Pacelli F, Doglietto GB, Bossola M. Expression of NF‐kappaB and IkappaB proteins in skeletal muscle of gastric cancer patients. Eur J Cancer 2010;46:191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Camargo RG, Riccardi DM, Ribeiro HQ, Carnevali LC Jr, de Matos‐Neto EM, Enjiu L, et al. NF‐kappaBp65 and expression of its pro‐inflammatory target genes are upregulated in the subcutaneous adipose tissue of cachectic cancer patients. Nutrients 2015;7:4465–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen C, Ju R, Zhu L, Li J, Chen W, Zhang DC, et al. Carboxyamidotriazole alleviates muscle atrophy in tumor‐bearing mice by inhibiting NF‐kappaB and activating SIRT1. Naunyn Schmiedebergs Arch Pharmacol 2017;390:423–433. [DOI] [PubMed] [Google Scholar]
- 26. Brown JL, Lee DE, Rosa‐Caldwell ME, Brown LA, Perry RA, Haynie WS, et al. Protein imbalance in the development of skeletal muscle wasting in tumour‐bearing mice. J Cachexia Sarcopenia Muscle 2018;9:987–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Quan‐Jun Y, Yan H, Yong‐Long H, Li‐Li W, Jie L, Jin‐Lu H, et al. Selumetinib attenuates skeletal muscle wasting in murine cachexia model through ERK inhibition and AKT activation. Mol Cancer Ther 2017;16:334–343. [DOI] [PubMed] [Google Scholar]
- 28. He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas‐Ahner J, et al. NF‐kappaB‐mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 2013;123:4821–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li Y, Yang L, Dong L, Yang ZW, Zhang J, Zhang SL, et al. Crosstalk between the Akt/mTORC1 and NF‐kappaB signaling pathways promotes hypoxia‐induced pulmonary hypertension by increasing DPP4 expression in PASMCs. Acta Pharmacol Sin 2019;40:1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL‐6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 2012;303:E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, et al. Adipose triglyceride lipase contributes to cancer‐associated cachexia. Science 2011;333:233–238. [DOI] [PubMed] [Google Scholar]
- 32. Kim SJ, Tang T, Abbott M, Viscarra JA, Wang Y, Sul HS. AMPK phosphorylates desnutrin/ATGL and hormone‐sensitive lipase to regulate lipolysis and fatty acid oxidation within adipose tissue. Mol Cell Biol 2016;36:1961–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ahmadian M, Abbott MJ, Tang T, Hudak CS, Kim Y, Bruss M, et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab 2011;13:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Batista ML Jr, Olivan M, Alcantara PS, Sandoval R, Peres SB, Neves RX, et al. Adipose tissue‐derived factors as potential biomarkers in cachectic cancer patients. Cytokine 2013;61:532–539. [DOI] [PubMed] [Google Scholar]
- 35. Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 2013;6:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Laurencikiene J, van Harmelen V, Arvidsson Nordstrom E, Dicker A, Blomqvist L, Naslund E, et al. NF‐kappaB is important for TNF‐alpha‐induced lipolysis in human adipocytes. J Lipid Res 2007;48:1069–1077. [DOI] [PubMed] [Google Scholar]
- 37. Lian KC, Chuang JJ, Hsieh CW, Wung BS, Huang GD, Jian TY, et al. Dual mechanisms of NF‐kappaB inhibition in carnosol‐treated endothelial cells. Toxicol Appl Pharmacol 2010;245:21–35. [DOI] [PubMed] [Google Scholar]
- 38. Yao H, Chen Y, Zhang L, He X, He X, Lian L, et al. Carnosol inhibits cell adhesion molecules and chemokine expression by tumor necrosis factor‐alpha in human umbilical vein endothelial cells through the nuclear factor‐kappaB and mitogen‐activated protein kinase pathways. Mol Med Rep 2014;9:476–480. [DOI] [PubMed] [Google Scholar]
- 39. Schwager J, Richard N, Fowler A, Seifert N, Raederstorff D. Carnosol and related substances modulate chemokine and cytokine production in macrophages and chondrocytes. Molecules 2016;21:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]