

Abstract
Background
Diesel exhaust (DE) emissions are a major contributor to ambient air pollution and are strongly associated with cardiovascular morbidity and mortality. Exposure to traffic‐related particulate matter is linked with acute adverse cardiovascular events; however, the mechanisms are not fully understood. We examined the role of the autonomic nervous system during exposure to DE that has previously only been indirectly investigated.
Methods and Results
Using microneurography, we measured muscle sympathetic nerve activity (MSNA) directly in the peroneal nerve of 16 healthy individuals. MSNA, heart rate, and respiration were recorded while subjects rested breathing filtered air, filtered air with an exposure mask, and standardized diluted DE (300 µg/m3) through the exposure mask. Heart rate variability was assessed from an ECG. DE inhalation rapidly causes an increase in number of MSNA bursts as well as the size of bursts within 10 minutes, peaking by 30 minutes (P<0.001), compared with baseline filtered air with an exposure mask. No significant changes occurred in heart rate variability indices during DE exposure; however, MSNA frequency correlated negatively with total power (r 2=0.294, P=0.03) and low frequency (r 2=0.258, P=0.045). Heart rate correlated positively with MSNA frequency (r 2=0.268, P=0.04) and the change in percentage of larger bursts (burst amplitude, height >50% of the maximum burst) from filtered air with an exposure mask (r 2=0.368, P=0.013).
Conclusions
Our study provides direct evidence for the rapid modulation of the autonomic nervous system after exposure to DE, with an increase in MSNA. The quick increase in sympathetic outflow may explain the strong epidemiological data associating traffic‐related particulate matter to acute adverse cardiovascular events such as myocardial infarction.
Registration
URL: https://www.clinicaltrials.gov; Unique identifier: NCT02892279.
Keywords: air pollution, autonomic nervous system, diesel, heart rate variability, muscle sympathetic nerve activity, sympatho‐excitation
Subject Categories: Autonomic Nervous System, Clinical Studies
Nonstandard Abbreviations and Acronyms
- ANS
autonomic nervous system
- DBP
diastolic blood pressure
- DE
diesel exhaust
- HF
high frequency
- HRV
heart rate variability
- LF
low frequency
- MSNA
muscle sympathetic nerve activity
- NOx
nitrogen oxides
- PM
particulate matter
- PM2.5
particulate matter with the mean geometric diameter <2.5 μm
- RMS
root mean squared
- RMSSD
root mean square of successive RR interval differences
- SBP
systolic blood pressure
- SDNN
standard deviation of NN intervals
- VLF
very low frequency
Clinical Perspective
What Is New?
Ambient air pollution, especially traffic‐derived particulate matter, is a major cause of global mortality and morbidity, with a large burden attributed to cardiovascular disease; however, the pathological mechanisms are not fully understood.
Exposure to diesel exhaust was shown to rapidly increase muscle sympathetic nervous activity in healthy individuals measured directly using microneurography and correlated with heart rate variability indices and heart rate.
What Are the Clinical Implications?
Inhalation of diluted diesel exhaust at levels encountered in an urban environment augments the autonomic nervous system, with a shift to sympathetic outflow.
The rapid sympatho‐excitation caused by diesel exhaust is a credible mechanism contributing to adverse cardiovascular events that occur shortly after acute exposure in at‐risk groups.
Chronic exposure to ambient air pollution may increase the risk of developing cardiovascular disease, such as hypertension, by increasing sympathetic outflow.
Ambient air pollution, especially the particulate matter (PM) constituents, is a critical global environmental problem and public health concern, with diesel exhaust emissions being a major contributor, particularly in urban environments. 1 The World Health Organization has estimated that approximately 90% of the world population living in cities are exposed to PM concentrations that exceed the World Health Organization’s air‐quality guidelines. 2 Recent calculations approximate a global annual mortality of 9.8 million attributable to ambient fine PM (PM2.5) air pollution, 3 and a mean loss of life expectancy of 2.9 years. 4 Epidemiological studies indicate that even small increases in PM exposure, both in the short and long term, are associated with an increase in cardiovascular morbidity and mortality. 5 , 6 A large proportion of the morbidity and mortality caused by air pollution is attributed to cardiovascular disease. 7 , 8 In particular, short‐term exposure to traffic‐related and ambient PM is associated with increased acute adverse cardiovascular events such as myocardial infarction, 9 , 10 stroke, 11 heart failure, 12 and cardiac arrhythmia. 13
The biological mechanisms eliciting the advancement of cardiovascular diseases as well as these subsequent adverse cardiovascular events seen after exposure to air pollution are not yet fully understood. The reactions seen in response to inhaled PM have been discussed in several informative reviews, 1 , 5 , 6 , 14 , 15 and include pulmonary and systemic oxidative stress and inflammation, vascular changes and endothelial dysfunction, an increase in thrombogenicity and decrease in fibrinolysis, changes in cardiac electrophysiological properties, and autonomic imbalance with a shift to an increase in sympathetic outflow. The time courses of these responses likely differ and overlap, and most probably act together to generate the clinical outcomes.
Changes in autonomic outflow have been postulated as a fast‐acting mechanism to explain the rapid onset of myocardial infarction and other cardiovascular clinical events in susceptible individuals (eg, people with underlying cardiovascular conditions and the elderly), where exposure to air pollution triggers pulmonary reflex arcs that tip the balance in favor of a pathological event. 16 , 17 Evidence of this effect comes from human exposure studies, where autonomic nervous system (ANS) imbalance has been suggested from changes in blood pressure, heart rate, and heart rate variability (HRV); however, the data are inconsistent. 5 , 18 In addition, these parameters provide only an indirect measure of assessing ANS balance and can be influenced by multiple factors. Despite this, exposure to PM has generally been associated with a reduction in HRV and a small elevation in diastolic blood pressure (DBP). 5 , 16
Direct measurements of sympathetic nerve activity have been obtained using intraneural microelectrode recordings from human peripheral nerve using microneurography, a well‐validated technique that can be used to record and quantify muscle sympathetic nerve activity (MSNA), as a direct measure of sympathetic nervous system outflow. Increases in MSNA lead to vasoconstriction in the skeletal muscle vascular bed, thereby increasing total peripheral resistance and hence systemic blood pressure. MSNA is increased in various forms of neurogenic hypertension, 19 as well as other cardiovascular diseases including congestive heart failure (CHF), 20 ischemic heart disease, and myocardial infarction. 21 To date, there have been no studies investigating the effect of diesel exhaust exposure on MSNA. The purpose of this study was to determine whether acute exposure to diesel exhaust increases MSNA, and directly address the theory that diesel exhaust PM can augment the ANS, as well as report changes in HRV, which has traditionally been used to study sympathetic/parasympathetic alterations. Because of the rapid onset of adverse cardiovascular events following exposure to air pollution, we hypothesized that diesel exhaust exposure causes a shift to increase sympathetic outflow, evidenced by an increase in MSNA, and thus could be a causal factor for cardiovascular events in at‐risk groups.
Methods
Data, Materials, and Code Disclosure Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Subjects
Sixteen healthy, nonsmoking men (aged 21–44 years) on no regular medications and with no history of cardiovascular, respiratory, or metabolic disease participated in this study. Ethics approval was obtained from the regional ethical review board at Umeå University. The study was performed in accordance with the Declaration of Helsinki, with the written informed consent of all volunteers. All volunteers had normal lung function, resting ECG, as well as normal results in a standard panel of blood tests. They reported no illness or respiratory tract infection for at least 2 weeks before the study, and were required to abstain from consuming alcohol and caffeine for at least 12 hours before the study visit.
Measurements
All measurements were performed at the SMP Swedish Machinery Testing site in Umeå, Sweden. Upon arrival and during measurements, subjects rested in the semisupine position on a comfortable bed in a room kept at an ambient temperature of around 23°C. Intraneural recordings of multiunit MSNA were recorded using microneurography and measured from muscle fascicles of the common peroneal nerve located at the fibular head. Briefly, an insulated tungsten microelectrode (FHC, Bowdoin, ME) (shaft diameter 200 μm, tapering to an uninsulated tip of 1–5 μm) was guided into the nerve fascicle, as previously described. 22 An uninsulated subdermal reference microelectrode was inserted 2 to 3 cm from the recording electrode, and a surface electrode on the leg acted as the ground electrode. Neural signals were amplified (20 000×) and filtered (300 Hz–3 kHz) using an isolated headstage (NeuroAmpEx; ADInstruments, Oxford, UK). Muscle fascicles were identified when intraneural stimulation evoked muscle twitches of the ankle or toe dorsiflexors, or foot evertor muscles, with no radiating paresthesia. Moreover, afferent activity could be recorded from muscle spindles during palpation of the muscle belly, percussion over the tendon or stretching the muscle, and no afferent response was generated by stroking the skin. Neural activity was recorded (10 kHz sampling) and root mean squared–processed (200 milliseconds moving average). MSNA detection was confirmed by listening to the neural activity for the characteristic bursts during rest for spontaneous burst activity, 23 as well as when the subject performed an inspiratory‐capacity apnea, which causes a sustained increase in MSNA. ECG, respiration, and pulsatile blood flow were recorded continuously using a 3‐lead ECG, a respiratory belt transducer wrapped around the chest, and an infrared plethysmograph finger clip, respectively (ADInstruments, Oxford, UK). All data were acquired using PowerLab 16/35 hardware and analyzed using LabChart 7 software (ADInstruments). Blood pressure was recorded intermittently using a portable blood pressure monitor (Boso Medicus, Jungingen, Germany).
Diesel Exhaust Exposure
Diesel exhaust was generated using a nonroad Volvo diesel engine (Volvo TD45, 4.5L, 4 cylinders, 680 rpm, manufactured in 1991) running on low sulphur SD10 (RF‐06‐03) diesel fuel (Preem AB, Stockholm, Sweden). The engine was connected to a dynamometer and running under control of a computer program using only the urban driving part of the European Transient Cycle. 24 Less than 10% of the generated exhaust was diluted with ambient filtered air, heated to 20°C (humidity ≈50%) and fed into an exposure chamber at a steady‐state concentration. 25 The air in the exposure chamber was continuously monitored for total hydrocarbons, nitrogen oxides (NOx, NO, NO2), and PM. Exposures were standardized at a particulate mass concentration (PM10) aimed at 300 μg/m3, based on online measurements of total nitrogen oxides (NOx), and PM mass concentration measured by tapered element oscillating microbalance (TEOM 1400; ThermoFisher Scientific, Waltham, MA), with repeat particle mass filter measurements. There was little variation in total hydrocarbons (0.78±0.05 parts per million), NOx (3.42±0.38 parts per million), NO2 (0.02±0.0), and PM (304±7.22 µg/m3) concentrations between exposures. To prevent investigator exposure, research participants were exposed to the standardized diesel exhaust/air mix just outside the chamber via a validated facemask (ComfortFull 2; Philips Healthcare, Best, the Netherlands), designed to have good flow to prevent rebreathing. 26 Similar particle concentrations within the exposure chamber and within the mask were confirmed using the Portacount Plus, TSI respirator fit tester (Comfort‐Control AB, Uppsala, Sweden).
Study Design
Once a suitable MSNA site had been obtained, the experiment commenced, and continuous measurements were started and assessed for 1 hour. A schematic of the experimental protocol is shown in Figure 1. Baseline data were recorded over the first initial 10‐minute period, in which subjects rested quietly with normal breathing and noise canceling headphones. The subject’s nose was then plugged to guarantee only mouth breathing, and the exposure mask was applied over their face. This was to facilitate particle deposition deep into the lungs that would more closely mimic previous studies that used intermittent exercise. 25 Measurements were then recorded for 10 minutes, with subjects breathing filtered air through the exposure mask (mask baseline). The air source was then switched to the diesel exhaust/air mix without the knowledge of the subject, and measurements recorded for a further 40 minutes (mask diesel exhaust exposure). Blood pressure was recorded at the start of baseline and mask baseline recordings, 15 minutes into the diesel exhaust exposure, and at the end of the experiment.
Figure 1. Experimental protocol. Subjects sat semisupine for 1 hour while parameters were recorded.
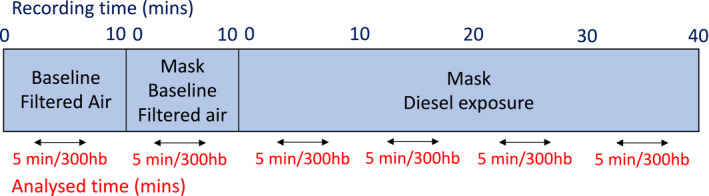
In the first 10 minutes, subjects were exposed to filtered air (baseline), after which nose plugs and the exposure masks were fitted, and they continued to breath filtered air for a further 10 minutes (mask baseline). The filtered air was then switched to the diluted diesel exhaust air mixture, and recordings were made for a further 40 minutes. For analysis, the experiment was broken down into 10‐minute blocks, and the median 5 minutes of each block were analyzed for muscle sympathetic nerve activity (MSNA) frequency (bursts/min), burst incidence (bursts/100 heart beats [hb]), and MSNA amplitude (burst height, µV) and area (µV/ms). Mins indicates minutes.
Data Analysis
The 1‐hour recordings were broken down into 10‐minute blocks, as shown in Figure 1. The median 5‐minute period of each block was analyzed using LabChart 7 software (ADInstruments) for MSNA burst frequency (bursts per minute), burst incidence (bursts per 100 heart beats), and burst amplitude (burst peak per height, microvolts) and burst area (microvolts per millisecond). Visual inspection and auditory recognition of the neural signal were used to verify MSNA bursts by a single experienced investigator. The baseline for amplitude and area analysis was defined manually in the root mean squared–processed signal and was determined as an average of the first 100 milliseconds of the selection. The peak amplitude was calculated by the computer as an average of 0.5 milliseconds on either side of the max point. Because burst amplitude and area are highly dependent on the proximity of the recording microelectrode to the sympathetic fibers, the burst amplitude and area were normalized to the largest recorded burst within the analyzed blocks to enable comparison between subjects. Heart rate, respiratory rate, respiratory cyclic height, and HRV were determined from the same 5‐minute analysis periods. Time‐domain and frequency‐domain methods were used to assess HRV and were analyzed using LabChart 8 software with HRV 2.0 module (ADInstruments), which uses the Lomb‐Scargle periodogram for spectral analysis. Time‐domain parameters were calculated for standard deviation of NN intervals (SDNN) and root mean square of successive RR interval differences. Frequency domain parameters were calculated for total power (≤0.4 Hz), very low frequency (0.003–0.04 Hz), low frequency (LF; 0.04–0.15 Hz), and high frequency (0.15–0.4 Hz) components of the power spectrum and expressed in absolute (milliseconds squared) and normalized units, as well as the LF/high‐frequency ratio. Ectopic beats were excluded from analysis.
Statistical Analysis
Group average data are reported as mean with 95% CI. Statistical analyses were performed with GraphPad Prism 6 for Mac OS (GraphPad Software, La Jolla, CA) using repeated‐measures ANOVA, with Geisser‐Greenhouse correction, and Tukey’s multiple comparison test, with individual variances computed for each comparison. Respiration depth, SDNN, root mean square of successive RR interval differences, total power, very low frequency, LF, and high‐frequency HRV parameters were log‐transformed before statistical analysis because they were not normally distributed. The relationship between variables was assessed using Pearson correlation tests. Unstandardized effect sizes are presented as the mean difference and reported with 95% CI. Hedges’ g is also presented as a standardized effect size and was calculated using an online calculator, 27 with mask baseline measurements used as the control group. Statistical significance was set at P<0.05.
Results
Sympathetic Neural Response to Diesel Exhaust Exposure
Inhalation of diesel exhaust quickly augmented sympathetic outflow to the muscle vascular bed. Experimental recordings of MSNA, heart rate, and respiration from one subject are shown in Figure 2. A statistically significant increase in MSNA burst frequency occurred within 10 minutes of diesel exhaust exposure and gradually increased as exposure continued, reaching a peak after 30 minutes (mean, 31.6; 95% CI, 27.8–35.5; Hedges’ g, 0.81; 95% CI, 0.09–1.53) (Figure 3A). MSNA incidence followed a similar pattern, with a significant increase following 20 minutes of diesel exhaust exposure (mean, 48.1; 95% CI, 42.4–53.9; Hedges’ g, 0.66; 95% CI, −0.05 to 1.37) (Figure 3B). When MSNA amplitude was normalized to enable comparison between individuals, a trend of increasing MSNA amplitude (burst height) and burst area was observed but was not statistically significant (Figure 3C). However, the largest variance occurred in bursts >50% of the maximum burst. These data were replotted as the percentage of bursts with an amplitude and area >50% of the maximum burst, and revealed a statistically significant increase in the frequency of larger bursts in amplitude (Figure 3D) and area (Figure 3E), after 20 minutes and 10 minutes, respectively, with the largest bursts occurring at the 30‐ to 40‐minute time point (mean burst amplitude, 20.8; 95% CI, 15.7–25.9; Hedges’ g, 1.02; 95% CI, 0.29–1.76; mean burst area, 13.3; 95% CI, 9.7–16.9; Hedges’ g, 1.16; 95% CI, 0.41–1.91). The mean differences for each time point compared with mask baseline for each of the MSNA outcomes are shown in Figure 3 and Figure 4.
Figure 2. Experimental recordings of muscle sympathetic nerve activity, ECG, and respiration in one representative subject at mask baseline (A) and after 25 minutes of breathing diluted diesel exhaust (B).
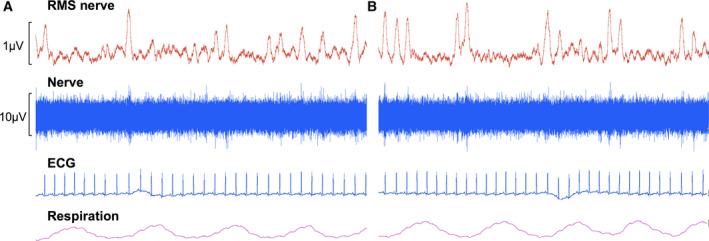
RMS indicates root mean squared.
Figure 3. Changes in muscle sympathetic nerve activity (MSNA) during exposure to diluted diesel exhaust.
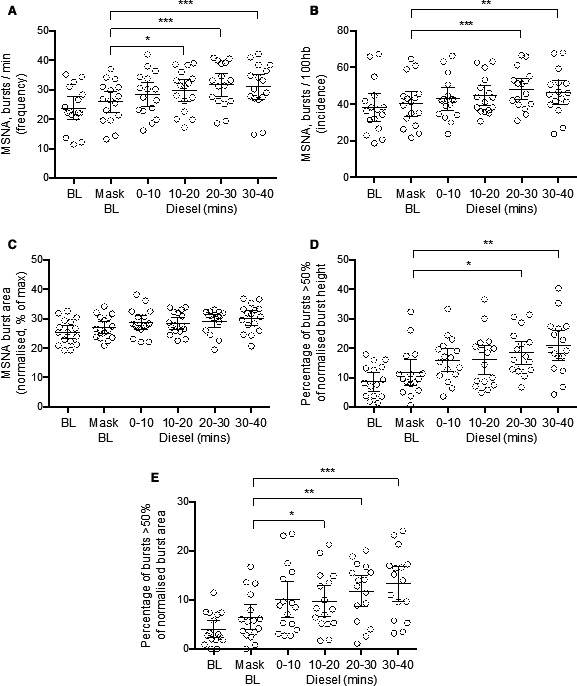
A, MSNA burst frequency. B, MSNA burst incidence. C, Average normalized MSNA burst area. D, MSNA burst amplitude (height). E, Burst area. For percentage of normalized bursts that were greater than half of the maximum burst for each individual subject. Bars represent mean with 95% CI. Significance ***P<0.001, **P<0.01, *P<0.05, repeated‐measures 1‐way ANOVA, with Geisser‐Greenhouse correction, and Tukey’s multiple comparison test. BL indicates baseline; and mins, minutes.
Figure 4. The mean difference with 95% CIs for muscle sympathetic nerve activity (MSNA) burst frequency (A).
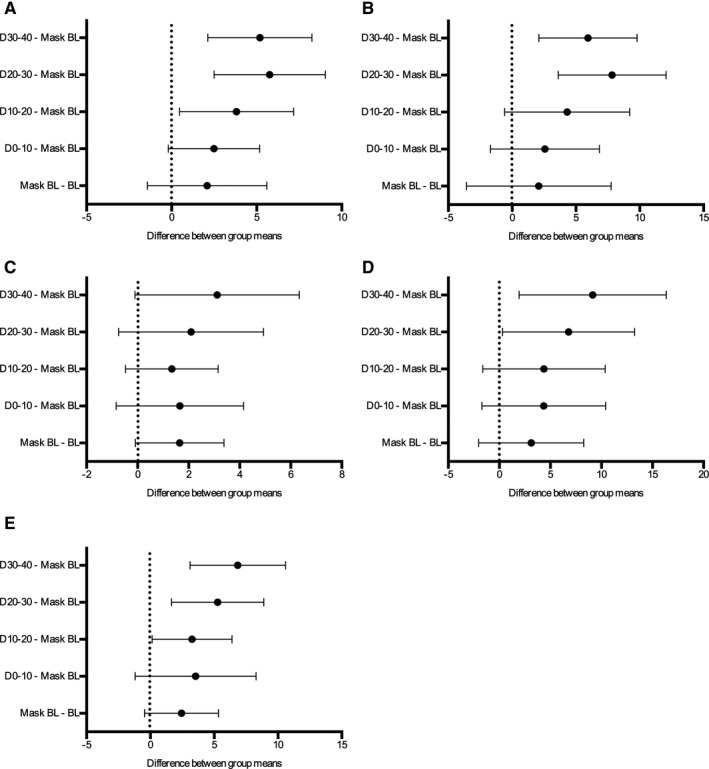
B, MSNA burst incidence. C, Average normalized MSNA burst area. D, MSNA burst amplitude (height). E, Burst area. For percentage of normalized bursts that were greater than half of the maximum burst for each individual subject. BL indicates baseline. D0–10 indicates DE exposure between 0 to 10 mins; D10–20 the DE exposure from 10 to 20 mins; D20–30 the DE exposure from 20 to 30 mins; and D30–40 the DE exposure from 30 to 40 mins.
To assess whether the increases in MSNA were consistent among study participants, we plotted the percentage difference of MSNA parameters from mask baseline, because this was deemed the more fitting baseline measurement for comparison with the filtered air exposure to diesel exhaust (Figure S1), given that participants were breathing through a mask with the nose occluded in both conditions. A consistent positive trend is seen in all subjects for MSNA frequency and incidence but was especially pronounced for measures of MSNA burst intensity, with a 400% to 600% increase in the number of bursts that were >50% of the maximum recorded burst, for burst amplitude and area, respectively (Figure S1C and S1D).
Because study subjects were in a static, semisupine position for a considerable length of time, we also assessed if this could affect MSNA by plotting the raw and percentage difference of MSNA variables against the length of time the subjects were required to be still (Figure S2). We found no relationship between any MSNA outcome and time, indicating that time was not a confounding factor.
There were also no statistically significant differences in MSNA burst frequency, incidence, amplitude, or area between baseline and mask baseline, suggesting that the main cause for change in sympathetic outflow was the diesel exhaust exposure (Figures 3 and 4).
Effect of Diesel Exhaust on Respiration, Heart Rate, Blood Pressure, and HRV
There were no differences in respiration or any of the frequency domain HRV parameters between either baseline measurements (with or without mask) or during the diesel exhaust exposure (Table). The results of the ANOVA for the SDNN component of HRV revealed that there was a difference between the mean values of the time points (ANOVA, P=0.047). However, when analyzing each individual time point using the post hoc test, no statistically significant differences (P<0.05) where found (group averages and mean differences are shown in Figure S3A and S3B, respectively). Heart rate, however, gradually increased as the experiment progressed, with a small but statistically significant increase in heart rate seen during mask baseline and the following time points during diesel exhaust exposure, compared with baseline. Even so, there was no statistically significant elevation in heart rate between mask baseline and following diesel exhaust exposure. A trend in increasing systolic blood pressure (SBP) and DBP was, however, observed when comparing mask baseline to diesel exhaust exposure measurements, although the ANOVA test did not indicate an overall statistically significant change. Nevertheless, after post hoc analysis, a significant increase in SBP from mask baseline levels was found after 40 minutes of diesel exhaust exposure (mean difference of 5.125; 95% CI, 0.37–9.9; Hedges’ g, 0.42; 95% CI, −0.29 to 1.12) (Figure S3).
Table 1.
Respiration, Heart Rate, and Heart Rate Variability Parameters
| Filtered Air | Diesel Exhaust Exposure With Exposure Mask | RM 1‐Way ANOVA P Value | |||||
|---|---|---|---|---|---|---|---|
| BL | Mask BL | 0–10 min | 10–20 min | 20–30 min | 30–40 min | ||
| Respiratory rate, breaths/min | 11.4 (9.8–13.1) | 10.2 (8.6–11.7) | 9.7 (8.3–11.1) | 10.6 (8.9–12.2) | 10.4 (9.0–11.8) | 10.8 (9.4–12.2) | 0.117 |
| Respiration depth, cyclic height, mV | 5.2 (4.1–6.2) | 5.4 (4.3–6.5) | 6.6 (4.9–8.3) | 6.2 (4.3–8.0) | 5.7 (4.4–7.0) | 6.0 (4.6–7.3) | 0.130* |
| Heart rate, beats/min | 64.3 (58.9–69.7) | 65.7 (60.2–71.3) | 66.7 (61.3–72.1) | 67.4 (61.5–72.1) | 67.2 (60.9–73.6) | 68.1 (62.2–74.0) | 0.001 |
| Systolic BP, mm Hg | 131.7 (126.4–136.9) | 127.6 (120.8–134.3) | 130.5 (124.7–136.3) | 132.7 (126.4–138.9) | 0.064 | ||
| Diastolic BP, mm Hg | 77.9 (74.6–81.3) | 74.5 (70.7–78.3) | 77.2 (73.1–81.3) | 76.9 (73.0–80.8) | 0.124 | ||
| Time domain | |||||||
| SDNN, ms | 58.1 (47.1–69.2) | 59.1 (48.6–69.6) | 67.6 (54.9–80.3) | 68.0 (52.7–83.3) | 67.4 (53.1–81.7) | 69.8 (53.0–86.7) | 0.047* |
| RMSSD, ms | 42.3 (31.5–53.1) | 42.1 (31.2–53.0) | 44.0 (33.1–54.9) | 42.8 (30.9–54.3) | 45.3 (30.9–59.7) | 43.7 (31.7–55.7) | 0.805* |
| Frequency domain | |||||||
| Total power, ms2 | 4417 (2524–6310) | 4348 (2348–63470) | 4900 (2982–6817) | 5383 (2588–8179) | 4892 (2979–6804) | 5839 (3106–8571) | 0.214* |
| VLF, ms2 | 1617 (554–2680) | 1613 (574–2653) | 1832 (754–2909) | 2536 (759–4313) | 1668 (934–2402) | 2273 (862–3685) | 0.601* |
| LF, ms2 | 1994 (1021–2966) | 1780 (844–2716) | 2201 (822–3580) | 1915 (936–2893) | 2057 (967–3147) | 2595 (811–4378) | 0.377* |
| HF, ms2 | 772 (440–1104) | 890 (392–1388) | 817 (430–1204) | 905 (501–1309) | 1095 (405–1785) | 942 (472–1412) | 0.444* |
| LF, nu | 68.5 (60.9–76.1) | 63.9 (51.3–76.6) | 68.0 (58.1–77.9) | 66.0 (58.7–73.3) | 63.2 (53.1–73.3) | 69.6 (60.5–78.8) | 0.537 |
| HF, nu | 30.9 (23.7–38.1) | 35.0 (23.3–46.7) | 31.9 (22.4–41.4) | 32.9 (25.8–40.1) | 35.3 (25.5–45.2) | 29.8 (21.0–38.7) | 0.684 |
| LF/HF ratio | 3.1 (1.7–4.5) | 3.2 (1.8–4.6) | 3.5 (1.8–5.3) | 2.7 (1.4–4.1) | 2.7 (1.4–4.0) | 3.7 (2.1–5.2) | 0.588 |
Values are presented as mean (95% CI). BL indicates baseline; BP, blood pressure; HF, high frequency; LF, low frequency; nu, normalized units; RM, repeated measures; RMSSD, root mean square of successive RR interval differences; SDNN, standard deviation of NN intervals; and VLF, very low frequency.
Statistical analysis performed on log values. RM 1‐way ANOVA, with Geisser‐Greenhouse correction, and Tukey’s multiple comparison test were performed unless otherwise stated.
Relationship Between Muscle Sympathetic Nerve Activity, Exposure, and HRV
Because the greatest group average of MSNA was seen between 20 and 30 minutes into the diesel exhaust exposure, we report here the main correlations observed at this time point. No statistically significant correlations were detected between total hydrocarbon, total nitrogen oxide (NOx) levels or PM concentration, and measurements of MSNA. Weak but statistically significant negative correlations were observed between MSNA frequency and several HRV indices including total power, LF, and SDNN, and a positive association with heart rate following diesel exhaust exposure (Figure 5). Furthermore, to see whether the degree of change in MSNA between subjects could relate to any measured variables, we also examined the difference in MSNA at the different time points using mask baseline levels as a reference. In this case, statistically significant correlations were only seen between MSNA amplitude (burst height) and heart rate (Figure 6).
Figure 5. Relationship between muscle sympathetic nerve activity (MSNA) frequency and total power (A), low frequency (LF) (B), standard deviation of NN intervals (SDNN) (C), and heart rate 20 to 30 minutes after diesel exhaust inhalation (D).
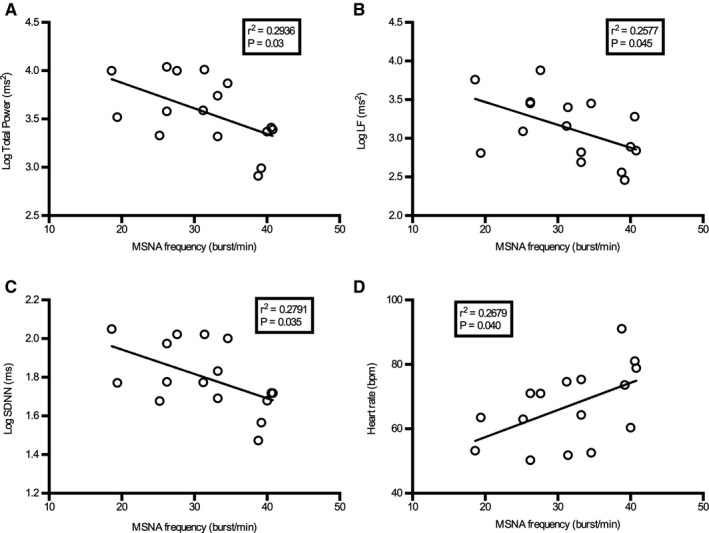
Pearson correlation coefficient tests were used and the line fitted using nonlinear regression with least squares fit.
Figure 6. Relationship between MSNA amplitude (burst height), the percentage of bursts >50% of the maximum burst amplitude compared with heart rate.
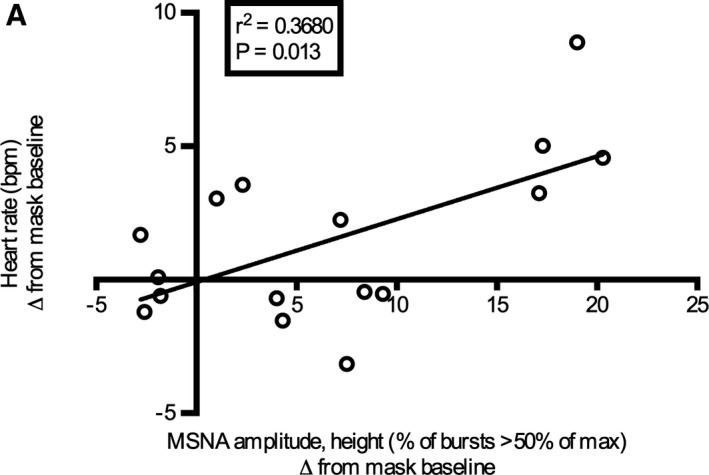
Data plotted are the differences from mask baseline for the 20‐ to 30‐minute time point values. Pearson correlation coefficient test was used and the line fitted using nonlinear regression with least squares fit.
DISCUSSION
This is the first study to demonstrate that short‐term acute exposure to diesel exhaust, a major contributor of urban PM air pollution, causes a rapid increase in muscle sympathetic nerve activity in humans. Direct quantification of MSNA was also related to HRV, a commonly used technique to assess autonomic balance. These important findings provide evidence for a change in the ANS to a shift in sympathetic outflow, as a plausible mechanism to explain the occurrence of acute adverse cardiovascular events seen shortly after exposure to air pollutants in at‐risk groups.
Increased sympathetic activity has been associated with several cardiovascular diseases including CHF, 20 hypertension, 19 , 28 ischemic heart disease, and myocardial infarction. 21 Human exposure studies provide a controlled and predictable environment in which to study pathophysiological mechanisms. In this study, we used exposure parameters that we have previously shown to elicit pathological responses in healthy human subjects such as endothelial dysfunction, 24 , 25 thrombus formation and platelet activation, 29 lung inflammation, 30 and reduction in airway antioxidants. 31 Using microneurography, we demonstrate that inhalation of diesel exhaust increases MSNA after just 10 minute of exposure. By 30 minutes of diesel exhaust exposure, the increase in MSNA frequency and incidence peaked, with a mean difference from mask baseline measurement of 5.8 (95% CI, 2.5–9.0; Hedges’ g, 0.81) and 7.8 (95% CI, 3.6–7.8; Hedges’ g, 0.66), respectively (Figure 4A and 4B). Despite this being a slight increase in MSNA, when compared with pathologies such as CHF for example, where the difference in MSNA frequency between disease and control groups can be up to 17 bursts, 20 in healthy individuals, diesel exhaust exposure still has a reasonably large effect on MSNA outflow, as shown by the calculated effect sizes. The rate at which this modulation occurs also adds weight to the hypothesis that activation of airway sensory nerves and autonomic reflex arcs by air pollutants can mediate the known acute cardiovascular effects. This complex comprehensive reflex system is responsible for maintaining homeostasis and acts in accordance on a moment‐to‐moment basis, responding to sensory activation/irritation (via pulmonary C‐fibers and rapidly adapting pulmonary receptors), changes in blood pressure (baroreceptors), as well as changes in levels of oxygen, carbon dioxide, pH, and temperature (central and peripheral chemoreceptors). 17 Multiple air pollutants, including components of diesel exhaust such as PM and polycyclic aromatic hydrocarbons have been shown to stimulate these pathways and are summarized in several excellent reviews. 1 , 15 , 17 , 32 Although MSNA is measured from vasoconstrictor neurons that innervate the skeletal muscle vascular bed, it has been proven to correlate strongly with cardiac noradrenaline spillover, and therefore is also a good indicator of sympathetic traffic to the heart. 33 The increase in sympathetic outflow, substantiated by an increase in MSNA, generated by diesel exhaust exposure in the present study is thus a credible mechanism to explain the recognized acute adverse cardiovascular events after short‐term exposure to ambient air pollution.
To our knowledge, only one other human exposure study has used microneurography to study the effects of air pollutants. 34 In contradiction to our results, they did not observe any difference in MSNA between controlled exposure conditions, ultrafine particle exposure by itself, or ultrafine particles in combination with ozone, as compared with filtered air. Our data indicate that changes in MSNA occur rapidly after exposure, and therefore, the window in which autonomic modulation occurs may have been missed in the study by Heusser et al, where they performed microneurography in healthy subjects 1.5 hours after a 3‐hour exposure. 34 It is possible that the change in sympathetic outflow, at least in healthy subjects, may be transient and may normalize sometime after eliminating the exposure. However, the opposite could also be the case and would need to be confirmed with longer recording durations during and after exposures. The time kinetics of this response would be of great interest to investigate further. Alternatively, the differences in air pollution exposure type could also explain the conflicting results. Diesel exhaust is a complex mixture made up of different particle‐size fractions, the chemical compounds adsorbed onto their surface, as well as noxious gaseous components, and therefore is fundamentally different from generated ultrafine particles (carbon black aerosol). 35 Importantly, although not directly measured in the current study, we and others have previously shown that levels of carbon monoxide generated in the diesel exhaust exposure used are low (≈3 parts per million) 18 and unlikely to cause the observed changes in MSNA. Furthermore, MSNA has been shown to be unaffected at carbon monoxide levels of up to 1000 parts per million. 36 Interestingly, Heusser et al reported a modest increase in, and reduced clearance of, norepinephrine, a neurotransmitter predominately released by postganglionic neurons of the sympathetic nervous system. 34 In regard to our results, it is likely that this increase in norepinephrine originated, at least in part, by the early increase in sympathetic vasoconstrictor activity, and remained elevated because of reduced norepinephrine turnover. Increased levels of norepinephrine, as well as other catecholamines, have also been shown to be associated with PM exposure. 37 Reduced catecholamine uptake and clearance, in addition to the increased sympathetic nerve activity could thus activate the sympathetic adrenal–medullary axis and neurohormonal stress response pathway, and thus be another potential underlying mechanism triggering the cardiovascular changes, as well as other adverse health outcomes seen in response to air pollution. 38
In addition to increased MSNA burst frequency and incidence, we also observed an increase in the size of bursts (burst amplitude and burst area), which is the first time this has been shown in response to air pollution exposure. This increase in burst amplitude and area, or burst intensity, likely either reflects multiple firing of already active nerve fibers and/or increased recruitment of fibers. However, this would need to be confirmed using single‐unit recording techniques that allow the investigation of firing characteristics of individual vasoconstrictor fibers. 39 , 40 In conditions in which cardiovascular morbidity and mortality are elevated, such as CHF, hypertension, obstructive sleep apnea, and obesity, the pattern of sympatho‐excitation has been shown to be an important factor, especially in regard to the firing frequency of individual fibers. 41 In CHF for example, where sympathetic bursts are increased and individual fibers have an increased firing frequency, 39 , 40 Rundqvist et al demonstrated using radiotracer techniques that there is marked cardiac norepinephrine spillover and reduced uptake. 42 Firing characteristics and increased firing frequency may therefore elevate neurotransmitter concentration at neuroeffector junctions resulting in amplified effector responses and pathology, depending on the extent of the sympatho‐excitation. 39 , 40 , 43
Although there was no overall statistically significant difference in the time point means for SBP or DBP over the course of the experiment indicated by the ANOVA analysis, a modest increase in SBP and DBP was observed over time during the diesel exhaust exposure compared with mask baseline, with a significant increase in SBP detected after 40 minutes of diesel exhaust exposure after post hoc analysis. A larger increase in blood pressure may have been observed at a later time point and if more frequent blood pressure measurements had been taken. Other controlled exposure studies to diesel exhaust 44 and concentrated ambient particles in combination with ozone 45 have also shown rapid increases in SBP and DBP, respectively. Moreover, in a crossover study conducted in Beijing, China, use of a highly efficient facemask significantly reduced SBP. 46 Together, the rapid modulation of blood pressure during and shortly after exposure to diesel exhaust and/or PM, supports our findings of increased sympathetic outflow and ANS disturbance.
Heart rate variability is the predominant methodology that has been used in air pollution research to assess the interaction between cardiac sympathetic and parasympathetic branches of the ANS. We previously have used HRV to assess autonomic dysfunction in healthy male subjects in a similar exposure scenario, and showed there were no changes in frequency or time domains at 2, 6, or 24 hours after exposure. 18 In the current study, we assessed HRV at a much earlier time point, but we still did not notice any statistically significant changes in any HRV indices. We did observe a gradual increase in heart rate. However, there was no statistically significant difference between diesel exhaust exposure and mask baseline. The gradual increase in SDNN, although not statistically significant, was an unexpected finding and reflects the complexity of the ANS. This could reflect baroreceptor‐mediated changes to counter the increases in blood pressure and MSNA. However, multiple integrative physiological mechanisms work to maintain balance. Nevertheless, some weak associations were noted between MSNA, HRV, and heart rate, such that a greater sympathetic outflow had a decreased total power, LF band, and SDNN, and greater heart rate. This is in accordance with a summary statement from the American Heart Association on HRV data and short‐term exposure to air pollution, where the general pattern is that heart rate increases and HRV indices decrease. 5 However, there are inconsistencies in the data, which may be attributable to multiple factors including different study populations (ie, old, young, underlying disease), different exposure methods and types (ie, diesel exhaust, concentrated ambient particles, ultrafine particles, real‐life exposures), study methodologies used (ie, different HRV measurements and analysis duration), and the time kinetics of the response (acute or chronic).
As mentioned previously, multiple factors can affect the ANS to regulate sympatho‐vagal activity and maintain balance. One factor to consider therefore is that the increase in MSNA observed could be underestimated because of compensatory mechanisms elicited after increases in blood pressure and heart rate, acting via the baroreceptor reflex system. If blood pressure and vasomotor tone were controlled and held steady at baseline levels, as has been done in studies of nitric oxide bioavailability, 47 greater increases in MSNA may be observed. Moreover, a decrease in baroreflex control has been shown in multiple cardiovascular pathologies, 48 as well as decreased sensitivity after smoking 49 and aging. 50 Decreased baroreflex sensitivity could thus also be a possible mechanism for the observed increases in MSNA during diesel exhaust exposure and could be addressed in future studies.
Strengths and Weaknesses
A major strength of this study is that we were able to measure muscle sympathetic outflow directly from muscle vasoconstrictor neurons throughout the study. The use of a verified exposure mask meant we could record continuous changes in autonomic tone during an ongoing diesel exhaust exposure. This has not been possible before using an exposure chamber because of the complexity of microneurography, which requires experimenters to be present throughout the measurements. The quantitative assessment of MSNA obtainable with microneurography is also superior to indirect measures of autonomic modulation such as HRV, and thus provides conclusive evidence of sympatho‐excitation following diesel exhaust inhalation. Furthermore, there are few studies that measure ANS effects during exposure. Because of the complexity of the ANS and modulatory mechanisms, such as baroreflex and respiratory modulation, even small changes such as the simple moving of subjects, or activity, can obscure small but relevant changes in autonomic tone and blood pressure, which may also be affected at different time points. This may explain why prior studies observe variable responses.
There are a few limitations to the study. In most human exposure models, a double‐blind crossover study design has been traditionally used in controlled air pollution studies. Because subjects only had one study visit and knew they would be exposed to diesel exhaust, we could only blind them to the time when they would be exposed. Although we did not undertake a separate recording session in which the same participants breathed filtered air for the entire experiment, we found no difference in levels of MSNA while breathing filtered air in the first baseline recording (no mask) and the second baseline recording (mask). Moreover, we know that MSNA remains constant in a given individual day to day, month to month, and year to year, 51 and that there is a genetic component to this; identical twins have similar levels of MSNA, whereas fraternal twins do not. 52 Furthermore, because burst amplitude was also one of our outcomes, multiple visits would not have been possible because burst amplitude is highly dependent on the position of the recording microelectrode in the nerve fascicle, which is impossible to replicate in multiple study sessions. Because burst amplitude can only be assessed within the same recording for intraindividual changes, we thus normalized burst amplitude to the maximum burst in each individual recording, giving us a relative measure of change in amplitude to compare between individuals.
A possible limitation is that stress or anxiety caused by the exposure could result in changes in MSNA. However, participants did not describe such feelings, and reported no change to mild symptoms after exposure compared with before exposure on a symptom scale. We also tried to reduce any external factors that could disturb the subject, supplying noise‐canceling headphones throughout the experiment and ensuring that the subject was comfortable. In addition, it has been shown that mental stress, rather than emotional stress, is a modifier of heart rate and blood pressure, and that neither stress response influenced MSNA. 53 A study examining subjects’ perceptions and reported symptoms in response to diesel exhaust exposure also found that blinding to exposure is generally effective, and that perceived exposure rather than true exposure is the dominant modifier of symptom reporting. 54 Another potential limitation is that by blocking the nose to ensure mouth breathing, we bypassed irritant mechanisms of the upper airways. The aim in doing this was to guarantee mouth breathing to ensure a good deposition of particles into the lungs, and to replicate breathing patterns in previous exposure studies, where subjects are exposed while performing intermittent exercise (predominantly mouth breathing). Because we saw a clear response, the loss of exposure in the upper airways appears negligible.
One major limitation to this study was the lack of continuous blood pressure recordings. Unfortunately, because of electrical interference with the recording microelectrode, our blood pressure apparatus for continuous measurements could not be used. Hence, we could only obtain sporadic blood pressure measurements using a standard blood pressure monitor. Another limitation was that we had only examined the response to diesel exhaust inhalation in young healthy men. This group was chosen as an initial subject group as the acute cardiovascular effects demonstrated following controlled diesel exhaust exposure have primarily been studied in male populations, as hormonal variations may influence those outcomes. Certainly, further research is required to determine if these effects are also seen in other groups such as women, the elderly, racial groups, and those with underlying conditions and disease. In addition, we only investigated effects during an acute exposure (40 minutes); future studies should aim to determine the time kinetics of this sympatho‐excitation response as well as in more chronic exposures.
Conclusions
Brief exposure to diluted diesel exhaust rapidly augments the autonomic nervous system, with a shift to increased sympathetic outflow. Because increased activation of the sympathetic nervous system has been shown to adversely affect cardiac rhythm and function, arterial tone, as well as blood pressure, this provides evidence linking the epidemiological observations for adverse cardiovascular events with acute air pollution exposure.
Sources of Funding
The authors acknowledge grant support from the Swedish Heart‐Lung Foundation, FORTE: Swedish Research Council for Health Working Life and Welfare, Umeå University, and County Council of Västerbotten (ALF and Spearhead grants).
Disclosures
None.
Supporting information
Figures S1–S3
Acknowledgments
The authors would like to thank the team at Svensk Maskinprovning for their expertise in setting up and monitoring exposure conditions.
(J Am Heart Assoc. 2021;10:e018448. DOI: 10.1161/JAHA.120.018448.)
See Editorial By Brook And Rajagopalan
For Sources of Funding and Disclosures, see page 13.
References
- 1. Ghelfi E. (2011). Air Pollution, Reactive Oxygen Species (ROS), and Autonomic Nervous System Interactions Modulate Cardiac Oxidative Stress and Electrophysiological Changes, Advanced Topics in Environmental Health and Air Pollution Case Studies, Prof. Anca Moldoveanu (Ed.), ISBN: 978‐953‐307‐525‐9, InTech, Available at: https://www.intechopen.com/books/advanced‐topics‐in‐environmental‐health‐and‐air‐pollution‐case‐studies/air‐pollution‐reactive‐oxygen‐species‐ros‐and‐autonomic‐nervous‐system‐interactions‐modulate‐cardiac. Accessed March, 2020.
- 2. World Health Organization . Global Health Observatory. Available at: https://www.who.int/gho/phe/air_pollution_pm25_concentrations/en/ (Accessed March 13, 2020).
- 3. Burnett R, Chen H, Szyszkowicz M, Fann N, Hubbell B, Pope CA, Apte JS, Brauer M, Cohen A, Weichenthal S, et al. Global estimates of mortality associated with long‐term exposure to outdoor fine particulate matter. Proc Natl Acad Sci USA. 2018;115:9592–9597. DOI: 10.1073/pnas.1803222115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lelieveld J, Pozzer A, Pöschl U, Fnais M, Haines A, Münzel T. Loss of life expectancy from air pollution compared to other risk factors: a worldwide perspective. Cardiovasc Res. 2020;116:1910–1917. DOI: 10.1093/cvr/cvaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brook RD, Rajagopalan S, Pope CA, Brook JR, Bhatnagar A, Diez‐Roux AV, Holguin F, Hong Y, Luepker RV, Mittleman MA, et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–2378. DOI: 10.1161/CIR.0b013e3181dbece1. [DOI] [PubMed] [Google Scholar]
- 6. Franchini M, Mannucci PM. Thrombogenicity and cardiovascular effects of ambient air pollution. Blood. 2011;118:2405–2412. DOI: 10.1182/blood-2011-04-343111. [DOI] [PubMed] [Google Scholar]
- 7. Rajagopalan S, Al‐Kindi SG, Brook RD. Air pollution and cardiovascular disease: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2018;72:2054–2070. DOI: 10.1016/j.jacc.2018.07.099. [DOI] [PubMed] [Google Scholar]
- 8. Lelieveld J, Klingmuller K, Pozzer A, Poschl U, Fnais M, Daiber A, Münzel T. Cardiovascular disease burden from ambient air pollution in Europe reassessed using novel hazard ratio functions. Eur Heart J. 2019;40:1590–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peters A, von Klot S, Heier M, Trentinaglia I, Hormann A, Wichmann HE, Löwel H. Exposure to traffic and the onset of myocardial infarction. N Engl J Med. 2004;351:1721–1730. DOI: 10.1056/NEJMoa040203. [DOI] [PubMed] [Google Scholar]
- 10. Mustafić H, Jabre P, Caussin C, Murad MH, Escolano S, Tafflet M, Périer M‐C, Marijon E, Vernerey D, Empana J‐P, et al. Main air pollutants and myocardial infarction: a systematic review and meta‐analysis. JAMA. 2012;307:713–721. DOI: 10.1001/jama.2012.126. [DOI] [PubMed] [Google Scholar]
- 11. Shah AS, Lee KK, McAllister DA, Hunter A, Nair H, Whiteley W, Langrish JP, Newby DE, Mills NL. Short term exposure to air pollution and stroke: systematic review and meta‐analysis. BMJ. 2015;350:h1295. DOI: 10.1136/bmj.h1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shah ASV, Langrish JP, Nair H, McAllister DA, Hunter AL, Donaldson K, Newby DE, Mills NL, et al. Global association of air pollution and heart failure: a systematic review and meta‐analysis. Lancet. 2013;382:1039–1048. DOI: 10.1016/S0140-6736(13)60898-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song X, Liu YU, Hu Y, Zhao X, Tian J, Ding G, Wang S. Short‐term exposure to air pollution and cardiac arrhythmia: A meta‐analysis and systematic review. Int J Environ Res Public Health. 2016;13:642– 10.3390/ijerph13070642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Langrish JP, Bosson J, Unosson J, Muala A, Newby DE, Mills NL, Blomerg A, Sandström T. Cardiovascular effects of particulate air pollution exposure: time course and underlying mechanisms. J Intern Med. 2012;272:224–239. DOI: 10.1111/j.1365-2796.2012.02566.x. [DOI] [PubMed] [Google Scholar]
- 15. Cascio WE. Proposed pathophysiologic framework to explain some excess cardiovascular death associated with ambient air particle pollution: Insights for public health translation. Biochim Biophys Acta. 2016;1860:2869–2879. DOI: 10.1016/j.bbagen.2016.07.016. [DOI] [PubMed] [Google Scholar]
- 16. Miller MR, Shaw CA, Langrish JP. From particles to patients: oxidative stress and the cardiovascular effects of air pollution. Future Cardiol. 2012;8:577–602. DOI: 10.2217/fca.12.43. [DOI] [PubMed] [Google Scholar]
- 17. Perez CM, Hazari MS, Farraj AK. Role of autonomic reflex arcs in cardiovascular responses to air pollution exposure. Cardiovasc Toxicol. 2015;15:69–78. DOI: 10.1007/s12012-014-9272-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mills NL, Finlayson AE, Gonzalez MC, Tornqvist H, Barath S, Vink E, Goudie C, Langrish JP, Soderberg S, Boon NA, et al. Diesel exhaust inhalation does not affect heart rhythm or heart rate variability. Heart. 2011;97:544–550. DOI: 10.1136/hrt.2010.199042. [DOI] [PubMed] [Google Scholar]
- 19. Schlaich MP, Lambert E, Kaye DM, Krozowski Z, Campbell DJ, Lambert G, Hastings J, Aggarwal A, Esler MD. Sympathetic augmentation in hypertension: role of nerve firing, norepinephrine reuptake, and Angiotensin neuromodulation. Hypertension. 2004;43:169–175. DOI: 10.1161/01.HYP.0000103160.35395.9E. [DOI] [PubMed] [Google Scholar]
- 20. Ando S, Dajani HR, Floras JS. Frequency domain characteristics of muscle sympathetic nerve activity in heart failure and healthy humans. Am J Physiol. 1997;273:R205–R212. DOI: 10.1152/ajpregu.1997.273.1.R205. [DOI] [PubMed] [Google Scholar]
- 21. Graham LN, Smith PA, Stoker JB, Mackintosh AF, Mary DA. Sympathetic neural hyperactivity and its normalization following unstable angina and acute myocardial infarction. Clin Sci (London). 2004;106:605–611. DOI: 10.1042/CS20030376. [DOI] [PubMed] [Google Scholar]
- 22. Brown R, Kemp U, Macefield V. Increases in muscle sympathetic nerve activity, heart rate, respiration, and skin blood flow during passive viewing of exercise. Front Neurosci. 2013;7:102. DOI: 10.3389/fnins.2013.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vallbo AB, Hagbarth KE, Wallin BG. Microneurography: how the technique developed and its role in the investigation of the sympathetic nervous system. J Appl Physiol. 1985;2004:1262–1269. DOI: 10.1152/japplphysiol.00470.2003. [DOI] [PubMed] [Google Scholar]
- 24. Barath S, Mills NL, Lundbäck M, Törnqvist H, Lucking AJ, Langrish JP, Söderberg S, Boman C, Westerholm R, Löndahl J, et al. Impaired vascular function after exposure to diesel exhaust generated at urban transient running conditions. Part Fibre Toxicol. 2010;7:19. 10.1186/1743-8977-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mills NL, Törnqvist H, Robinson SD, Gonzalez M, Darnley K, MacNee W, Boon NA, Donaldson K, Blomberg A, Sandstrom T, et al. Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation. 2005;112:3930–3936. DOI: 10.1161/CIRCULATIONAHA.105.588962. [DOI] [PubMed] [Google Scholar]
- 26. Miller MR, Raftis JB, Langrish JP, McLean SG, Samutrtai P, Connell SP, Wilson S, Vesey AT, Fokkens PHB, Boere AJF, et al. Inhaled nanoparticles accumulate at sites of vascular disease. ACS Nano. 2017;11:4542–4552. DOI: 10.1021/acsnano.6b08551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lenhard W, Lenhard A. (2016) Calculation of Effect Sizes. Available at: https://www.psychometrica.de/effect_size.html. Dettelbach (Germany): Psychometrica. Accessed October 20, 2020. [Google Scholar]
- 28. Grassi G, Seravalle G, Brambilla G, Pini C, Alimento M, Facchetti R, Spaziani D, Cuspidi C, Mancia G. Marked sympathetic activation and baroreflex dysfunction in true resistant hypertension. Int J Cardiol. 2014;177:1020–1025. DOI: 10.1016/j.ijcard.2014.09.138. [DOI] [PubMed] [Google Scholar]
- 29. Lucking AJ, Lundback M, Mills NL, Faratian D, Barath SL, Pourazar J, Cassee FR, Donaldson K, Boon NA, Badimon JJ, et al. Diesel exhaust inhalation increases thrombus formation in man. Eur Heart J. 2008;29:3043–3051. DOI: 10.1093/eurheartj/ehn464. [DOI] [PubMed] [Google Scholar]
- 30. Salvi S, Blomberg A, Rudell B, Kelly F, Sandstrom T, Holgate ST, Frew A. Acute inflammatory responses in the airways and peripheral blood after short‐term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med. 1999;159:702–709. DOI: 10.1164/ajrccm.159.3.9709083. [DOI] [PubMed] [Google Scholar]
- 31. Behndig AF, Mudway IS, Brown JL, Stenfors N, Helleday R, Duggan ST, Wilson SJ, Boman C, Cassee FR, Frew AJ, et al. Airway antioxidant and inflammatory responses to diesel exhaust exposure in healthy humans. Eur Respir J. 2006;27:359–365. DOI: 10.1183/09031936.06.00136904. [DOI] [PubMed] [Google Scholar]
- 32. Widdicombe J, Lee LY. Airway reflexes, autonomic function, and cardiovascular responses. Environ Health Perspect. 2001;109(Suppl 4):579–584. DOI: 10.1289/ehp.01109s4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wallin BG, Esler M, Dorward P, Eisenhofer G, Ferrier C, Westerman R, Jennings G, et al. Simultaneous measurements of cardiac noradrenaline spillover and sympathetic outflow to skeletal muscle in humans. J Physiol. 1992;453:45–58. DOI: 10.1113/jphysiol.1992.sp019217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heusser K, Tank J, Holz O, May M, Brinkmann J, Engeli S, Diedrich A, Framke T, Koch A, Großhennig A, et al. Ultrafine particles and ozone perturb norepinephrine clearance rather than centrally generated sympathetic activity in humans. Sci Rep. 2019;9:3641. DOI: 10.1038/s41598-019-40343-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Steiner S, Bisig C, Petri‐Fink A, Rothen‐Rutishauser B. Diesel exhaust: current knowledge of adverse effects and underlying cellular mechanisms. Arch Toxicol. 2016;90:1541–1553. DOI: 10.1007/s00204-016-1736-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hausberg M, Somers VK. Neural circulatory responses to carbon monoxide in healthy humans. Hypertension. 1997;29:1114–1118. DOI: 10.1161/01.HYP.29.5.1114. [DOI] [PubMed] [Google Scholar]
- 37. Li H, Cai J, Chen R, Zhao Z, Ying Z, Wang L, Chen J, Hao KE, Kinney PL, Chen H, et al. Particulate matter exposure and stress hormone levels: a randomized, double‐blind. Crossover Trial of Air Purification. Circulation. 2017;136:618–627. DOI: 10.1161/CIRCULATIONAHA.116.026796. [DOI] [PubMed] [Google Scholar]
- 38. Kodavanti UP. Stretching the stress boundary: Linking air pollution health effects to a neurohormonal stress response. Biochim Biophys Acta. 2016;1860:2880–2890. DOI: 10.1016/j.bbagen.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 39. Elam M, Sverrisdottir YB, Rundqvist B, McKenzie D, Wallin BG, Macefield VG. Pathological sympathoexcitation: how is it achieved? Acta Physiol Scand. 2003;177:405–411. DOI: 10.1046/j.1365-201X.2003.01080.x. [DOI] [PubMed] [Google Scholar]
- 40. Macefield VG, Wallin BG. Physiological and pathophysiological firing properties of single postganglionic sympathetic neurons in humans. J Neurophysiol. 2018;119:944–956. DOI: 10.1152/jn.00004.2017. [DOI] [PubMed] [Google Scholar]
- 41. Lambert E, Dawood T, Schlaich M, Straznicky N, Esler M, Lambert G. Single‐unit sympathetic discharge pattern in pathological conditions associated with elevated cardiovascular risk. Clin Exp Pharmacol Physiol. 2008;35:503–507. DOI: 10.1111/j.1440-1681.2008.04905.x. [DOI] [PubMed] [Google Scholar]
- 42. Rundqvist B, Elam M, Bergmann‐Sverrisdottir Y, Eisenhofer G, Friberg P. Increased cardiac adrenergic drive precedes generalized sympathetic activation in human heart failure. Circulation. 1997;95:169–175. DOI: 10.1161/01.CIR.95.1.169. [DOI] [PubMed] [Google Scholar]
- 43. Lambert EA, Schlaich MP, Dawood T, Sari C, Chopra R, Barton DA, Kaye DM, Elam M, Esler MD, Lambert GW. Single‐unit muscle sympathetic nervous activity and its relation to cardiac noradrenaline spillover. J Physiol. 2011;589:2597–2605. DOI: 10.1113/jphysiol.2011.205351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cosselman KE, Krishnan RM, Oron AP, Jansen K, Peretz A, Sullivan JH, Larsson TV, Kaufman JD. Blood pressure response to controlled diesel exhaust exposure in human subjects. Hypertension. 2012;59:943–948. DOI: 10.1161/HYPERTENSIONAHA.111.186593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Urch B, Silverman F, Corey P, Brook JR, Lukic KZ, Rajagopalan S, Brook RD. Acute blood pressure responses in healthy adults during controlled air pollution exposures. Environ Health Perspect. 2005;113:1052–1055. DOI: 10.1289/ehp.7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Langrish JP, Mills NL, Chan JK, Leseman DL, Aitken RJ, Fokkens PH, Cassee FR, Li J, Donaldson K, Newby DE, et al. Beneficial cardiovascular effects of reducing exposure to particulate air pollution with a simple facemask. Part Fibre Toxicol. 2009;6:8. DOI: 10.1186/1743-8977-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Langrish JP, Unosson J, Bosson J, Barath S, Muala A, Blackwell S, Söderberg S, Pourazar J, Megson IL, Treweeke A, et al. Altered nitric oxide bioavailability contributes to diesel exhaust inhalation‐induced cardiovascular dysfunction in man. J Am Heart Assoc. 2013;2:e004309. DOI: 10.1161/JAHA.112.004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. La Rovere MT, Pinna GD, Raczak G. Baroreflex sensitivity: measurement and clinical implications. Ann Noninvasive Electrocardiol. 2008;13:191–207. DOI: 10.1111/j.1542-474X.2008.00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grassi G, Seravalle G, Calhoun DA, Bolla GB, Giannattasio C, Marabini M, Del Bo A, Mancia G. Mechanisms responsible for sympathetic activation by cigarette smoking in humans. Circulation. 1994;90:248–253. [DOI] [PubMed] [Google Scholar]
- 50. Jones PP, Christou DD, Jordan J, Seals DR. Baroreflex buffering is reduced with age in healthy men. Circulation. 2003;107:1770–1774. DOI: 10.1161/01.CIR.0000057811.86187.88. [DOI] [PubMed] [Google Scholar]
- 51. Fagius J, Wallin BG. Long‐term variability and reproducibility of resting human muscle nerve sympathetic activity at rest, as reassessed after a decade. Clin Auton Res. 1993;3:201–205. DOI: 10.1007/BF01826234. [DOI] [PubMed] [Google Scholar]
- 52. Wallin BG, Kunimoto MM, Sellgren J. Possible genetic influence on the strength of human muscle nerve sympathetic activity at rest. Hypertension. 1993;22:282–284. DOI: 10.1161/01.HYP.22.3.282. [DOI] [PubMed] [Google Scholar]
- 53. Carter JR, Durocher JJ, Kern RP. Neural and cardiovascular responses to emotional stress in humans. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1898–R1903. DOI: 10.1152/ajpregu.90646.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carlsten C, Oron AP, Curtiss H, Jarvis S, Daniell W, Kaufman JD. Symptoms in response to controlled diesel exhaust more closely reflect exposure perception than true exposure. PLoS One. 2013;8:e83573. DOI: 10.1371/journal.pone.0083573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S3