

A large body of evidence shows that air pollution, most notably particulate matter (PM), is a serious threat to cardiovascular health at a global scale. 1 , 2 Fine PM <2.5 µm increases the risk for myocardial infarctions, strokes, heart failure, arrhythmias, and cardiovascular death after both short‐term (hours‐to‐days) and long‐term (years) exposures. Although PM levels have generally decreased in North America and Europe, air quality remains extremely poor and is actually worsening across much of the developing world (Asia and Africa). 2 One major source of exposure faced by billions of people on a daily basis (eg, commuting, near roadways, and urban settings) is traffic‐related and diesel exhaust pollution (DEP), a mixture of gases (eg, nitrogen oxides), primary (hydrocarbons, ultrafine PM <1 µm) and secondary (fine PM) aerosols, as well as nontailpipe pollutants. 3 Several studies have shown that the risk for myocardial infarction increases within only a few hours after being in traffic or following exposure to vehicular pollutants. 4 , 5 Given this heightened risk and the omnipresence of traffic, it is not surprisingly that it is the leading global risk factor for the triggering of myocardial infarctions (population‐attributable factor of 7.4%). 6
Several effector pathways (eg, vascular dysfunction, enhanced thrombogenicity, and elevated blood pressure [BP]) have been demonstrated that can explain the triggering of acute cardiovascular events by ambient PM and DEP. 1 Several mediating pathways, whereby pollutants elicit these adverse responses remote from the site of inhalation in the lungs, have also been proposed. The most long‐standing hypothesis, the genesis of systemic inflammation, has been borne out by solid experimental evidence. 1 There is also some evidence in humans that nanoparticles and/or certain constituents (eg, metals) may reach the circulation and thereby directly impart tissue damage. 1 A third and commonly implicated pathway is autonomic imbalance: augmented sympathetic nervous system (SNS) and/or decreased parasympathetic activity. 1 However, most of the evidence in humans has come from extrapolation from secondhand smoke studies 7 or from indirect inference in relation to certain responses following air pollution exposure (eg, acute vasoconstriction, increased heart rate and BP, and altered heart rate variability). 1
In this regard, the study by Rankin et al in this issue of the Journal of the American Heart Association (JAHA) should be considered a landmark investigation. 8 It is the first experiment to provide direct biological evidence that air pollutants can acutely elevate SNS activity in humans. Sixteen healthy young male nonsmokers were exposed to 10 minutes of filtered air followed by 10 minutes of filtered air and 40 minutes of high concentrations of DEP (PM, 304±7.22 µg/m3; nitrogen oxides, 3.4±0.4 ppm) by oral inhalation via facemask using a well‐validated experimental platform. Peripheral muscle sympathetic nerve activity (MSNA) was quantified real‐time during exposures by the time‐honored approach of microneurography of multiunit postganglionic fibers in the peroneal nerve. 9 DEP caused a significant elevation within 10 to 30 minutes during exposures in several MSNA metrics (burst frequency and incidence and percentage of bursts >50% of normalized height or area) compared with filtered air. Heart rate significantly increased, while there were trends for elevations in systolic and diastolic BP, likely hemodynamic consequences of SNS activation. Finally, there were no effects of DEP on respiration and heart rate variability parameters, except for a small unexpected (perhaps reflex) increase in the SD of normal‐to‐normal intervals.
There are several strengths to this study. SNS activity is notoriously difficult to characterize in humans because of its mercurial nature, multiple input determinants, discordant outflow between organs, baroreflex compensations, and changes in autonomic reflex and receptor sensitivities over time. 9 There are also limitations in most methods: some give only a blunt whole‐body assessment (eg, plasma or 24‐hour urine catecholamines), and others give an indirect or incomplete picture of complex integrated direct plus reflex responses (eg, heart rate, heart rate variability, BP changes and variability, and hemodynamic responses to autonomic blockade). Norepinephrine “spillover” techniques at the whole body or organ level have unique advantages but would be difficult to incorporate into short‐term exposure studies. Most likely this method could only be used with considerable effort to compare long‐term SNS activity levels between patients facing differences in long‐term air pollution exposures. As such, MSNA is the best available method to characterize rapid changes in autonomic activity in response to short‐term exposure. It has been well validated to represent peripheral SNS activity and is an accepted surrogate that typically mirrors autonomic outflow to cardiovascular organs. 9 The investigators should be lauded for the meticulous protocol that served to mitigate many pitfalls of this challenging technique, including reducing factors (eg, stress and activity) that could have obfuscated small MSNA changes attributable to DEP.
The importance of another key study design aspect cannot be overstated: modifications to the exposure facility, allowing for the quantification of MSNA in real‐time concomitant with DEP inhalation. In a previous double‐blind crossover study of 18 healthy older adults, MSNA was not increased by 3‐hour exposures to ultrafine carbon±ozone versus filtered air. 10 However, patients were instrumented for microneurography and examined only starting 1.5 hours after exposures. Plasma norepinephrine levels were elevated, but this was likely attributable to reduced neuronal reuptake at this remote time point. Although several factors may account for the negative results (eg, reduced toxicity of carbon ultrafine particles, older patients, exercise, or a meal), the principal explanation is likely that MSNA was not quantified during the actual exposure. Even if the air pollutants did trigger an increase in SNS outflow, it would have likely already resolved and thus gone undetected 1.5 to 2 hours later. Our own series of controlled exposures to ambient fine and coarse PM 11 , 12 supports this contention and thus the necessity to quantify autonomically mediated responses concomitant with the inhalation of pollutants. 8 We have observed short‐term changes in heart rate variability and consistent elevations in BP during the inhalation of PM, which rapidly abate or are overwhelmed by the effects of other factors, within minutes after exposure. Other researchers have also reported rapid elevations in BP during DEP inhalation, most likely reflecting SNS activation (albeit altered endothelin and NO bioavailability may also play roles). 13 , 14 , 15 Similarly, less consistent elevations in BP or serum catecholamines have been reported during postexposure periods. Standing, walking to exit the exposure facility, changing environments and ambient temperatures, noise, pain, anxiety from study procedures (eg, microneurography and blood draws), and even minor activities (eg, talking, urinating, eating, and laughing) could all influence highly labile autonomic balance enough, to obfuscate modest and transient effects of air pollution. This likely explains why the study design by Rankin et al 8 was the first to definitively show that air pollutants can elicit a short‐term increase in SNS activity in humans. Altogether, the available data support that the autonomic response to short‐term exposure is most robust (or only detectable by current technology) during the inhalation of pollutants. It also appears to be transient in nature (rapidly resolving after exposure). This is not unexpected as it follows basic physiological principles of a rapid onset and offset (ie, within seconds) of SNS activity above baseline tone in response to short‐term stimuli. 9
There were also a few limitations in the study by Rankin et al, 8 most notably the lack of a double‐blind crossover placebo (filtered air) design. However, this and other shortcomings were well discussed by the researchers and do not require elaboration here. 8 Conversely, several unresolved questions deserve further attention. First, the role of the baroreflex in buffering versus potentiating the autonomic hyperactivity (eg, attributable to blunted sensitivity, as can occur with secondhand smoke 7 ) remains unknown. Second, patient susceptibility factors, such as age, diet and fitness, smoking, medications (eg, α or β blockers), and disorders associated with baseline SNS activation (obesity, hypertension, and heart failure) are not known. Third, the dose response‐relationship and the culprit pollutants (PM constituents and gases) need to be elucidated. Likely because DEP concentrations were held relatively constant by Rankin et al, no correlations were observed between any pollutant(s) and MSNA. 8 Prior studies by members of this same group have shown that PM components of DEP were solely responsible for other adverse cardiovascular responses (eg, vascular dysfunction). 16 Coupled with our own findings, 11 , 12 we believe this will likely also prove true for MSNA.
Another important issue that cannot be addressed by any short‐term study is whether SNS hyperactivity persists over longer‐term exposures. Indirect evidence from one study of the impact of months of harvesting burnt sugar cane in Brazil showed trends toward long‐term elevations in MSNA. 17 However, the effects attributable solely to air pollutants could not be established. Exposures to higher year‐long fine PM levels in 2 urban US locations have been linked to small elevations in overnight urinary epinephrine levels in 1002 adults. 18 Reductions in PM exposures using indoor air filters among students residing in Shanghai led to decreases in serum catecholamines, along with other stress hormones, within a few days. 19 Although only a few studies have been published with suggestive but mixed results, the overall evidence to date tends to support that SNS activation endures (at least to some degree) over long‐term periods, so long as ongoing exposures persist. More definitive evaluations are warranted to verify this contention for several reasons beyond advancing scientific knowledge alone. Persistent SNS activation could be playing a mechanistic role in the development of chronic diseases linked to air pollution (hypertension, diabetes mellitus, and heart failure). 1 It is therefore possible that approaches known to reduce chronic SNS activity (eg, weight loss, exercise, and meditation) might prove to be viable long‐term preventive strategies and/or antisympathomimetic medications might be helpful when used prophylactically, such as for high‐risk people during travel to regions with poor air quality. 20
A useful construct to visualize the mechanisms responsible for activation of the SNS in response to air pollution may be to consider that the relevant pathways for initiation and maintenance likely differ according to the temporal windows of exposure (Figure). Although some of pathways are well validated, others remain largely speculative. 1 Experimental animal models have shown that the short‐term triggering of autonomic reflex arcs may follow the inhalation of pollutants and chemicals by activation of a variety of irritant receptors in the lung. 1 Long‐term PM exposure, akin to a high‐fat diet, may lead to persistent activation of the SNS through inflammation in the arcuate nucleus of the hypothalamus via nuclear factor‐κB pathways and presynaptic α 2A receptors in the brain, resulting in increased efferent sympathetic outflow and persistence of hypertension. 1 Air pollutants have also been shown to increase endothelin‐1, angiotensin II, as well as circulating adrenal catecholamine and corticosteroid levels by several mechanisms. 1 Each in turn is known to potentiate sympathetic neural activity at varied anatomic levels and could theoretically play a role in PM‐induced SNS hyperactivity or its maintenance over subacute periods. Recent data suggest that short‐term ultrafine PM and ozone exposure may raise plasma norepinephrine levels by reducing presynaptic neuronal reuptake and degradation. 10 It is possible that inhaled nanoparticles or metals that reach the systemic circulation may be responsible for this as they have been shown to alter ion channels and receptor signaling. Recent data also show that air pollutants can unfavorably alter the gastrointestinal microbiome. There are well‐described enteric‐to‐central nervous system efferent nerves, and it is conceivable that air pollutants might chronically increase SNS activity in this manner. We also cannot exclude the possibility of other undiscovered pathways. Further research is warranted to better understand the complex biological relationships between air pollution and the SNS. More important, failing to account for acute and possibly chronic autonomic effects of ubiquitous air pollutants could confound investigations into the biology of the SNS in many other scientific fields.
Figure 1. Potential pathways responsible for air pollution–induced autonomic imbalance and the health consequences.
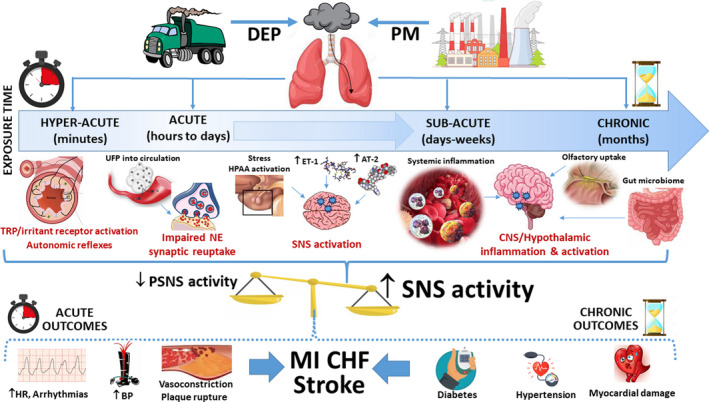
AT‐2 indicates angiotensin‐2; BP, blood pressure; CHF, congestive heart failure; CNS, central nervous system; DEP, diesel exhaust pollution; ET‐1, endothelin‐1; HPAA, hypothalamic pituitary adrenal axis; HR, heart rate; MI, myocardial infarction; NE, norepinephrine; PM, particulate matter; PSNS, parasympathetic nervous system; SNS, sympathetic nervous system; TRP, transient receptor potential; and UFP, ultrafine particle.
In closing, the fact that DEP can activate the SNS within minutes of exposure has important public health and regulatory implications as it further bolsters the mechanistic foundation for many epidemiologic studies that have linked traffic exposure 4 , 5 and/or inhalation of DEP (eg, ultrafine PM) 5 , 16 with adverse cardiovascular events. 11 , 13 , 14 , 15 , 16 This study may thus serve as an exhortation for reconsideration of current air pollution standards that target only daily and annual levels of fine PM, do not regulate ultrafine particles, and only consider subdaily windows of exposure (1–8 hours long) for gaseous pollutants (nitrogen oxides, ozone, and sulfur dioxide), largely to protect against pulmonary health effects. Regulation of ultrafine particle concentrations and/or shorter temporal windows of fine PM averages have been matters of debate in the past, including society‐wide logistical difficulties of monitoring and achieving improvements. However, mounting evidence, 1 , 4 , 5 , 11 , 12 , 13 , 14 , 15 , 16 including the new findings by Rankin et al, 8 supports that some action in this regard may be required to optimally protect the most susceptible and vulnerable individuals from the health effects of air pollution.
Disclosures
None.
(J Am Heart Assoc. 2021;10:e021675. DOI: 10.1161/JAHA.121.021675.)
The opinions expressed in this article are not necessarily those of the editors or of the American Heart Association.
For Disclosures, see page 4.
See Article by Rankin et al.
References
- 1. Rajagopalan S, Al‐Kindi SG, Brook RD. Air pollution and cardiovascular disease: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2018;72:2054–2070. DOI: 10.1016/j.jacc.2018.07.099. [DOI] [PubMed] [Google Scholar]
- 2. Risk Factors Collaborators global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396:1223–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.HEI Panel on the Health Effects of Traffic‐Related Air Pollution. 2010. Traffic‐related air pollution: a critical review of the literature on emissions, exposure, and health effects. HEI Special Report 17. https://www.healtheffects.org/publication/traffic‐related‐air‐pollution‐critical‐review‐literature‐emissions‐exposure‐and‐health. Accessed March 24, 2021.
- 4. Peters A, von Lot S, Heier M, Trentingalia I, Hormann A, Wichmann E, Lowel H. Exposure to traffic and the onset of myocardial infarction. N Engl J Med. 2004;351:1721–1730. DOI: 10.1056/NEJMoa040203. [DOI] [PubMed] [Google Scholar]
- 5. Chen K, Schneider A, Cyrys J, Wolf K, Meisinger C, Heier M, von Scheidt W, Kuch B, Pitz M, Peters A, et al. Hourly exposure to ultrafine particle metrics and the onset of myocardial infarction in Augsburg, Germany. Environ Health Perspect. 2020;128:17003. DOI: 10.1289/EHP5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nawrot TS, Perez L, Kunzli N, Munters E, Nemery B. Public health importance of triggers of myocardial infarction: a comparative risk assessment. Lancet. 2011;377:732–740. DOI: 10.1016/S0140-6736(10)62296-9. [DOI] [PubMed] [Google Scholar]
- 7. Middlekauff HR, Park J, Moheimani RS. Adverse effects of cigarette and noncigarette smoke exposure on the autonomic nervous system: mechanisms and implications for cardiovascular risk. J Am Coll Cardiol. 2014;64:1740–1750. DOI: 10.1016/j.jacc.2014.06.1201. [DOI] [PubMed] [Google Scholar]
- 8. Rankin GD, Kabele M, Brown R, Macefield V, Sanstrom T, Bosson JA. Acute exposure to diesel exhaust increases muscle sympathetic nerve activity in humans. J Am Heart Assoc. 2021;10:e018448. DOI: 10.1161/JAHA.120.018448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grassi G. Assessment of sympathetic cardiovascular drive in human hypertension: achievements and perspectives. Hypertension. 2009;54:690–697. DOI: 10.1161/HYPERTENSIONAHA.108.119883. [DOI] [PubMed] [Google Scholar]
- 10. Heusser K, Tank J, Holz O, May M, Brinkmann J, Engeli S, Diedrich A, Framke T, Koch A, Großhennig A, et al. Ultrafine particles and ozone perturb norepinephrine clearance rather than centrally generated sympathetic activity in humans. Sci Rep. 2019;9:3641. DOI: 10.1038/s41598-019-40343-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brook RD, Urch B, Dvonch JT, Bard RL, Speck M, Keeler G, Morishita M, Marsik FJ, Kamal AS, Kaciroti N, et al. Insights into the mechanisms and mediators of the effects of air pollution exposure on blood pressure and vascular function in healthy humans. Hypertension. 2009;54:659–667. DOI: 10.1161/HYPERTENSIONAHA.109.130237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morishita M, Bard RL, Wang LU, Das R, Dvonch JT, Spino C, Mukherjee B, Sun Q, Harkema JR, Rajagopalan S, et al. The characteristics of coarse particulate matter air pollution associated with alterations in blood pressure and heart rate during controlled exposures. J Expo Sci Environ Epidemiol. 2015;25:153–159. DOI: 10.1038/jes.2014.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peretz A, Sullivan JH, Leotta DF, Trenga CA, Sands FN, Allen J, Carlsten C, Wilkinson CW, Gill EA, Kaufman JD. Diesel exhaust inhalation elicits acute vasoconstriction in vivo. Environ Health Perspect. 2008;116:937–942. DOI: 10.1289/ehp.11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cosselman KE, Krishnan RM, Oron AP, Jansen K, Peretz A, Sullivan JH, Larson TV, Kaufman JD. Blood pressure response to controlled diesel exhaust exposure in human subjects. Hypertension. 2012;59:943–948. DOI: 10.1161/HYPERTENSIONAHA.111.186593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Langrish JP, Unosson J, Bosson J, Barath S, Muala A, Blackwell S, Söderberg S, Pourazar J, Megson IL, Treweeke A, et al. Altered nitric oxide bioavailability contributes to diesel exhaust inhalation‐induced cardiovascular dysfunction in man. J Am Heart Assoc. 2013;2:e004309. DOI: 10.1161/JAHA.112.004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lucking AJ, Lundbäck M, Barath SL, Mills NL, Sidhu MK, Langrish JP, Boon NA, Pourazar J, Badimon JJ, Gerlofs‐Nijland ME, et al. Particle traps prevent adverse vascular and prothrombotic effects of diesel engine exhaust inhalation in men. Circulation. 2011;123:1721–1728. DOI: 10.1161/CIRCULATIONAHA.110.987263. [DOI] [PubMed] [Google Scholar]
- 17. Barbosa CMG, Terra‐Filho M, de Albuquerque ALP, Di Giorgi D, Gripi C, Negrao CE, Rondon MUPB, Martinez DG, Marcourakis T, dos Santon FA, et al. Burnt sugarcane harvesting—cardiovascular effects on a group of health workers, Brazil. PLoS One. 2012;7:e46142. DOI: 10.1371/journal.pone.0046142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hajat A, Diez Roux AV, Vastro‐Diehl C, Cosselman K, Golden SH, Haslehurst MF, Szpiro A, Vedal S, Kaufman JD. The association between long‐term air pollution and urinary catecholamines: evidence from the Multi‐Ethnic Study of Atherosclerosis. Environ Health Perspect. 2019;127:57007. DOI: 10.1289/EHP3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H, Cai J, Chen R, Zhao Z, Ying Z, Wang L, Chen J, Hao KE, Kinney PL, Chen H, et al. Particulate matter exposure and stress hormone levels: a randomized, double‐blind, crossover trial of air purification. Circulation. 2017;136:618–627. DOI: 10.1161/CIRCULATIONAHA.116.026796. [DOI] [PubMed] [Google Scholar]
- 20. Rajagopalan S, Brauer M, Bhatnagar A, Bhatt DL, Brook JR, Huang W, Munzel T, Newby D, Siegel J, Brook RD. Personal‐level protective actions against particulate matter air pollution exposure: a scientific statement from the American Heart Association. Circulation. 2020;142:e411–e431. DOI: 10.1161/CIR.0000000000000931. [DOI] [PubMed] [Google Scholar]