

Abstract
Cardiac amyloidosis (CA) is an increasingly recognized cause of heart failure, arrhythmias, and sudden cardiac death. While CA was previously rapidly fatal, recent advances in diagnosis and treatment have significantly improved outcomes. Advances in cardiac imaging and biomarkers have critically improved the accuracy and efficiency with which CA is diagnosed, even allowing for the noninvasive diagnosis of transthyretin CA. Cardiac magnetic resonance imaging, technetium nuclear imaging, echocardiography, and blood‐based biomarkers have established important and complementary roles in the management and advancement of care. At the same time, the development of novel targeted amyloid therapies has allowed patients with CA to live longer and potentially achieve better quality of life. Still, despite this significant progress, there remain critical ongoing questions in the field. Accordingly, within this review we will highlight recent advances in cardiac imaging and therapeutics for CA, while focusing on key opportunities for further optimization of care and outcomes among this growing population. Specifically, we will discuss ongoing debates in the diagnosis of CA, including the interpretation of indeterminate cardiac imaging findings, the best technique to screen asymptomatic transthyretin amyloidosis gene mutation carriers for cardiac involvement, and the ideal method for monitoring response to CA treatment. We will additionally focus on recent advances in treatment for transthyretin amyloidosis‐CA, including a discussion of available agents as well as highlighting ongoing clinical trials. Together, these data will allow clinicians to emerge with a greater understanding of the present and future of diagnosis, management, and potentially enhanced outcomes in this rapidly advancing field.
Keywords: cardiac amyloidosis, cardiac imaging, cardio‐oncology, targeted therapies, transthyretin
Nonstandard Abbreviations and Acronyms
- AL‐CM
immunoglobulin light chain cardiomyopathy
- ATTR
transthyretin amyloidosis
- ATTR‐CM
transthyretin cardiomyopathy
- ATTRv
variant transthyretin amyloidosis
- CA
cardiac amyloid
- ECV
extracellular volume
- KCCQ
Kansas City Cardiomyopathy Questionnaire
- LGE
late gadolinium enhancement
- Tc‐DPD
technetium 3,3‐diphosphono‐1,2‐propanodicarboxylic acid
- Tc‐PyP
technetium pyrophosphate
Amyloidosis is a pathological disease process in which abnormally folded proteins deposit within various tissues, leading to marked interstitial expansion, end organ dysfunction, and damage. When it involves the heart, amyloidosis associates with particularly high morbidity and mortality. 1 , 2 , 3 The 2 most common causes of cardiac amyloidosis (CA) are transthyretin and immunoglobulin light chain proteins, which combined account for 95% of cases of CA. TTR amyloidosis (ATTR) can occur either spontaneously (ATTR wild‐type) or because of a pathologic germline mutation (ATTR variant). While previously thought to be rare, the presence of CA is increasingly recognized to be more common, with an estimated prevalence of 13% to 16% among patients presenting with heart failure with preserved ejection fraction and severe aortic stenosis. 4 , 5 , 6 Recently, the development of several drugs, which stabilize or prevent the production of the TTR protein, have dramatically improved the outcomes of patients with ATTR. 1 , 2 , 3 With effective therapies now available for ATTR, it is becoming imperative to make an early diagnosis of ATTR cardiomyopathy (ATTR‐CM) in order to streamline effective therapies. Significant advances in both cardiac imaging and blood‐based biomarkers in the field of CA have helped make this possible.
Yet, despite the recent strides there remain several key unanswered questions in the rapidly evolving field of ATTR‐CM. These include:
The evaluation of ATTR gene mutation carriers with early (preclinical) forms of CA,
The interpretation of indeterminate diagnostic findings in the presence of clinically manifested disease,
The efficacy of applying novel therapies approved for noncardiac ATTR for the control of cardiac symptoms and pathology,
The optimal methods for screening asymptomatic variant transthyretin amyloidosis (ATTRv) carriers, and
The potential (bio)markers for monitoring response to therapy in ATTR‐CM.
Accordingly, within this review we will highlight recent advances in cardiac imaging and therapeutics for CA, while focusing on key opportunities for further optimization of care and outcomes among this growing population. We will also highlight ongoing clinical trials and areas of active research within this emerging field.
Current Diagnostic and Treatment Strategies
Diagnosis of ATTR‐CM
Currently, the screening and diagnosis of CA requires a comprehensive approach with biomarkers, multimodality imaging, and invasive tissue biopsy often playing complementary roles (Table 1). Echocardiography is frequently used as an initial screening test in patients suspected to have CA. Echocardiography typically reveals increased left and right ventricular wall thickness, bi‐atrial enlargement, valvular thickening, speckled appearance of the myocardium, and/or reduced mitral annular tissue velocities (Figure 1A). 6 , 7 Two‐dimensional speckle tracking may reveal impairment in global longitudinal strain with or without relative apical sparing, a finding that is somewhat specific for CA. 8 This classical strain pattern appears to be predominantly related to regional variability in amyloid mass rather than concentration when correlated with advanced imaging. 9 Because of its tissue characterization capabilities, cardiac magnetic resonance (CMR) is particularly well suited to assess CA. Late gadolinium enhancement imaging (LGE) and extracellular volume (ECV) mapping allow for the visual and quantitative assessment of myocardial amyloid infiltration (Figure 1B). 10 In CA, LGE typically begins in a subendocardial pattern and progresses to transmural involvement in the later stages of disease. 11 The use of pre‐ and postgadolinium T1 mapping allows for the quantification of ECV—the percentage of myocardium composed of the extracellular space. ECV is markedly elevated in CA and correlates with histologic amyloid burden. 12 , 13
Table 1.
Practical Considerations in the Diagnosis and Management of Cardiac Amyloidosis
| Modality | Screening | Diagnosis/Subtyping | Staging | Hallmark Findings | Advantages | Disadvantages |
|---|---|---|---|---|---|---|
| Electrocardiography | + | − | + |
|
|
|
| Echocardiography | + | − | + |
|
|
|
| Cardiac magnetic resonance | + | − | + |
|
|
|
| Serum/urine light chain analysis | − | + | + |
|
|
|
| Tc‐PyP/DPD | + | + | − |
|
|
|
| Cardiac biomarkers (troponin, NT‐proBNP, etc) | +/− | − | + |
|
|
|
| Tissue biopsy | − | + | − |
|
|
|
AL‐CM, immunoglobulin light chain amyloidosis; ATTR, transthyretin amyloidosis; DPD, 3,3‐diphosphono‐1,2‐propanodicarboxylic acid; ECV, extracellular volume; EF, ejection fraction; LGE, late gadolinium enhancement; NT‐proBNP, N‐terminal pro‐brain‐type natriuretic peptide; PyP, pyrophosphate; and Tc, technetium.
Figure 1. Cardiac imaging features of cardiac amyloidosis.
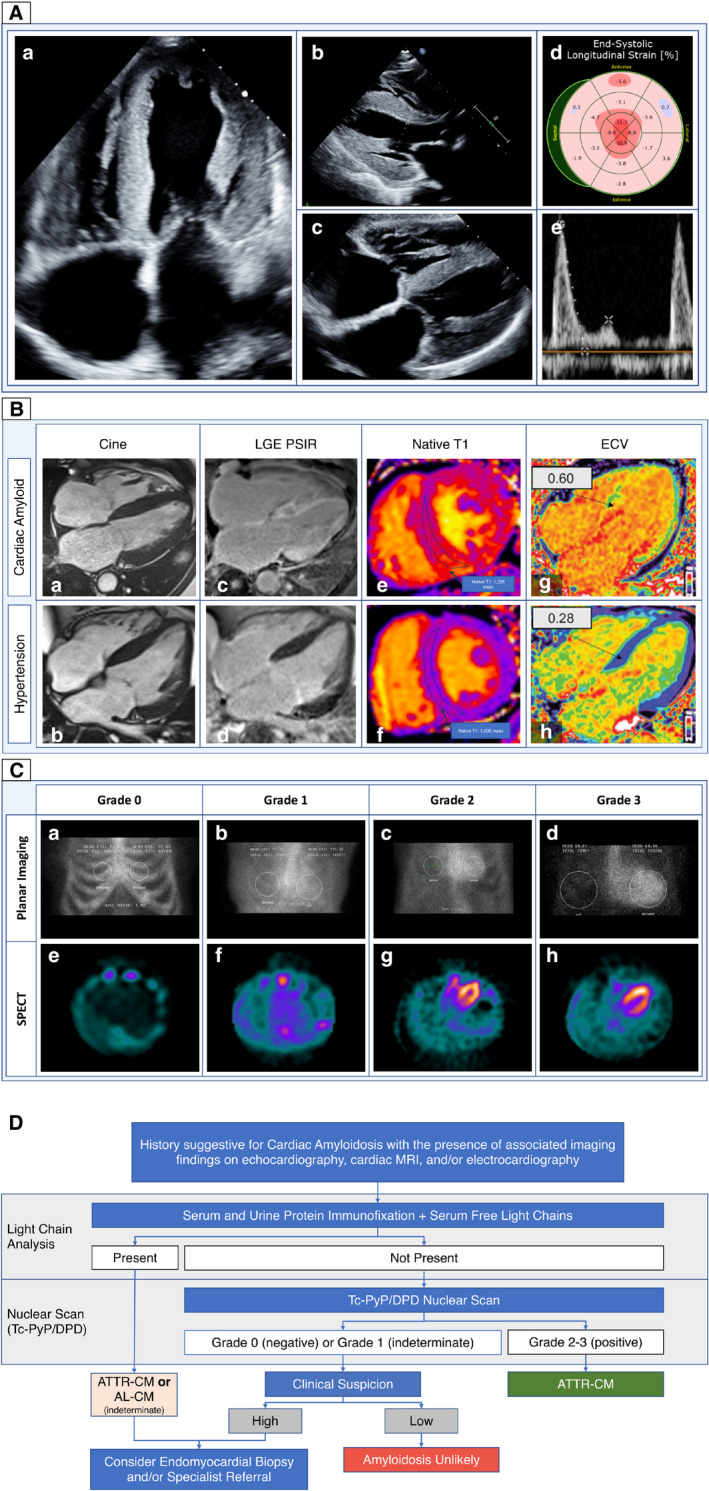
A, Echocardiographic features in cardiac amyloidosis. a and b, Classic 2‐dimensional images demonstrating increased left ventricular wall thickness with biatrial enlargement. c, Increased thickness of the right ventricle and interatrial septum. d, Globally impaired longitudinal strain with relative apical sparing (“cherry on top” pattern). e, Marked impairment in diastolic function with diminutive A‐wave suggestive of atrial contractile dysfunction. B, CMR in CA: CMR in a patient with CA ( top panel) and HHD (bottom panel). a and b, Cine images showing increased biventricular wall thickness with biatrial enlargement in both patients. With (c and d) LGE imaging demonstrating diffuse, transmural fibrosis in CA as compared with minimal replacement fibrosis in HHD. As well as (e through H) T1 mapping before (e and f) gadolinium contrast administration and ECV mapping (g and h) demonstrate markedly elevated precontrast T1 with very high ECV in CA as compared with HHD (CA indicates cardiac amyloidosis; CMR, cardiac magnetic resonance; ECV, extracellular volume; HHD, hypertensive heart disease; LGE, late gadolinium enhancement). Reprinted from Myocardial Edema and Prognosis in Amyloidosis, 71(25):2919–2931, Kotecha et al, ©2018 with permission from Elsevier. C, Technetium pyrophosphate (Tc‐PyP) in CA: planar (a through d) and SPECT (e through h) imaging in Tc‐PyP demonstrating examples of Grades 0, 1, 2, and 3 uptake. In grade 0 uptake, there is no cardiac uptake by either planar or SPECT imaging. In grade 1 uptake, there is slight cardiac uptake on planar and SPECT imaging that is exceeded by bone uptake. In grade 2, cardiac uptake exceeds bony uptake. In grade 3 uptake, there is diffuse cardiac uptake on planar and SPECT imaging with diminished bone uptake (SPECT indicates single photon emission computed tomography). D, Proposed diagnostic algorithm for suspected cardiac amyloidosis: this figure depicts the proposed algorithm for diagnosis of cardiac amyloidosis in patients suspected to have cardiac amyloidosis on the basis of suggestive clinical features and/or cardiac imaging. AL‐CM indicates immunoglobulin light chain cardiac amyloidosis; ATTR‐CM, transthyretin amyloidosis‐; and TcPyP/DPD, technetium pyrophosphate/technetium 3,3‐diphosphono‐1,2‐propanodicarboxylic acid. Reprinted from ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis: Part 1 of 2—Evidence Base and Standardized Methods of Imaging, 25(11):e1–e39, Dorbala et al, ©2019, with permission from Elsevier.
When CA is suspected on the basis of clinical or imaging findings, patients should undergo evaluation for a monoclonal gammopathy to screen for immunoglobulin light chain cardiac amyloidosis (AL‐CM). If there is no monoclonal gammopathy detected, cardiac scintigraphy with a bone‐avid radiotracer, either technetium pyrophosphate in the United States, or 3,3‐diphosphono‐1,2‐propanodicarboxylic acid in Europe, or 99mTc‐hydroxymethylene diphosphonate, can confirm the diagnosis of ATTR‐CM. If the technetium pyrophosphate or technetium 3,3‐diphosphono‐1,2‐propanodicarboxylic acid scan is equivocal or there is a monoclonal gammopathy, an invasive biopsy is required (Figure 1C). 14 A diagnosis of CA can be made on invasive biopsy when amyloid fibrils are present on Congo Red staining. 15 Amyloid subtype is then determined invasively with either laser dissection with mass spectroscopy or immunohistochemistry, with the former method being the criterion standard. 16 If ATTR is detected, genetic testing is required to evaluate for the presence of a pathologic TTR gene mutation. Recently, it has been demonstrated that a reliable diagnosis of ATTR‐CM can also be made noninvasively with 99% specificity when grade ≥2 uptake on Tc‐PyP/DPD imaging is present in the absence of a serum or urine monoclonal light chain protein (Figure 1D). 14 The widespread adoption of Tc‐PyP/DPD has greatly increased our ability to diagnose ATTR‐CM and has consequently improved patient access to potentially life‐prolonging ATTR therapies. While ATTR‐CM may be diagnosed by either nuclear scan or invasive biopsy, a diagnosis of AL‐CM requires tissue biopsy. 14 , 15 , 17 A comprehensive list of currently available diagnostics in CA is summarized in Table 1. Since prevalence of amyloidosis seldom exceeds 15% in various populations, a “Bayesian” approach might suggest that CMR is an appropriate first‐line test given its diagnostic versatility to identify relevant cardiac disease when amyloidosis is not present, and ability of ECV by CMR to quantify disease burden. Bone scintigraphy scanning does not appear well suited for tracking quantitative measures of amyloid burden serially over time; however, this is an area of growing interest. 18 , 19 , 20 Further research into optimal diagnostic pathways is needed.
Transthyretin Amyloidosis Treatment
In 2018, 3 randomized controlled clinical trials transformed the landscape of ATTR treatment. 1 , 2 , 3 On the basis of these compelling data, the US Food and Drug Administration approved tafamidis for the treatment of ATTR‐CM and both patisiran and inotersen for hereditary ATTR polyneuropathy within the year. 21 Patisiran—currently approved to treat hereditary transthyretin polyneuropathy—is currently under investigation for ATTR‐CM. These drugs act at multiple sites along the pathway of amyloid fibrilogenesis (Figure 2). Before these therapies, treatments available for transthyretin amyloidosis were primarily limited to off‐label medications, supportive care for symptoms, and/or therapies related to specific organ dysfunction. 14 However, each novel ATTR amyloidosis therapy is delivered differently with unique requirements affecting access and ease of use. 21
Figure 2. Basic mechanisms of amyloid fibril deposition, and targets of novel therapeutic interventions.
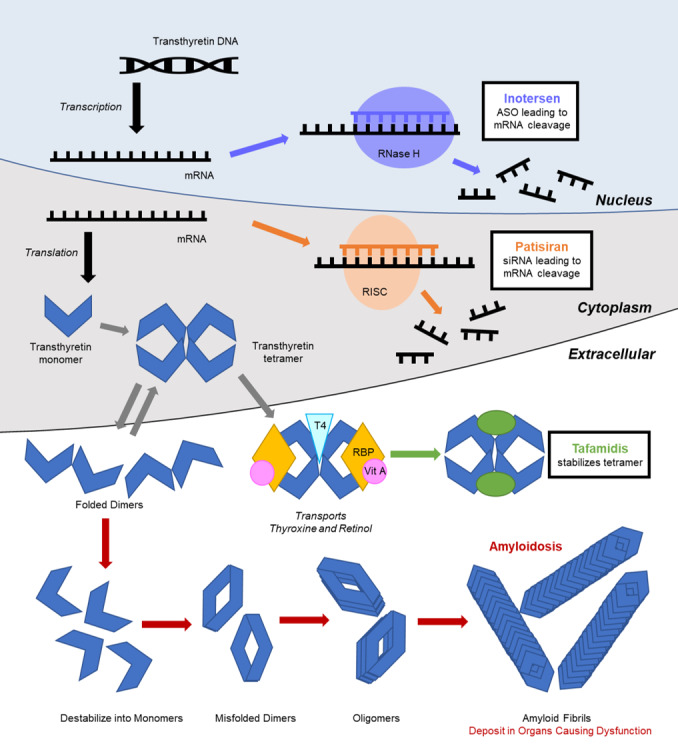
Transthyretin Stabilizing Molecule Therapies
Tafamidis—a benzoxazole derivative—is an analogue of diflunisal that lacks nonsteroidal anti‐inflammatory drug activity. It acts by binding the thyroxine binding site of transthyretin, stabilizing the tetramer and preventing the dissociation into monomers, which can subsequently misfold into amyloid fibrils (Figure 2). Tafamidis was inspired by the discovery of a stabilizing mutation in transthyretin, Thr119Met, which protects Val30Met mutation carriers from developing amyloidosis. 22 Tafamidis is taken as a daily oral medication. The ATTR‐ACT (The Transthyretin Amyloidosis Cardiomyopathy Clinical Trial) enrolled 441 patients with ATTR‐CM (≈75% wild‐type), randomizing 264 patients to tafamidis and 177 patients to placebo. 1 Patients on tafamidis had significantly lower all‐cause mortality, lower rate of cardiovascular‐related hospitalizations, and reduced decline in functional capacity/quality of life compared with placebo‐treated patients. Improvements in biomarkers were noted starting at 9 months. After 18 months, survival benefit was observed. No significant adverse events were observed. 1 In our clinical experience, some patients prefer to take the tafamidis at night because of perceptions of abdominal discomfort or lethargy associated with morning dosing. No laboratory monitoring is required.
Investigational Transthyretin‐Stabilizing Molecule Therapies
Other drugs that stabilize the transthyretin tetramer similar to tafamidis are also currently being tested. A large, phase 3 trial is currently under way to assess the effects of orally administered acoramidis (AG‐10) in preventing major adverse cardiovascular events in ATTR‐CM (Table 2). 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 In the phase 2 clinical trial, acoramidis was well tolerated and successfully reduced serum transthyretin levels. 34
Table 2.
Summary of Ongoing Clinical Trials and Investigational Drugs in Familial Amyloid Polyneuropathy and Transthyretin Cardiac Amyloidosis
| Mechanism of Action | Drug (Route of Administration) | FDA Approval | Strength of Supportive Evidence | Active Clinical Trials | |
|---|---|---|---|---|---|
| Cardiomyopathy | Neuropathy | ||||
| SiRNA | Patisiran (IV) | … | ✓ | Large RCT |
|
| |||||
| Vutrisiran (SQ) | … | … | Ongoing RCT | ||
| ASO | Inotersen (IV) | … | ✓ | Large RCT |
|
| AKCEA‐TTR‐LRx (SQ) | … | … | Ongoing RCT |
|
|
| Transthyretin stabilizer | Tafamidis (PO) | ✓ | … | Large RCT |
|
| AG‐10 (PO) | … | … | Ongoing RCT |
|
|
| |||||
| Diflunisal (PO) | … | … | Observational |
|
|
| Amyloid fibril disassembly | Doxycycline (PO) | … | … | Observational |
|
| Ursodiol (TUDCA, PO) | … | … | Observational | ||
| Green tea extract (PO) | … | … | Observational |
|
|
ASO indicates antisense oligonucleotide; ATTR, transthyretin amyloidosis; CM, cardiomyopathy; FDA, US Food and Drug Administration; hATTR, hereditary ATTR; siRNA, small interfering ribonucleic acid; N/A , Not Applicable; RCT, randomized clinical trial; and TUDCA, tauroursodeoxycholic acid.
Inhibitory Oligonucleotide Therapies: Patisiran and Inotersen
Patisiran and inotersen are both oligo‐nucleotide‐based therapeutic agents that target the 3' untranslated region of the transthyretin mRNA. 35 Patisiran originally circulates as a double‐stranded RNA molecule, subsequently dissociating into single‐stranded molecules in the cytoplasm. The sense strand then functions as a small interfering RNA, binding the transthyretin mRNA to form an RNA‐induced silencing complex, which is subsequently cleaved, inhibiting synthesis of the transthyretin protein (Figure 2). These agents result in decreased levels of both normal and mutant transthyretin proteins. Inotersen is an antisense oligonucleotide that binds to the target mRNA in the nucleus. This complex is then degraded by ribonuclease H (Figure 2).
Patisiran is an intravenous medication delivered every 3 weeks. To reduce the risk of an infusion reaction, patients are premedicated with a corticosteroid, acetaminophen, and antihistamines (H1 and H2 blockers). The effects of patisiran in hereditary amyloid neuropathy were studied in the APOLLO (The Study of an Investigational Drug, Patisiran [ALN‐TTR02], for the Treatment of Transthyretin [TTR]‐Mediated Amyloidosis) trial, which randomized 148 patients to patisiran, and 77 patients to placebo. 2 Patisiran‐treated patients had significant improvements in both neuropathy scores and gait speed compared with baseline, while the control arm progressively worsened in both respects. 2 In a subgroup analysis of patients with ATTR‐CM, patisiran use was associated with reductions in left ventricular wall thickness, NT‐proBNP (N‐terminal B‐type natriuretic peptide), and even a composite end point of cardiovascular hospitalization or mortality. 36
The most common adverse event in this study was an infusion reaction, which occurred in 19% of patients receiving patisiran as compared with 9% in the placebo group. 2 Therefore, after receiving the drug, patients must be monitored for symptoms of infusion reactions, which can include flushing, back pain, nausea, abdominal pain, shortness of breath, or headache. While some insurances cover home infusions, most patients must travel to an infusion center to receive the medication, limiting its widespread availability. Additionally, transthyretin aids in vitamin K transport via a complex with vitamin A. Therefore, all patients treated with patisiran should be prescribed vitamin A supplementation to avoid deficiencies, which could result in ocular toxicity. No laboratory monitoring is required with this medication.
Inotersen is administered as a weekly subcutaneous injection. After observed administration by a healthcare professional for the first dose, patients can self‐administer the injection at home. Inotersen was also studied in the NEURO‐TTR (Efficacy and Safety of Inotersen in Familial Amyloid Polyneuropathy) trial, which randomized 172 patients with hereditary transthyretin polyneuropathy to inotersen versus placebo. 3 Inotersen use was associated with greater stabilization in neuropathy scores as compared with the placebo. In the inotersen group, 3% of patients developed glomerulonephritis and 3% developed thrombocytopenia. Because of adverse events in this clinical trial, patients taking inotersen require serial monitoring of platelet counts and renal and hepatic function. In a subsequent study of patients with ATTR‐CM, inotersen was shown to halt disease progression and even led to reductions in left ventricular mass on echocardiography in some patients. 3 Unfortunately, a recent trial of inotersen in patients with ATTR‐CM is currently on hold because of side effects of glomerulonephritis and thrombocytopenia. 27
Investigational Small Inhibitory RNA Molecule Therapies
In addition to intravenous small interfering RNA, several clinical trials are currently under way to study small interfering RNA vutrisiran for treatment of ATTR. The HELIOS‐A (A Study of Vutrisiran [ALN‐TTRSC02] in Patients With Hereditary Transthyretin Amyloidosis [hATTR Amyloidosis]) and HELIOS‐B are 2 ongoing clinical trials studying the safety and efficacy of vutrisiran in patients with hereditary transthyretin polyneuropathy and ATTR‐CM (Table 2). 25 , 26 A major advantage of vutrisiran is that it only requires quarterly injections in comparison to inotersen, which must be administered weekly. In addition, the phase 3 CARDIO‐TTRansform (A Study to Evaluate the Efficacy and Safety of AKCEA‐TTR‐LRx in Participants With Transthyretin‐Mediated Amyloid Cardiomyopathy [ATTR CM]) trial is currently assessing the effects of the antisense oligonucleotide molecule ION‐682884 (AKCEA‐TTR‐LRx) on survival and major adverse cardiovascular events in patients with ATTR‐CM (Table 2). 28 The ION 682884 compound shares the same parent compound as inotersen but is instead conjugated to GalNAc. Unfortunately, another clinical trial of the small interfering RNA drug, revusiran, was stopped early because of increased observed mortality in the treatment arm. 37
Off‐Label Amyloidosis Therapies
In addition to the aforementioned novel drugs, several off‐label therapies have also been studied in ATTR. Expert support for these agents is mixed and their use is currently at the discretion of the treating physician.
Diflunisal is a generic nonsteroidal anti‐inflammatory drug that can stabilize TTR tetramers. A randomized placebo‐controlled trial demonstrated efficacy of diflunisal in reducing the rate of progression of hereditary transthyretin polyneuropathy. 38 A retrospective study in patients with ATTR‐CM demonstrated some evidence of cardiac stabilization. 39
Doxycycline and ursodiol (tauroursodeoxycholic acid) were shown to disaggregate formed amyloid fibrils in in vitro studies. 40 In an open label study, no cardiomyopathy or neuropathy progression was observed in 7 patients after 1 year of receiving this combination. 41 A retrospective study of 53 patients with ATTR‐CM on this therapy showed no significant cardiac disease progression over 22 months. 42 Larger prospective, randomized control trials are currently under way to assess the safety and efficacy of combined doxycycline and tauroursodeoxycholic acid in ATTR and ATTR‐CM (Table 2). 32 , 33
Epigallocatechin 3‐gallate, the most abundant catechin in green tea, demonstrated disruption of transthyretin amyloid fibrils in mouse study. 43 One study of 25 patients with wild‐type transthyretin amyloidosis‐CM treated with 600 mg epigallocatechin 3‐gallate daily for 1 year showed a decrease in left ventricular mass without change in left ventricular wall thickness by CMR, 44 but ECV was not reported.
Affordability of Targeted Amyloid Therapies
With the current and expected future availability of therapies for ATTR‐CM that are both efficacious and expensive, the question of whether these treatments are cost effective is highly important. Currently, the average annual costs of tafamidis, patisiran, and inotersen are a staggering $225 000, $350 000, and $450 000, respectively. In cost–benefit analysis of the ATTR‐ACT trial, tafamidis was not found to be cost effective at its current price with a cost per adjusted quality‐of‐life year of $1 135 000. 45 Although the efficacies of patisiran and inotersen for ATTR‐CM are not yet known, similar concerns regarding cost effectiveness are likely to occur at their current prices. This creates an ethical dilemma wherein the equitable delivery of potentially life‐prolonging therapies may be limited by drug costs. This has profound implications when one accounts for the increasing recognition of the high prevalence of ATTR‐CM. 4 , 5 , 6 It is therefore likely that either reductions in drug prices or the development of less expensive novel agents will be necessary before targeted ATTR‐CM therapies can become widely available.
Emerging Challenges and Dilemmas
CA is diagnosed and staged using a multimodality approach combining cardiac imaging, biomarkers, and occasionally invasive tissue biopsy. Echocardiography and CMR have an important role in screening patients suspected to have CA based on clinical features. ECV by CMR is highly reproducible and can track amyloidosis disease burden serially to assess therapeutic response. 10 , 46 , 47 Tc‐PyP/DPD and light chain analysis are important in screening for CA and in helping to determine amyloid subtype. Invasive tissue biopsy is required in immunoglobulin light chain; however, in ATTR, invasive biopsy is generally reserved when the initial testing is inconclusive. While this approach successfully guides therapy for most patients, there remain key gaps in our current understanding. These include growing awareness that indeterminate or discrepant findings can be seen with either cardiac imaging studies or blood biomarkers, and can complicate the diagnosis of CA. Additionally, the optimal method of screening asymptomatic ATTRv carriers and monitoring response to amyloid treatment is currently poorly understood.
Interpreting Indeterminate Cardiac Imaging
While echocardiography and CMR are highly valuable modalities in the evaluation of suspected CA, each has its limitations. Many of the studies that established the high diagnostic accuracy of CMR and echocardiography were performed at referral centers or in patients with classic CA, introducing the possibility of referral bias and questions on sensitivity among more heterogeneous populations and community‐based healthcare systems. It is unclear whether these tests would be as accurate in routine clinical practice where the prevalence of CA is much lower. Echocardiography in particular can be negative in early‐stage CA. In a small study of 12 asymptomatic ATTRv carriers without evidence of CA on echocardiogram, Tc‐PyP uptake was still seen in 84% of cases. 48 Additionally, echocardiography cannot reliably differentiate CA from other causes of pathologic left ventricular hypertrophy such as hypertensive heart disease, hypertrophic cardiomyopathy, and other infiltrative disorder that must be carefully excluded.
While CMR is more sensitive than echocardiography given its tissue characterization capabilities, there are currently a lack of definitive data comparing these 2 modalities in ATTR. Although elevated myocardial ECV and diffuse LGE on CMR are classical features of CA, these may not be present in early‐stage disease. In early CA, only faint LGE and mild elevations in ECV may be present, which may sometimes make it difficult to differentiate from myocardial fibrosis (eg, from hypertensive heart disease). Mild elevations in ECV and LGE may also be seen in hypertensive heart disease because of the presence of diffuse and focal fibrosis. 49
In our clinical practice, we find echocardiography and CMR to be highly useful but not infallible screening tools in patients suspected to have CA. When positive, these tests can reliably guide further testing. When pre‐test probability is low, an unremarkable echocardiogram and/or CMR is often sufficient to exclude CA. However, because of the potential for lack of sensitivity, when pre‐test probability is high, we frequently pursue Tc‐PyP/DPD and light chain analysis, even when CMR or echocardiography appear normal to detect the earliest stages of disease where early treatment may matter most.
Interpreting Indeterminate Findings in Amyloid Subtyping
In some cases, it may be difficult to determine amyloid subtype based on the results of the initial testing. For example, ATTR‐CM cannot be diagnosed noninvasively with Tc‐PyP/DPD when a monoclonal serum or urine light chain is present. Up to 20% of patients with AL‐CM may have false‐positive uptake on Tc‐PyP/DPD. 14 In fact, the most common error in diagnosing CA is failure to exclude AL‐CM when Tc‐PyP/DPD uptake is present. 50 Serum protein electrophoresis alone is insufficient to rule out AL‐CM and may be negative in more than half of cases. 51 Light chain analysis should include serum free light chains and both serum and urine protein electrophoresis with immunofixation. This combination can reliably exclude AL‐CM with 99.8% certainty. 51 Additionally, 10% of patients with ATTR‐CM also have an incidental monoclonal gammopathy of undetermined significance unrelated to their CA diagnosis. 52 Therefore, when both grade ≥2 uptake on Tc‐PyP/DPD and monoclonal light chain are both present, invasive biopsy is required to establish subtype.
One must also be careful to avoid pitfalls in interpreting Tc‐PyP/DPD scans. A common cause of false‐positive Tc‐PyP/DPD and referrals to amyloid centers is blood pool uptake. High concentrations of Tc‐PyP/DPD in the blood pool can mimic myocardial uptake on planar imaging and may occur particularly in low cardiac output states. 50 This can be excluded with the use of single photon emission computed tomography imaging to confirm that uptake is of myocardial and not blood pool origin. False‐positive Tc‐PyP scans have also been reported to occur with cardiotoxicity from hydroxychloroquine. 53 While the sensitivity for Tc‐PyP appears to be superior to echocardiography for detecting ATTR‐CM, false negatives can also occur. 48 For example, of the 111 patients with grade 0/1 uptake on Tc‐PyP/DPD in the Gilmore et al study, 18% were ultimately found to have ATTR‐CM on biopsy. 14 Among patients with just grade 1 uptake on Tc‐PyP/DPD, the prevalence of ATTR‐CM was even higher at 46%. False‐negative scans can also occur, particularly in patients with earlier cardiac involvement or in those with specific mutations such as Phe64Leu and Val30Met. 50 , 54 Therefore, one should be somewhat careful in excluding ATTR‐CM solely on the basis of Tc‐PyP/DPD, particularly when grade 1 uptake is present and a high clinical suspicion remains.
At our institution, when findings on Tc‐PyP and light chain analysis are indeterminate, we generally proceed with endomyocardial biopsy. When pre‐test probability is high, we may sometimes proceed with endomyocardial biopsy even despite the absence of Tc‐PyP uptake, given the possibility of false‐negative results, particularly with grade 1 uptake. In some instances, we may also opt to follow patients with a repeat CMR or Tc‐PyP scan 6 to 12 months later to evaluate for the subsequent development of cardiac involvement.
Surveillance of the Asymptomatic ATTRv Carrier
Genetic testing is indicated in all patients diagnosed with ATTR‐CM to screen for a pathologic gene mutation (ATTRv). The most commonly identified pathologic ATTR gene mutations worldwide are Val30Met, Val122Ile, and Thr60Ala. 55 Genetic testing has implications not just for the patient but also for family members in whom identification of a pathologic ATTRv mutation can help facilitate early diagnosis and treatment. In patients with ATTR‐CM, initiation of therapy at an earlier stage of illness is associated with better outcomes. In subgroup analysis of the ATTR‐ACT study, tafamidis demonstrated net benefit in both survival and cardiac hospitalizations among patients with baseline New York Heart Association class I–II heart failure symptoms. 1 Conversely, in those with New York Heart Association Class III symptoms, increased rates of hospitalizations were observed in the tafamidis‐treated group. Based on these data, the US Food and Drug Administration has approved the use of tafamidis for all patients with ATTR‐CM regardless of symptoms. Based on this evidence, a large multimodality imaging consensus statement considered it reasonable to screen asymptomatic carriers of pathologic ATTRv gene mutations for cardiac involvement. 15
Currently, the ideal method and intervals for screening ATTRv carriers remain unknown. Yet, it may be reasonable to screen ATTRV carriers before the expected age of onset of cardiomyopathy for their particular mutation. For patients with the Val122Ile, Val30Met, and Th60Ala mutations, onset of cardiomyopathy is typically beyond 65, 50, and 45 years, respectively. 56 In comparison, cardiomyopathy usually occurs later in wild‐type transthyretin amyloidosis beyond the sixth decade of life.
ATTRv carriers may be screened for cardiac involvement with serum biomarkers, electrocardiography, echocardiography, CMR, and/or Tc‐PyP/DPD. Biomarkers such as NT‐proBNP and troponin are an attractive choice because they are inexpensive and widely available. NT‐proBNP levels appear to be higher in ATTRv carriers with cardiac involvement as compared with asymptomatic ATTRv carriers. 48 , 57 An ECG may be obtained to screen for low‐voltage pattern. However, both biomarkers and ECG lack sensitivity and may be negative, particularly in early‐stage disease. 48 , 58
Echocardiography and CMR have been demonstrated to have excellent sensitivity and specificity in symptomatic patients; however, there is a paucity of data assessing their use in screening ATTRv carriers. The modality with the most robust evidence in support of screening in ATTRv carriers appears to be Tc‐PyP/DPD. Among ATTRv carriers, cardiac involvement is frequently observed using Tc‐PyP and may precede echocardiographic evidence of disease by 1 to 2 years. 48 , 59 One concern with repeated Tc‐PyP scans is the theoretical risk of cancer related to repeated exposure to ionizing radiation. While the risk associated with a single Tc‐PyP scan appears to be low, the effects of serial scans—particularly in younger patients—is currently unknown. 60
Despite this, based on the available evidence, we generally begin screening ATTRv carriers 5 to 10 years before the expected age of onset of cardiomyopathy based on their gene mutation and family history. We typically screen all ATTRv carriers initially referred to our clinic with troponin, NT‐proBNP, ECG, a Tc‐PyP scan, an echocardiogram, and frequently a CMR. When cardiac involvement is not present, we often repeat biomarkers, an ECG, and either an echocardiogram or a Tc‐PyP scan annually. When a strong family history of cardiomyopathy or indeterminate imaging findings such as grade I uptake on Tc‐PyP are present, we preferentially perform surveillance with a Tc‐PyP/DPD scan given its superior sensitivity when compared with echocardiography. In our comprehensive amyloid clinic, we also work collaboratively with hematology, nephrology, and neurology departments to monitor for signs of other end‐organ manifestations such as neuropathy and nephropathy.
Monitoring Response to Therapy
Another current gap in our understanding is how best to monitor response to therapy in patients receiving treatment for CA. Multiple markers may be used for this purpose including clinical parameters, cardiac biomarkers, and cardiac imaging. There is evidence from recent clinical trials to support all of these methods.
Functional capacity and patient quality of life appear to be impacted by treatment of ATTR‐CM. 1 In the ATTR‐ACT study, tafamidis was found to attenuate declines in both 6‐minute walk test and Kansas City Cardiomyopathy Questionnaire as compared with placebo. 1 One criticism of patient‐centered outcomes such as the Kansas City Cardiomyopathy Questionnaire is that they are nonspecific. It may therefore be difficult to determine whether improvements in these markers are because of changes in cardiac, nerve, or other end‐organ functioning. As a result, their role in clinical practice is currently uncertain. Improvements or stabilization in cardiac biomarkers such as NT‐proBNP have also been observed in patients with ATTR‐CM treated with patisiran and tafamidis. 1 , 36
With respect to cardiac imaging, there is evidence that echocardiography, CMR, and positron emission tomography (PET) imaging may all potentially be useful. In large randomized studies, echocardiographic parameters including left ventricular mass and wall thickness have been shown to differ between patients with ATTR‐CM treated with patisiran and inotersen as compared with placebo. 36 , 61 However, these measurements may be less reproducible because of the lower spatial resolution of echocardiography and the reliance on geometric assumptions to calculate left ventricular mass. CMR‐derived ECV is a useful marker for trending treatment response in CA since ECV quantifies the degree of expansion of the interstitium where amyloid protein resides. In patients with ATTR‐CM, ECV has been shown to decrease in response to patisiran treatment. 47 Similarly, in AL‐CM, reductions in ECV have been demonstrated in response to chemotherapy and are associated with favorable treatment response. 10 , 46 For these reasons, CMR tissue characterization parameters (eg ECV) are increasingly being incorporated in therapeutic clinical trial outcomes. Because it is the most quantitative, highly reproducible marker available, the use of ECV should be strongly considered to monitor responses to therapies, particularly in the context of research studies.
Although there is a growing body of literature regarding the quantification of bone‐avid radiotracers using calcium zinc telluride–single photon emission computed tomography, serial Tc‐PyP/DPD are not appropriate for monitoring response to therapy. 19 , 20 In a study of patients with ATTR‐CM, neither heart‐to‐contralateral lung ratio nor visual uptake changed significantly over time despite obvious clinical deterioration. 18 More recently, novel PET tracers have been developed, which have been shown to bind with high avidity to both ATTR and immunoglobulin light chain deposits including the 11c‐Pittsburgh B (11C‐PiB) and 18F‐Florbetapir compounds. 62 , 63 Unlike Tc‐PyP/DPD, PET tracer uptake can be quantified and correlates closely with amyloid burden. 62 Because of this, PET uptake with 11c‐Pittsburgh B (11C‐PiB) and 18F‐Florbetapir may be a promising way to quantitate response to therapy in CA. The sensitivity of 18F Florbetapir for detecting early cardiac involvement in immunoglobulin light chain amyloidosis may even exceed that of CMR. 64 However, the higher cost and radiation exposure with cardiac PET as compared with CMR are potential limitations to its widespread use. A prospective clinical trial is already under way to track changes in 18F‐Florbetapir uptake in patients receiving chemotherapy for AL‐CM. 65
To date, the optimal marker for assessing response to treatment in CA remains unclear. Aside from the use of ECV in AL‐CM treatment, it is also currently unknown whether changes in serial markers can predict treatment response. Future studies are needed to identify noninvasive markers, which can predict response to treatment in order to best tailor pharmacotherapy to each individual patient with CA. In the clinical setting, we generally follow patients with annual troponin, NT‐proBNP, ECG, and an echocardiogram while being treated for ATTR‐CM because they are affordable and pose no risk to the patient.
Methods to Address Diagnostic Challenges in the Field
While echocardiography, CMR, and Tc‐PyP/DPD have an established role in imaging CA, there remain gaps in our understanding of how best to apply these modalities. For example,
while the diagnostic efficacies of echocardiography, CMR, and Tc‐PyP/DPD have been well studied in patients with suspected CA, few studies have directly compared them. Further multimodality studies are needed to determine the cardiac imaging modality of choice for screening patients with suspected CA and asymptomatic ATTRv carriers. Additionally, further studies are needed to better understand the role of novel cardiac imaging techniques such as 18F‐Florbetafir PET, and hybrid PET/CMR. 65 , 66 , 67 , 68 , 69 Studies are also currently under way to assess whether serum misfolded ATTR oligomer levels can be used to noninvasively screen for cardiac involvement in ATTR. 70 Whether these contemporary techniques have a role in clinical practice or are better suited to the research setting is currently unknown.
Moreover, further research is needed to determine the ideal cardiac imaging and blood‐based biomarkers for assessing response to therapy in CA both in clinical practice as well as clinical trials. While ongoing clinical trials dedicated to this question are limited, cardiac imaging markers are now increasingly being utilized as end points in large pharmaceutical trials. 26 The increased inclusion of echocardiography, CMR, and PET parameters in amyloid clinical trials will help us to better understand the additive role of cardiac imaging in monitoring treatment response in CA.
Other Ongoing Pharmaceutical Trials
In addition to tafamidis, there are now a number of active clinical trials testing both new drugs and novel applications of existing treatments for ATTR‐CM. As discussed above, multiple studies—including the APOLLO‐B, HELIOS‐B, and CARDIO‐TTRansform trials—are currently under way to assess the efficacy of inhibitory oligonucleotide therapies including patisiran, vutrisiran, and ION‐682884 (AKCEA‐TTR‐LRx) in patients with ATTR‐CM. 23 , 26 , 28 Additionally, Phase III clinical trials are currently under way to test the novel stabilizing molecule acoramidis (AG‐10) in patients with ATTR‐CM. 31 A comprehensive list of active drug trials is summarized in Table 2. Hopefully, trials such as these will result in the availability of additional, effective therapies to treat patients with ATTR‐CM. These trials, combined with advancements in cardiac imaging and clinical experience, will enhance our ability to effectively identify and treat even less advanced forms of CA.
Conclusions
The development and approval of multiple agents that stabilize or prevent production of transthyretin protein has greatly improved outcomes in both hereditary transthyretin polyneuropathy and cardiomyopathy. Over the same period, diagnostics in the field of CA have also substantially improved. Echocardiography, CMR, and Tc‐PyP have become established tools in the diagnosis and management of CA. Novel cardiac imaging techniques including myocardial PET may also have an evolving role in CA. Hopefully in the future, we will have a better understanding of how each of the biomarkers and cardiac imaging modalities can best complement each other in the diagnosis, risk stratification, and monitoring of treatment in ATTR‐CM. While it is difficult to imagine how the speed of recent monumental gains made in the field of ATTR‐CM could be matched in the future, with an increasing number of new drugs in development as well as novel applications of cardiac imaging and biomarkers, the future appears similarly promising. With the inclusion of more precise strategies for the diagnosis, prognostication, and management of ATTR‐CM, we are poised for dramatic alterations to the care of these patients.
Sources of Funding
This work was supported in part by NIH grants P30 CA016058 and K12‐CA133250 (Addison) and by National Center for Advancing Translational Sciences TL1TR002735 grant (Campbell).
Disclosures
Dr. Addison received support from the NCI K12‐CA133250, NCI P30 CA016058, and the American Heart Association‐Robert Wood Johnson Foundation Faculty Development Program grants. Dr. Campbell receives support from the National Center For Advancing Translational Sciences (TL1TR002735). The authors have no relevant conflicts of interest to disclose.
Acknowledgments
The article's content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
(J Am Heart Assoc. 2021;10:e019840. DOI: 10.1161/JAHA.120.019840.)
This manuscript was sent to Carol Ann Remme, MD, PhD, Senior Guest Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 12.
References
- 1. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington‐Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007–1016. DOI: 10.1056/NEJMoa1805689. [DOI] [PubMed] [Google Scholar]
- 2. Adams D, Gonzalez‐Duarte A, O’Riordan WD, Yang C‐C, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:11–21. DOI: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 3. Benson MD, Waddington‐Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, Planté‐Bordeneuve V, Barroso FA, Merlini G, Obici L, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:22–31. DOI: 10.1056/NEJMoa1716793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. González‐López E, Gallego‐Delgado M, Guzzo‐Merello G, de Haro‐del Moral FJ, Cobo‐Marcos M, Robles C, Bornstein B, Salas C, Lara‐Pezzi E, Alonso‐Pulpon L, et al. Wild‐type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36:2585–2594. DOI: 10.1093/eurheartj/ehv338. [DOI] [PubMed] [Google Scholar]
- 5. Bennani Smires Y, Victor G, Ribes D, Berry M, Cognet T, Méjean S, Huart A, Roussel M, Petermann A, Roncalli J, et al. Pilot study for left ventricular imaging phenotype of patients over 65 years old with heart failure and preserved ejection fraction: the high prevalence of amyloid cardiomyopathy. Int J Cardiovasc Imaging. 2016;32:1403–1413. DOI: 10.1007/s10554-016-0915-z. [DOI] [PubMed] [Google Scholar]
- 6. Castaño A, Narotsky DL, Hamid N, Khalique OK, Morgenstern R, DeLuca A, Rubin J, Chiuzan C, Nazif T, Vahl T, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38:2879–2887. DOI: 10.1093/eurheartj/ehx350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dorbala S, Cuddy S, Falk RH. How to image cardiac amyloidosis: a practical approach. JACC Cardiovasc Imaging. 2020;13:1368–1383. DOI: 10.1016/j.jcmg.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phelan D, Collier P, Thavendiranathan P, Popović ZB, Hanna M, Plana JC, Marwick TH, Thomas JD. Relative apical sparing of longitudinal strain using two‐dimensional speckle‐tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart. 2012;98:1442–1448. DOI: 10.1136/heartjnl-2012-302353. [DOI] [PubMed] [Google Scholar]
- 9. Bravo PE, Fujikura K, Kijewski MF, Jerosch‐Herold M, Jacob S, El‐Sady MS, Sticka W, Dubey S, Belanger A, Park M‐A, et al. Relative apical sparing of myocardial longitudinal strain is explained by regional differences in total amyloid mass rather than the proportion of amyloid deposits. JACC Cardiovasc Imaging. 2019;12:1165–1173. DOI: 10.1016/j.jcmg.2018.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martinez‐Naharro A, Abdel‐Gadir A, Treibel TA, Zumbo G, Knight DS, Rosmini S, Lane T, Mahmood S, Sachchithanantham S, Whelan CJ, et al. CMR‐verified regression of cardiac AL amyloid after chemotherapy. JACC Cardiovasc Imaging. 2018;11:152–154. DOI: 10.1016/j.jcmg.2017.02.012. [DOI] [PubMed] [Google Scholar]
- 11. Fontana M, Pica S, Reant P, Abdel‐Gadir A, Treibel TA, Banypersad SM, Maestrini V, Barcella W, Rosmini S, Bulluck H, et al. Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2015;132:1570–1579. DOI: 10.1161/CIRCULATIONAHA.115.016567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Banypersad SM, Sado DM, Flett AS, Gibbs SDJ, Pinney JH, Maestrini V, Cox AT, Fontana M, Whelan CJ, Wechalekar AD, et al. Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: an equilibrium contrast cardiovascular magnetic resonance study. Circ Cardiovasc Imaging. 2013;6:34–39. DOI: 10.1161/CIRCIMAGING.112.978627. [DOI] [PubMed] [Google Scholar]
- 13. Duca F, Kammerlander AA, Panzenböck A, Binder C, Aschauer S, Loewe C, Agis H, Kain R, Hengstenberg C, Bonderman D, et al. Cardiac magnetic resonance T1 mapping in cardiac amyloidosis. JACC Cardiovasc Imaging. 2018;11:1924–1926. DOI: 10.1016/j.jcmg.2018.06.010. [DOI] [PubMed] [Google Scholar]
- 14. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, Wechalekar AD, Berk JL, Quarta CC, Grogan M, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404–2412. DOI: 10.1161/CIRCULATIONAHA.116.021612. [DOI] [PubMed] [Google Scholar]
- 15. Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, Fontana M, Gheysens O, Gillmore JD, Glaudemans AWJM, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 1 of 2‐evidence base and standardized methods of imaging. J Card Fail. 2019;25:e1–e39. [DOI] [PubMed] [Google Scholar]
- 16. Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry‐based proteomic analysis in clinical biopsy specimens. Blood. 2009;114:4957–4959. DOI: 10.1182/blood-2009-07-230722. [DOI] [PubMed] [Google Scholar]
- 17. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2019;73:2872–2891. DOI: 10.1016/j.jacc.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Castaño A, DeLuca A, Weinberg R, Pozniakoff T, Blaner WS, Pirmohamed A, Bettencourt B, Gollob J, Karsten V, Vest JA, et al. Serial scanning with technetium pyrophosphate (99m Tc‐PYP) in advanced ATTR cardiac amyloidosis. J Nucl Cardiol. 2016;23:1355–1363. DOI: 10.1007/s12350-015-0261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dorbala S, Park MA, Cuddy S, Singh V, Sullivan K, Kim S, Falk RH, Taqueti V, Skali H, Blankstein R, et al. Absolute quantitation of cardiac 99m Tc‐pyrophosphate using cadmium zinc telluride‐based SPECT/CT. J Nucl Med. 2020:jnumed.120.247312. Sep 4. DOI: 10.2967/jnumed.120.247312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bellevre D, Bailliez A, Delelis F, Blaire T, Agostini D, Mouquet F, Maréchaux S, Manrique A. Quantitation of myocardial 99m Tc‐HMDP uptake with new SPECT/CT cadmium zinc telluride (CZT) camera in patients with transthyretin‐related cardiac amyloidosis: ready for clinical use? J Nucl Cardiol. 2020. DOI: 10.1007/s12350-020-02274-2. [DOI] [PubMed] [Google Scholar]
- 21. FDA approves new treatments for heart disease caused by a serious rare disease, transthyretin mediated amyloidosis. Available at: https://www.fda.gov/news‐events/press‐announcements/fda‐approves‐new‐treatments‐heart‐disease‐caused‐serious‐rare‐disease‐transthyretin‐mediated#:~:text=On%20May%203%2C%20the%20U.S.,approved%20treatments%20for%20ATTR%2DCM. Accessed February 11, 2020.
- 22. Hammarström P, Schneider F, Kelly JW. Trans‐suppression of misfolding in an amyloid disease. Science. 2001;293:2459–2462. DOI: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 23. APOLLO‐B: a study to evaluate Patisiran in participants with transthyretin amyloidosis with cardiomyopathy (ATTR amyloidosis with cardiomyopathy). 2019. Available at: https://clinicaltrials.gov/ct2/show/NCT03997383. Accessed April 11, 2020. (identification No. NCT03997383).
- 24. Patisiran in patients with hereditary transthyretin‐mediated amyloidosis (hATTR amyloidosis ) disease progression post‐liver transplant. Available at: https://clinicaltrials.gov/ct2/show/NCT03862807. Accessed April 11, 2020. (Study No. NCT03862807).
- 25. HELIOS‐A: a study of vutrisiran (ALN‐TTRSC02) in patients with hereditary transthyretin amyloidosis (hATTR amyloidosis). Available at: https://clinicaltrials.gov/ct2/show/NCT03759379. Accessed April 11, 2020. (Study No. NCT03759379).
- 26. HELIOS‐B: a study to evaluate vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy. 2019. Available at: https://clinicaltrials.gov/ct2/show/NCT04153149. Accessed April 11, 2020. (identification No. NCT04153149).
- 27. 24 month open label study of the tolerability and efficacy of Inotersen in TTR amyloid cardiomyopathy patients. Available at: https://clinicaltrials.gov/ct2/show/NCT03702829. Accessed April 11, 2020. (Study No. NCT03702829).
- 28. CARDIO‐TTRansform: a study to evaluate the efficacy and safety of AKCEA‐TTR‐LRx in participants with transthyretin‑mediated amyloid cardiomyopathy (ATTR CM). Available at: https://clinicaltrials.gov/ct2/show/NCT04136171?cond=cardiac+amyloidosis&draw=2&rank=26. Accessed April 11, 2020. (Study No. NCT04136171).
- 29. Long‐term safety of Tafamidis in subjects with transthyretin cardiomyopathy. Available at: https://clinicaltrials.gov/ct2/show/NCT02791230. Accessed April 11, 2020. (Study No. NCT02791230).
- 30. Open‐label study of AG10 in patients with cardiomyopathy. Available at: https://clinicaltrials.gov/ct2/show/NCT03536767. Accessed February 1, 2021. (Study No. NCT03536767).
- 31. Efficacy and safety of AG10 in subjects with transthyretin amyloid cardiomyopathy (ATTRIBUTE‐CM). Available at: https://clinicaltrials.gov/ct2/show/NCT03860935. Accessed May 11, 2020. (Study No. NCT03860935).
- 32. Tolerability and efficacy of a combination of Doxycycline and TUDCA in patients with transthyretin amyloid cardiomyopathy. Available at: https://clinicaltrials.gov/ct2/show/NCT01855360?cond=cardiac+amyloidosis&draw=2&rank=19. Accessed May 11, 2020. (Study No. NCT01855360).
- 33. A study of Doxycycline and tauroursodeoxycholic acid (Doxy/TUDCA) plus standard supportive therapy versus standard supportive therapy alone in cardiac amyloidosis caused by transthyretin. Available at: https://clinicaltrials.gov/ct2/show/NCT03481972?cond=cardiac+amyloidosis&draw=2&rank=10. Accessed May 11, 2020. (Study No. NCT03481972).
- 34. Judge DP, Heitner SB, Falk RH, Maurer MS, Shah SJ, Witteles RM, Grogan M, Selby VN, Jacoby D, Hanna M, et al. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol. 2019;74:285–295. DOI: 10.1016/j.jacc.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 35. Buxbaum JN. Oligonucleotide drugs for transthyretin amyloidosis. N Engl J Med. 2018;379:82–85. DOI: 10.1056/NEJMe1805499. [DOI] [PubMed] [Google Scholar]
- 36. Solomon SD, Adams D, Kristen A, Grogan M, González‐Duarte A, Maurer MS, Merlini G, Damy T, Slama MS, Brannagan TH, et al. Effects of Patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin‐mediated amyloidosis. Circulation. 2019;139:431–443. DOI: 10.1161/CIRCULATIONAHA.118.035831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Judge DP, Kristen AV, Grogan M, Maurer MS, Falk RH, Hanna M, Gillmore J, Garg P, Vaishnaw AK, Harrop J, et al. Phase 3 multicenter study of revusiran in patients with hereditary transthyretin‐mediated (hATTR) amyloidosis with cardiomyopathy (ENDEAVOUR). Cardiovasc Drugs Ther. 2020;34:357–370. DOI: 10.1007/s10557-019-06919-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, Heneghan MA, Gorevic PD, Litchy WJ, Wiesman JF, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658–2667. DOI: 10.1001/jama.2013.283815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lohrmann G, Pipilas A, Mussinelli R, Gopal DM, Berk JL, Connors LH, Vellanki N, Hellawell J, Siddiqi OK, Fox J, et al. Stabilization of cardiac function with diflunisal in transthyretin (ATTR) cardiac amyloidosis. J Card Fail. 2020;26:753–759. DOI: 10.1016/j.cardfail.2019.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ward JE, Ren R, Toraldo G, SooHoo P, Guan J, O'Hara C, Jasuja R, Trinkaus‐Randall V, Liao R, Connors LH, et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood. 2011;118:6610–6617. DOI: 10.1182/blood-2011-04-351643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Obici L, Cortese A, Lozza A, Lucchetti J, Gobbi M, Palladini G, Perlini S, Saraiva MJ, Merlini G. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid. 2012;19(suppl 1):34–36. DOI: 10.3109/13506129.2012.678508. [DOI] [PubMed] [Google Scholar]
- 42. Karlstedt E, Jimenez‐Zepeda V, Howlett JG, White JA, Fine NM. Clinical experience with the use of doxycycline and ursodeoxycholic acid for the treatment of transthyretin cardiac amyloidosis. J Card Fail. 2019;25:147–153. DOI: 10.1016/j.cardfail.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 43. Ferreira N, Saraiva MJ, Almeida MR. Epigallocatechin‐3‐gallate as a potential therapeutic drug for TTR‐related amyloidosis: "in vivo" evidence from FAP mice models. PLoS One. 2012;7:e29933. DOI: 10.1371/journal.pone.0029933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. aus dem Siepen F, Bauer R, Aurich M, Buss S, Steen H, Altland K, Kristen A, Katus HA. Green tea extract as a treatment for patients with wild‐type transthyretin amyloidosis: an observational study. Drug Des Devel Ther. 2015;9:6319–6325. DOI: 10.2147/DDDT.S96893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kazi DS, Bellows BK, Baron SJ, Shen C, Cohen DJ, Spertus JA, Yeh RW, Arnold SV, Sperry BW, Maurer MS, et al. Cost‐effectiveness of tafamidis therapy for transthyretin amyloid cardiomyopathy. Circulation. 2020;141:1214–1224. DOI: 10.1161/CIRCULATIONAHA.119.045093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brahmanandam V, McGraw S, Mirza O, Desai AA, Farzaneh‐Far A. Regression of cardiac amyloidosis after stem cell transplantation assessed by cardiovascular magnetic resonance imaging. Circulation. 2014;129:2326–2328. DOI: 10.1161/CIRCULATIONAHA.114.009135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fontana M, Martinez‐Naharro A, Chacko L, Rowczenio D, Gilbertson JA, Whelan CJ, Strehina S, Lane T, Moon J, Hutt DF, et al. Reduction in CMR derived extracellular volume with patisiran indicates cardiac amyloid regression. JACC Cardiovasc Imaging. 2021;14:189–199. DOI: 10.1016/j.jcmg.2020.07.043. [DOI] [PubMed] [Google Scholar]
- 48. Haq M, Pawar S, Berk JL, Miller EJ, Ruberg FL. Can 99m ‐Tc‐pyrophosphate aid in early detection of cardiac involvement in asymptomatic variant TTR amyloidosis? JACC Cardiovasc Imaging. 2017;10:713–714. DOI: 10.1016/j.jcmg.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rudolph A, Abdel‐Aty H, Bohl S, Boyé P, Zagrosek A, Dietz R, Schulz‐Menger J. Noninvasive detection of fibrosis applying contrast‐enhanced cardiac magnetic resonance in different forms of left ventricular hypertrophy relation to remodeling. J Am Coll Cardiol. 2009;53:284–291. DOI: 10.1016/j.jacc.2008.08.064. [DOI] [PubMed] [Google Scholar]
- 50. Hanna M, Ruberg FL, Maurer MS, Dispenzieri A, Dorbala S, Falk RH, Hoffman J, Jaber W, Soman P, Witteles RM, et al. Cardiac scintigraphy with technetium‐99m‐labeled bone‐seeking tracers for suspected amyloidosis: JACC review topic of the week. J Am Coll Cardiol. 2020;75:2851–2862. DOI: 10.1016/j.jacc.2020.04.022. [DOI] [PubMed] [Google Scholar]
- 51. Muchtar E, Gertz MA, Kyle RA, Lacy MQ, Dingli D, Leung N, Buadi FK, Hayman SR, Kapoor P, Hwa YL, et al. A modern primer on light chain amyloidosis in 592 patients with mass spectrometry‐verified typing. Mayo Clin Proc. 2019;94:472–483. DOI: 10.1016/j.mayocp.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 52. Connors LH, Sam F, Skinner M, Salinaro F, Sun F, Ruberg FL, Berk JL, Seldin DC. Heart failure resulting from age‐related cardiac amyloid disease associated with wild‐type transthyretin: a prospective, observational cohort study. Circulation. 2016;133:282–290. DOI: 10.1161/CIRCULATIONAHA.115.018852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chang ICY, Bois JP, Bois MC, Maleszewski JJ, Johnson GB, Grogan M. Hydroxychloroquine‐mediated cardiotoxicity with a false‐positive. Circ Cardiovasc Imaging. 2018;11:e007059. DOI: 10.1161/CIRCIMAGING.117.007059. [DOI] [PubMed] [Google Scholar]
- 54. Musumeci MB, Cappelli F, Russo D, Tini G, Canepa M, Milandri A, Bonfiglioli R, Di Bella G, My F, Luigetti M, et al. Low sensitivity of bone scintigraphy in detecting Phe64Leu mutation‐related transthyretin cardiac amyloidosis. JACC Cardiovasc Imaging. 2020;13:1314–1321. DOI: 10.1016/j.jcmg.2019.10.015. [DOI] [PubMed] [Google Scholar]
- 55. Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, Judge DP, Lenihan DJ, Gottlieb SS, Shah SJ, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (transthyretin amyloid outcome survey). J Am Coll Cardiol. 2016;68:161–172. DOI: 10.1016/j.jacc.2016.03.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126:1286–1300. DOI: 10.1161/CIRCULATIONAHA.111.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Damy T, Deux J‐F, Moutereau S, Guendouz S, Mohty D, Rappeneau S, Guellich A, Hittinger L, Loric S, Lefaucheur J‐P, et al. Role of natriuretic peptide to predict cardiac abnormalities in patients with hereditary transthyretin amyloidosis. Amyloid. 2013;20:212–220. DOI: 10.3109/13506129.2013.825240. [DOI] [PubMed] [Google Scholar]
- 58. Rahman JE, Helou EF, Gelzer‐Bell R, Thompson RE, Kuo C, Rodriguez ER, Hare JM, Baughman KL, Kasper EK. Noninvasive diagnosis of biopsy‐proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43:410–415. DOI: 10.1016/j.jacc.2003.08.043. [DOI] [PubMed] [Google Scholar]
- 59. Russo M, Mazzeo A, Stancanelli C, Di Leo R, Gentile L, Di Bella G, Minutoli F, Baldari S, Vita G. Transthyretin‐related familial amyloidotic polyneuropathy: description of a cohort of patients with Leu64 mutation and late onset. J Peripher Nerv Syst. 2012;17:385–390. DOI: 10.1111/j.1529-8027.2012.00436.x. [DOI] [PubMed] [Google Scholar]
- 60. Einstein AJ, Shuryak I, Castaño A, Mintz A, Maurer MS, Bokhari S. Estimating cancer risk from 99m Tc pyrophosphate imaging for transthyretin cardiac amyloidosis. J Nucl Cardiol. 2020;27:215–224. DOI: 10.1007/s12350-018-1307-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dasgupta NR, Rissing SM, Smith J, Jung J, Benson MD. Inotersen therapy of transthyretin amyloid cardiomyopathy. Amyloid. 2020;27:52–58. DOI: 10.1080/13506129.2019.1685487. [DOI] [PubMed] [Google Scholar]
- 62. Lee SP, Suh HY, Park S, Oh S, Kwak SG, Kim HM, Koh Y, Park JB, Kim HK, Cho HJ, et al. Pittsburgh B compound positron emission tomography in patients with AL cardiac amyloidosis. J Am Coll Cardiol. 2020;75:380–390. DOI: 10.1016/j.jacc.2019.11.037. [DOI] [PubMed] [Google Scholar]
- 63. Pilebro B, Arvidsson S, Lindqvist P, Sundström T, Westermark P, Antoni G, Suhr O, Sörensen J. Positron emission tomography (PET) utilizing Pittsburgh compound B (PIB) for detection of amyloid heart deposits in hereditary transthyretin amyloidosis (ATTR). J Nucl Cardiol. 2018;25:240–248. DOI: 10.1007/s12350-016-0638-5. [DOI] [PubMed] [Google Scholar]
- 64. Cuddy SAM, Bravo PE, Falk RH, El‐Sady S, Kijewski MF, Park MA, Ruberg FL, Sanchorawala V, Landau H, Yee AJ, et al. Improved quantification of cardiac amyloid burden in systemic light chain amyloidosis: redefining early disease? JACC Cardiovasc Imaging. 2020;13:1325–1336. DOI: 10.1016/j.jcmg.2020.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cardiac uptake of 18F florbetapir in patients undergoing chemotherapy. Available at: https://clinicaltrials.gov/ct2/show/NCT03333551. Accessed May 11, 2020. (Study No. NCT03333551).
- 66. Abulizi M, Sifaoui I, Wuliya‐Gariepy M, Kharoubi M, Israël JM, Emsen B, Bodez D, Monnet A, Didierlaurent D, Tacher V, et al. F‐sodium fluoride PET/MRI myocardial imaging in patients with suspected cardiac amyloidosis. J Nucl Cardiol. Sep 2019. DOI: 10.1007/s12350-019-01885-8. [DOI] [PubMed] [Google Scholar]
- 67. The potential role of 18F‐NaF PET/CT in diagnosing cardiac amyloidosis. Available at: https://clinicaltrials.gov/ct2/show/NCT04146480. Accessed May 11, 2020. (Study No. NCT04146480).
- 68. PET/MR imaging in patients with cardiac amyloidosis. Available at: https://clinicaltrials.gov/ct2/show/NCT03626584. Accessed May 11, 2020. (study No. NCT03626584).
- 69. Imaging cardiac amyloidosis: a pilot study using F‐18 florbetapir positron emission tomography. Available at: https://clinicaltrials.gov/ct2/show/NCT01683825. Accessed May 11, 2020. (Study No. NCT01683825).
- 70. Monitoring of early disease progression in hereditary transthyretin amyloidosis (MED‐hATTR). Available at: https://clinicaltrials.gov/ct2/show/NCT03431896?cond=cardiac+amyloidosis&draw=2&rank=46. Accessed May 11, 2020. (Study No. NCT03431896).