

Abstract
Background
To investigate the therapeutic potential of combined therapy with polyethylene glycol‐20k (PEG‐20k) and MCC950 on post‐resuscitation myocardial function in a rat model of cardiac arrest.
Methods and Results
Thirty rats were randomized into 5 groups: Sham, Control, PEG‐20k, MCC950, PEG‐20k+ MCC950. Except for sham, animals were subjected to 6 minutes of ventricular fibrillation followed by 8 minutes cardiopulmonary resuscitation. Two milliliters PEG‐20k was administered by intravenous injection coincident with the start of cardiopulmonary resuscitation; MCC950 (10 mg/kg), a highly selective NLRP3 inflammasome inhibitor, was delivered immediately after restoration of spontaneous circulation. Myocardial function, sublingual microcirculation, mitochondrial function, plasma cardiac troponin I, and interleukin‐1β, expression of proteins in SIRT1 (sirtuin 1)/PGC‐1α (peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha) and NLRP3 (the NOD‐like receptor family protein 3) inflammasome pathways were evaluated. Following cardiopulmonary resuscitation, myocardial function was compromised with a significantly decreased cardiac output, ejection fraction, and increased myocardial performance index, cardiac troponin I. Sublingual microcirculation was disturbed with impaired perfused vessel density and microvascular flow index. Cardiac arrest reduced mitochondrial routine respiration, Complex I‐linked respiration, respiratory control rates and oxidative phosphorylation coupling efficiency. PEG‐20k or MCC950 alone restored mitochondrial respiratory function, restituted sublingual microcirculation, and preserved myocardial function, whereas a combination of PEG‐20k and MCC950 further improved these aspects. PEG‐20k restored the expression of SIRT1 and PGC‐1α, and blunted activation of NLRP3 inflammasomes. MCC950 suppressed expression of cleaved‐caspase‐1/pro‐caspase‐1, ASC (apoptosis‐associated speck‐like protein), GSDMD [gasdermin d], and interleukin‐1β.
Conclusions
Combined therapy with PEG‐20k and MCC950 is superior to either therapy alone for preserving post‐resuscitated myocardial function, restituting sublingual microcirculation at restoration of spontaneous circulation at 6 hours. The responsible mechanisms involve upregulated expression of SIRT1/PGC1‐α in tandem with inhibition of NLRP3 inflammasomes.
Keywords: cardiac arrest, mitochondrial dysfunction, myocardial dysfunction, NLRP3 inflammasomes, pyroptosis
Subject Categories: Cardiopulmonary Arrest, Cardiopulmonary Resuscitation and Emergency Cardiac Care, Animal Models of Human Disease
Nonstandard Abbreviations and Acronyms
- CA
cardiac arrest
- CI
complex I‐linked respiration
- CII
complex II‐linked respiration
- CIL
the non‐phosphorylating LEAK‐respiration
- CIP
complex I‐linked phoshorylating capacity
- CIV
complex IV‐linked respiration
- CO
cardiac output
- cTnI
cardiac troponin I
- G
glutamate 10 μmol/L
- GSDMD
gasdermin D
- I/R
ischemia‐reperfusion
- M
malate 2 mmol/L
- MFI
microvascular flow index
- NLRP3
the NOD‐like receptor family protein 3
- OXPHOS
oxidative phosphorylation
- P
pyruvate 5 mmol/L
- PEG‐20k
polyethylene glycol‐20k
- PVD
perfused vessel density
- RCR
respiratory control rates
- ROSC
restoration of spontaneous circulation
- SIRT1
sirtuin 1
- TMPD
N,N,N 0,N 0‐Tetramethyl‐p‐phenylenediamine dihydrochloride
- TTFA
thenoyltrifluoroacetone
Clinical Perspective
What Is New?
To our knowledge, this is the first study to evaluate the therapeutic potential of combined therapy with polyethylene glycol‐20k and MCC950 on post‐resuscitation myocardial function in a rat model of cardiac arrest.
Polyethylene glycol‐20k improved myocardial mitochondrial function via SIRT1 [sirtuin 1]/PGC‐1α [peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha] pathway, subsequently inhibited activation of NLRP3 (the NOD‐like receptor family protein 3) inflammasomes.
In turn, NLRP3 inflammasomes inhibition by MCC950 also improved mitochondrial function. Polyethylene glycol‐20k ‐MCC950 combination upregulates expression of SIRT1/PGC1‐α and inhibits activation of NLRP3 inflammasomes.
What Are the Clinical Implications?
Our findings support and highlight the perspective on protection of complex I of the electron transport chain and a positive microcirculatory effect leading to better post‐resuscitation myocardial function.
Multi‐pattern and multi‐level horizontal linkage pathways is the main barrier to improving survival after cardiac arrest.
Abovementioned multiple molecular‐cellular perturbations in the current study are blocked by polyethylene glycol‐20k‒MCC950 combination; this research is also translational in nature since the results of the proposed studies will potentially lead to a novel means of active protection of the heart and lead to an improvement in survival following sudden cardiac arrest.
One major barrier to improving survival after cardiac arrest (CA) is global myocardial ischemia‐reperfusion (I/R) injury, which subsequently drives mitochondrial damage and leads to myocardial dysfunction. Studies have suggested that mitochondria are pivotal components of I/R injury, both as targets and effectors.1, 2, 3 Damaged mitochondria release various substrates like reactive oxygen species, mitochondrial DNA, and cardiolipin, which are responsible for direct activation of the NLRP3 (NOD‐like receptor family protein 3) inflammasomes and ensuing pyroptosis. Our preliminary data have confirmed the protective effect of MCC950, a highly selective NLRP3 inflammasomes inhibitor, in preventing myocardial dysfunction in a rat model of CA by quenching pyroptosis. However, whether selective NLPR3 inflammasomes inhibition with MCC950 is mechanistically linked to restoration of post‐resuscitated mitochondrial function remains to be elucidated.
Moreover, microvascular obstruction or no‐reflow phenomenon is a major contributor to I/R injury and mitochondrial dysfunction.4, 5, 6 The no‐reflow phenomenon is characterized by swollen vascular endothelial and parenchymal cells, which are primarily contributed to ATP depletion. In this regard, mitochondria are victim and inflicter within I/R cells. Our previous study 7 demonstrated that polyethylene glycol‐20k (PEG‐20k) prevented no‐flow phenomenon, improved microcirculation through abolishing ischemia‐induced cell swelling and interstitial edema and preserved post‐resuscitated myocardial dysfunction.
In the present study, we evaluated the therapeutic potential of combined therapy with PEG‐20k and MCC950 in post‐resuscitated myocardial function in a rat model of CA and cardiopulmonary resuscitation (CPR) and the potential mechanisms.
Methods
The data, analytic methods, and study materials that support the findings of this study are available from the corresponding author upon reasonable request to other researchers for purposes of reproducing the results or replicating the procedure. This study was performed under a protocol (AD10001396, specific aim 19) approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University. All animals were handled in accordance with the G uide for the Care and Use of Laboratory Animals published by the National Institute of Health.
Animal Preparation
Animal preparation and establishment of rat model of CA and CPR were performed according to our previous studies.7, 8 Male Sprague‐Dawley rats weighing 450 to 550 g were used in the present study. Animals were kept under standard conditions with ad libitum access to food and water and maintained at a 12‐hour day and night cycle. Following rendering unconsciousness and recumbency with inhalation of CO2 (50% CO2, 50% O2) for 30 seconds, animals were anesthetized by intraperitoneal injection of pentobarbital (45 mg/kg). Additional doses (10 mg/kg) were administered at intervals of 1 hour or when appropriate to maintain anesthesia. Shaved rats were orally tracheal intubated with a 14‐G cannula mounted on a blunt needle (Abbocath‐T; Abbott Hospital Products Division, North Chicago, IL, USA) with a 145°‐angled tip. End‐tidal CO2 was continuously monitored with a side‐stream infrared CO2 analyzer (Capstar‐100 Carbon Dioxide Analyzer; CWE, Ardmore, PA) interposed between the tracheal cannula and the ventilator. A conventional lead II ECG was also continuously monitored. Through the left external jugular vein, a polyethylene catheter (PE‐50, Becton Dickinson, Sparks, MD) was advanced into the right atrium for right atrial pressures measurement. Another PE‐50 catheter was advanced into the descending aorta from the left femoral artery for measurement of arterial pressure, and arterial blood withdrawal. Drug administration was given through a PE‐50 catheter placed in the right femoral vein. A thermocouple microprobe (IT‐18; Physitemp Instruments Inc., NJ, USA) was inserted into the left femoral vein for measurement of core body temperature. The core body temperature (venous blood temperature) was maintained at 37±0.5°C by heated surgical board and lamp. Finally, through the right external jugular vein, a 3‐F catheter (Model C‐PMS‐301J; Cook Critical Care, Bloomington, IN) was advanced into the right atrium for facilitating guidewire insertion. After all catheters were placed, a pre‐curved guidewire was advanced through the right external jugular vein catheter into the right ventricle confirmed by transthoracic ECG. All catheters were flushed intermittently with saline containing 2.5 IU/mL of crystalline bovine heparin.
CA and CPR Model
Fifteen minutes before inducing ventricular fibrillation (VF), baseline hemodynamics, sublingual microcirculation and echocardiography measurement were performed. Mechanical ventilation was established at a tidal volume of 0.60 mL/100 g of body weight, a frequency of 100 breaths per minute and an inspired O2 fraction of 21%. Mechanical ventilation was discontinued after onset of VF. VF was induced through a guidewire advanced from the abovementioned right jugular catheter into the right ventricle. A progressive increase in 60‐Hz current to a maximum of 3.5 mA was then delivered to the right ventricular endocardium. The current flow was continued for 3 minutes to prevent spontaneous defibrillation. Precordial chest compression, together with mechanical ventilation (tidal volume 0.60 mL/100 g body weight, frequency 100 breaths per minute, inspired O2 fraction of 100%), was initiated after 6 minutes of untreated VF with a pneumatically driven mechanical chest compressor. Precordial chest compressions were maintained at a rate of 200/min and synchronized to provide a compression: ventilation ratio of 2:1 with equal compression‐relaxation for 8 minutes. The depth of compression was initially adjusted to maintain coronary perfusion pressure at 25±2 mm Hg, and final depth of compression was recorded. Defibrillation was attempted with up to 3 4‐J counter shocks after 8 minutes of CPR. Restoration of spontaneous circulation (ROSC) was defined as the return of supraventricular rhythm with a mean aortic pressure >50 mm Hg for 5 minutes. In the case, ROSC was not achieved after the first defibrillation attempt, a 30‐second interval of CPR was performed before the next defibrillation attempt (up to 3 attempts). The rats were continuously monitored for 6 hours, and fraction of inspired O2 stepwise adjusted (100% for the first hour, 50% for the second hour and 21% thereafter).
Experimental Protocol
Thirty rats were randomized with the Sealed Envelope Method into 5 groups (Figure S1): (1) Sham (S), (2) Control (C), (3) PEG‐20k (P), (4) MCC950 (M), (5) Combination (PEG‐20k+ MCC950). Except for sham, animals were subjected to 6 minutes of untreated VF followed by 8 minutes CPR. Two‐milliliter PEG‐20k (10% weight/volume) was continuously administered by intravenous injection coincident with the start of CPR, meanwhile MCC950 (10 mg/kg, dissolved in sterile saline) was delivered immediately after ROSC in the PEG‐20k+ MCC950 group. Rats in the PEG‐20k group received 2‐mL PEG‐20k and same volume of sterile saline at corresponding timepoint to MCC950, while those in the MCC950 group received the same dose of MCC950 after ROSC and 2‐mL sterile saline at corresponding timepoint to PEG‐20k. Moreover, 2‐mL sterile saline and another comparable volume of vesicle were given after recording baseline and 8 minutes after observation in the sham group, respectively.
At ROSC 6 hour, the rats were euthanized with an intravenous injection of Euthasol (150 mg/kg). Then the heart was rapidly excised, ≈200 mg fresh heart tissue was used for high‐resolution respirometry, remaining tissue was frozen in liquid nitrogen for further assay. A necropsy was routinely performed to inspect for gross abnormalities, including evidences of traumatic injuries consequent to cannulation, airway management, or precordial compression.
Measurements
The histological materials and blood samples were numbered with blinded material numbers, and measured blinded and always by the same person.
Hemodynamics and Myocardial Function
Electrocardiogram, heart rate, aortic and right atrial pressures, end‐tidal CO2, and core body temperature values were continuously recorded on a personal computer‐based data‐acquisition system supported by WINDAQ software (DATAQ, Akron, OH). Myocardial function including ejection fraction, cardiac output, and myocardial performance index was measured by echocardiography (HD11XE; Philips Medical Systems, Eindhoven, Netherlands, USA).9, 10 Cardiac output and ejection fraction were adopted to estimate the myocardial contractility; myocardial performance index combines time intervals related to systolic and diastolic functions and reflects the global cardiac function calculated using the formula (a−b)/b, where a=mitral closure‐to‐opening interval (time interval from cessation to onset of mitral inflow) and b=ET (aortic flow ejection time, obtained at the left ventricle outflow tract). All echocardiography recordings were reviewed by 2 independent observers blinded to the groups, as well as sublingual microcirculation.
Sublingual Microcirculation
Sublingual microcirculation was assessed using a sidestream dark‐field imaging device (MicroScan; MicroVision Medical Inc., Amsterdam, Netherlands) with a 5× imaging objective, resulting in an on‐screen magnification of ×276. Microvascular flow index (MFI) and perfused vessel density (PVD) were calculated according to previous study.11, 12
Assessing the Function of Mitochondria With High‐Resolution Respirometry
Preparation of Homogenate
All steps of the preparation were performed on ice. Fresh left ventricular tissue was rapidly but gently dissected and immediately transferred into 5 mL of ice‐cold buffer solution (K medium), stored on ice. Then tissue samples were transported to mitochondrial laboratory in the same building within 5 minutes and processed immediately. According to modified protocol for human skeletal muscle homogenates 13 connective and adipose tissue were removed using pre‐cold scissors and anatomic forceps to ensure only heart muscle tissue was homogenized. After gentle blotting by a piece of sterile gauze, heart muscle tissue was weighted using analytical scale (1‐μg readability) and cut into fine fragments. Then tissue fragments were scraped into a pre‐chilled Dounce Tissue Grinder (glass construction; Wheaton, Millville, USA), 1 mL of K medium was added for each 100 mg of muscle to obtain 10% homogenate. Minced cardiac tissues were then manually homogenized by 8 to 10 initial strokes with a glass pestle. Raw homogenate was then filtered through cheesecloth. The whole procedure took 5 to 10 minutes. Notably, the same operator prepared all the samples for measurement in all animals to minimize variability.
Buffers and Reagents
All reagents used for homogenate preparation and high‐resolution respirometry were obtained from Sigma‐Aldrich. K medium contains TrisHCl (10 mmol/L), KCl (80 mmol/L), MgCl2 (3 mmol/L), KH2PO4 (5 mmol/L), EDTA (1 mmol/L), BSA (0.5 mg/mL), pH 7.4 stored at 4°C. Respiratory medium MiR05 is composed of EGTA (0.5 mmol/L), MgCl2 (3 mmol/L), lactobionic acid (60 mmol/L), taurine (20 mmol/L), KH2PO4 (10 mmol/L), HEPES (20 mmol/L), sucrose (110 mmol/L), and 1 g/L of BSA (pH 7.1). The final concentrations of reagents in each chamber were used as following: glutamate 10 μmol/L (G), pyruvate 5 mmol/L (P) and malate 2 mmol/L (M) (GPM, substrates for complex I), digitonin 10 μg/mL (a permeabilizer), adenosine diphosphate 1 mmol/L, rotenone 0.05 μmol/L (Rot, Complex I inhibitor), succinate 10 mmol/L (succinate, substrate for complex II), thenoyltrifluoroacetone 80 μmol/L (TTFA, complex II inhibitor), ascorbic acid 2 mmol/L (antioxidant) and N,N,N 0,N 0‐Tetramethyl‐p‐phenylenediamine dihydrochloride 0.5 mmol/L (TMPD, artificial substrate for complex IV), sodium azide (complex IV inhibitor). All buffers and reagents were stored at 4°C and ice‐cold before use.
Oxygraph‐2k Fluorometry
Mitochondrial respiration was measured using high‐resolution respirometry Oxygraph‐2k (Oroboros Instruments, Innsbruck, Austria) with a polarographic oxygen electrode and two 2‐mL chambers allowing for parallel measurements. 14 Experiments were performed at a controlled constant temperature of 37°C. Oxygen solubility factor used for calibration was 0.93 for K medium and 0.92 for MiR05, respectively. Initial O2 concentration was kept at 200 to 220 μmol/L and reagents were added into the closed chamber through a small capillary tube by using Hamilton syringes (Oroboros Instruments, Innsbruck, Austria). Oxygen concentration (μmol/L) and oxygen flux [pmol/(s×mL)] were simultaneously recorded real‐time using DatLab software (Datlab Version 7.3.0.3, Oroboros Instruments, Innsbruck, Austria).
Another 2 rats were used to determine the optimal amount of homogenate in each chamber for well detectable mitochondria respiration, but avoiding too low oxygen consumption rate or rapid exhaustion of oxygen with too much homogenate. 25 μL, 50 μL, and 100 μL 10% homogenate were separately added into a chamber prefilled with MiR05, measurements were performed in duplicate. Subsequently, stepwise addition of G/P/M, digitonin, ADP, Rot, succinate, TTFA, ascorbic acid+TMPD, and azide were used in the present study. Re‐oxygenation by opening chamber was performed, as appropriate to avoid oxygen concentration <100 μmol/L, since too‐low oxyfgen concentration results in oxygen flux becomes oxygen‐dependent. Based on the results of optimization experiments, 50‐μL homogenate was used as the final amount in present study.
This experimental protocol allowed for calculation of following mitochondrial function indices (Figure 1A) 15 : Routine respiration, representing oxygen consumption rate in the physiological coupling and is controlled by cellular energy demand, energy turnover, and the degree of coupling to phosphorylation of ADP. The non‐phosphorylating LEAK‐respiration (CIL ) was measured after addition of the CI‐linked substrates G/P/M and digitonin. Subsequently, CI‐linked phoshorylating capacity (CIP ) is oxygen consumption rate in the presence of a saturating concentration of ADP and substrates for complex I. Complex I‐linked respiration (CI) was calculated by subtracting oxygen consumption rate of rotenone from CIP . Similarly, Complex II‐linked respiration (CII) was quantified by subtracting oxygen consumption rate of TTFA from CIIP , which was measured by subsequent adding substrates for complex II, succinate. Finally, Complex IV‐linked respiration (CIV)=TMPD‐azide. Furthermore, the respiratory control rates (RCR) were measured as indicators of the mitochondrial coupling state (RCR=CIP /CIL ratio), RCR was also transformed to its corresponding flux control factors, which is the oxidative phosphorylation (OXPHOS) coupling efficiency calculated as (CIP −CIL )/CIp=1−CIL /CIP. All oxygen flux was corrected and expressed based on tissue wet weight, pmol/(min×mg) for statistical analysis.
Figure 1. Polyethylene glycol‐20k and MCC950 restored myocardial mitochondrial function after cardiac arrest and cardiopulmonary resuscitation.
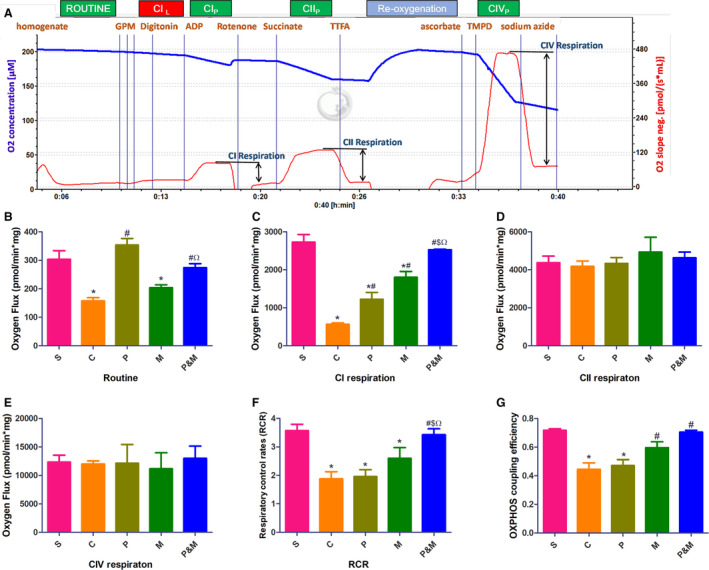
Exemplary respirometric traces outlining the protocol applied in the high‐resolution respirometry experiment (A). Blue line indicates O2 concentration (μmol/L, left y‐axis), red line indicates O2 flux in pmol/(s×mL), right y‐axis. B, Routine respiration; (C), complex I‐linked respiration; (D), complex II‐linked respiration; (E), complex IV‐linked respiration; (F), respiratory control rates; (G), oxidative phosphorylation coupling efficiency. C indicates cardiac arrest control group; CI, complex I‐linked respiration; CII, complex II‐linked respiration; CIL , the non‐phosphorylating LEAK‐respiration; CIP , complex I‐linked phoshorylating capacity; CIIP , complex II‐linked phoshorylating capacity; CIVP , complex IV‐linked phoshorylating capacity; CIV, complex IV‐linked respiration; M, MCC950 group; MAP, mean artery pressure; OXPHOS, oxidative phosphorylation; P&M, polyethylene glycol‐20k‒MCC950 group, a highly selective NLRP3 inflammasome inhibitor; P, polyethylene glycol‐20k group; RCR, respiratory control rates; and S, sham control group without cardiac arrest. *P<0.05 vs S, # P<0.05 vs C, $ P<0.05 vs P, Ω P<0.05 vs M, respectively. All statistical analysis was performed by 1‐way ANOVA, followed by Tukey post‐hoc analysis (n=6 for each group). Each sample was measured in duplicate. Data are expressed as the mean±SD.
ELISA
Plasma cardiac troponin I (cTnI) and interleukin‐1β were detected using commercial ELISA kits according to the manufacturer's instructions.
Immunoblotting Analysis
Thirty‐microgram protein samples were separated and transferred to low‐fluorescence polyvinylidene fluoride (PVDF) membranes. After blocking at room temperature for 1 hour, the membranes were incubated with primary antibodies against SIRT1 (sirtuin 1; 1:1000, ab189494, Abcam, USA), PGC1α (peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha; 1:1000, ab54481, Abcam, USA), NLRP3 (1:500, ab214185, Abcam, USA), pro‐caspase‐1+p10+p12 (1:1000, ab179515, Abcam, USA), ASC (1:500, sc‐514414, Santa Cruz, CA, USA), GSDMD (gasdermin D; 1:1000, ab219800, Abcam, USA), interleukin‐1β (IL‐1β; 1:2500; ab9722, Abcam, MA, USA), or GAPDH (1:2000, ab9482, Abcam, USA) overnight at 4°C, respectively. After washing 5 times, the membranes incubated with corresponding HRP‐conjugated secondary antibodies (1:5000, Abcam, MA, USA). Finally, protein bands were detected after incubating with Clarity Max Western ECL Substrate using the ChemiDoc MP Imaging System. GAPDH was used for total protein loading control. The densities of protein blots were quantified by using Image Lab Software (1709690, BIO‐RAD, Irvine, CA) and normalized to the sham.
Statistical Analysis
All continuous variables are presented as mean±SD after confirming for normality of distribution by Kolmogorov‒Smirnov test. Data were analyzed by 1‐way analysis of variance (1‐way ANOVA) or 2‐way ANOVA, when appropriate, followed by Tukey post‐hoc tests for comparisons between multiple groups. Data failed the test for normality of distribution were analyzed by the Kruskal–Wallis. Statistical significance was assumed at P<0.05. The statistical analyses were performed using SPSS version 23.0 for Windows (SPSS, Chicago, IL). Coefficient of variation was also calculated as coefficient of variation (%)=100×SD/mean.
Results
Baseline Characteristics
A total of 35 rats were used; 2 rats were used for determining the optimal amount of homogenate, and 3 rats were excluded because of instrumentation or technical failure during animal preparation. The remaining rats were randomized with the Sealed Envelope Method into 5 groups (Figure S1). There were no significant differences in physiological and hemodynamic parameters among 5 groups (Table).
Table 1.
Baseline Characteristics of All Animals
| Variables | S (n=6) | C (n=6) | P (n=6) | M (n=6) | P&M (n=6) |
|---|---|---|---|---|---|
| Body weights, g | 480±12 | 486±29 | 482±19 | 478±11 | 479±20 |
| Surgery time, min | 28±5 | 28±3 | 26±4 | 29±6 | 24±3 |
| Heart rate, bpm | 359±29 | 355±17 | 344±29 | 363±20 | 359±29 |
| MAP, mm Hg | 137±6 | 137±5 | 140±11 | 144±2 | 141±13 |
| Temperature, °C | 36.7±0.1 | 36.6±0.1 | 36.7±0.1 | 36.8±0.3 | 36.6±0.1 |
| PC depth, mm | … | 12±1 | 13±1 | 12±1 | 12±1 |
| No. of shocks | … | 1.5±0.5 | 2.2±0.8 | 1.3±1.0 | 1.7±0.8 |
| PC3 CPP, mm Hg | … | 27.83±2.31 | 27.83±2.71 | 27.33±2.33 | 26.46±2.43 |
| PC5 CPP, mm Hg | … | 27.00±1.67 | 27.17±1.83 | 26.50±2.07 | 27.17±0.98 |
| PC7 CPP, mm Hg | … | 26.83±1.72 | 26.33±2.07 | 27.33±2.42 | 25.83±1.47 |
| ETCO2, mm Hg | |||||
| Baseline | 37.7±5.8 | 39.3±7.1 | 41.2±4.3 | 40.2±5.0 | 41.4±6.1 |
| PC3 | … | 17.33±1.75 | 17.50±2.66 | 18.17±1.72 | 17.50±2.66 |
| PC5 | … | 18.00±1.79 | 17.67±1.97 | 18.33±1.63 | 18.50±1.87 |
| PC7 | … | 18.50±2.42 | 17.33±1.75 | 18.83±1.94 | 18.00±1.41 |
Data are presented as means±SD. All variables 1‐way ANOVA, P>0.05. C indicates cardiac arrest control group; CPP, coronary perfusion pressure; ETCO2, end‐tidal CO2; M, MCC950 group; MAP, mean artery pressure; P&M, polyethylene glycol‐20k ‒MCC950 group; P, polyethylene glycol‐20k group; PC, precordial compression; PCn, n minute after PC; and S, sham control group without cardiac arrest.
Combined Therapy With PEG20‐k and MCC950 Retrieved Myocardial Function After CA
CA and I/R injury significantly impaired myocardial function, as indicated by ejection fraction, cardiac output, and myocardial performance index. However, myocardial function was all significantly retrieved either in the PEG‐20k or MCC950 group at each time point when compared with controls. As expected, ejection fraction, cardiac output, and myocardial performance index were further improved in the PEG‐20k+ MCC950 group (Figure 2A through 2C). Plasma cTnI was significantly elevated after CA (1774.10±428.83 versus 155.55±17.70 pg/mL, P<0.001). However, either PEG‐20k or MCC950 significantly reduced the release of cTnI. PEG‐20k‒MCC950 combination further decreased the release of cTnI (473.47±80.05 versus 1774.10±428.83 pg/mL, P<0.001, Figure 2D). The combined therapy with PEG20‐k and MCC950 further reduced the severity of post‐resuscitation myocardial dysfunction as compared with PEG‐20k or MCC950.
Figure 2. Combined therapy with polyethylene glycol‐20k and MCC950 retrieved myocardial function after cardiac arrest and cardiopulmonary resuscitation.
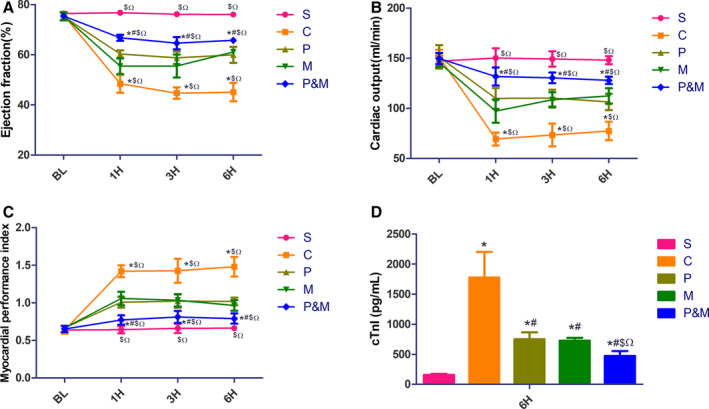
A, Ejection fraction; (B), cardiac output; (C), myocardial performance index; (D), plasma cardiac troponin I concentration. BL indicates baseline; C, the cardiac arrest control group; cTnI, cardiac troponin I; H, hours post‐resuscitation; M, the MCC950 group; P&M, polyethylene glycol‐20k‒MCC950 group; P, polyethylene glycol‐20k group; and S, the sham group. *P<0.05 vs S, # P<0.05 vs C, $ P<0.05 vs P, Ω P<0.05 vs M, respectively. All statistical analysis was performed by 2‐way ANOVA for ejection fraction, CO, and myocardial performance index, and by 1‐way ANOVA for cardiac troponin I, followed by Tukey post‐hoc analysis (n=6 for each group). Data are expressed as the mean±SD.
PEG‐20k and MCC950 Prevented Post‐Resuscitated No‐Reflow by Restituting Sublingual Microcirculation
Both PVD and MFI were dramatically reduced in the control group when compared with the sham after ROSC (Figure 3). PEG‐20k prevented the decrease of PVD and MFI as early as ROSC 2‐hour. Intriguingly, significant restitution of PVD and MFI was also observed in the MCC95 group except at ROSC ‐2‐hour. More importantly, both PVD and MFI were significantly improved in the PEG‐20k+ MCC950 group.
Figure 3. Polyethylene glycol‐20k and MCC950 both reduced post‐resuscitated no‐reflow by restituting sublingual microcirculation.
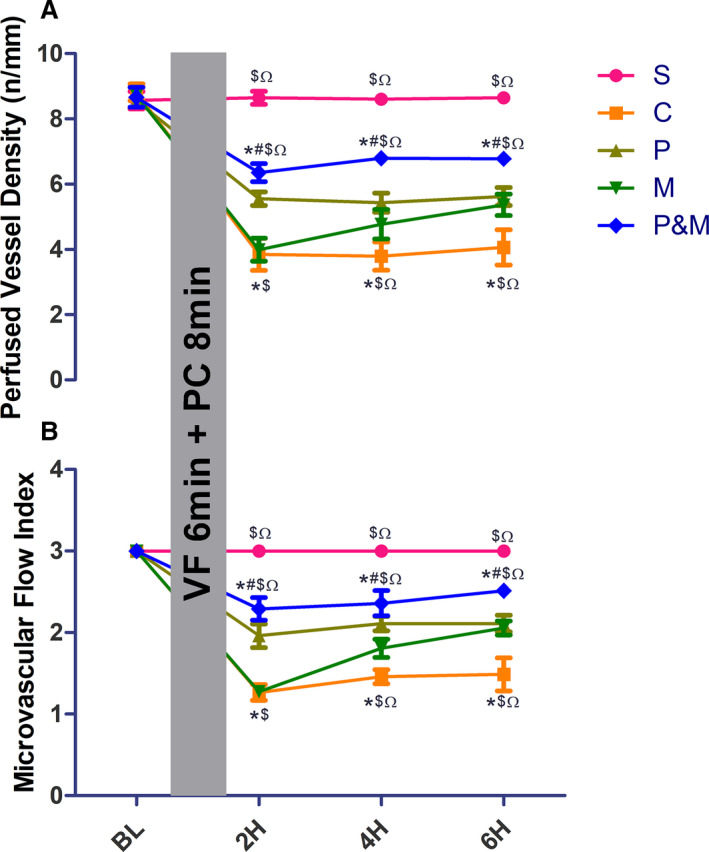
A, perfumed vessel density; (B), microcirculatory flow index. C indicates cardiac arrest control group; H, hours post resuscitation; M, the MCC950 group; P&M, polyethylene glycol‐20k group ‒MCC950 group; P, polyethylene glycol‐20k group; PC, precordial compression; S, the sham group; and VF, ventricular fibrillation. *P<0.05 vs S, # P<0.05 vs C, $ P<0.05 vs P, Ω P<0.05 vs M, respectively. All statistical analysis was performed by 2‐way ANOVA , followed by Tukey post‐hoc analysis (n=6 for each group). Data are expressed as the mean±SD.
PEG‐20k and MCC950 Restored Myocardial Mitochondrial Function After CA
Myocardial mitochondrial function compromised ROSC 6‐hour, and PEG‐20k and MCC950 restored myocardial mitochondrial function. In detail, routine respiration was significantly lower in the control and MCC950 group when compared with sham (157.87±21.62 and 203.91±20.26 versus 303.61±50.64 pmol/min×mg, all P<0.05), whereas routine respiration was comparable among the PEG‐20k, PEG‐20k+ MCC950 group and sham group (Figure 1B). In addition, PEG‐20k, MCC950, and the combination significantly enhanced CIL (767.05±99.30, 859.48±188.99, 828.95±101.05 pmol/min×mg, all P<0.001) when compared with the control group (Figure S2). Notably, the only complex susceptible to CA and I/R injury was Complex I‐linked respiration (CI) (564.04±159.97 in the control group versus 2729.52±339.45 pmol/min×mg in the sham group, P<0.001), whereas CI respiration was higher in the PEG‐20k and MCC950 group as compared with the control group and the combination further restored CI (Figure 1C). We found no significant differences in CII and CIV among all groups (Figure 1D and 1E). I/R injury decreased RCR (1.88±0.46 in the control group versus 3.57±0.38 in the sham group, P<0.001). RCR was strikingly increased in the PEG‐20k+ MCC950 group (3.43±0.33, P<0.001, Figure 1F). Similarly, OXPHOS coupling efficiency was also restored in the MCC950 and PEG‐20k+ MCC950 group (Figure 1G).
PEG‐20k Improved Myocardial Mitochondrial Function via SIRT1/PGC‐1α Pathway
I/R injury downregulated expression of SIRT1 and PGC‐1αin the control group when compared with the sham group (Figure 4A through 4C). Nevertheless, the expression of both SIRT1 and PGC‐1α were significantly upregulated by PEG‐20k rather than MCC95.
Figure 4. Polyethylene glycol‐20k and MCC950 upregulated the expression of SIRT1 (sirtuin 1)/PGC1‐α (peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha) in tandem with inhibition of NLRP3 (the NOD‐like receptor family protein 3) inflammasomes.
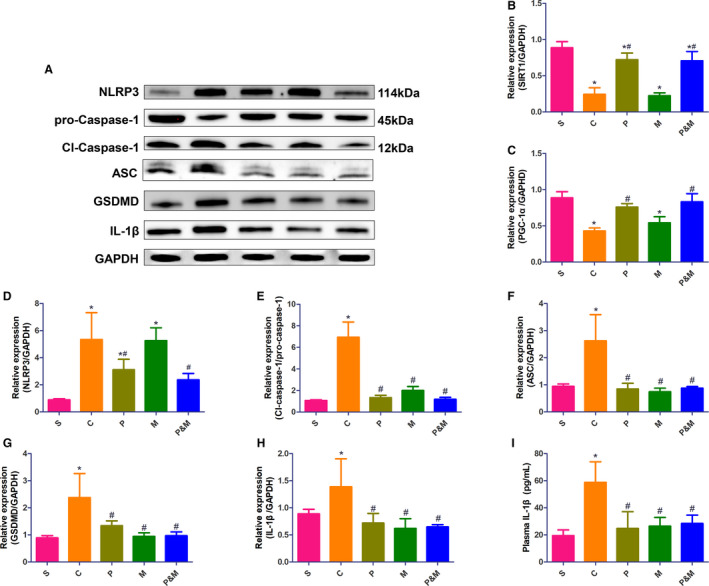
A, Representative western blot analysis of SIRT1/PGC1‐α and NLRP3 inflammasomes pathway; (B), Relative expression of SIRT1; (C), Relative expression of PGC‐1α; (D), Relative expression of NLRP3; (E), Relative expression of cleaved‐caspase‐1/pro‐caspase‐1; (F), Relative expression of ASC; (G), Relative expression of GSDMD; (H), Relative expression of interleukin‐1β; (I), plasma IL‐1β concentration. Relative expression values were normalized to GAPDH internal control. C indicates cardiac arrest control group; GSDMD, gasdermin D; IL‐1β, interleukin‐1β; NLRP3, the NOD‐like receptor family protein 3; M, the MCC950 group; P, polyethylene glycol‐20k group; P&M, the polyethylene glycol‐20k group‒MCC950 group; PGC1‐α, peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha; ASC, apoptosis‐associated speck‐like protein; S, the sham group; and SIRT1, sirtuin 1. *P<0.05 vs S, # P<0.05 vs C. All statistical analysis was performed by 1‐way ANOVA, followed by Tukey post‐hoc analysis (n=6 for each group). Data are expressed as mean±SD.
PEG‐20k and MCC950 Both Inhibited Activation of NLRP3 Inflammasomes
I/R triggered activation of NLRP3 inflammasomes, evidenced by significantly higher expression of NLRP3, cleaved‐caspase‐1/pro‐caspase‐1, ASC, IL‐1β, and GSDMD (Figure 4D through 4H, all P<0.05), facilitating the release of IL‐1β to the plasma (58.74±15.26 versus 19.47±4.17 pg/mL in the sham group, P<0.001, Figure 4I). Expression of NLRP3 was markedly downregulated in the PEG‐20k and PEG‐20k+ MCC950 group as compared with the control group (P<0.05 and P<0.001, respectively), while expression of NLRP3 did not change between the MCC950 and control group. Other components of NLRP3 inflammasomes, including ASC and caspase‐1 and its downstream molecular IL‐Iβ and GSDMD, were concurrently quenched by PEG20‐k, MCC950, and the combination.
Discussion
In the present study, we investigated the effectiveness of the combination PEG‐20k‒MCC950 treatment on post‐resuscitation myocardial function in a rat model of CA and CRP. Our results demonstrated that global I/R injury following CA and CPR elicited myocardial dysfunction, leading to no‐reflow occurrence and impaired mitochondrial function. The underlying mechanisms were associated with multiple molecular‐cellular perturbations, including downregulated SIRT1/PGC1‐α‐expression, activation of NLRP3 inflammasomes, and ensuing pyroptosis. PEG‐20k improved post‐resuscitation myocardial function by preventing no‐reflow phenomenon and restoring mitochondrial function, as with MCC950. Most importantly, combined therapy with PEG20‐k and MCC950 further improved myocardial function at ROSC 6‐hour.
Preclinical and clinical studies have demonstrated that post‐resuscitated myocardial dysfunction is a major contributor to mortality and disability.16, 17, 18 In agreement with our previous studies and others' studies,7, 19, 20 the most important findings in the present study were that compromised myocardial function were retrieved after receiving either PEG‐20k or MCC950 treatment and further improved in the combined PEG‐20k‒MCC950 group.
It has been well documented that tissue and cell swelling results from I/R injury can cause the “no‐reflow phenomenon”, which limits positive resuscitation outcomes and amplifies the ischemic cycle.5, 21 Cell swelling responding to I/R injury is primarily attributed to failure of the energy‐dependent cell volume control mechanism. Mitochondria play the central role in this mechanism. Of note, swollen mitochondria, in turn, lead to decreased ATP production, setting up a vicious circle, finally resulting in cardiomyocytes death. 22 PEG‐20k, a linear polymer composed of repeating ethylene glycol units, has been well characterized by significantly improving outcomes via reducing I/R cell swelling, increasing tolerance to low volume state, improving capillary blood flow, oxygen exchange, and lactate clearance.23, 24, 25, 26 Our results demonstrated that the CA animals presented with significantly lower PVD and MFI as described by previous study, 7 PEG‐20k largely restituted both PVD and MFI as early at ROSC 2‐hour. Surprisingly, MCC950 also significantly improved post‐resuscitated sublingual microcirculation, although the underlying mechanism may need further clarification. Importantly, no‐reflow phenomenon was further reversed in the PEG‐20k+ MCC950 group.
In the present study, mitochondrial respiratory function was measured using high‐resolution respirometry Oxygraph‐2k, which has been widely used to assess oxygen flux.15, 27, 28 Heart homogenates prepared by the same operator to minimize variability were used in the current study.13, 29 Our results demonstrated that reproducibility measured as intra‐assay coefficient of variation of duplicate measurement of oxygen flux was acceptable (ranging from 6.9% to 15.0%), in keeping with Talise E. Müller results. 30 Herein, complex I‐linked respiration, routine respiration, RCR, and OXPHOS coupling efficiency were significantly compromised following CA and I/R injury, while complex II and IV‐linked respiration did not alter. Consistent with our results, Han et al reported that complex I activity progressively deteriorated after 8 minutes of cardiac arrest, but complex II and III activity were relatively resistant to I/R injury in mice cardiomyocytes. 31 Conversely, Chen et al reported complex II activity was significantly decreased in rat myocardium after 30 minutes of ischemia and 24 hours of reperfusion. 32 Possible explanations for this dissimilarity include differences in the degree of ischemia (global ischemia versus focal ischemia with some degree of collateral flow) and ischemia or reperfusion duration. Collectively, our findings reinforced the conception of post‐resuscitated mitochondrial dysfunction, and complex I is highly sensitive to I/R injury.
We evaluated whether PEG20k and MCC950 restored myocardial function and prevented no‐reflow phenomenon is through improving mitochondrial function. As expected, PEG‐20k triggered routine and complex I‐linked respiration, while MCC950 only increased complex I‐linked respiration. Most importantly, PEG‐20k‒MCC950 combination further preserved complex I‐linked respiration, as well as with RCR and OXPHOS coupling efficiency.
The SIRT1 gene is activated in response to stress, starvation, and caloric restriction. 33 SIRT1 is mechanistically linked to protecting against I/R injury by modulating mitochondrial function. 34 Activation of SIRT1 promotes the transcription of genes in regulation of mitochondrial biogenesis and bioenergetics to maintain energy and metabolic homeostasis. A major finding of our study is that SIRT1 was significantly downregulated, concurrent with impaired mitochondrial function following CA. Similarly, Biel et al reported that SIRT1expression was significantly decreased after I/R, and loss of SIRT1 caused a sequential chain of defective autophagy, mitochondrial dysfunction, and hepatocyte death. 34 It has been well documented that SIRT1 deacetylates the PGC‐1α and further increases its transcriptional activity. 35 PGC‐1α subsequently induces the expression of several downstream genes, like Nuclear respiratory factor‐1 and ‐2, mitochondrial transcription factor A, and estrogen‐related receptors. These genes play pivotal roles in the regulation of mitochondrial biogenesis and bioenergetics.36, 37 Herein, PEG‐20k upregulated protein levels of SIRTI and PGC‐1αconcurrent with restoration of mitochondrial function, while suppressing activation of NLRP3 inflammasomes by MCC950 did not affect the expression of SIRT1 and PGC‐1α. Thus, it is conceivable that PEG‐20k retrieves mitochondrial function, subsequently restores post‐resuscitated myocardial function and restitutes systemic microcirculation via the SIRT1/PGC‐1α pathway.
NLRP3 inflammasomes activation mediates excessive sterile systemic inflammatory responses and ensuing pyroptosis account for the pathogenesis of a variety of diseases.38, 39 NLRP3 inflammasomes are activated by assembling of NLRP3, apoptosis‐associated speck‐like protein containing a CARD domain (ASC), and pro‐caspase‐1. Subsequently, autocatalytic cleavage of pro‐caspase‐1 generates its activated subunits p10 and p20, which further cleaves proinflammatory cytokines IL‐1β and IL‐18 into their mature and secreted forms. Activated caspase‐1 also cleaves the pore‐forming protein GSDMD, promoting polymerization of GSDMD. Finally, GSDMD together with IL‐1β and IL‐18 result in pyroptosis. The high level of inflammatory cytokines and increased pyroptosis of vascular endothelial cells and cardiomyocytes are highly and inversely associated with aggravation of post‐resuscitated myocardial dysfunction. 40
Previous studies have demonstrated that there is a dramatic increase in activation of NLRP3 inflammasomes and release of IL‐1β into plasma following I/R injury.19, 41 In the present study, we demonstrated that the selective NLPR3‐inflammasomes inhibitor MCC950 largely abrogated NLRP3 inflammasomes activation and quenched pyroptosis. Moreover, NLRP3 inflammasomes inhibition was associated with improvement of mitochondrial function. Most likely, GSDMD could bind to and induce permeabilization of mitochondria membrane, 42 where reduction of GSDMD by MCC950 prevents further mitochondrial damage.
We also found NLRP3 inflammasomes activation was largely suppressed by PEG‐20k. This anti‐inflammatory property of PEG‐20k could be attributed to various reasons,43, 44, 45 among which the SIRT1/PGC‐1α/mitochondria pathway plays a key role. Reactive oxygen species production from damaged mitochondria and the release of mitochondrial danger‐associated molecular patterns, such as DNA and cardiolipin are responsible for direct NLRP3 inflammasomes activation. 45 In support of this, we demonstrated that improved mitochondria by PEG‐20k via the SIRT1/PGC‐1αpathway was associated with quenching of NLRP3 inflammasomes. Since the combined therapy with PEG‐20k and MCC950 was superior in preserving myocardial function, restituting microcirculation, rescuing mitochondrial function when compared with either therapy alone, it is reasonable to extrapolate that PEG‐20k‒MCC950 combination exerts a protective effect by interrupting the mitochondrial vicious circle between mitochondrial permeabilization, release of mitochondrial danger‐associated molecular patterns, reactive oxygen species production, NLRP3 inflammasomes, and further mitochondrial damage. The proposed mechanisms are summarized in Figure 5. This research is also translational in nature since the results of the proposed studies will potentially lead to a novel means of active protection of the heart and lead to an improvement in survival following sudden cardiac arrest.
Figure 5. Proposed mechanisms underlying the positive therapeutic effects of combined therapy with polyethylene glycol‐20k and MCC950 preserved myocardial function in a rat model of cardiac arrest and cardiopulmonary resuscitation.
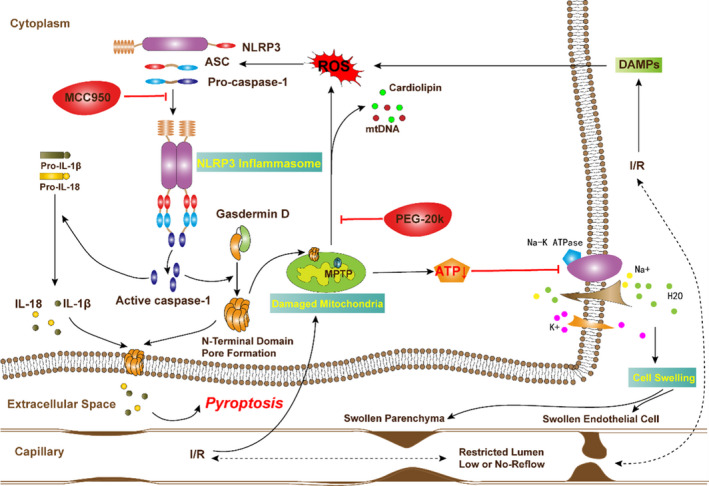
Polyethylene glycol‐20k‒MCC950 combination exerts a protective effect by interrupting the mitochondrial vicious circle between mitochondrial permeabilization, release of mitochondrial danger‐associated molecular patterns, reactive oxygen species production, NLRP3 (the NOD‐like receptor family protein 3) inflammasome, cell swelling, and pyroptosis, and further mitochondrial damage. MCC950 is a highly selective NLRP3 inflammasomes inhibitor. I/R indicates ischemia‐reperfusion; IL‐1β, interleukin‐1β; IL‐18, interleukin‐18; MPTP, mitochondrial permeability transition pore; NLRP3, the NOD‐like receptor family protein 3; PEG‐20k, olyethylene glycol‐20k; PGC1‐α, peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha; ASC, apoptosis‐associated speck‐like protein; ROS, reactive oxygen species; S, the sham group; and SIRT1, sirtuin 1.
There are several limitations needed to be addressed. Firstly, we did not measure other markers of mitochondrial function, such as ATP generation, reactive oxygen species production, mitochondrial permeability transition pore opening. Although mitochondrial respiration measured by high‐resolution respirometry is currently widely used for assessing mitochondrial function independently. Secondly, pyroptosis of vascular endothelial cell, cardiomyocytes, or other cells were not directly visualized and quantified. Instead, we evaluated the degree of cardiomyocytes injury with plasma cTnI level and inflammatory response with IL‐1β. Dynamical microcirculatory condition was visualized by the sidestream dark‐field imaging device. Lastly, on the multifactorial nature of I/R injury, further studies should elucidate the precise mechanisms of how PEG‐20k and MCC950 work in tandem to improve myocardial function in the CA and CPR animals, although we have initially provided molecular mechanistic insights into protective effect of PEG‐20k‒MCC950 combination on post‐resuscitated myocardial function.
Conclusions
Combined therapy with PEG‐20k and MCC950 is superior to either therapy alone for preserving post‐resuscitated myocardial function, restituting sublingual microcirculation in a rat model of cardiac arrest. The responsible mechanisms may involve the upregulated expression of SIRT1/PGC1‐α in tandem with inhibition of NLRP3 inflammasomes.
Source of Funding
This work was supported, in part, by the Weil Family Foundation, California. The funding agency had no role in study design, data collection, or analysis.
Disclosures
None.
Supporting information
Figures S1–S2
Acknowledgments
All the work was performed at the Weil Institute of Emergency and Critical Care Research at Virginia Commonwealth University. All authors have made contributions to the research.
(J Am Heart Assoc. 2021;10:e019177. DOI: 10.1161/JAHA.120.019177.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.019177
For Disclosures, see page 12.
References
- 1. Wiberg S, Stride N, Bro‐Jeppesen J, Holmberg MJ, Kjaergaard J, Larsen S, Donnino MW, Hassager C, Dela F. Mitochondrial dysfunction in adults after out‐of‐hospital cardiac arrest. Eur Heart J Acute Cardiovasc Care. 2019;S138–S144. DOI: 10.1177/2048872618814700. [DOI] [PubMed] [Google Scholar]
- 2. Kim J, Villarroel JP, Zhang W, Yin T, Shinozaki K, Hong A, Lampe JW, Becker LB. The responses of tissues from the brain, heart, kidney, and liver to resuscitation following prolonged cardiac arrest by examining mitochondrial respiration in rats. Oxid Med Cell Longev. 2016;7463407. DOI: 10.1155/2016/7463407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dey S, DeMazumder D, Sidor A, Foster DB, O'Rourke B. Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ Res. 2018;356–371. DOI: 10.1161/CIRCRESAHA.118.312708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang J, Toan S, Zhou H. Mitochondrial quality control in cardiac microvascular ischemia‐reperfusion injury: new insights into the mechanisms and therapeutic potentials. Pharmacol Res. 2020;104771. DOI: 10.1016/j.phrs.2020.104771. [DOI] [PubMed] [Google Scholar]
- 5. Oikonomou E, Mourouzis K, Vogiatzi G, Siasos G, Deftereos S, Papaioannou S, Latsios G, Tsalamandris S, Tousoulis D. Coronary microcirculation and the no‐reflow phenomenon. Curr Pharm Des. 2018;2934–2942. DOI: 10.2174/1381612824666180911122230. [DOI] [PubMed] [Google Scholar]
- 6. Wang J, Toan S, Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis. 2020;299–314. DOI: 10.1007/s10456-020-09720-2. [DOI] [PubMed] [Google Scholar]
- 7. Yang J, Xiao Y, Quan EY, Hu Z, Guo Q, Miao C, Bradley JL, Peberdy MA, Ornato JP, Mangino MJ, et al. Effects of polyethylene Glycol‐20k on postresuscitation myocardial and cerebral function in a rat model of cardiopulmonary resuscitation. Crit Care Med. 2018;e1190–e1195. DOI: 10.1097/CCM.0000000000003415. [DOI] [PubMed] [Google Scholar]
- 8. Wang W, Hua T, Li H, Wu X, Bradley J, Peberdy MA, Ornato JP, Tang W. Decreased cAMP level and decreased downregulation of beta1‐adrenoceptor expression in therapeutic hypothermia‐resuscitated myocardium are associated with improved post‐resuscitation myocardial function. J Am Heart Assoc. 2018;e006573. DOI: 10.1161/JAHA.117.006573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ye S, Weng Y, Sun S, Chen W, Wu X, Li Z, Weil MH, Tang W. Comparison of the durations of mild therapeutic hypothermia on outcome after cardiopulmonary resuscitation in the rat. Circulation. 2012;123–129. DOI: 10.1161/CIRCULATIONAHA.111.062257. [DOI] [PubMed] [Google Scholar]
- 10. Arnlov J, Ingelsson E, Riserus U, Andren B, Lind L. Myocardial performance index, a Doppler‐derived index of global left ventricular function, predicts congestive heart failure in elderly men. Eur Heart J. 2004;2220–2225. DOI: 10.1016/j.ehj.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 11. Pozo MO, KanooreEdul VS, Ince C, Dubin A. Comparison of different methods for the calculation of the microvascular flow index. Crit Care Res Pract. 2012;102483. DOI: 10.1155/2012/102483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Backer D, Hollenberg S, Boerma C, Goedhart P, Buchele G, Ospina‐Tascon G, Dobbe I, Ince C. How to evaluate the microcirculation: report of a round table conference. Crit Care. 2007;R101. DOI: 10.1186/cc6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ziak J, Krajcova A, Jiroutkova K, Nemcova V, Dzupa V, Duska F. Assessing the function of mitochondria in cytosolic context in human skeletal muscle: adopting high‐resolution respirometry to homogenate of needle biopsy tissue samples. Mitochondrion. 2015;106–112. DOI: 10.1016/j.mito.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 14. Doerrier C, Garcia‐Souza LF, Krumschnabel G, Wohlfarter Y, MeszarosAT GE. High‐resolution FluoRespirometry and OXPHOS protocols for human cells, permeabilized fibers from small biopsies of muscle, and isolated mitochondria. Methods Mol Biol. 2018;31–70. DOI: 10.1007/978-1-4939-7831-1_3. [DOI] [PubMed] [Google Scholar]
- 15. Makrecka‐Kuka M, Krumschnabel G, Gnaiger E. High‐resolution respirometry for simultaneous measurement of oxygen and hydrogen peroxide fluxes in permeabilized cells, Tissue Homogenate and Isolated Mitochondria. Biomolecules. 2015;1319–1338. DOI: 10.3390/biom5031319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jentzer JC, Chonde MD, Dezfulian C. Myocardial dysfunction and shock after cardiac arrest. Biomed Res Int. 2015;314796. DOI: 10.1155/2015/314796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cour M, Turc J, Madelaine T, Argaud L. Risk factors for progression toward brain death after out‐of‐hospital cardiac arrest. Ann Intensive Care. 2019;45. DOI: 10.1186/s13613-019-0520-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Piao L, Fang YH, Hamanaka RB, Mutlu GM, Dezfulian C, Archer SL, Sharp WW. Suppression of superoxide‐hydrogen peroxide production at site IQ of mitochondrial complex I attenuates myocardial stunning and improves postcardiac arrest outcomes. Crit Care Med. 2020;e133–e140. DOI: 10.1097/CCM.0000000000004095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Hout GP, Ellenbroek GH, de Haan JJ, van Solinge WW, Cooper MA, Arslan F, de Jager SC, Robertson AA, Pasterkamp G. The selective NLRP3‐inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J. 2017;828–836. DOI: 10.1093/eurheartj/ehw247. [DOI] [PubMed] [Google Scholar]
- 20. Toldo S, Marchetti C, Mauro AG, Chojnacki J, Mezzaroma E, Carbone S, Zhang S, Van Tassell B, Salloum FN, Abbate A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia‐reperfusion in the mouse. Int J Cardiol. 2016;215–220. DOI: 10.1016/j.ijcard.2016.02.043. [DOI] [PubMed] [Google Scholar]
- 21. van Genderen ME, Lima A, Akkerhuis M, Bakker J, van Bommel J. Persistent peripheral and microcirculatory perfusion alterations after out‐of‐hospital cardiac arrest are associated with poor survival. Crit Care Med. 2012;2287–2294. DOI: 10.1097/CCM.0b013e31825333b2. [DOI] [PubMed] [Google Scholar]
- 22. Petit PX, Goubern M, Diolez P, Susin SA, Zamzami N, Kroemer G. Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: the impact of irreversible permeability transition. FEBS Lett. 1998;111–116. DOI: 10.1016/S0014-5793(98)00318-4. [DOI] [PubMed] [Google Scholar]
- 23. Parrish D, Plant V, Lindell SL, Limkemann A, Reichstetter H, Aboutanos M, Mangino MJ. New low‐volume resuscitation solutions containing PEG‐20k. J Trauma Acute Care Surg. 2015;22–29. DOI: 10.1097/TA.0000000000000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Parrish D, Lindell SL, Reichstetter H, Aboutanos M, Mangino MJ. Cell impermeant‐based low‐volume resuscitation in hemorrhagic shock: a biological basis for injury involving cell swelling. Ann Surg. 2016;565–572. DOI: 10.1097/SLA.0000000000001049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Plant V, Limkemann A, Liebrecht L, Blocher C, Ferrada P, Aboutanos M, Mangino MJ. Low‐volume resuscitation using polyethylene glycol‐20k in a preclinical porcine model of hemorrhagic shock. J Trauma Acute Care Surg. 2016;1056–1062. DOI: 10.1097/TA.0000000000001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Plant V, Parrish DW, Limkemann A, Ferrada P, Aboutanos M, Mangino MJ. Low‐volume resuscitation for hemorrhagic shock: understanding the mechanism of PEG‐20k. J Pharmacol Exp Ther. 2017;334–340. DOI: 10.1124/jpet.116.239822. [DOI] [PubMed] [Google Scholar]
- 27. Gnaiger E. Mitochondrial Pathways and Respiratory Control. An Introduction in OXPHOS Analysis. Innsbruck, Austria: Oroboros MiPNet Publications; 2014. [Google Scholar]
- 28. Karlsson M, Hara N, Morata S, Sjovall F, Kilbaugh T, Hansson MJ, Uchino H, Elmer E. Diverse and tissue‐specific mitochondrial respiratory response in a mouse model of sepsis‐induced multiple organ failure. Shock. 2016;404–410. DOI: 10.1097/SHK.0000000000000525. [DOI] [PubMed] [Google Scholar]
- 29. Pecinova A, Drahota Z, Nuskova H, Pecina P, Houstek J. Evaluation of basic mitochondrial functions using rat tissue homogenates. Mitochondrion. 2011;722–728. DOI: 10.1016/j.mito.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 30. Muller TE, Nunes MEM, Rodrigues NR, Fontana BD, Hartmann DD, Franco JL, Rosemberg DB. Neurochemical mechanisms underlying acute and chronic ethanol‐mediated responses in zebrafish: the role of mitochondrial bioenergetics. Neurochem Int. 2019;104584. DOI: 10.1016/j.neuint.2019.104584. [DOI] [PubMed] [Google Scholar]
- 31. Han F, Da T, Riobo NA, Becker LB. Early mitochondrial dysfunction in electron transfer activity and reactive oxygen species generation after cardiac arrest. Crit Care Med. 2008;S447–S453. DOI: 10.1097/CCM.0b013e31818a8a51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen YR, Chen CL, Pfeiffer DR, Zweier JL. Mitochondrial complex II in the post‐ischemic heart: oxidative injury and the role of protein S‐glutathionylation. J Biol Chem. 2007;32640–32654. DOI: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- 33. Fan H, Yang HC, You L, Wang YY, He WJ, Hao CM. The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion‐induced acute kidney injury. Kidney Int. 2013;404–413. DOI: 10.1038/ki.2012.394. [DOI] [PubMed] [Google Scholar]
- 34. Biel TG, Lee S, Flores‐Toro JA, Dean JW, Go KL, Lee M‐H, Law BK, Law ME, Dunn WA Jr, Zendejas I, et al. Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2‐dependent manner. Cell Death Differ. 2016;279–290. DOI: 10.1038/cdd.2015.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Long J, Badal SS, Ye Z, Wang Y, Ayanga BA, Galvan DL, Green NH, Chang BH, Overbeek PA, Danesh FR. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Invest. 2016;4205–4218. DOI: 10.1172/JCI87927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Di W, Lv J, Jiang S, Lu C, Yang Z, Ma Z, Hu W, Yang Y, Xu B. PGC‐1: the energetic regulator in cardiac metabolism. Curr Issues Mol Biol. 2018;29–46. DOI: 10.21775/cimb.028.029. [DOI] [PubMed] [Google Scholar]
- 37. Yuan Y, Cruzat VF, Newsholme P, Cheng J, Chen Y, Lu Y. Regulation of SIRT1 in aging: roles in mitochondrial function and biogenesis. Mech Ageing Dev. 2016;10–21. DOI: 10.1016/j.mad.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 38. Qiu Z, Lei S, Zhao B, Wu Y, Su W, Liu M, Meng Q, Zhou B, Leng Y, Xia ZY. NLRP3 inflammasome activation‐mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats. Oxid Med Cell Longev. 2017;9743280. DOI: 10.1155/2017/9743280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anders H‐J, Suarez‐Alvarez B, Grigorescu M, Foresto‐Neto O, Steiger S, Desai J, Marschner JA, Honarpisheh M, Shi C, Jordan J, et al. The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis‐related chronic kidney disease independent from IL‐1‐mediated tissue injury. Kidney Int. 2018;656–669. DOI: 10.1016/j.kint.2017.09.022. [DOI] [PubMed] [Google Scholar]
- 40. Niemann JT, Rosborough JP, Youngquist S, Shah AP, Lewis RJ, Phan QT, Filler SG. Cardiac function and the proinflammatory cytokine response after recovery from cardiac arrest in swine. J Interferon Cytokine Res. 2009;749–758. DOI: 10.1089/jir.2009.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu Z, Lu X, Zhou X, Chen Z, Zhao Y. The role of inflammation in postresuscitation myocardial dysfunction. Int J Cardiol. 2013;e47–e48. DOI: 10.1016/j.ijcard.2013.08.124. [DOI] [PubMed] [Google Scholar]
- 42. Platnich JM, Chung H, Lau A, Sandall CF, Bondzi‐Simpson A, Chen H‐M, Komada T, Trotman‐Grant AC, Brandelli JR, Chun J, et al. Shiga toxin/lipopolysaccharide activates caspase‐4 and gasdermin D to trigger mitochondrial reactive oxygen species upstream of the NLRP3 inflammasome. Cell Rep. 2018;1525–1536.e1527. DOI: 10.1016/j.celrep.2018.09.071. [DOI] [PubMed] [Google Scholar]
- 43. Compan V, Baroja‐Mazo A, López‐Castejón G, Gomez A, Martínez C, Angosto D, Montero María T, Herranz A, Bazán E, Reimers D, et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity. 2012;487–500. DOI: 10.1016/j.immuni.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 44. Li Y, Yang X, He Y, Wang W, Zhang J, Zhang W, Jing T, Wang B, Lin R. Negative regulation of NLRP3 inflammasome by SIRT1 in vascular endothelial cells. Immunobiology. 2017;552–561. DOI: 10.1016/j.imbio.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 45. Platnich JM, Muruve DA. NOD‐like receptors and inflammasomes: a review of their canonical and non‐canonical signaling pathways. Arch Biochem Biophys. 2019;4–14. DOI: 10.1016/j.abb.2019.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S2