

Abstract
Background
Pediatric cardiomyopathy is a genetically heterogeneous disease with substantial morbidity and mortality. Current guidelines recommend genetic testing in children with hypertrophic, dilated, or restrictive cardiomyopathy, but practice variations exist. Robust data on clinical testing practices and diagnostic yield in children are lacking. This study aimed to identify the genetic causes of cardiomyopathy in children and to investigate clinical genetic testing practices.
Methods and Results
Children with familial or idiopathic cardiomyopathy were enrolled from 14 institutions in North America. Probands underwent exome sequencing. Rare sequence variants in 37 known cardiomyopathy genes were assessed for pathogenicity using consensus clinical interpretation guidelines. Of the 152 enrolled probands, 41% had a family history of cardiomyopathy. Of 81 (53%) who had undergone clinical genetic testing for cardiomyopathy before enrollment, 39 (48%) had a positive result. Genetic testing rates varied from 0% to 97% between sites. A positive family history and hypertrophic cardiomyopathy subtype were associated with increased likelihood of genetic testing (P=0.005 and P=0.03, respectively). A molecular cause was identified in an additional 21% of the 63 children who did not undergo clinical testing, with positive results identified in both familial and idiopathic cases and across all phenotypic subtypes.
Conclusions
A definitive molecular genetic diagnosis can be made in a substantial proportion of children for whom the cause and heritable nature of their cardiomyopathy was previously unknown. Practice variations in genetic testing are great and should be reduced. Improvements can be made in comprehensive cardiac screening and predictive genetic testing in first‐degree relatives. Overall, our results support use of routine genetic testing in cases of both familial and idiopathic cardiomyopathy.
Registration
URL: https://www.clinicaltrials.gov; Unique identifier: NCT01873963.
Keywords: exome, heart failure, infant, molecular, mutation
Subject Categories: Cardiomyopathy, Genetics
Nonstandard Abbreviations and Acronyms
- DCM
dilated cardiomyopathy
- HCM
hypertrophic cardiomyopathy
- LVNC
left ventricular noncompaction
- PCMR
pediatric cardiomyopathy registry
- RCM
restrictive cardiomyopathy
- VUS
variant of uncertain significance
Clinical Perspective
What Is New?
There is significant practice variation in ordering genetic testing for pediatric patients with cardiomyopathy.
In a pediatric cardiomyopathy cohort in which ≈50% had clinical genetic testing, 21% of nontested patients had a diagnostic genetic finding with research‐based testing of known cardiomyopathy genes.
What Are the Clinical Implications?
Routine use of genetic testing in children with familial or idiopathic cardiomyopathy is indicated.
Cardiomyopathy is a clinically and genetically heterogeneous form of heart muscle disease with substantial morbidity and mortality in children. There are 5 phenotypes: hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM), left ventricular noncompaction cardiomyopathy (LVNC), and arrhythmogenic ventricular cardiomyopathy. 1 Current consensus guidelines recommend genetic testing in children and adults with cardiomyopathy. Cardiac surveillance is recommended in first‐degree relatives, and genetic testing for a known familial variant is indicated. 2 , 3 , 4 Despite these recommendations, financial factors related to reimbursement, the family's understanding and perception of genetic testing, and limited data to guide physician practice with respect to pediatric cardiomyopathies are all barriers to genetic testing. In addition, variations in genetic testing practices in this age group are not well studied. 5
Epidemiologic studies indicate that although the causes of cardiomyopathy in children are more diverse than in adults, 6 , 7 , 8 genes encoding sarcomeric, cytoskeletal, or desmosomal proteins are important causes of cardiomyopathy across all ages. 9 , 10 , 11 , 12 , 13 However, data regarding the diagnostic yield of genetic testing in children with cardiomyopathy are limited and conflicting. For example, molecular analysis in children with HCM at a single institution found a higher prevalence of pathogenic variants than in adults, 14 but results from a large clinical genetic testing laboratory found a lower prevalence. 15 Case reports have prompted speculation that children with ≥2 pathogenic variants may have early‐onset severe disease. A recent retrospective review of sarcomeric mutation carriers showed that 25% of patients <18 years of age had ≥2 pathogenic or likely pathogenic variants (versus 4.8% for adults), but the number of children was small (n=24) and potentially confounded by ascertainment bias. 16 The frequency of 2 mutations in adults with HCM was recently found to be much lower than previously indicated. 17 Conflicting literature on genetic findings may contribute to practice variation in the genetic evaluation of children with cardiomyopathy.
Two centers have recently published retrospective evaluations of their patients with pediatric cardiomyopathy who underwent clinical genetic testing, 18 , 19 identifying a diagnostic yield of 26% in 151 pediatric patients with mixed cardiomyopathy types 19 and 40% in 70 patients without HCM. 18 Patients with pediatric cardiomyopathy who did not undergo clinical testing were not included. In a 2020 retrospective analysis of clinical genetic testing in pediatric patients with DCM (n=73) at a single institution from 2008 to 2018, 86% underwent clinical genetic testing, with 30% having positive findings and a high de novo variant rate. 20 A recent prospective study in 60 pediatric patients with all types of cardiomyopathy provided excellent information on clinical outcome and interrogated 89 genes but considered variants of uncertain significance (VUSs), likely pathogenic, and pathogenic variants all as positive genetic findings. 21 These studies identified challenges inherent in the study of pediatric cardiomyopathy such as the small patient numbers at a single institution and lack of comprehensive genetic evaluation or testing. We therefore initiated the PCM Genes (Pediatric Cardiomyopathy Genes study) to perform exome sequencing in a large prospectively recruited cohort. The overall goals of the study are to develop approaches to exome analysis for autosomal dominant disease, to determine genotype‐phenotype correlations and to identify genetic modifiers that influence the long‐term clinical course of children with cardiomyopathy. Here, we present the first results of the study, in which we determine the prevalence of pathogenic variants in known cardiomyopathy genes; investigate the associations between phenotype and age of onset, sex, race, and ethnic correlates; and identify practice variation in genetic testing of these children.
Methods
Study Design
The study used the network established by the PCMR (Pediatric Cardiomyopathy Registry). 22 , 23 The University of Miami (2012–2014), Wayne State University (2014–2018), and the University at Buffalo (2018–2020) served as the Administrative Coordinating Centers, which were responsible for regulatory and clinical adherence, study implementation, protocol guidance, data collection (Appendix S1), and analysis planning. The New England Research Institute was the Data and Statistical Coordinating Center. Cincinnati Children's Hospital Medical Center (2012–2014) and Indiana University (2014–2020) served as Genetic Coordinating Centers. All analytic and study materials are available within this article and its online supplementary files. Deidentified genomic data will be publicly available within the database of Genotypes and Phenotypes.
Patients
Enrollment for the study began April 12, 2013 and continued through February 29, 2016. Children were eligible if they had a diagnosis of idiopathic or primary cardiomyopathy with a phenotype of DCM, HCM, RCM, or LVNC or presumed myocarditis before 18 years of age. Diagnoses were ascertained by chart review and confirmed by echocardiography (Table S1) or cardiac magnetic resonance imaging. Children with cardiomyopathy secondary to another condition such as neuromuscular disease or a genetic syndrome associated with cardiomyopathy were excluded (Table S2).
Biological parents and affected siblings or relatives were approached for enrollment. After written informed consent or assent, blood was drawn for genetic testing and a 3‐generation pedigree was obtained. Saliva samples were collected when blood collection failed. A positive family history was defined as at least 1 additional biological family member with a diagnosis of cardiomyopathy or with positive genetic testing results for cardiomyopathy if phenotype negative. If clinical genetic testing was already performed, results from the testing laboratory were uploaded to the database. All clinical genetic results were manually reviewed.
For this report, clinical genetic testing refers to molecular testing for cardiomyopathy, typically with a next‐generation sequencing cardiomyopathy panel; other types of clinical genetic testing, such as chromosomes, were excluded.
Cardiac phenotypic data were collected for 3 years before enrollment until 2 years after unless the child died, underwent heart transplant, or withdrew from the study. Data collected at study visits included demographic information, anthropometric measurements, family history with pedigree, eligibility data, heart failure class, the results of cardiac studies (echocardiograms, cardiac magnetic resonance imaging, ECGs, Holter monitoring, and endomyocardial biopsies), hospitalizations, cardiac transplant status, and cause of death. Study data were entered into a web‐based eClinicalOS system through dedicated, password‐protected study computers. The Institutional Review Boards at the Administrative Coordinating Center and participating institutions approved this study.
Sample Collection, Preparation, and Sequencing
Blood or saliva samples were collected at each site and shipped to the Genetic Coordinating Center. DNA was extracted on a Maxwell RSC instrument (Promega, Madison, WI) according to the manufacturer's protocol. Exome sequencing was performed using sequence capture (SeqCap EZ Human Exome 2.0; Nimblegen, Madison, WI) and a HiSeq2500 sequencer (Illumina, San Diego, CA) at Cincinnati Children's Hospital Medical Center in a Clinical Laboratory Improvement Amendments–approved lab. All samples were sequenced to a mean coverage of 72× (36× to 148×) (Table S3). Alignment was performed to Genome Reference Consortium Human Build 37 GRCh37 with BWA aln/sampe version 0.5.9 using default parameters. Genome Analysis Toolkit version 3.3 best practices were followed for variant calling, 24 and variants were annotated using a Cincinnati Children's Hospital Medical Center in‐house script, according to their predicted impact on University of California–Santa Cruz Known Genes (hg19).
Variant Assessment
At study initiation, 37 genes from clinical cardiomyopathy genetic testing panels were selected for interrogation (Table 1). These genes were selected at study initiation based on panels that were orderable from commercial clinical laboratories. In addition, they had been shown clinically or in research testing in children to cause disease. The one exception was the TTR gene, which is not associated with cardiomyopathy in children but was included as a potential modifier. Results were returned to participants for all genes (n=36) except TTR if return of results was elected upon consent. Among 152 children, 6 (4%) declined to receive genetic results. A positive result was defined as ≥1 pathogenic or likely pathogenic variants known to cause cardiomyopathy in children. Results that confirmed previous clinical testing results were not returned to participants; thus, only new positive findings were returned.
Table 1.
Cardiomyopathy‐Causing Genes Analyzed in the Pediatric Cardiomyopathy Genes Study
| ABCC9 | LAMP2 | NEXN | TNNC1 |
| ACTC1 | LDB3 | PLN | TNNI3 |
| ACTN2 | LMNA | PRKAG2 | TNNT2 |
| ANKRD1 | MYBPC3 | RBM20 | TPM1 |
| BAG3 | MYH6 | SCN5A | TTN |
| CAV3 | MYH7 | SCO2 | TTR |
| CRYAB | MYL2 | SGCD | VCL |
| CSRP3 | MYL3 | SURF1 | |
| DES | MYPN | TAZ | |
| EMD | NEBL | TCAP |
Filtering was performed (single‐nucleotide polymorphism database 144, minor allele frequency <5%) at low stringency to retain potential modifying variants for future studies. Retained variants were predicted to alter the protein (missense, frameshift, insertion/deletion, stop gained or lost, and splice‐site mutations). Because of the large number of variants in the TTN gene encoding the protein titin and good evidence for truncating variants in TTN causing DCM, only nonsense and frameshift variants located within the A‐band region of the protein were interpreted. 25 , 26
For all 37 genes, potential protein‐altering rare variants were interpreted according to the American College of Medical Genetics and Genomics standards and guidelines for the interpretation of sequence variants to assess pathogenicity 27 (Table S4). All variant interpretations were concordant with ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) as of December 1, 2018. Variants were interpreted as pathogenic, likely pathogenic, VUS, likely benign, or benign (Table S4). All novel variants are being deposited into ClinVar.
Statistical Analysis
Categorical variables are summarized with frequencies, and age at diagnosis is summarized with medians with interquartile ranges. To identify differences between the cardiomyopathy phenotypes, we first combined LVNC and mixed phenotypes because of the small sample size for each group (3 and 13, respectively). Fisher's exact test and chi‐squared test were used to assess associations between phenotype and categorical variables, and the Wilcoxon rank sum test (given the non‐normal distribution) was used to assess age at diagnosis and enrollment.
To identify factors associated with the decision to undergo clinical genetic testing, we used contingency tables, chi‐squared, or Fisher's exact tests, as appropriate, for categorical variables (sex, race/ethnicity [races from East Asian or South Asian superpopulations or non‐Hispanic White ethnicity], family history) and the Wilcoxon rank‐sum test for age at diagnosis and enrollment. To evaluate whether factors independently contributed, logistic regression was performed, with performance of clinical genetic testing as the dependent variable and age at diagnosis, sex, race/ethnicity, family history, and cardiomyopathy type as predictor variables. Chi‐squared tests evaluated whether cardiomyopathy phenotype was associated with positive genetic findings in children with clinical test results. Alpha was set at 0.05 and all tests were 2‐tailed. Significance thresholds were not adjusted for multiplicity. Data were analyzed with JMP v13.1.0 (SAS Institute, Cary, NC).
Results
PCM Genes Participants
Demographics for the first 152 probands enrolled in the PCM Genes, representing 9 of the 14 participating sites, are shown in Table 2. The cohort was 51% male and predominantly non‐Hispanic White. Median age (interquartile range) at diagnosis was 4.5 (0.5–13.2) years (Table 2). Median age (interquartile range) at enrollment was 10.5 (3.3–15.7) years. The ratio of DCM to HCM was about 2:1, which is consistent with other epidemiologic studies 6 , 8 , 20 , 26 , 27 , 28 , 29 (Figure 1). Children with RCM represented 9% of the cohort, slightly higher than the reported frequency of 5% among all cardiomyopathies. 30 Three children (2%) had isolated LVNC, and 13 (9%) had a mixed phenotype that frequently included LVNC (9/13; 69%).
Table 2.
Characteristics of Children With Cardiomyopathy in the Pediatric Cardiomyopathy Genes Study, by Phenotype
| Characteristic | All, N=152 | Type of Cardiomyopathy | |||
|---|---|---|---|---|---|
| Dilated, n=80 | Hypertrophic, n=42 | Restrictive, n=14 | Left Ventricular Noncompaction/Mixed, n=16 | ||
| Male, % | 51.3 | 43.8 | 71.4 | 42.9 | 43.8 |
| Race/ethnicity, % | |||||
| White | 73.7 | 66.3 | 90.5 | 78.6 | 62.5 |
| Black | 13.2 | 16.3 | 4.8 | 7.1 | 25.0 |
| East Asian, South Asian | 13.2 | 17.5 | 4.8 | 14.3 | 12.5 |
| Non‐Hispanic | 81.6 | 83.8 | 83.3 | 78.6 | 68.8 |
| Median (IQR) age at diagnosis, y | 4.5 (0.5–13.2) | 1.9 (0.3–11.1) | 12.0 (6.0–15.0) | 9.7 (1.0–14.4) | 0.84 (0.09–8.5) |
| Median (IQR) age at enrollment, y | 11.2 (4.8–16.8) | 10.5 (3.3–15.7) | 16.1 (10.1–18.8) | 11.9 (4.5–17.7) | 5.3 (2.0–9.1) |
| Median (IQR) time from diagnosis to enrollment, y | 2.7 (0.5–6.9) | 3.8 (0.6–7.6) | 3.0 (0.5–7.6) | 2.2 (1.0–2.9) | 1.4 (0.4–3.5) |
| Family history, % positive | 40.8 | 28.8 | 66.7 | 28.6 | 43.8 |
IQR indicates interquartile range.
Figure 1. Characteristics of 152 children in the pediatric cardiomyopathy genes study.
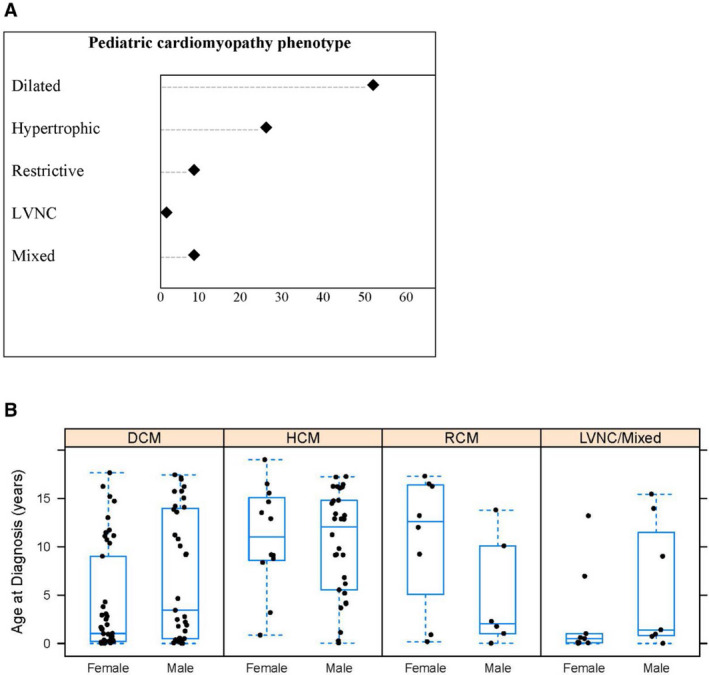
A, Distribution of cardiomyopathy phenotypes; B, Median (interquartile range) age at cardiomyopathy diagnosis by sex and cardiomyopathy phenotype. DCM indicates dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LVNC, left ventricular noncompaction; and RCM, restrictive cardiomyopathy.
Differences by Cardiomyopathy Phenotype
Age at diagnosis differed significantly between the cardiomyopathy phenotypes (P<0.0001; Table 2 and Figure 1). These differences were largely driven by the higher median age at diagnosis for HCM than for DCM (P<0.0001) and LVNC/Mixed (P=0.0004). The phenotypes also differed by sex (P=0.02), with HCM having more males (71%) than the other phenotypes, and by race or ethnicity (P=0.02), with HCM and RCM having higher proportions of non‐Hispanic White individuals (81% and 71%, respectively) than DCM and LVNC/Mixed (58% and 44%, respectively). Children with HCM had the highest frequency of a positive family history (67%), with DCM and RCM each having 29% (P<0.001).
Clinical Genetic Testing Practices and Outcomes
Figure 2 shows the proportion of patients who underwent clinical genetic testing. Overall, 89 children (59%) had results available at the time of exome analysis. Of these, 81 (53%) had a cardiomyopathy gene panel, and another 8 had other genetic testing (targeted testing for a known familial variant [6], exome testing [1], and unspecified cardiomyopathy genetic testing [1]). These 8 children were excluded when the frequency and yield of clinical panel testing was calculated (Figure 2).
Figure 2. Frequency of clinical genetic cardiomyopathy panel testing by cardiomyopathy subtype.
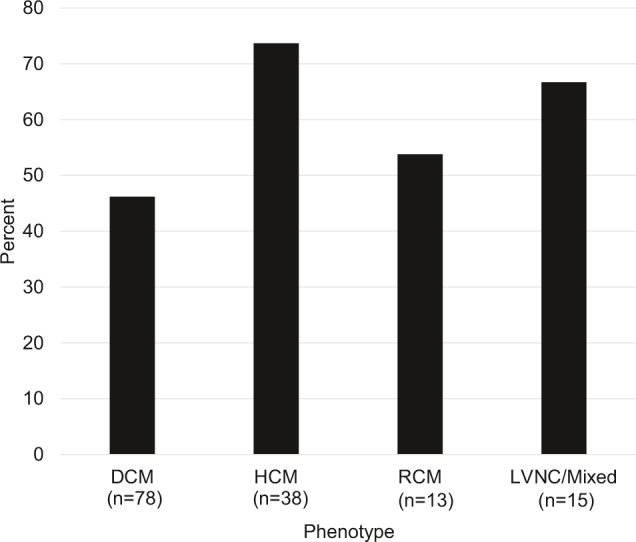
DCM indicates dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LVNC, left ventricular noncompaction; and RCM, restrictive cardiomyopathy.
We evaluated factors associated with cardiomyopathy gene panel testing between those who did and did not have previous clinical test results (Table 3). Children who had undergone testing had a higher rate of positive family history (51% versus 22%; P=0.005). Age of enrollment, sex, and race were not associated with clinical genetic testing, but cardiomyopathy phenotype was (P=0.03; Figure 2). Specifically, children with HCM were more likely than children with DCM to undergo clinical testing (74% versus 46%, P=0.005). In a logistic regression model, only family history retained significance (OR=3.1 [1.4–6.8]; P=0.0044). The lack of significance for type of CM was likely attributable to the strong association between type of cardiomyopathy and family history with HCM having a much higher rate of positive family history than DCM (67% versus 29%, respectively; P<0.0001). Variation across the sites was marked (Figure S1). Of sites contributing at least 10 cases, 3 sites performed testing in less than one‐third of children (range, 19%–32%), 1 site performed testing in about half (47%), and 3 sites performed testing in at least two‐thirds of children (range, 68%–97%).
Table 3.
Factors Associated With Clinical Genetic Testing in Children With a Cardiomyopathy Diagnosis
| Factor | Clinical Genetic Testing | P Value | |
|---|---|---|---|
| Yes*, n=81 | No, n=63 | ||
| Median (IQR) age at diagnosis, y | 5.5 (0.7–13.2) | 2.9 (0.4–13.2) | 0.59 |
| Male, % | 49.4 | 49.2 | 0.98 |
| Race, non‐Hispanic White, % | 65.4 | 58.7 | 0.41 |
| Family history, % positive | 50.6 | 22.2 | 0.005 |
| Cardiomyopathy type, % | 0.03 | ||
| Hypertrophic | 34.6 | 15.9 | |
| Dilated | 44.4 | 66.7 | |
| Restrictive | 8.6 | 9.5 | |
| Left ventricular noncompaction/mixed | 12.4 | 7.9 | |
IQR indicates interquartile range.
Eight children with genetic testing other than cardiomyopathy panel testing (eg, testing for known familial variant) were excluded.
Of 81 children who underwent clinical cardiomyopathy gene panel testing, 39 (48%) had a positive result; 19 (24%) had a VUS as the primary finding; and 23 (28%) had negative genetic testing results (Figure 3). Overall, the frequency of positive results was higher in children with HCM (68%) than with DCM (31%; P=0.0008).
Figure 3. Clinical genetic testing results in 81 cardiomyopathy patients by phenotype.
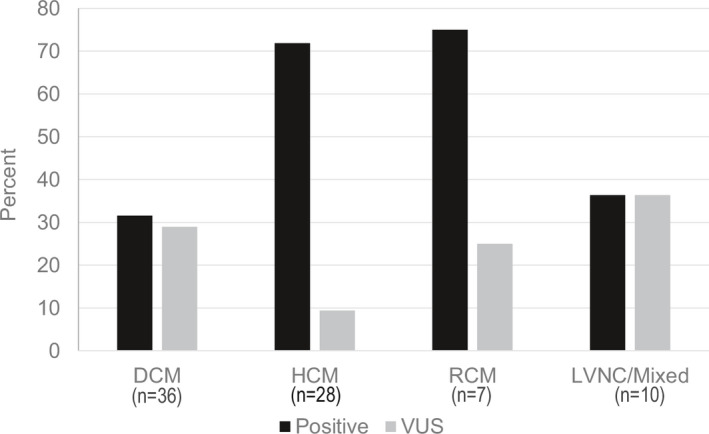
Results interpreted as pathogenic or likely pathogenic are considered positive. Patients who did not have cardiomyopathy panel testing were excluded. DCM indicates dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LVNC, left ventricular noncompaction; RCM, restrictive cardiomyopathy; and VUS, variant of uncertain significance.
Sequencing Results
Of the variants reported for children who had already undergone clinical testing, 89% were also identified by exome sequencing, with indel variants in TTN accounting for most of the variants missed by exome. In the 63 children who had not previously undergone clinical genetic testing, a group predicted to have a lower diagnostic yield because of the increased number of children with DCM and children without a family history of disease, 21% (13/63) had an identifiable pathogenic or likely pathogenic variant. Four had DCM, 6 had HCM, 2 had RCM, and 1 had LVNC/Mixed. Of these 13 children, 12 had a single variant identified as pathogenic or likely pathogenic, and 1 child had 2 variants.
Based on exome sequencing of 37 known cardiomyopathy genes, 32% (49/152) had a positive pathogenic or likely pathogenic finding. Five children (3%) had 2 pathogenic or likely pathogenic variants; no child had >2. Positive results (Table S4) differed by cardiomyopathy type and family history (Figure 4). Diagnostic yields for familial DCM, HCM, RCM, and LVNC/Mixed cases were 35%, 68%, 75%, and 43%, respectively. Although idiopathic cases of HCM or RCM also had high diagnostic yields of 36% and 50%, respectively, idiopathic DCM cases tested positive less frequently (9%). None of the 3 children with isolated LVNC had positive findings, although 2 of the 3 had a positive family history of cardiomyopathy.
Figure 4. Frequency of positive exome testing results in children with cardiomyopathy by phenotype and family history status.
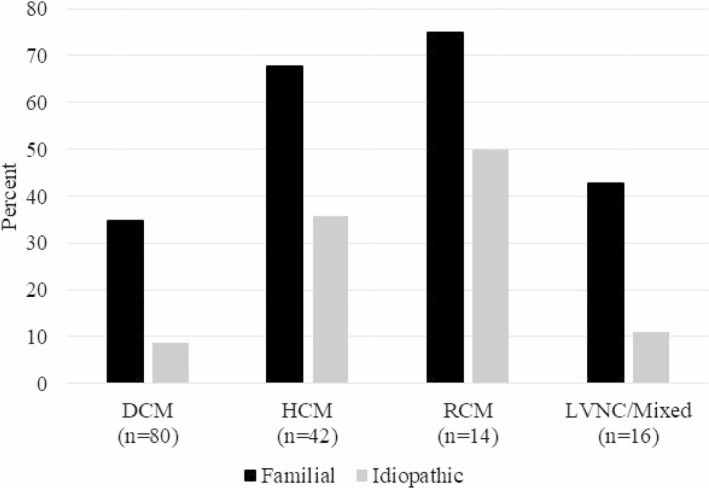
DCM indicates dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LVNC, left ventricular noncompaction; and RCM, restrictive cardiomyopathy.
Cardiac Surveillance and Genetic Testing in Family Members
Cardiac surveillance in first‐degree relatives of a child with cardiomyopathy is recommended. Similarly, predictive genetic testing is indicated in first‐degree relatives of those with a pathogenic or likely pathogenic variant. Among our patients with a positive family history of cardiomyopathy, 89% (55/72 familial cases) had cardiac surveillance in first‐degree relatives, whereas in those without a family history, 42% (38/90 idiopathic cases) had cardiac surveillance of first‐degree relatives. When focusing on the subset of probands who did not have clinical genetic testing (n=63), 10 of 14 familial cases (71%) had cardiac imaging in first‐degree relatives, whereas 16 of 49 idiopathic cases (41%) had cardiac screening in first‐degree relatives. Overall, predictive genetic testing was performed in 58% of first‐degree relatives of familial cases. In probands with no documented family history who underwent clinical genetic testing, 3 had presumed de novo pathogenic variants (both parents documented as negative for the pathogenic variant), 4 had VUS results that were inherited from an unaffected parent, and 1 had clinical exome testing performed as a quad test with mother, father, and brother, which was negative. Of note, this study was not longitudinal in nature and therefore additional family members may have undergone cardiac screening or predictive genetic testing since study enrollment. Importantly, 77% (10/13) of children with positive exome results who had not previously had clinical testing also had no family history of disease. For the entire cohort, the 14 children with new positive molecular findings had 44 at‐risk first‐degree relatives (parents and siblings).
Discussion
We conducted a multicenter study to better understand the genetic basis of familial and idiopathic pediatric cardiomyopathy. Information collected on 152 children provided important new data about clinical genetic testing practices in North America. With research sequencing on 152 children, we identified minimum diagnostic yields in known cardiomyopathy genes for broadly inclusive genetic testing in children with either familial or idiopathic cardiomyopathy.
Previous studies of the PCMR have provided information on the epidemiologic features and clinical outcomes of children with cardiomyopathy or heart failure, 8 , 22 , 29 , 30 , 31 , 32 and an understanding of the causal categories 7 , 33 , 34 and their impact on risk stratification and outcome, 32 , 35 , 36 , 37 but information on genetic causes specifically in the pediatric population has been limited. The PCM Genes study was intended to establish disease‐specific genotype‐phenotype correlations in pediatric cardiomyopathy and to provide a more sophisticated understanding of the molecular basis of disease and its association with presentation, clinical course, and outcome.
Another PCMR study 8 found that 51% of children had a diagnosis of DCM, a finding similar to the 53% (80/152) of our cohort. However, HCM (n=42) was less common in our cohort (28% versus 42%), and, as anticipated, this group had the highest proportion of children with positive family histories of cardiomyopathy. Somewhat surprisingly, the frequency of family history of cardiomyopathy in the LVNC/Mixed cardiomyopathy group was 44% (7/16). Two recent publications recommend genetic testing when HCM or DCM is seen in conjunction with LVNC and do not recommend testing for isolated LVNC in the absence of a family history. 3 , 38 An additional publication of LVNC patients of all ages that included 52 patients <18 years of age also found a low genetic diagnostic yield in sporadic cases of isolated LVNC and determined that left ventricular systolic dysfunction at presentation and long‐term outcome were related to genetics. 39 All positive cases in our LVNC/Mixed group would have been identified clinically following these guidelines. However, with only 16 children, our LVNC/Mixed group was small, and additional studies are required to better understand the genetic basis of mixed cardiomyopathy phenotypes.
Our results show that although genetic testing is being widely used it is not yet universal, and variations in practice are great. The frequency of testing ranged from none to 97% of patients. In previous studies, factors influencing decisions to order or recommend genetic testing have included perceived ability to obtain testing, knowledge and comfort level with understanding and discussing results, and perception of benefits or clinical utility. 40 , 41 , 42 At the time of study initiation, no clinical guidelines existed that specifically recommended genetic testing for cardiomyopathy in children, which may account for some of the variability. The availability and integration of geneticist and genetic counselor expertise as well as the cardiologists' acceptance of the importance of cardiac genetics may also have played roles, although this study was not designed to assess this directly. A recent study has shown that genetic testing is less used in index patients with HCM than in long QT syndrome. 43 At‐risk first‐degree family members of a child with cardiomyopathy or long QT syndrome are more likely to participate in recommended cardiac screening than genetic testing, 5 , 43 although screening participation was higher in families of gene‐positive children. In the current study, family history status and cardiomyopathy type were significantly associated with performing testing. However, family history status is at least partially dependent on comprehensive cardiac surveillance in first‐degree relatives. First‐degree relatives underwent cardiac screening in 42% of idiopathic cases, potentially leading to an underestimation of the prevalence of familial disease in affected asymptomatic family members. In the field of cardiovascular genetics, barriers to cardiac screening or genetic testing centered on patient concerns have not been well studied, but at least 1 study suggests that family decisions are more frequently the cause than insurance. 43 Further studies investigating motivation and barriers to genetic testing in children with cardiomyopathy will be necessary to delineate provider‐ and family‐specific factors.
We identified pathogenic or likely pathogenic variants in 32% of children (49/152). Among tested patients, HCM and RCM both had a high diagnostic yield and a low frequency of VUS. Because RCM is quite rare, yields of genetic testing have not been widely reported, although a 2008 study previously reported 4 of 12 RCM patients with mutations in sarcomeric genes including TNNI3, ACTC, and TNNT2. 44 In this study, 8 of 14 (57%) had pathogenic or likely pathogenic variants identified, suggesting that more widespread use of testing in this population should be considered. In contrast, the positive and VUS frequencies were nearly equal in children with DCM and LVNC/Mixed phenotypes, perhaps reflecting a greater genetic heterogeneity in these subtypes. Of 63 children without previous genetic testing, positive results were identified in 13 (21%), as well as in 1 child whose previous test results were negative. These missed opportunities for a molecular diagnosis by clinical testing have important implications for first‐degree relatives for whom targeted familial testing can be offered, allowing risk stratification and targeted cascade screening. Ongoing cardiac surveillance is indicated in first‐degree relatives of a child with cardiomyopathy when the cause is unknown. 2 , 3 , 4 For these 14 families with a new molecular diagnosis, testing for the known familial pathogenic variant in the 44 first‐degree relatives can identify individuals who require ongoing surveillance and those who do not and is cost‐effective by limiting ongoing evaluations and cardiac imaging to at‐risk family members. 15 , 45 Importantly, these missed genetic diagnoses occurred across all types of cardiomyopathy, indicating that all functional phenotypes of pediatric cardiomyopathy have a potential molecular cause identifiable through genetic testing.
A family history of cardiomyopathy indicates a genetic cause, and therefore it was not surprising to find a significant association with clinical genetic testing in this study. Interestingly, we also identified pathogenic genetic variants in 36% of children with idiopathic HCM and 50% of children with idiopathic RCM. Although DCM typically has a lower diagnostic yield than that in other types of cardiomyopathy (20%–30%), we found that 35% with familial DCM had positive results. In idiopathic DCM, the rate was lower (9%). Nevertheless, reclassifying 9% of idiopathic DCM cases as heritable substantially alters the care for family members. Thus, family history status may be useful to estimate prior probability of a positive genetic test but cannot be used to rule out the need for genetic testing. Overall, our results support use of genetic testing in cases of both familial and idiopathic cardiomyopathy.
Strengths and Limitations of the Study
Children were enrolled at centers with known expertise in pediatric cardiomyopathy; therefore, we cannot exclude ascertainment bias for more severe or atypical cases. All eligible patients with cardiomyopathy were approached, but we cannot completely eliminate the possibility of a survivor bias. This study was not longitudinal in nature and family history is dynamic. Cardiac surveillance and predictive cascade clinical genetic testing in family members may have occurred on a clinical basis after study data capture. Because we tested for 37 known cardiomyopathy genes, the diagnostic yield for each phenotype should be viewed as a minimum estimate.
Variant interpretation continues to be a challenge in clinical genetics. 46 , 47 , 48 Recent studies on HCM have called for more stringent interpretation especially in research studies. 15 , 49 , 50 We interpreted variants conservatively using currently accepted American College of Medical Genetics and Genomics guidelines. 27 A subset of VUS identified in this study may be upgraded to likely pathogenic or pathogenic variants with further investigation with family‐based segregation studies, which were not performed in this proband only study.
New approaches to better predict variant effects are needed to increase their clinical utility and are particularly important in the context of autosomal dominant diseases such as cardiomyopathy. Our results indicate that additional genetic causes remain to be discovered in pediatric cardiomyopathy.
Conclusions
Our study highlights that a definitive molecular genetic diagnosis can be made in a substantial proportion of children for whom the cause and heritable nature of their cardiomyopathy have been previously unknown. Overall, our results support use of genetic testing in cases of both familial and idiopathic cardiomyopathy. The diagnostic yields of genetic testing we found can serve as minimum yields for future studies. Additional genetic causes of pediatric cardiomyopathy remain to be discovered. The wide variation in genetic testing practices should be addressed, and improvements can be made in comprehensive cardiac screening and predictive genetic testing in first‐degree relatives. Barriers to genetic testing for pediatric cardiomyopathy should be identified and removed to make testing more widely available.
Sources of Funding
This work was supported by grants from the National Heart, Lung, and Blood Institute (HL 111459) and the Children's Cardiomyopathy Foundation (Tenafly, NJ). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Heart, Lung, and Blood Institute or Children's Cardiomyopathy Foundation.
Disclosures
Dr Steven E. Lipshultz is a consultant for Tenaya Therapeutics, Bayer, and on an advisory board for Myokardia.
Supporting information
Appendix S1
Tables S1–S4
Figure S1
Acknowledgments
We thank the children and their families for their participation. We also thank the Children's Cardiomyopathy Foundation for their ongoing support of the Pediatric Cardiomyopathy Registry's research efforts.
(J Am Heart Assoc. 2021;10:e017731. DOI: 10.1161/JAHA.120.017731.)
For Sources of Funding and Disclosures, see page 9.
References
- 1. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB; American Heart Association, Council on Clinical Cardiology Heart Failure, and Transplantation Committee, Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Group; Council on Epidemiology and Prevention . Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. DOI: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 2. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2011;58:2703–2738. DOI: 10.1016/j.jacc.2011.10.825. [DOI] [PubMed] [Google Scholar]
- 3. Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, Morales A, Taylor MRG, Vatta M, Ware SM. Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J Card Fail. 2018;24:281–302. DOI: 10.1016/j.cardfail.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8:1308–1339. DOI: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 5. Miller EM, Wang Y, Ware SM. Uptake of cardiac screening and genetic testing among hypertrophic and dilated cardiomyopathy families. J Genet Couns. 2013;22:258–267. DOI: 10.1007/s10897-012-9544-4. [DOI] [PubMed] [Google Scholar]
- 6. Nugent AW, Daubeney PEF, Chondros P, Carlin JB, Cheung M, Wilkinson LC, Davis AM, Kahler SG, Chow CW, Wilkinson JL, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348:1639–1646. DOI: 10.1056/NEJMoa021737. [DOI] [PubMed] [Google Scholar]
- 7. Kindel SJ, Miller EM, Gupta R, Cripe LH, Hinton RB, Spicer RL, Towbin JA, Ware SM. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail. 2012;18:396–403. DOI: 10.1016/j.cardfail.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, Cox GF, Lurie PR, McCoy KL, McDonald MA, Messere JE, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003;348:1647–1655. DOI: 10.1056/NEJMoa021715. [DOI] [PubMed] [Google Scholar]
- 9. Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, Towbin JA, Seidman JG, Seidman CE. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358:1899–1908. DOI: 10.1056/NEJMoa075463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tariq M, Ware SM. Importance of genetic evaluation and testing in pediatric cardiomyopathy. World J Card. 2014;6:1156–1165. DOI: 10.4330/wjc.v6.i11.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee TM, Hsu DT, Kantor P, Towbin JA, Ware SM, Colan SD, Chung WK, Jefferies JL, Rossano JW, Castleberry CD, et al. Pediatric cardiomyopathies. Circ Res. 2017;121:855–873. DOI: 10.1161/CIRCRESAHA.116.309386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ware SM. Genetics of paediatric cardiomyopathies. Curr Opin Pediatr. 2017;29:534–540. DOI: 10.1097/MOP.0000000000000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaski JP, Syrris P, Tome Esteban MT, Jenkins S, Pantazis A, Deanfield JE, McKenna WJ, Elliott PM. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:436–441. DOI: 10.1161/CIRCGENETICS.108.821314. [DOI] [PubMed] [Google Scholar]
- 14. Loar RW, Bos JM, Will ML, Ommen SR, Ackerman MJ. Genotype‐phenotype correlations of hypertrophic cardiomyopathy when diagnosed in children, adolescents, and young adults. Congenit Heart Dis. 2015;10:529–536. DOI: 10.1111/chd.12280. [DOI] [PubMed] [Google Scholar]
- 15. Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, Shen J, McLaughlin HM, Clark EH, Babb LJ, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880–888. DOI: 10.1038/gim.2014.205. [DOI] [PubMed] [Google Scholar]
- 16. Bales ND, Johnson NM, Judge DP, Murphy AM. Comprehensive versus targeted genetic testing in children with hypertrophic cardiomyopathy. Pediatr Cardiol. 2016;37:845–851. DOI: 10.1007/s00246-016-1358-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fourey D, Care M, Siminovitch KA, Weissler‐Snir A, Hindieh W, Chan RH, Gollob MH, Rakowski H, Adler A. Prevalence and clinical implication of double mutations in hypertrophic cardiomyopathy: revisiting the gene‐dose effect. Circ Cardiovasc Genet. 2017;10:e001685. DOI: 10.1161/CIRCGENETICS.116.001685. [DOI] [PubMed] [Google Scholar]
- 18. Ellepola CD, Knight LM, Fischbach P, Deshpande SR. Genetic testing in pediatric cardiomyopathy. Pediatr Cardiol. 2018;39:491–500. DOI: 10.1007/s00246-017-1779-2. [DOI] [PubMed] [Google Scholar]
- 19. Ouellette AC, Mathew J, Manickaraj AK, Manase G, Zahavich L, Wilson J, George K, Benson L, Bowdin S, Mital S. Clinical genetic testing in pediatric cardiomyopathy: is bigger better? Clin Genet. 2018;93:33–40. DOI: 10.1111/cge.13024. [DOI] [PubMed] [Google Scholar]
- 20. Quiat D, Witkowski L, Zouk H, Daly KP, Roberts AE. Retrospective analysis of clinical genetic testing in pediatric primary dilated cardiomyopathy: testing outcomes and the effects of variant reclassification. J Am Heart Assoc. 2020;9:e016195. DOI: 10.1161/JAHA.120.016195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Al‐Wakeel‐Marquard N, Degener F, Herbst C, Kühnisch J, Dartsch J, Schmitt B, Kuehne T, Messroghli D, Berger F, Klaassen S. RIKADA study reveals risk factors in pediatric primary cardiomyopathy. J Am Heart Assoc. 2019;8:e012531. DOI: 10.1161/JAHA.119.012531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grenier MA, Osganian SK, Cox GF, Towbin JA, Colan SD, Lurie PR, Sleeper LA, Orav EJ, Lipshultz SE. Design and implementation of the North American Pediatric Cardiomyopathy Registry. Am Heart J. 2000;139:S86–S95. DOI: 10.1067/mhj.2000.103933. [DOI] [PubMed] [Google Scholar]
- 23. Wilkinson JD, Sleeper LA, Alvarez JA, Bublik N, Lipshultz SE; the Pediatric Cardiomyopathy Registry Group . The Pediatric Cardiomyopathy Registry: 1995‐2007. Prog Pediatr Cardiol. 2008;25:31–36. DOI: 10.1016/j.ppedcard.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20:1297–1303. DOI: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herman DS, Lam L, Taylor MRG, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. DOI: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave‐Castillo LM, Wolfgeher D, McNally EM. Population‐based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–399. DOI: 10.1161/CIRCGENETICS.112.962928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. DOI: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arola A, Jokinen E, Ruuskanen O, Saraste M, Pesonen E, Kuusela AL, Tikanoja T, Paavilainen T, Simell O. Epidemiology of idiopathic cardiomyopathies in children and adolescents. A nationwide study in Finland. Am J Epidemiol. 1997;146:385–393. DOI: 10.1093/oxfordjournals.aje.a009291. [DOI] [PubMed] [Google Scholar]
- 29. Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, Cox GF, Lurie PR, Orav EJ, Towbin JA. Epidemiology and cause‐specific outcome of hypertrophic cardiomyopathy in children: findings from the Pediatric Cardiomyopathy Registry. Circulation. 2007;115:773–781. DOI: 10.1161/CIRCULATIONAHA.106.621185. [DOI] [PubMed] [Google Scholar]
- 30. Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876. DOI: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 31. Wilkinson JD, Landy DC, Colan SD, Towbin JA, Sleeper LA, Orav EJ, Cox GF, Canter CE, Hsu DT, Webber SA, et al. The pediatric cardiomyopathy registry and heart failure: key results from the first 15 years. Heart Fail Clin. 2010;6:401–413, vii. DOI: 10.1016/j.hfc.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Webber SA, Lipshultz SE, Sleeper LA, Lu M, Wilkinson JD, Addonizio LJ, Canter CE, Colan SD, Everitt MD, Jefferies JL, et al. Outcomes of restrictive cardiomyopathy in childhood and the influence of phenotype: a report from the Pediatric Cardiomyopathy Registry. Circulation. 2012;126:1237–1244. DOI: 10.1161/CIRCULATIONAHA.112.104638. [DOI] [PubMed] [Google Scholar]
- 33. Cox GF, Sleeper LA, Lowe AM, Towbin JA, Colan SD, Orav EJ, Lurie PR, Messere JE, Wilkinson JD, Lipshultz SE. Factors associated with establishing a causal diagnosis for children with cardiomyopathy. Pediatrics. 2006;118:1519–1531. DOI: 10.1542/peds.2006-0163. [DOI] [PubMed] [Google Scholar]
- 34. Ware SM. Evaluation of genetic causes of cardiomyopathy in childhood. Cardiol Young. 2015;25(suppl 2):43–50. DOI: 10.1017/S1047951115000827. [DOI] [PubMed] [Google Scholar]
- 35. Lipshultz SE, Orav EJ, Wilkinson JD, Towbin JA, Messere JE, Lowe AM, Sleeper LA, Cox GF, Hsu DT, Canter CE, et al. Risk stratification at diagnosis for children with hypertrophic cardiomyopathy: an analysis of data from the Pediatric Cardiomyopathy Registry. Lancet. 2013;382:1889–1897. DOI: 10.1016/S0140-6736(13)61685-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rusconi P, Wilkinson JD, Sleeper LA, Lu M, Cox GF, Towbin JA, Colan SD, Webber SA, Canter CE, Ware SM, et al. Differences in presentation and outcomes between children with familial dilated cardiomyopathy and children with idiopathic dilated cardiomyopathy: a report from the Pediatric Cardiomyopathy Registry Study Group. Circ Heart Fail. 2017;10:e002637. DOI: 10.1161/CIRCHEARTFAILURE.115.002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilkinson JD, Lowe AM, Salbert BA, Sleeper LA, Colan SD, Cox GF, Towbin JA, Connuck DM, Messere JE, Lipshultz SE. Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: a study from the Pediatric Cardiomyopathy Registry. Am Heart J. 2012;164:442–448. DOI: 10.1016/j.ahj.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 38. Miller EM, Hinton RB, Czosek R, Lorts A, Parrott A, Shikany AR, Ittenbach RF, Ware SM. Genetic testing in pediatric left ventricular noncompaction. Circ Cardiovasc Genet. 2017;10:e001735. DOI: 10.1161/CIRCGENETICS.117.001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Waning JI, Caliskan K, Hoedemaekers YM, van Spaendonck‐Zwarts KY, Baas AF, Boekholdt SM, van Melle JP, Teske AJ, Asselbergs FW, Backx Ad PCM, et al. Genetics, clinical features, and long‐term outcome of noncompaction cardiomyopathy. J Am Coll Cardiol. 2018;71:711–722. DOI: 10.1016/j.jacc.2017.12.019. [DOI] [PubMed] [Google Scholar]
- 40. McPherson E. Genetic diagnosis and testing in clinical practice. Clin Med Res. 2006;4:123–129. DOI: 10.3121/cmr.4.2.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Spiech KM, Tripathy PR, Woodcock AM, Sheth NA, Collins KS, Kannegolla K, Sinha AD, Sharfuddin AA, Pratt VM, Khalid M, et al. Implementation of a renal precision medicine program: clinician attitudes and acceptance. Life. 2020;10:32. DOI: 10.3390/life10040032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hendricks‐Sturrup MKM, Sturm AC, Lu CY. Barriers and facilitators to genetic testing for familial hypercholesterolemia in the United States: a review. J Pers Med. 2019;9:32. DOI: 10.3390/jpm9030032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Knight LM, Miller E, Kovach J, Arscott P, von Alvensleben JC, Bradley D, Valdes SO, Ware SM, Meyers L, Travers CD, et al. Genetic testing and cascade screening in pediatric long QT syndrome and hypertrophic cardiomyopathy. Heart Rhythm. 2020;17:106–112. DOI: 10.1016/j.hrthm.2019.06.015. [DOI] [PubMed] [Google Scholar]
- 44. Kaski JP, Syrris P, Burch M, Tome‐Esteban M‐T, Fenton M, Christiansen M, Andersen PS, Sebire N, Ashworth M, Deanfield JE, et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008;94:1478–1484. DOI: 10.1136/hrt.2007.134684. [DOI] [PubMed] [Google Scholar]
- 45. Ingles J, McGaughran J, Scuffham PA, Atherton J, Semsarian C. A cost‐effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart. 2012;98:625–630. DOI: 10.1136/heartjnl-2011-300368. [DOI] [PubMed] [Google Scholar]
- 46. Hershberger RE, Morales A. Dilated cardiomyopathy overview. In: Adam MP, Editor‐in‐Chief. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle; 2007. [updated August 23, 2018]. [Google Scholar]
- 47. Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH, Murray ML, Tokita MJ, Gallego CJ, Kim DS, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res. 2015;25:305–315. DOI: 10.1101/gr.183483.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Amendola L, Jarvik G, Leo M, McLaughlin H, Akkari Y, Amaral M, Berg J, Biswas S, Bowling K, Conlin L, et al. Performance of ACMG‐AMP Variant‐interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;99:247. DOI: 10.1016/j.ajhg.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Walsh R, Buchan R, Wilk A, John S, Felkin LE, Thomson KL, Chiaw TH, Loong CCW, Pua CJ, Raphael C, et al. Defining the genetic architecture of hypertrophic cardiomyopathy: re‐evaluating the role of non‐sarcomeric genes. Eur Heart J. 2017;38:3461–3468. DOI: 10.1093/eurheartj/ehw603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19:192–203. DOI: 10.1038/gim.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Tables S1–S4
Figure S1