

Abstract
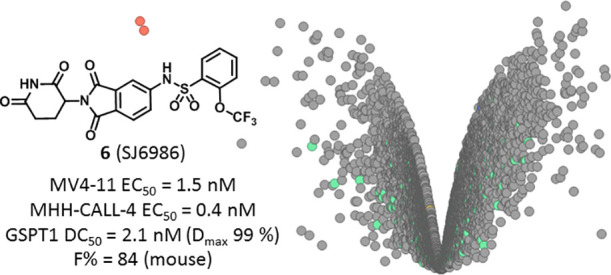
Whereas the PROTAC approach to target protein degradation greatly benefits from rational design, the discovery of small-molecule degraders relies mostly on phenotypic screening and retrospective target identification efforts. Here, we describe the design, synthesis, and screening of a large diverse library of thalidomide analogues against a panel of patient-derived leukemia and medulloblastoma cell lines. These efforts led to the discovery of potent and novel GSPT1/2 degraders displaying selectivity over classical IMiD neosubstrates, such as IKZF1/3, and high oral bioavailability in mice. Taken together, this study offers compound 6 (SJ6986) as a valuable chemical probe for studying the role of GSPT1/2 in vitro and in vivo, and it supports the utility of a diverse library of CRBN binders in the pursuit of targeting undruggable oncoproteins.
Introduction
Targeted protein degradation (TPD) is a novel chemical biology approach with potential profound effects on fundamental biology and drug discovery research by providing opportunities toward drugging undruggable targets.1,2 The TPD paradigm includes two main approaches of differing molecular design that generate small molecules with a similar proteasome-dependent mechanism of action, namely, proteolysis targeting chimeras (PROTACs)3,4 and molecular glues (MGs).5−8 MGs are small molecules capable of binding to an E3 ligase and altering its surface and specificity, leading to the recruitment, ubiquitination, and subsequent degradation of substrates that are normally not targeted by the ligase (neosubstrates). Recognition of the neosubstrate is governed by a protein–ligase surface interaction (a structural degron motif) and does not require a ligandable pocket. This provides an unprecedented opportunity to degrade hitherto undruggable targets, such as fusion oncoproteins and transcription factors.9,10 Immunomodulatory imide drugs (IMiDs), thalidomide and its close analogues pomalidomide and lenalidomide, are the “original” molecular glues providing the mechanistic and clinical validation of this approach.7,11
Interestingly, despite close structural similarity, IMiDs display different protein degradation profiles. Both pomalidomide (1) and lenalidomide (2) degrade the transcription factors IKZF1/3 but only lenalidomide induces degradation of CSNK1A1 (CK1α), illustrating how a small change in the molecular structure can significantly alter the specificity for the neosubstrate (Figure 1).12 Moreover, diversification around the IMiD scaffold has been shown to influence the potency and kinetics of neosubstrate degradation, as exemplified by CC-220 (3), which is 10-fold more potent in cells than lenalidomide,13 or specificity, as demonstrated by the discovery of novel neosubstrate degraders, such as the GSPT1 (G1 to S phase transition 1) degraders CC-885 and CC-90009 (Figure 1).14−16 These chemical modifications lead to considerable changes in cellular responses, creating new clinical translation opportunities.
Figure 1.

Analogues of thalidomide induce degradation of distinct disease-relevant proteins.
For IMiDs and closely related analogues, an increasing number of neosubstrates containing the common C2H2 zinc finger recognition degron motif have been discovered (IKZF2/4, SALL4, RNF166, ZFP91, ZNF692, ZNF276, ZNF653, and ZNF827).11,15 Each IMiD was found to display distinct patterns of substrate specificity, supporting the notion that neosubstrate diversity can be modulated by structural alterations of the ligand and is not limited to traditionally known targets. It also suggests that achieving selective protein degradation is a challenge and that understanding the structural basis of how ligand modification alters the interaction of the neosubstrate at the cereblon (CRBN) interface is important.17 Several recently reported studies have shown how simple structural modifications can result in an unexpected conversion of a PROTAC into a GSPT1 molecular glue degrader.18,19
In high-risk cancers like childhood acute leukemia (AL) and medulloblastoma (MB), aberrant activation, dysregulation, and/or mutation of C2H2 zinc finger transcription factors are prevalent, with limited targeted treatments. Representative examples include ZNF384 fusion oncoproteins observed in B-ALL and lineage ambiguous acute leukemia,20 IKZF1 mutations in acute lymphoblastic leukemia (ALL),21 deregulated MECOM in high-risk acute myeloid leukemia (AML), and enhancer-hijacking-dependent activation of GFI1, GFI1B, and PRDM6 in high-risk Group 3 and Group 4 MB subgroups.22−25 This provides an ideal opportunity to explore a molecular glue approach. We hypothesize that a chemically diverse library of CRBN modulators will be a valuable tool to screen against a range of representative cell line models of pediatric cancers to uncover novel CRBN-dependent vulnerabilities. In this work, we provide further evidence for the merit of such a MG library.26 We describe the design and synthesis strategy of a focused library of thalidomide derivatives and the cell-based phenotypic screening results against five cell lines. These efforts have led to the discovery of structurally novel, potent, selective, and orally bioavailable GSPT1/2 degraders.
Results and Discussion
Library Design and Properties
Fundamental to our library design was to leverage the rich structure–activity knowledge of the IMiD scaffold and utilize a combination of modern medicinal chemistry principles, structure-based drug design (SBDD), and structure–activity relationships (SAR) to preserve the minimum thalidomide pharmacophore features necessary for CRBN engagement while maximizing the three-dimensionality of chemical diversity displayed at the CRBN substrate-binding surface. The glutarimide ring of thalidomide (Figure 2A) is known to engage in a hydrogen-bonding network inside the tritryptophan hydrophobic pocket of CRBN and is essential for binding.17 The phthalimide protrudes toward a solvent-exposed region making van der Waals interactions with the β-hairpin loop of the neosubstrate and is critical for substrate recognition. Decoration around the phthalimide ring has been shown to influence the neosubstrate interaction and specificity with broader opportunities for modification. Based on this knowledge, we implemented an approach centered around structural and spatial diversity at the extremities of the phthalimide ring to broadly exploit the surface and plasticity of CRBN at the neomorphic interface.27
Figure 2.

Molecular glue library (MGL) design and properties. (A) Design strategy around the IMiD core scaffold. (B) Docking poses of selected library scaffolds in the binding pocket of Cereblon (PDB: 4TZ4) illustrating 3D spatial coverage. The IMiD core scaffold is highlighted in green. (C) Physicochemical properties of the library compounds: A log P vs molecular weight colored by polar surface area.
Accordingly, we acquired and synthesized IMiD-derived scaffolds containing functional groups at different positions of the phthalimide or isoindolinone cores to enable a diverse set of synthetic transformations, including acylations, sulfonylamidations, nucleophilic aromatic substitutions, reductive aminations, alkylations, amidations, and Suzuki and Buchwald reactions. For each scaffold, we performed chemical feasibility studies prior to library production to probe the scope and robustness of the scaffold under reaction conditions and purification methods used. Building block selection was based on a diverse set of electron-rich and electron-poor aromatic and heteroaromatic rings and cyclic and linear aliphatics to probe both electronic and steric diversification while accounting for a balanced profile of important physicochemical properties such as lipophilicity (A log P), molecular weight (MW), polar surface area (PSA), and H-bond donors and acceptors. Library production was executed in a parallel fashion either in 24 or 48 reactions at a time, and purification was accomplished via automated preparative HPLC. Compounds passing a purity criterion of greater than 90% were plated in a 384-well format for screening. In this fashion, we synthesized a molecular glue library (MGL) of 415 compounds containing analogues of 30 different thalidomide-derived scaffolds.
The landscape of the three-dimensional chemical diversity covered by the library was evaluated by the scaffold docking poses to the DDB1–CRBN crystal structure (PDB: 4TZ4). Figure 2B illustrates the positional scanning and spatial coverage of representative scaffolds docked into the binding cavity of cereblon. Physicochemical descriptors were calculated to characterize the overall library and demonstrated a reasonable distribution of A log P (1.3 avg), MW (445 avg), PSA (119 avg), HBD (1.8 avg), and HBA (8.7 avg), well within the drug-like property chemical space (Figure 2C and Supp. Figure S1).28
Since all compounds in the library contained the conserved IMiD motif, we anticipated comparable binding affinities to the CRBN–DDB1 complex. This was verified by testing a subset of the library in a competitive fluorescence polarization (FP) assay using the Cy5-conjugated lenalidomide analogue (Cy5-O-Len) as the fluorescent probe. In this assay, CC-885, lenalidomide, and thalidomide were used as controls and displayed IC50 values of 0.018 (±0.001), 0.286 (±0.109), and 1.38 μM (±0.268), respectively. Upon testing the subset of the library (238 compounds), 90% of the compounds displayed binding potencies to CRBN equal to or less than 5 μM and only 3.4% of the tested compounds showed the lack of activities up to 10 μM, confirming CRBN affinity for the majority of the library members (Supp. Figure S2).
Library Screening
We postulated that cell-based phenotypic screening of the library in multiple cell lines would enable triaging and prioritizing hits on the basis of their cell line specificity. Hence, we screened the library compounds in full dose response cell viability assays, using a diverse set of pediatric cancer cell lines. The screening panel consisted of five patient-derived cell lines representing acute leukemia (AL) and medulloblastoma (MB), two of the leading causes of childhood cancer death. The panel included two AL cell lines, MV4-11 (KMT2A-rearranged acute myeloid leukemia) and MHH-CALL4 (CRLF2-rearranged JAK2-mutated ALL),29 and three Group 3 MB cell lines, HD-MB03 (TP53 wild type),30 MB002 (TP53 mutated at codon 141 TGC---TTC), and MB004 (TP53 mutated at codon 274 GTT---TGT),31 all displaying MYC amplification. The MB lines were derived from a patient primary cerebellar tumor (HD-MB03) or patient recurrent tumors located either in the primary site (MB004) or in the leptomeningeal compartment (MB002).32 Importantly, all five cell lines were confirmed to be insensitive to the prototypical IMiDs (thalidomide, lenalidomide, and pomalidomide) so that any hits that may result from the library screening are likely to display nonclassical IMiD mechanisms of action.
The library screening was performed by incubating cells with compounds at 10 point concentration range over 3 days, after which cell viability was assessed using a CellTiter-Glo assay kit (Promega). The compounds’ EC50 values were determined using the proprietary software RISE (Robust Investigation of Screening Experiments), developed in-house on the Pipeline Pilot platform (Biovia, v. 17.2.0). This effort resulted in identification of a number of potent hits (EC50 < 1 μM) across the five cell lines, while, as expected, the IMiDs showed no effect (EC50 > 10 μM). Interestingly, whereas some of the screening hits were active in all five cell lines, others showed more selective profiles, suggesting different mechanisms of action. As illustrated by the heat map (Figure 3A) and the Venn diagram (Figure 3B), screening in the MHH-CALL-4 cell line produced the highest number of hits displaying antiproliferative effects <5 μM (25, 6% hit rate) and the greatest number of cell-type specific compounds (17 hits). The screening of the MGL against the HD-MB03 cells resulted in identification of 11 hits in total (2.6% hit rate) with 7 compounds demonstrating cell line specificity, followed by MV4-11 with 10 total and 3 nonoverlapping hits (2.1% hit rate). The MB002 and MB004 cell lines were similar and showed the lowest hit rate (0.7%, 3 hits) with no cell-selective hits. While elucidating the mechanism of action for cell-type specific hits is the ultimately desirable goal, we initially focused on the characterization of the two potent hits shared among all five cell lines to understand and annotate compounds that could be functioning through general cytotoxic mechanisms. The two hits sulfonamides 6 (SJ6986) and 7 (SJ7023) demonstrated antiproliferative activities across the panel and especially in the two leukemia cell lines, as shown in Table 1. The subsequent hit characterization and mechanistic studies described here were carried out in MV4-11 and MHH-CALL-4 cell lines.
Figure 3.
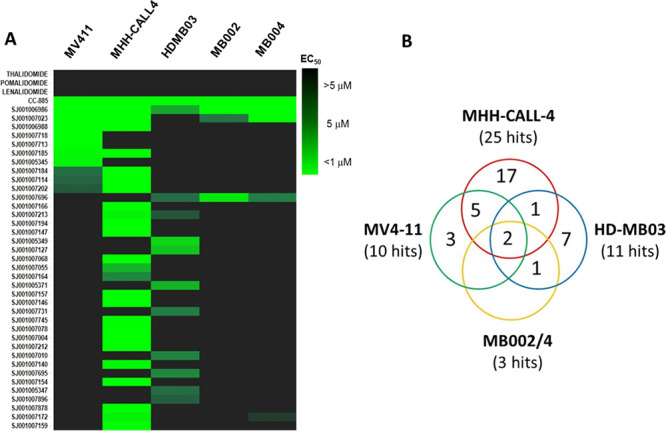
Phenotypic profiling of library. (A) Heat map of 41 hits with IMiD controls screened against five different cell lines (MV4-11, MHH-CALL-4, HD-MB03, MB002, and MB004) for antiproliferative effects. (B) Venn diagram showing the distribution of screening hits across the different cell lines.
Table 1. In Vitro Activity Profiles of Lead Compounds 6 and 7.

| EC50 (nM)a |
|||||||
|---|---|---|---|---|---|---|---|
| cmpd | R | MV4-11 | MHH-CALL-4 | MB002 | MB004 | HD-MB03 | CRBN IC50 (nM)a |
| 6 (SJ6986) | 2-OCF3 | 1.5 | 0.4 | 726 | 336 | 3583 | 15 |
| 7 (SJ7023) | 2-Me, 3-Cl | 52 | 4.4 | 5009 | 1104 | >8 | 16 |
All values are the mean of ≥2 independent experiments.
Hit Profiling and Target Identification
Having identified compounds with potent antiproliferative effects in cells, we next examined whether the observed effect was CRBN-dependent. We first carried out ligand competition experiments. MV4-11 cells were treated with 6 or 7 in dose-response in the presence of high concentrations of lenalidomide 2 (10 μM). Excess lenalidomide results in saturation of the CRBN binding site, thus making it inaccessible for other CRBN modulators to bind and thereby abrogating the antiproliferative effects of 6 and 7 as shown in Figure 4A. In an orthogonal approach, we tested compounds 6 and 7 in wild-type and CRBN knockout MV4-11 cells in a cell viability assay (Figure 4B, Supp. Figure S3). Our results unambiguously showed that, in the absence of CRBN, the antiproliferative activities of the compounds were abolished, further confirming a CRBN-dependent mechanism.
Figure 4.

Effect of the treatment with increasing concentrations of 6 and 7 over 3 days on viability of MV4-11 cells: (A) in the absence and presence of 2 (lenalidomide, 10 μM); (B) wild type vs CRBN–/– MV4-11 cells.
Next, we proceeded with immunoblot experiments to determine if the cytotoxicity was associated with degradation of known neosubstrates. Among the reference compounds included in the screen, 4 (CC-885) was the only control that displayed antiproliferative effects in all five cell lines, so we sought to determine whether our compounds similarly degraded GSPT1 and IKZF1. We performed immunoblotting experiments after 4 h of drug exposure based on literature evidence that neosubstrate degradation occurs rapidly, typically by 4 to 8 h of IMiD treatment.7,9 As illustrated in Figure 5A, compound 6 displayed a dose-dependent decrease in GSPT1 protein abundance with a half-maximal degradation concentration (DC50) of 9.7 nM and nearly complete protein degradation (90%) at 100 nM in MV4-11 cells. The less potent analogue 7 induced only a partial depletion of GSPT1 at 4 h, and residual levels of GSPT1 protein remained even at the highest concentration (10 μM, maximum degradation Dmax = 60%). However, after 24 h of incubation, compound 7 achieved 90% of the GSPT1 protein degradation at 100 nM, with a DC50 value of 10 nM (Figure 5B). The longer incubation also resulted in a slightly improved GSPT1 degradation by compound 6 (DC50 = 2.1 nM). Therefore, both compounds reached a similar extent of GSPT1 degradation but at different rates.
Figure 5.

Immunoblots for GSPT1 and IKZF1 proteins after the treatment with increasing concentrations of compounds 6 and 7 of: MV4-11 cells over 4 (A) and 24 h (B) and MHH-CALL-4 cells over 4 (C) and 24 h (D). Degradation values were calculated using quantified band intensities from immunoblots and DC50 values calculated based on the average of at least two independent experiments.
It has been reported that depletion of GSPT1 triggers the activation of the integrated stress response pathway, which is associated with the induction of apoptosis and inhibition of proliferation.33−35 To correlate the degradation profiles of compounds 6 and 7 to their MV4-11 antiproliferative activities, we tested them in a luminescent Caspase-Glo assay in dose-response at three timepoints (4, 8, and 24 h).14 In the case of compound 6, caspase activation was measurable at 8 h at both concentrations tested (10 and 100 nM), whereas caspase activity for 7 was delayed and only observed at the 24 h timepoint (Supp. Figure S4). Collectively, these experiments demonstrate that the faster and more efficient depletion of GSPT1 by compound 6 relative to 7 results in earlier induction of apoptosis and correlates with higher antiproliferative potency. Indeed, a similar trend was observed in MHH-CALL-4 cells, where the more potent GSPT1 degrader 6 also displayed greater cytotoxicity in this cell line when compared to compound 7 (Table 1). We speculate that the difference in sensitivity between these two cell lines is a consequence of the more sensitive MHH-CALL-4 expressing over 200% higher levels of CRBN compared to the less sensitive MV4-11 cells, while the levels of GSPT1 protein were found to be similar (Supp. Figure S5). It is also worth noting that the two lead compounds displayed similar GSPT1 degradation profiles in the medulloblastoma MB004 cell line (Supp. Figure S6).
Interestingly, compounds 6 and 7 did not significantly degrade IKZF1 after 4 h at any of the concentrations tested, providing a wide window of selectivity for GSPT1 in the two leukemia cell lines, as shown in Figure 5A,C. However, the DC50 values for GSPT1 and IKZF1 obtained after a 24 h treatment of MV4-11 cells showed only a modest selectivity window for compound 6 (14-fold), whereas from a chemical probe perspective, a more optimal36 30-fold selectivity profile was obtained for compound 7 (Figure 5B). The loss of the IKZF1 protein after 24 h was less pronounced in MHH-CALL-4 cells, with neither of the two compounds producing Dmax > 50% at the highest concentration tested (Figure 5D). This delayed reduction in IKZF1 protein levels was particularly notable since all IMiDs are known to degrade IKZF1 shortly after drug treatment (as early as 3 h).7 Potential interpretations of these results are that the compounds either directly recruit IKZF1 to CRBN, which ultimately results in its degradation, but with slower kinetics than GSPT1, or that the rapid degradation of GSPT1, which has an essential role in global protein synthesis, indirectly affects abundance of IKZF1 at later timepoints.37
It has been previously established by Sievers et al.11 that zinc finger 2 (ZF2) of IKZF1 is directly involved in IMiD-induced CRBN binding in vitro and is required for CRBN-dependent degradation in cells. To determine if our GSPT1 degraders induce similar ternary complex formation, we employed AlphaScreen technology to measure luminescence arising from the proximity of CRBN/DDB1-bound acceptor beads and IKZF1(ZF2)-bound donor beads. As expected, CC-220,1313 one of the most recently reported IKZF1 degraders,13 displayed a strong dose response in this assay, similar to IMiDs previously reported by Sievers et al.11 However, neither 6 nor 7 showed a signal even at the highest concentration (Figure 6). These data suggest that compounds 6 and 7 do not induce IKZF1-CRBN/DDB1 ternary complex formation and that the observed loss of IKZF1 protein after 24 h of incubation in the leukemia cell lines is an indirect consequence of the GSPT1 degradation induced by these compounds.
Figure 6.
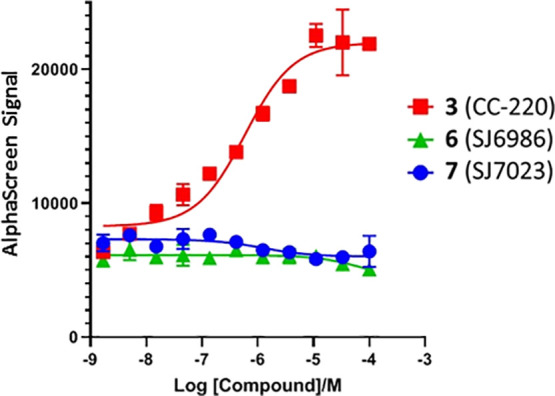
IKZF1(ZF2)-CRBN/DDB1 complex formation: While IKZF1 degrader CC-220 displayed a dose-dependent AlphaScreen signal, compounds 6 and 7 showed no effect even at the highest concentration.
To establish whether the CRBN-dependent degradation of GSPT1 was the mechanism solely responsible for the cytotoxic effects of compounds 6 and 7, we introduced a FLAG-tagged degradation-resistant G575N mutant of GSPT1 into MV4-11 cells by lentiviral transduction.14,35 Indeed, we found that expression of the resistant variant GSPT1(G575N) completely abrogated compound 6- and 7-induced antiproliferation effects in MV4-11 cells (Figure 7A). As expected, in contrast to the wild type protein, the GSPT1(G575N) mutant was not degraded following 24 h of incubation with compounds 6 and 7 at 1 and 10 μM concentrations (Figure 7B). It is interesting to point out that levels of the IKZF1 protein were also found unaffected in the GSPT1(G575N) mutant cell line (Figure 7B). This finding further supports the notion that the loss of IKZF1 protein observed after 24 h of incubation of the wild type MV4-11 cells is a downstream consequence of the GSPT1 degradation rather than a result of the direct interaction with these compounds.
Figure 7.

MV4-11 parental cell line and cells stably expressing degradation-resistant GSPT1(G575N)-FLAG: (A) effect of the treatment with increasing concentrations of 6 and 7 over 3 days on proliferation of MV4-11 parental cells or cells stably expressing GSPT1(G575N)-FLAG; (B) immunoblots for GSPT1 and IKZF1 proteins after the treatment with increasing concentrations of compounds 6 and 7 of MV-411 parental cells or cells stably expressing GSPT1(G575N)-FLAG over 24 h.
The library design allowed us to have a rapid access to several direct analogues of the screening hits and generation of preliminary structure–activity relationship (SAR) information. The analogues were generally found to display high aqueous solubilities with modest cell permeabilities and potent CRBN affinities with no apparent correlation between CRBN binding and cellular potency (Table 2). For example, compounds 6 and 10 shared similar CRBN binding affinities (IC50 = 0.015 and 0.010 μM, respectively) but significantly different MV4-11 potencies (EC50 = 0.0015 and >10 μM, respectively) despite similar physicochemical properties such as aqueous solubility and permeability.
Table 2. CRBN, GSPT1, and MV4-11 Structure–Activity Relationship (SAR).
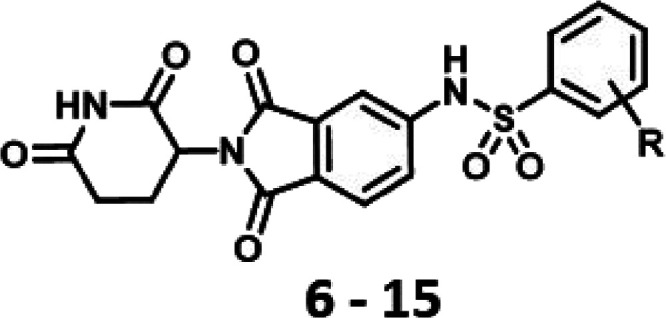
| cmpd | R | CRBN IC50 (μM)a | MV4-11 EC50 (μM)a | HEK293 hGSPT1-HiBit EC50 (μM)a | sol. (μM)a,b | Papp (nm/s)a,c |
|---|---|---|---|---|---|---|
| 5 (CC-90009) | 0.824 | 0.062 | 0.0292 | 77 | 49 | |
| 6 (SJ6986) | 2-OCF3 | 0.015 | 0.0015 | 0.0001 | 67 | 23 |
| 7 (SJ7023) | 2-Me, 3-Cl | 0.016 | 0.052 | 0.108 | 64 | 36 |
| 8 | H | 0.003 | >10 | >10 | 71 | 36 |
| 9 | 4-Me, 3-Cl | 0.001 | >10 | >10 | ND | 46 |
| 10 | 3-OCF3 | 0.010 | >10 | >10 | 64 | 40 |
| 11 | 4-OCF3 | 0.009 | >10 | >10 | 76 | 11 |
| 12 | 2-OMe | 0.612 | >10 | >10 | 85 | 6 |
| 13 | 2-Me | 0.011 | 7.4 | 9.32 | 84 | 68 |
| 14 | 2-CF3 | 0.337 | 0.089 | 0.052 | 66 | 14 |
| 15 | 3-Cl | 0.023 | >10 | >10 | 74 | 17 |
All values are the mean of ≥2 independent experiments.
Aqueous solubility measured at pH = 7.4 in triplicate.
Permeability in MDCK cells. ND = not determined.
To assess GSPT1 protein degradation at higher throughput, we developed an hGSPT1-HiBit assay in HEK293T cells.38 Compounds were screened in dose-response at the 4 h timepoint, and a good correlation was observed between GSPT1 protein degradation and the antiproliferative effect in MV4-11 cells.38
What transpired from the SAR data was the critical importance of the ortho-substitution for cellular potency. For example, the regioisomer of hit compound 7, 4-Me-3-Cl-phenyl sulfonamide 9, was a potent CRBN binder (IC50 = 0.001 μM), but it showed no effect in cellular assays. A lack of potent cell activity was also observed for analogues 13 and 15, suggesting that, in addition to ortho-substitution, hydrophobicity or electronic factors were also contributors to cellular potency. In a similar fashion, whereas unsubstituted phenylsulfonamide 8 was inactive, 2-OCF3 derivative 6 was the most potent compound in the series, while 3-OCF3 (10) and 4-OCF3 (11) analogues showed no activity in cells (Table 2). We confirmed that compound 10 had no effects on the protein levels of GSPT1 and IKZF1 by immunoblotting (Supp. Figure S7). To probe the contribution of the trifluoromethyl group, the 2-OMe (12) analogue was synthesized and found to be inactive in cells, whereas analogue 14 was a potent degrader of GSPT1, supporting a strong CF3 influence on cell activity.
To rationalize the observed SAR, we carried out docking and molecular dynamics (MD) simulations using the previously reported DDB1–CRBN–CC-885–GSPT1 complex structure (PDB: 5HXB).14 Compound 6 was initially docked into the tritryptophan pocket of CRBN, using an InducedFit protocol (Schrödinger suite release 2019-4) where it was predicted to bind in a similar fashion to 4 (CC-885). However, due to its small size compared to 4, a large unoccupied space remained between the compound and CRBN loop Glu147-Glu153 (Figure 8A). This loop contains the Phe150 residue that was previously shown to be critical for the GSPT1 recruitment by CC-855,14 and it is rather flexible as indicated by a considerable shift in the CC-220–CRBN complex (PDB: 5V3O).13 To allow stabilization of the CRBN–GSPT1–compound 6 ternary complex and relaxation of the loop, we performed a series of MD simulations totaling 2 μs that ultimately revealed a narrow hydrophobic pocket created by Val570, Val590, Val536, Gln534, and Lys628 of GSPT1 and Phe150 of the CRBN Glu147-Glu153 loop to which the aryl ring of compound 6 was predicted to bind (Figure 8B). The sulfonamide moiety is positioned between CRBN residues Glu377 and His353 and engaged in hydrogen bonding with both residues, in a similar fashion to the urea of CC-885. This narrow pocket requires the phenyl ring to be in a staggered conformation with respect to the sulfonamide oxygens, which is a favored ground-state conformation for analogues featuring ortho-substitution, such as compound 6 (Supp. Figure S9). In this binding mode, the 2-OCF3 group is projected toward a hydrophobic environment forming a van der Waals contact with Phe150, which may also account for the superior GSPT1 degradation and cellular activity of compound 6, when compared to 2-CF3 analogue 14. Compound 7 adopted a similar binding mode, with the aryl ring in the staggered conformation and the Cl atom buried in the narrow hydrophobic pocket (Supp. Figure 8A). In contrast, sulfonamides lacking the ortho-substitution adopt an eclipsed conformation, which is stabilized by the aryl ring hyperconjugation with the S=O group39 by around 3 kcal/mol compared to the staggered conformation (Supp. Figure S9). Therefore, these results suggest that the bioactive staggered conformation is energetically disfavored by aryl sulfonamides lacking an ortho-substitution, prevents recruitment of GSPT1, and therefore shows reduced cytotoxicity, as demonstrated by compounds 8–11 (Table 2).
Figure 8.

In silico modeling of the lead compound 6 in the DDB1–CRBN–CC-885–GSPT1 complex (PDB: 5HXB). (A) Compound 4 (gray sticks) relative position to 6 (green sticks) docked into the CRBN structure before (gray cartoon) and after 2 μs of MD simulation (green cartoon); (B) MD simulations predict compound 6 to be accommodated in a narrow hydrophobic pocket formed by CRBN residues (gold sticks) and GSPT1 residues (gray sticks).
We have also attempted to rationalize the GSPT1 over IKZF1 degradation selectivity displayed by our lead compounds. Unfortunately, in contrast to GSPT1, the only available crystal structure in complex with CRBN and pomalidomide is its zinc finger 2 (ZF2) domain of IKZF1 (PDB: 6H0F),11 just a short (38 amino acid) peptide out of the 519 amino acid containing full-length protein. We superimposed this structure on compound 6 docked into the reported structure of the CRBN–CC-885–GSPT1 ternary complex (PDB: 5HXB).14 The only observed difference in predicted ligand–protein interactions was Gln146 of IKZF1 replacing Val570 in GSPT1 in close proximity (2.3 Å) to the phenyl ring of compound 6 (Figure 8B, and Supp. Figure S8B). We speculate that the difference in polarity between the two residues may contribute to a weaker interaction with the ZF2 and consequent degradation selectivity of our lead compounds over IKZF1.
Next, we were interested in benchmarking the degradation profiles of our hits against CC-90009 (5, Table 2), which is the first and currently the only GSPT1 degrader that entered the clinic and has recently successfully completed Phase 1 clinical trials in patients with relapsed or refractory AML (NCT02848001).16 We observed that, after 4 h in MV4-11 cells, compound 5, similar to 7, only partially degraded GSPT1 (Dmax = 74% at 10 μM). However, unlike either 6 or 7, after 4 h of treatment, it also degraded the IKZF1 protein with a Dmax of 55% at 10 μM (Supp. Figure S10A). Interestingly, at the 24 h timepoint, the degradation profile of 5 was more similar to that of compound 6, achieving Dmax > 90% for both GSPT1 and IKZF1 proteins with DC50 values of 1.6 and 10 nM, respectively (Table 1 and Supp. Figure S10B). Lopez-Girona et al. have recently reported tandem mass tag quantitative mass spectrometry analysis in AML cells showing that 5 selectively reduced the abundance of GSPT1 with little effect on the rest of the proteome.35
To investigate the effects of compound 6 on cellular protein levels more broadly and evaluate its overall proteome-wide selectivity, we performed multiplexed mass spectrometry-based proteomic analysis in MV4-11 cells. Upon 4 h of drug or DMSO treatment, proteins were extracted and digested into peptides. The resulting peptides were differentially labeled with tandem mass tag (TMT) isobaric reagents, equally pooled, and analyzed by liquid chromatography and mass spectrometry,40 leading to the quantification of unique proteins. The resulting data was filtered to include only proteins that had a minimum of three unique peptides to reduce false positives. This experiment showed that, out of the 8927 quantified proteins, compound 6 reduced most potently and selectively abundance of GSPT1 and GSPT2 (Figure 9). The observation of GSPT2 as a target of downregulation is not surprising, given the high sequence homology (87%) between the two proteins.37 While the prototypical IMiD neosubstrates such as IKZF1/3 and CK1α (CSNKA1) were not affected, less pronounced but statistically significant reductions in abundance of two unrelated proteins were observed: contactin-associated protein-like 4 (CNTNAP4; UniProtKB-Q9C0A0) and pigment epithelium-derived factor (SERPINF1; UniProtKB-P36955).
Figure 9.

Quantitative TMT-proteomics in MV4-11 cells after 4 h of treatment with compound 6 at 1 μM concentration. Dataset represents average of n = 4 replicates. Proteins downregulated more than 2-fold (dotted line on Log2 = −1 on X-axis) with the p-value less than 0.001 (dotted line on −Log10P value = 3 on Y-axis) are shown in the upper left region of the plot.
Next, we examined the effect of compounds 4–7 on the viability of peripheral blood mononuclear cells (PBMCs) from healthy donors and normal human fibroblast BJ cells. Perhaps unsurprisingly, considering the essential role of GSPT1 for normal cell function, all four degraders displayed high PBMC toxicities, as shown in Figure 10 and Supp. Table S1. Owing to a higher sensitivity of MHH-CALL-4 cells to the GSPT1 degradation, all four compounds exhibited higher PBMC versus MHH-CALL-4 therapeutic index (TI) than MV4-11 cells. The highest PBMC versus MHH-CALL-4 therapeutic index was observed for compound 6 (TI = 21), followed by 7 (TI = 14), both of which favorably compared to 5 (TI = 7), the only GSPT1 degrader currently being evaluated in human patients. Interestingly, BJ cells were found to be considerably less sensitive to the GSPT1 degradation, and consequently, all four compounds displayed high TI with respect to this cell line. The trend was very similar to the one observed in PBMCs, with compound 6 showing the highest BJ versus MHH-CALL-4 therapeutic index, TI = 205,262 (Figure 10).
Figure 10.
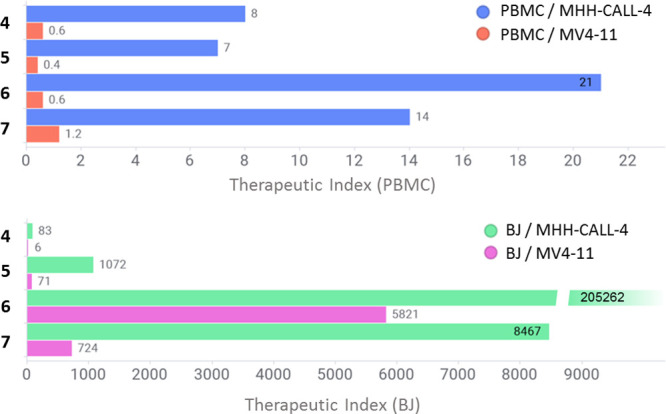
In vitro therapeutic index of compounds 4–7: the bars represent the ratio of EC50 values in noncancerous and leukemia cells: PBMC and MHH-CALL-4 (blue), PBMC and MV4-11 (red), BJ and MHH-CALL-4 (green), and BJ and MV4-11 (magenta).
Having identified potent and selective GSPT1 degraders, we obtained in vitro ADME data for compound 6 to evaluate its suitability for in vivo studies. Compound 6 was found to be stable in mouse and human liver microsomes (t1/2 = 4.7 and 5.7 h, respectively) with low intrinsic clearance (Clint = 12 and 4 mL/min/kg). Plasma protein binding was high and measured to be 99.3 and 99.1% for the mouse and human, respectively.
With its ADME profile established, we evaluated the pharmacokinetics of compound 6 in mice (Figure 11). Upon dosing at 3 mg/kg IV, we observed low clearance (0.46 mL/min/kg), in agreement with the liver microsome stability data. The terminal elimination half-life (t1/2) was calculated to be 3.4 h, and the volume of distribution was low at 0.15 L/kg, indicating minimal extravascular distribution with compound restriction mainly in the blood compartment. A single oral dose of 6 at 10 mg/kg gave peak plasma concentrations at 0.25 h, suggesting rapid absorption, and the oral bioavailability was calculated to be 84% (Figure 11). This PK profile warrants further exploration of 6 in animal disease models and supports the prospect of this novel chemical series of molecular glues.
Figure 11.

Pharmacokinetics data of compound 6 in the CD1 mouse following a single intravenous (IV) and oral administration (PO). Dose: 3 mg/kg, IV; 10 mg/kg, PO. Formulation vehicle: 5% v/v NMP: 5% v/v solute HS-15 and 90% v/v normal saline. The measured plasma free fraction used to calculate unbound concentration was 0.00744 ± 0.001. NA = not applicable.
Conclusions
In summary, we described our approach toward building a chemical library of CRBN modulators to degrade “undruggable” targets in pediatric cancers. Our methods included the design and synthesis of a thalidomide-based library covering broad diversity in chemical and physicochemical properties and three-dimensional space. Phenotypic screening in a diverse set of pediatric cell lines enabled us to triage compounds based on their antiproliferative cellular specificity. Whereas some of the screening hits proved to be potent in all five cell lines, others showed more selective profiles, suggesting different mechanisms of action. We initially focused on the characterization of the hits that demonstrated potent antiproliferative activity across the panel of cell lines. This led to the discovery of sulfonamide 6 (SJ6986), a novel and potent GSPT1 degrader displaying selectivity over ∼9000 proteins, including the classical IMiD neosubstrates such as IKZF1. To our best knowledge, this is also the first GSPT1 degrader with reported oral bioavailability (F = 84% in mice).
GSPT1 and GSPT2 are small GTPases initially found essential for the G1 to S phase transition of the cell cycle and later reported to function as a polypeptide chain release factor 3 (eRF3). In this role, GSPT1/2 associates with eRF1 to mediate stop codon recognition and nascent protein release from the ribosome.41 GSPT1 has also been shown to play several additional roles in critical cellular processes such as cell cycle regulation, cytoskeleton organization, and apoptosis.42 Changes in the expression pattern of translation factors can lead to several changes in the tumor cells, such as an increase in the overall rate of protein synthesis and/or overexpression of proteins involved in cell growth and proliferation. Indeed, GSPT1 has been found to be overexpressed and oncogenic in a number of cancers,43 including gastric,44 colorectal,45 lung,46 and breast cancers.47 To investigate GSPT1 expression in pediatric cancer, we turned to St. Jude’s PeCAN Portal,48 a web application, which provides RNA sequencing (RNA-seq) data for 928 pediatric tumors.49 This analysis showed that GSPT1 is overexpressed in many pediatric cancers, but mostly in hematopoietic malignancies (71%) including B-ALL, T-ALL, and AML (Supp. Figure S11). As shown in the present study, and as previously reported by others,35 acute leukemia cells are highly sensitive to GSPT1 degradation, strongly supporting further evaluation of this mechanism as a potential therapeutic approach to the treatment of hematological cancers.16 Specifically, the high potency in MHH-CALL-4 cells and therapeutic index against PBMCs of compound 6 observed in this study raise a possibility of pursuing this approach for high-risk pediatric ALLs, which will be indeed the focus of our future research in this area. Compound 6 is a particularly suitable candidate for in vitro/vivo proof-of-concept studies in preclinical models of ALL not only because of its high in vitro potency and oral bioavailability but also due to its broader selectivity, especially over the classical IMiD neosubstrates such as IKZF1. Such a profile is desirable for two main reasons. First, all three classical IMiDs were found to be inactive (EC50 > 10 μM) in both MV4-11 and MHH-CALL-4 cell lines (Figure 3), showing that IKZF1 degradation had no effect on viability of acute leukemia cells. Second, IKZF1 degradation may be associated with undesirable effects since this transcriptional factor is an essential regulator in hematopoiesis.50 Its critical importance is underlined by the fact that loss-of-function or dominant negative genetic alterations of IKZF1 contribute to the pathogenesis and poor prognosis of both mouse and human leukemia.51 IKZF1 deletion occurs in 15% of the pediatric B-cell precursor ALL, the most common malignancy in children, and is associated with significantly increased risk of relapse and a poor outcome.21,52IKZF1 was also found to be a tumor suppressor gene in pediatric AML, and its deletion is a strong determinant of oncogenesis in AML.53 Taken together, these data strongly suggest that IKZF1 degradation is not only unnecessary but also an undesirable off-target effect in the development of a therapeutic agent for pediatric leukemias.
More generally, this study supports the utility of a diverse library of CRBN binders in the pursuit of targeting undruggable oncoproteins. It is important to point out that the neosubstrate scope of IMiD-based molecular glue molecules is likely limited to substrates harboring the specific glycine-containing beta hairpin degron.11 Consequently, expanding the library with structurally novel cereblon binders and ligand binding to other E3 ligases are critical for unlocking the full potential of this approach.
Experimental Section
Chemistry: General Methods and Synthesis
All reagents and solvents were obtained from commercially available sources and were used without further purification. Reactions were set up in air and carried out under a nitrogen atmosphere. Parallel synthesis was accomplished using MiniBlock XT synthesizers purchased from Mettler-Toledo AutoChem placed on a stirring hot plate. Library compounds were filtered using a 96-well Thomson 2 mL filter plate (25 μm) containing Celite 545 into a Waters 96-well 2 mL receiving plate prior to purification. Automated weighing was done using a Bohdan balance automator (Mettler-Toledo AutoChem), and parallel evaporations were carried out using a Genevac HT-24. HPLC analyses were accomplished using a UPLC/UV/ELSD/SQD (single quadrupole detector) with the stationary phase: BEH C18, 1.7 μm, solvents: A: 0.1% formic acid in water, B: 0.1% formic acid in acetonitrile, detector types: PDA (210 to 400 nm) and ELSD. Library purification was performed on the Waters purification/analytical LC/UV/ELSD system. Column: Gemini Aixia packed C18 50 mm × 30 mm, 5 μm. Collection: UV @ 214 nm and/or ELSD. Gradients: water/acetonitrile/0.1% formic acid, the beginning acetonitrile percentage varied based on retention times determined in prepurification analysis on the UPLC. Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker NMR spectrometer at 500 MHz for 1H NMR spectra and 125 MHz for 13C NMR spectra. Chemical shifts (ppm) are reported relative to the solvent peak. Signals are designated as follows: s, singlet; d, doublet; dd, doublet of doublet; t, triplet; q, quadruplet; m, multiplet. Coupling constants (J) are expressed in hertz (Hz). High-resolution mass spectral data were obtained on a Waters Xevo G2 QTof mass spectrometer. The purity of final compounds was determined using an Acquity UPLC BEH C18 1.7 μm, 2.1 × 50 mm column (Waters Corporation, Milford, MA) and Acquity ultraperformance liquid chromatography system. The flow rate was 0.7 mL/min. The sample injection volume was 3 μL. The UPLC column was maintained at 50 °C, and the gradient program started at 90% A (0.1% formic acid in MilliQ H2O), changed to 95% B (0.1% formic acid in acetonitrile) over 2.5 min, and held for 0.35 min and then to 90% A over 0.05 min. All final compounds were >95% pure by this method.
General Procedure for the Syntheses of Sulfonamides
In a 48 position Mettler Toledo XT reaction block containing 11.5 × 110 mm test tubes equipped with a stir bar were added 0.1 mmol of 5-amino-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione and the corresponding sulfonyl chloride (2.0 equiv, 0.2 mmol). Anhydrous pyridine (500 μL) was added, and the reactions were heated in a reaction block to 80 °C and stirred overnight under nitrogen. The reactions were checked by UPLC the next morning, concentrated to dryness, and diluted with 1 mL of DMSO. Prepurification analysis in the UPLC was done with water/acetonitrile/0.1% formic acid. Library purification was performed on the Waters purification/analytical LC/UV/ELSD system, and parallel evaporations were carried out using a Genevac HT-24. Compounds 6–15 were obtained using methods described below and are shown in Supp. Scheme S1.
N-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)-2-(trifluoromethoxy)benzenesulfonamide (6)
Yield: 34 mg, 68%. Purity >95%. 1H NMR (500 MHz, methanol-d4) δ 8.17 (dd, J = 7.9, 1.7 Hz, 1H), 7.77–7.71 (m, 2H), 7.62 (d, J = 2.0 Hz, 1H), 7.56–7.48 (m, 3H), 5.10 (dd, J = 12.8, 5.5 Hz, 1H), 2.86 (ddd, J = 17.7, 14.0, 5.4 Hz, 1H), 2.80–2.62 (m, 2H), 2.15–2.05 (m, 1H). 13C NMR (125 MHz, MeOD) δ 173.14, 170.03, 166.78, 166.76, 145.96, 143.90, 135.31, 133.50, 131.43, 131.20, 126.81, 126.01, 124.40, 123.35, 123.42–117.24 (q), 120.53, 112.45, 49.19, 30.72, 22.18. LCMS (m/z) M + H = 498.5. HRMS: calcd for C20H14F3N3O7S + H: 498.0583; found: 498.0582 [M + H].
3-Chloro-N-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)-2-methylbenzenesulfonamide (7)
Yield: 26 mg, 56%. Purity >95%. 1H NMR (500 MHz, methanol-d4) δ 8.08 (dd, J = 8.1, 1.3 Hz, 1H), 7.75 (d, J = 8.2 Hz, 1H), 7.67 (dd, J = 8.1, 1.3 Hz, 1H), 7.57 (d, J = 2.0 Hz, 1H), 7.47 (dd, J = 8.2, 2.0 Hz, 1H), 7.38 (t, J = 8.0 Hz, 1H), 5.09 (dd, J = 12.7, 5.4 Hz, 1H), 2.86 (ddd, J = 17.7, 14.0, 5.4 Hz, 1H), 2.74 (s, 3H), 2.79–2.63 (m, 2H), 2.15–2.04 (m, 1H). 13C NMR (125 MHz, MeOD) δ 173.13, 170.07, 166.78, 166.74, 143.83, 139.52, 136.84, 134.97, 134.03, 133.61, 128.67, 126.86, 125.82, 124.53, 122.69, 111.80, 49.18, 30.72, 22.17, 15.52. LCMS (m/z) M + H = 462.5. HRMS: calcd for C20H16ClN3O6S + H: 462.0527; found: 462.0515 [M + H].
N-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)benzenesulfonamide (8)
Yield: 25 mg, 61%. Purity >95%. 1H NMR (500 MHz, DMSO-d6) δ 11.31 (s, 1H), 11.10 (s, 1H), 7.87 (dd, J = 7.4, 1.7 Hz, 2H), 7.82 (d, J = 8.1 Hz, 1H), 7.66 (t, J = 7.3 Hz, 1H), 7.62–7.59 (m, 2H), 7.56–7.48 (m, 2H), 5.09 (dd, J = 12.9, 5.4 Hz, 1H), 2.86 (ddd, J = 17.2, 13.9, 5.4 Hz, 1H), 2.60–2.56 (m, 1H), 2.51–2.43 (m, 1H), 2.04–1.99 (m, 1H). LCMS (m/z) M + H = 414.4. HRMS: calcd for C19H15N3O6S + H: 414.0760; found: 414.0750 [M + H].
3-Chloro-N-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)-4-methylbenzenesulfonamide (9)
Yield: 29 mg, 62%. Purity >95%. 1H NMR (500 MHz, DMSO-d6) δ 11.33 (s, 1H), 11.11 (s, 1H), 7.88–7.81 (m, 2H), 7.71 (d, J = 8.1 Hz, 1H), 7.59 (d, J = 8.2 Hz, 1H), 7.53–7.52 (m, 2H), 5.10 (dd, J = 12.8, 5.4 Hz, 1H), 2.87 (ddd, J = 18.0, 14.0, 5.4 Hz, 2H), 2.62–2.55 (m, 1H), 2.49–2.45 (m, 1H), 2.37 (s, 3H), 2.10–1.99 (m, 1H). LCMS (m/z) M + H = 462.4. HRMS: calcd for C20H16ClN3O6S + H: 462.0527; found: 462.0519 [M + H].
N-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)-3-(trifluoromethoxy)benzenesulfonamide (10)
Yield: 16 mg, 32%. Purity >95%. 1H NMR (500 MHz, DMSO-d6) δ 11.42 (s, 1H), 11.11 (s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.84 (d, J = 8.8 Hz, 1H), 7.80–7.73 (m, 2H), 7.71 (dd, J = 8.1, 2.3 Hz, 1H), 7.58–7.51 (m, 2H), 5.10 (dd, J = 12.9, 5.4 Hz, 1H), 2.87 (ddd, J = 17.2, 13.9, 5.5 Hz, 1H), 2.58 (dt, J = 17.2, 3.4 Hz, 1H), 2.51–2.42 (m, 1H), 2.01 (dtd, J = 13.1, 5.4, 2.3 Hz, 1H). LCMS (m/z) M + H = 498.2. HRMS: calcd for C20H14F3N3O7S + H: 498.0583; found: 498.0578 [M + H].
N-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)-4-(trifluoromethoxy)benzenesulfonamide (11)
Yield: 4.5 mg, 12%. Purity >95%. 1H NMR (500 MHz, DMSO-d6) δ 11.41 (s, 1H), 11.09 (s, 1H), 7.97 (d, J = 8.5 Hz, 2H), 7.77 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 8.4 Hz, 2H), 7.51 (s, 1H), 7.44 (s, 1H), 5.08 (dd, J = 12.9, 5.4 Hz, 1H), 2.86 (ddd, J = 17.8, 14.1, 5.5 Hz, 1H), 2.61–2.53 (m, 1H), 2.52–2.46 (m, 1H), 2.00 (dd, J = 10.3, 4.4 Hz, 1H). LCMS (m/z) M + H = 498.2. HRMS: calcd for C20H14F3N3O7S + H: 498.0583; found: 498.0586 [M + H].
N-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)-2-methoxybenzenesulfonamide (12)
Yield: 17 mg, 42%. Purity >95%. 1H NMR (500 MHz, DMSO-d6) δ 11.10 (s, 1H), 11.07 (s, 1H), 7.90 (dd, J = 7.9, 1.7 Hz, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.62 (ddd, J = 8.8, 7.4, 1.7 Hz, 1H), 7.55–7.47 (m, 2H), 7.22–7.16 (m, 1H), 7.11 (td, J = 7.6, 0.9 Hz, 1H), 5.07 (dd, J = 12.9, 5.4 Hz, 1H), 3.86 (s, 3H), 2.86 (ddd, J = 17.1, 13.9, 5.4 Hz, 1H), 2.57 (dt, J = 18.1, 4.1 Hz, 1H), 2.45 (td, J = 13.3, 4.4 Hz, 1H), 2.00 (dtd, J = 13.0, 5.4, 2.4 Hz, 1H). LCMS (m/z) M + H = 444.5. HRMS: calcd for C20H17N3O7S + H: 444.0865; found: 444.0866 [M + H].
N-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)-2-methylbenzenesulfonamide (13)
Yield: 9.9 mg, 23%. Purity >95%. 1H NMR (500 MHz, DMSO-d6) δ 11.45 (s, 1H), 11.09 (s, 1H), 7.99 (d, J = 7.9 Hz, 1H), 7.79 (d, J = 8.6 Hz, 1H), 7.55 (t, J = 7.4 Hz, 1H), 7.48–7.39 (m, 4H), 5.07 (dd, J = 12.9, 5.4 Hz, 1H), 2.86 (ddd, J = 17.2, 13.9, 5.5 Hz, 1H), 2.62 (s, 3H), 2.60–2.56 (m, 1H), 2.50–2.42 (m, 1H), 2.04–1.96 (m, 1H). LCMS (m/z) M + H = 428.4. HRMS: calcd for C20H17N3O6S + H: 428.0916; found: 428.0911 [M + H].
N-(2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)-2-(trifluoromethyl)benzenesulfonamide (14)
Yield: 13 mg, 30%. Purity >95%. 1H NMR (500 MHz, DMSO-d6) δ 11.10 (s, 1H), 8.19–8.12 (m, 1H), 8.03 (dd, J = 7.4, 1.8 Hz, 1H), 7.91–7.82 (m, 3H), 7.51–7.46 (m, 2H), 5.07 (dd, J = 12.9, 5.4 Hz, 1H), 2.85 (ddd, J = 17.2, 13.9, 5.5 Hz, 1H), 2.62–2.52 (m, 1H), 2.49–2.42 (m, 1H), 2.01 (dtd, J = 12.9, 5.4, 2.5 Hz, 1H). LCMS (m/z) M + H = 482.4. HRMS: calcd for C20H14F3N3O6S + H: 482.0634; found: 482.0627.
3-Chloro-N-(2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)benzenesulfonamide (15)
Yield: 9 mg, 28%. Purity >95%. 1H NMR (500 MHz, DMSO-d6) δ 11.39 (s, 1H), 11.10 (s, 1H), 7.86 (s, 1H), 7.80 (dd, J = 8.0, 5.1 Hz, 2H), 7.73 (dd, J = 8.0, 2.0 Hz, 1H), 7.62 (t, J = 8.0 Hz, 1H), 7.53–7.46 (m, 2H), 5.09 (dd, J = 12.9, 5.4 Hz, 1H), 2.86 (ddd, J = 18.1, 13.9, 5.4 Hz, 1H), 2.62–2.53 (m, 1H), 2.50–2.42 (m, 1H), 2.01 (ddd, J = 12.0, 7.0, 4.4 Hz, 1H). LCMS (m/z) M + H = 448.4. HRMS: calcd for C20H14ClN3O6S + H: 448.0370; found: 448.0370 [M + H].
CRBN Fluorescence Polarization Assay
In this competitive fluorescence polarization assay, Cy5-conjugated lenalidomide analogue (Cy5-O-Len)13 was used as a fluorescent probe. The 6XHis-CRBN-DDB1 protein (200 nM) and Cy5-O-Len probe (30 nM) were combined in 20 mM HEPES pH 7, 150 mM NaCl, and 0.005% Tween-20 assay buffer. An amount of 20 μL of this assay cocktail was dispensed into wells of Corning 3821 black 384-well plates. Compounds were transferred to the assay plate from a dose-response plate using a Pintool on a Biomek FXP Laboratory Automation Workstation (Beckman Coulter). The plates were incubated in the dark for 1 h at room temperature and then read on an EnVision plate reader (PerkinElmer, Massachusetts, USA). IC50 values were determined using a proprietary software RISE (Robust Investigation of Screening Experiments), developed in house on the Pipeline Pilot platform (Biovia, v. 17.2.0). Data represent the mean of three independent determinations.
Cell Proliferation and Lenalidomide Competition Assay
To screen the library in human cancer cell lines, each cell line was cultured in the complete medium recommended by the vendor and seeded in Corning 8804 BC white 384-well assay plates at densities of 1000, 1000, 1000, 1500, and 7500 cells per well, for MV4-11, HD-MB03, MB004, MB002, and MHH-CALL4 cells, respectively. After overnight incubation at 37 °C in a humidified 5% CO2 incubator, cells were treated with compounds in dose-response format using a Pintool on a Biomek FXP Laboratory Automation Workstation (Beckman Coulter). For the lenalidomide competition assay, the MV4-11 cell line was co-treated with 10 μM lenalidomide and DMSO or the respective compounds. After 72 h of incubation, cell proliferation was assessed using a Cell Titer-Glo (CTG) luminescent cell viability assay (Promega) according to the manufacturer’s instruction. Luminescence signal was measured using an EnVision plate reader (PerkinElmer).
MV4-11 CRBN KO Cell Line
CRBN-deficient MV4:11 cells were generated using CRISPR-Cas9 technology. Briefly, 400,000 MV4:11 cells were transiently co-transfected with precomplexed ribonuclear proteins (RNPs) consisting of 100 pmol of chemically modified sgRNA (5′-UGUAUGUGAUGUCGGCAGAC-3′, Synthego) and 35 pmol of Cas9 protein (St. Jude Protein Production Core) via nucleofection (Lonza, 4D-Nucleofector X-unit) using solution SF and program DJ-100 in small (20 μL) cuvettes according to the manufacturer’s recommended protocol. Five days post nucleofection, cells were single-cell sorted by FACs to enrich for GFP+ (transfected) cells, clonally selected, and verified for the desired targeted modification via targeted deep sequencing using gene-specific primers with partial Illumina adapter overhangs (hCRBN.F – 5′-gcagagagtgaggaagaagatga-3′ and hCRBN.R – 5′-gcccatgtcctcatccacaa-3′, overhangs not shown) and analyzed using CRIS.py.54 Two hCRBN knockout clones were identified. Protein knockout was confirmed by immunoblotting (Supp. Figure S3). Antibodies were rabbit anti-human eRF3/GSPT1 pAb (ab126090, Abcam) and mouse antihuman Ikaros mAb (sc-398265, Santa Cruz Biotechnology). The genotype of each clone is indicated below. Upper case base pairs are insertions and (−) are deletions.WT 5′-ttttgacaccagtctgccgacatcacatacagtatgtaattta-3′ Clones 1B6 −1bp 5′-ttttgacaccagtctg-cgacatcacatacagtatgtaattta-3′ +1bp 5′-ttttgacaccagtctTgccgacatcacatacagtatgtaattta-3′ 4B12 −1bp 5′-ttttgacaccagtc-gccgacatcacatacagtatgtaattta-3′ −1bp 5′-ttttgacaccag-ctgccgacatcacatacagtatgtaattta-3′
Immunoblot Analysis (MV4-11 and MHH-CALL4 Cells)
Cells were seeded in 6-well plates (1 × 106 cells per well). After overnight incubation, the cells were treated with indicated concentrations for specific timepoints. The harvested cells were spinned down, washed with PBS, and lysed with 1× LDS loading buffer followed by sonication and heated at 70 °C. The prepared samples were loaded on NuPAGE 4–12% Bis–Tris protein gels and transferred to the nitrocellulose membrane. After blocking the membrane with LI-COR TBS blocking buffer and incubation in primary antibody overnight, the corresponding protein signals were detected using IRDye secondary antibodies and an Odyssey imaging system. Image Studio Lite Ver 5.2 was used for blot quantification. Rabbit anti-human eRF3/GSPT1 pAb (ab126090, Abcam), mouse anti-human Ikaros mAb (sc-398265, Santa Cruz Biotechnology), mouse anti-GAPDH mAb (sc-47724, Santa Cruz Biotechnology) were used as primary antibodies. The IRDye 800CW Goat anti-Mouse IgG Secondary Antibody and IRDye 680RD Goat anti-Rabbit IgG Secondary Antibody were used as secondary antibodies.
AlphaScreen Assay (IKZF1(ZF2)-CRBN/DDB1)
Biotin-IKZF1(ZF2) protein was prepared following the procedure reported by Sievers et al.11 The His-tagged CRBN-DDB1 protein was prepared following the procedure reported by Matyskiela et al.14 The reaction mixture contained 240 nM His-tagged CRBN/DDB1, 40 nM biotin-tagged IKZF1(ZF2), 20 μg/mL nickel chelate AlphaScreen acceptor, and streptavidin donor beads (PerkinElmer), and serially diluted test compounds in a buffer comprising 25 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% BSA, 0.05% Tween 20, and 0.005% ProClin-300. Briefly, a 96-well plate containing 5× test compound, 5× His-tagged CRBN/DDB1, and 5× biotin-tagged IKZF1(ZF2) was incubated at rt for 90 min. After the incubation, 5 μL of the solution was transferred to a 384-well white OptiPlate (PerkinElmer) in duplicate followed by a 10 μL nickel chelate AlphaScreen acceptor (1:100) and 10 μL streptavidin donor beads (1:100). The plate was sealed and mixed on a MixMate (Eppendorf) for 60 min at rt, and then luminescence detection was performed on an EnVision plate reader (PerkinElmer).
MV4-11 Cell Line Expressing GSPT1(G575N)-FLAG
Human full-length GSPT1 cDNA was obtained from Origene (SC327953), and G575N mutation was introduced using the Q5 site-directed mutagenesis kit (New England Biolabs) using the following primers: CAA AAA ATC AAA CGA AAA AAG TAA GAC CCG AC (forward) and TCT ACC AAG CAG ATT AAG (reverse). To distinguish the GSPT1-G575N from endogenous wildtype GSPT1, the FLAG tag was inserted to the C-terminus of mutant GSPT1 cDNA by PCR and cloned into the lentiviral vector pCL20-MSCV-IRES-GFP using the following primers: GCG AAT TCA CCA TGG ATC CGG GCA GTG (forward) and GCG AAT TCT TAC TTA TCG TCA TCG TCT TTG TAA TCG TCT TTC TCT GGA ACC AG (reverse). Lentivirus pseudotyped with the BaEV-Rless (baboon endogenous retrovirus R-less envelope protein) was generated55 and used to transduce MV4-11 cells using RetroNectin (Takara, Cat#T100B)-coated plate in the presence of Lentiboost A and B (1:200, respectively) from SIRON Biotech. GFP expressing cells were sorted and expanded for the experiments.
hGSPT1 HiBiT Cell Line
HEK293_hGSPT1_HiBiT-tagged cells were generated using CRISPR-Cas12a technology. Briefly, ∼400,000 HEK293 cells were transiently co-transfecting with precomplexed ribonuclear proteins (RNPs) consisting of 80 pmol of crRNA (IDT), 62 pmol of Cas12a protein (IDT), 3 μg of ssODN donor (IDT; AltRTM modifications), 78 pmol of electroporation enhancer (IDT), and 200 ng of pMaxGFP(Lonza). The transfection was performed via nucleofection (Lonza, 4D-Nucleofector X-unit) using solution P3 and program CM-130 in a small (20 μL) cuvette according to the manufacturer’s recommended protocol. Five days post-nucleofection, cells were single-cell-sorted for GFP+ (transfected) cells by FACs in 96-well plates and clonally selected. Clones were screened and verified for the desired modification via targeted deep sequencing using gene-specific primers with partial Illumina adapter overhangs as previously described.56 In brief, clonal cell pellets were harvested, lysed, and used to generate gene-specific amplicons with partial Illumina adapters in PCR#1. Amplicons were indexed in PCR#2 and pooled with other targeted amplicons for other loci to create sequence diversity. Additionally, 10% PhiX sequencing control V3 (Illumina) was added to the pooled amplicon library prior to running the sample on a Miseq Sequencer System (Illumina) to generate paired 2 × 250 bp reads. Samples were demultiplexed using the index sequences, fastq files were generated, and NGS analysis was performed using CRIS.py.54 Final clones were authenticated using the PowerPlex fusion system (Promega) performed at the Hartwell Center (St. Jude) and tested negative for mycoplasma by the MycoAlertTMPlus mycoplasma detection kit (Lonza). Editing construct sequences and screening primers are outlined below (sequence from 5′ to 3′).
hGSPT1Cas12acrRNA, CAGE635.GSPT1.g1 (TTTCTCTGGAACCAGTTTCAGAACT); CAGE635.g1.anti.ssODN: ttcctcacagtattgtgcagggtcatcaagaaaatgcttaGCTAATCTTCTTGAACAGCCGC CAGCCGCTCACgtcCttctctggaaccagtttcagaacttttccaattgcaatggtcttacctagaaatgaaattttaa (HiBiT tag and silent blocking modifications to prevent Cas12a recutting after integration are in upper case); CAGE635.hGSPT1.DS.F: GGTTTGGCAGTAAAGCTAGTTAAT; CAGE635.hGSPT1.DS.R (GTGAA GTAGGCTTCTGCAGTC).
hGSPT1 HiBiT Assay
HEK293 hGSPT1 HiBiT tag cells were seeded in white 384-well assay plates at a density of 800 cells per well in triplicates. After overnight incubation, cells were treated with compounds in a dose-response format using a Pintool on a Biomek FXP Laboratory Automation Workstation (Beckman Coulter). After 4 h of incubation, the level of GSPT1 HiBiT tag protein was evaluated using the Nano-Glo HiBiT lytic detection system (Promega) according to the manufacturer’s instruction. The luminescence signal was measured using an EnVision plate reader (PerkinElmer).
Molecular Dynamics Simulations
Compound 6 was modeled into the ligand binding pocket in the DDB1–CRBN–CC-885–GSPT1 complex (PDB: 5HXB)14 by restraining the glutaramide and phthalimide interactions with CRBN. This was followed by a series of microsecond-scale MD simulations that allowed slow relaxation of the Cereblon loop Glu147 to Glu153. The final ternary compound CRBN–6–GSPT1 complex was allowed to further evolve in a 1 μs molecular dynamics simulation to confirm the stability of the final bound pose. All molecular dynamics simulations were carried out using the GPU implementation of Amber18.57 Periodic boundary conditions, particle-mesh, Ewald treatment of the long-term electrostatics, and SHAKE-enabled 2 fs time steps were employed. Compound 6 was docked into this optimized receptor complex using induced fit docking in Schrödinger Maestro.58
Protein Digestion and Peptide Isobaric Labeling by TMT Reagents
The experiment was performed with a previously optimized protocol59 with slight modification. Cell pellets were lysed in lysis buffer (50 mM HEPES, pH 8.5, 8 M urea, and 0.5% sodium deoxycholate). To profile the whole proteome, the protein lysates (approximately 100 μg of protein per sample) were proteolyzed with LysC (Wako) at an enzyme-to-substrate ratio of 1:100 (w/w) for 2 h at 21 °C. Following this, the samples were diluted to a final 2 M urea concentration and further digested with trypsin (Promega) at an enzyme-to-substrate ratio of 1:50 (w/w) for at least 3 h. The peptides were reduced by adding 1 mM DTT for 30 min at 21 °C followed by alkylation with 10 mM iodoacetamide for 30 min in the dark. The unreacted IAA was quenched with 30 mM DTT for 30 min. Finally, the digestion was stopped by adding trifluoroacetic acid (TFA) to 1%, desalted using C18 cartridges (Harvard Apparatus), and dried by SpeedVac. The purified peptides were resuspended in 50 mM HEPES (pH 8.5) and labeled with TMT reagents (Thermo Scientific). The differentially labeled samples were pooled equally, desalted, and dried for the subsequent peptide fractionation. Peptide analysis by two-dimensional liquid chromatography–tandem mass spectrometry (LC/LC–MS/MS) and MS data analysis are described in the Supporting Information.
In Vivo Pharmacokinetic Study
Healthy female CD1 mice (8–12 weeks old) weighing between 20 and 30 g were procured from Global, India. Three mice were housed in each cage. Temperature and humidity were maintained at 22 ± 3 °C and 30–70%, respectively, and illumination was controlled to give a sequence of 12 h light and 12 h dark cycle. Temperature and humidity were recorded by an auto-controlled data logger system. All the animals were provided a laboratory rodent diet (Envigo Research private Ltd., Hyderabad). Reverse osmosis water treated with ultraviolet light was provided ad libitum. Formulation vehicle: 5% v/v NMP: 5% v/v solute HS-15 and 90% v/v normal saline. Animals in Group 1 were administered intravenously as slow bolus injection through the tail vein, with solution formulation of compound 6 at 3 mg/kg dose. Animals in Group 2 were administered through the oral route with a solution formulation of compound 6 at 10 mg/kg dose. The dosing volume for intravenous was 5 mL/kg, and for oral administration, it was 10 mL/kg. Blood samples (approximately 60 μL) were collected under light isoflurane anesthesia (Surgivet) from the retro orbital plexus from a set of three mice at 0.08 (for IV only), 0.25, 0.5, 1, 2, 4, 6 (for PO only), 8, 12, and 24 h. Immediately after blood collection, plasma was harvested by centrifugation at 4000 rpm, 10 min at 40 °C, and samples were stored at −70 ± 10 °C until bioanalysis. Concentrations of compound 6 in mouse plasma samples were determined by the fit-for-purpose LC–MS/MS method. The Non-Compartmental-Analysis tool of Phoenix WinNonlin (Version 8.0) was used to assess the pharmacokinetic parameters. The peak plasma concentration (Cmax) and time for the peak plasma concentration (Tmax) were the observed values. The areas under the concentration time curve (AUClast and AUCinf) were calculated by the linear trapezoidal rule. The terminal elimination rate constant, ke, was determined by regression analysis of the linear terminal portion of the log plasma concentration–time curve. Clearance was estimated as Dose/AUCinf and Vss as CL × MRT. The oral bioavailability was calculated as the ratio of the dose-normalized AUCs of the oral and intravenous groups multiplied by 100.
The PK study was conducted at the AAALAC accredited facility of Sai Life Sciences Limited, Pune, India, in accordance with the Study Protocol SAIDMPK/PK-20-08-538. All animals were maintained in standard animal cages under conventional laboratory conditions (12 h/12 h light/dark cycle, 22 °C) with ad libitum access to food and water. All procedures of the present study were performed in accordance with the guidelines provided by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) as published in The Gazette of India, December 15, 1998. Prior approval of the Institutional Animal Ethics Committee (IAEC) was obtained before initiation of the study.
Acknowledgments
We are grateful for the support of the American Lebanese Syrian Associated Charities (ALSAC) and St. Jude Children’s Research Hospital, and we would like to thank the patients, their families, and the staff at our institution. We thank the compound management team at the Department of Chemical Biology and Therapeutics for performing general QC and compound plate reformatting for screening. We are grateful to Dr. Benjamin Ebert (DFCI) for providing the plasmid for IKZF1(ZF2) and Drs. Munan Shaik and Ravi Kalathur at the Department of Structural Biology for producing IKZF1(ZF2) protein.
Glossary
Abbreviations Used
- ADME
absorption, distribution, metabolism, excretion
- CK1α
casein kinase 1α
- CL
clearance
- CRBN
cereblon
- F
bioavailability
- GSPT1
G1 to S phase transition 1
- HBA
hydrogen-bond acceptors
- HBD
hydrogen-bond donors
- IMiDs
immunomodulatory imide drugs
- IKZF
Ikaros family zinc finger
- IV
intravenous
- LCMS
liquid chromatography mass spectrometry
- PPB
plasma protein binding
- PK
pharmacokinetics
- PSA
polar surface area
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01313.
Library physicochemical properties and CRBN fluorescence polarization assay data; immunoblots for compounds 5 (MV4-11), 6, and 7 (MB004); molecular modeling methods and coordinates; GSPT1 expression in pediatric cancer patients; H NMR, C NMR, and UPLC/MS spectra for lead compounds 6 and 7; cell culture and materials for caspase 3/7 activity; and ADME assays (PDF)
Molecular formula strings (CSV)
Author Contributions
◆ G.N., F.K., and J.M. contributed equally. Z.R. conceived the study. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
NCI P01 CA096832 and Core Grant P30 CA021765 (to M.F.R.). Leukemia and Lymphoma Society Translational Research Program, NCI R35 CA197695, and CA012765 Cancer Center Support Grant (to C.G.M.). Alex’s Lemonade Stand Foundation (ALSF) Crazy 8 Initiative.
The authors declare no competing financial interest.
Supplementary Material
References
- Chamberlain P. P.; Hamann L. G. Development of Targeted Protein Degradation Therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- Hanzl A.; Winter G. E. Targeted Protein Degradation: Current and Future Challenges. Curr. Opin. Chem. Biol. 2020, 56, 35–41. 10.1016/j.cbpa.2019.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toure M.; Crews C. M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem., Int. Ed. 2016, 55, 1966–1973. 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- Schapira M.; Calabrese M. F.; Bullock A. N.; Crews C. M. Targeted Protein Degradation: Expanding the Toolbox. Nat. Rev. Drug Discovery 2019, 18, 949–963. 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- Baek K.; Schulman B. A. Molecular Glue Concept Solidifies. Nat. Chem. Biol. 2020, 16, 2–3. 10.1038/s41589-019-0414-3. [DOI] [PubMed] [Google Scholar]
- Fink E. C.; Ebert B. L. The Novel Mechanism of Lenalidomide Activity. Blood 2015, 126, 2366–2369. 10.1182/blood-2015-07-567958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke J.; Udeshi N. D.; Narla A.; Grauman P.; Hurst S. N.; McConkey M.; Svinkina T.; Heckl D.; Comer E.; Li X.; Ciarlo C.; Hartman E.; Munshi N.; Schenone M.; Schreiber S. L.; Carr S. A.; Ebert B. L. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T.; Goralski M.; Gaskill N.; Capota E.; Kim J.; Ting T. C.; Xie Y.; Williams N. S.; Nijhawan D. Anticancer Sulfonamides Target Splicing by Inducing RBM39 Degradation via Recruitment to DCAF15. Science 2017, 356, eaal3755. 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- Knott M. M. L.; Hölting T. L. B.; Ohmura S.; Kirchner T.; Cidre-Aranaz F.; Grünewald T. G. P. Targeting the Undruggable: Exploiting Neomorphic Features of Fusion Oncoproteins in Childhood Sarcomas for Innovative Therapies. Cancer Metastasis Rev. 2019, 38, 625–642. 10.1007/s10555-019-09839-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushweller J. H. Targeting Transcription Factors in Cancer — from Undruggable to Reality. Nat. Rev. Cancer 2019, 19, 611–624. 10.1038/s41568-019-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers Q. L.; Petzold G.; Bunker R. D.; Renneville A.; Słabicki M.; Liddicoat B. J.; Abdulrahman W.; Mikkelsen T.; Ebert B. L.; Thomä N. H. Defining the Human C2H2 Zinc Finger Degrome Targeted by Thalidomide Analogs through CRBN. Science 2018, 362, eaat0572. 10.1126/science.aat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krönke J.; Fink E. C.; Hollenbach P. W.; MacBeth K. J.; Hurst S. N.; Udeshi N. D.; Chamberlain P. P.; Mani D. R.; Man H. W.; Gandhi A. K.; Svinkina T.; Schneider R. K.; McConkey M.; Järås M.; Griffiths E.; Wetzler M.; Bullinger L.; Cathers B. E.; Carr S. A.; Chopra R.; Ebert B. L. Lenalidomide Induces Ubiquitination and Degradation of CK1α in Del(5q) MDS. Nature 2015, 523, 183–188. 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyskiela M. E.; Zhang W.; Man H.-W.; Muller G.; Khambatta G.; Baculi F.; Hickman M.; LeBrun L.; Pagarigan B.; Carmel G.; Lu C.-C.; Lu G.; Riley M.; Satoh Y.; Schafer P.; Daniel T. O.; Carmichael J.; Cathers B. E.; Chamberlain P. P. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J. Med. Chem. 2018, 61, 535–542. 10.1021/acs.jmedchem.6b01921. [DOI] [PubMed] [Google Scholar]
- Matyskiela M. E.; Lu G.; Ito T.; Pagarigan B.; Lu C.-C.; Miller K.; Fang W.; Wang N.-Y.; Nguyen D.; Houston J.; Carmel G.; Tran T.; Riley M.; Nosaka L.; Lander G. C.; Gaidarova S.; Xu S.; Ruchelman A. L.; Handa H.; Carmichael J.; Daniel T. O.; Cathers B. E.; Lopez-Girona A.; Chamberlain P. P. A Novel Cereblon Modulator Recruits GSPT1 to the CRL4CRBN Ubiquitin Ligase. Nature 2016, 535, 252–257. 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- Sperling A. S.; Burgess M.; Keshishian H.; Gasser J. A.; Bhatt S.; Jan M.; Słabicki M.; Sellar R. S.; Fink E. C.; Miller P. G.; Liddicoat B. J.; Sievers Q. L.; Sharma R.; Adams D. N.; Olesinski E. A.; Fulciniti M.; Udeshi N. D.; Kuhn E.; Letai A.; Munshi N. C.; Carr S. A.; Ebert B. L. Patterns of Substrate Affinity, Competition, and Degradation Kinetics Underlie Biological Activity of Thalidomide Analogs. Blood 2019, 134, 160–170. 10.1182/blood.2019000789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J. D.; Correa M.; Alexander M.; Nagy M.; Huang D.; Sapienza J.; Lu G.; LeBrun L. A.; Cathers B. E.; Zhang W.; Tang Y.; Ammirante M.; Narla R. K.; Piccotti J. R.; Pourdehnad M.; Lopez-Girona A. CC-90009: A Cereblon E3 Ligase Modulating Drug That Promotes Selective Degradation of GSPT1 for the Treatment of Acute Myeloid Leukemia. J. Med. Chem. 2021, 64, 1835–1843. 10.1021/acs.jmedchem.0c01489. [DOI] [PubMed] [Google Scholar]
- Matyskiela M. E.; Clayton T.; Zheng X.; Mayne C.; Tran E.; Carpenter A.; Pagarigan B.; McDonald J.; Rolfe M.; Hamann L. G.; Lu G.; Chamberlain P. P. Crystal Structure of the SALL4–Pomalidomide–Cereblon–DDB1 Complex. Nat. Struct. Mol. Biol. 2020, 27, 319–322. 10.1038/s41594-020-0405-9. [DOI] [PubMed] [Google Scholar]
- Yang J.; Li Y.; Aguilar A.; Liu Z.; Yang C.-Y.; Wang S. Simple Structural Modifications Converting a Bona Fide MDM2 PROTAC Degrader into a Molecular Glue Molecule: A Cautionary Tale in the Design of PROTAC Degraders. J. Med. Chem. 2019, 62, 9471–9487. 10.1021/acs.jmedchem.9b00846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishoey M.; Chorn S.; Singh N.; Jaeger M. G.; Brand M.; Paulk J.; Bauer S.; Erb M. A.; Parapatics K.; Müller A. C.; Bennett K. L.; Ecker G. F.; Bradner J. E.; Winter G. E. Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders. ACS Chem. Biol. 2018, 13, 553–560. 10.1021/acschembio.7b00969. [DOI] [PubMed] [Google Scholar]
- Alexander T. B.; Gu Z.; Iacobucci I.; Dickerson K.; Choi J. K.; Xu B.; Payne-Turner D.; Yoshihara H.; Loh M. L.; Horan J.; Buldini B.; Basso G.; Elitzur S.; de Haas V.; Zwaan C. M.; Yeoh A.; Reinhardt D.; Tomizawa D.; Kiyokawa N.; Lammens T.; De Moerloose B.; Catchpoole D.; Hori H.; Moorman A.; Moore A. S.; Hrusak O.; Meshinchi S.; Orgel E.; Devidas M.; Borowitz M.; Wood B.; Heerema N. A.; Carrol A.; Yang Y.-L.; Smith M. A.; Davidsen T. M.; Hermida L. C.; Gesuwan P.; Marra M. A.; Ma Y.; Mungall A. J.; Moore R. A.; Jones S. J. M.; Valentine M.; Janke L. J.; Rubnitz J. E.; Pui C.-H.; Ding L.; Liu Y.; Zhang J.; Nichols K. E.; Downing J. R.; Cao X.; Shi L.; Pounds S.; Newman S.; Pei D.; Guidry Auvil J. M.; Gerhard D. S.; Hunger S. P.; Inaba H.; Mullighan C. G. The Genetic Basis and Cell of Origin of Mixed Phenotype Acute Leukaemia. Nature 2018, 562, 373–379. 10.1038/s41586-018-0436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.-F.; Dai Y.-T.; Lilljebjörn H.; Shen S.-H.; Cui B.-W.; Bai L.; Liu Y.-F.; Qian M.-X.; Kubota Y.; Kiyoi H.; Matsumura I.; Miyazaki Y.; Olsson L.; Tan A. M.; Ariffin H.; Chen J.; Takita J.; Yasuda T.; Mano H.; Johansson B.; Yang J. J.; Yeoh A. E.-J.; Hayakawa F.; Chen Z.; Pui C.-H.; Fioretos T.; Chen S.-J.; Huang J.-Y. Transcriptional Landscape of B Cell Precursor Acute Lymphoblastic Leukemia Based on an International Study of 1,223 Cases. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E11711–E11720. 10.1073/pnas.1814397115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz J. R.; Ma J.; Walsh M. P.; Chen X.; Lamprecht T.; Kamens J.; Newman S.; Zhang J.; Gruber T. A.; Ma X.; Klco J. M. Comprehensive Genomic Profiling of Pediatric Therapy-Related Myeloid Neoplasms Identifies Mecom Dysregulation to Be Associated with Poor Outcome. Blood 2019, 134, 1394–1394. 10.1182/blood-2019-128665. [DOI] [Google Scholar]
- Hinai A. A.; Valk P. J. M. Review: Aberrant EVI1 Expression in Acute Myeloid Leukaemia. Br. J. Haematol. 2016, 172, 870–878. 10.1111/bjh.13898. [DOI] [PubMed] [Google Scholar]
- Northcott P. A.; Buchhalter I.; Morrissy A. S.; Hovestadt V.; Weischenfeldt J.; Ehrenberger T.; Gröbner S.; Segura-Wang M.; Zichner T.; Rudneva V. A.; Warnatz H.-J.; Sidiropoulos N.; Phillips A. H.; Schumacher S.; Kleinheinz K.; Waszak S. M.; Erkek S.; Jones D. T. W.; Worst B. C.; Kool M.; Zapatka M.; Jäger N.; Chavez L.; Hutter B.; Bieg M.; Paramasivam N.; Heinold M.; Gu Z.; Ishaque N.; Jäger-Schmidt C.; Imbusch C. D.; Jugold A.; Hübschmann D.; Risch T.; Amstislavskiy V.; Gonzalez F. G. R.; Weber U. D.; Wolf S.; Robinson G. W.; Zhou X.; Wu G.; Finkelstein D.; Liu Y.; Cavalli F. M. G.; Luu B.; Ramaswamy V.; Wu X.; Koster J.; Ryzhova M.; Cho Y.-J.; Pomeroy S. L.; Herold-Mende C.; Schuhmann M.; Ebinger M.; Liau L. M.; Mora J.; McLendon R. E.; Jabado N.; Kumabe T.; Chuah E.; Ma Y.; Moore R. A.; Mungall A. J.; Mungall K. L.; Thiessen N.; Tse K.; Wong T.; Jones S. J. M.; Witt O.; Milde T.; Von Deimling A.; Capper D.; Korshunov A.; Yaspo M.-L.; Kriwacki R.; Gajjar A.; Zhang J.; Beroukhim R.; Fraenkel E.; Korbel J. O.; Brors B.; Schlesner M.; Eils R.; Marra M. A.; Pfister S. M.; Taylor M. D.; Lichter P. The Whole-Genome Landscape of Medulloblastoma Subtypes. Nature 2017, 547, 311–317. 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott P. A.; Lee C.; Zichner T.; Stütz A. M.; Erkek S.; Kawauchi D.; Shih D. J. H.; Hovestadt V.; Zapatka M.; Sturm D.; Jones D. T. W.; Kool M.; Remke M.; Cavalli F. M. G.; Zuyderduyn S.; Bader G. D.; VandenBerg S.; Esparza L. A.; Ryzhova M.; Wang W.; Wittmann A.; Stark S.; Sieber L.; Seker-Cin H.; Linke L.; Kratochwil F.; Jäger N.; Buchhalter I.; Imbusch C. D.; Zipprich G.; Raeder B.; Schmidt S.; Diessl N.; Wolf S.; Wiemann S.; Brors B.; Lawerenz C.; Eils J.; Warnatz H.-J.; Risch T.; Yaspo M.-L.; Weber U. D.; Bartholomae C. C.; von Kalle C.; Turányi E.; Hauser P.; Sanden E.; Darabi A.; Siesjö P.; Sterba J.; Zitterbart K.; Sumerauer D.; van Sluis P.; Versteeg R.; Volckmann R.; Koster J.; Schuhmann M. U.; Ebinger M.; Grimes H. L.; Robinson G. W.; Gajjar A.; Mynarek M.; von Hoff K.; Rutkowski S.; Pietsch T.; Scheurlen W.; Felsberg J.; Reifenberger G.; Kulozik A. E.; von Deimling A.; Witt O.; Eils R.; Gilbertson R. J.; Korshunov A.; Taylor M. D.; Lichter P.; Korbel J. O.; Wechsler-Reya R. J.; Pfister S. M. Enhancer Hijacking Activates GFI1 Family Oncogenes in Medulloblastoma. Nature 2014, 511, 428–434. 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J. D.; Condroski K.; Correa M.; Muller G.; Man H.-W.; Ruchelman A.; Zhang W.; Vocanson F.; Crea T.; Liu W.; Lu G.; Baculi F.; LeBrun L.; Mahmoudi A.; Carmel G.; Hickman M.; Lu C.-C. Protein Degradation via CRL4CRBN Ubiquitin Ligase: Discovery and Structure–Activity Relationships of Novel Glutarimide Analogs That Promote Degradation of Aiolos and/or GSPT1. J. Med. Chem. 2018, 61, 492–503. 10.1021/acs.jmedchem.6b01911. [DOI] [PubMed] [Google Scholar]
- Nowak R. P.; DeAngelo S. L.; Buckley D.; He Z.; Donovan K. A.; An J.; Safaee N.; Jedrychowski M. P.; Ponthier C. M.; Ishoey M.; Zhang T.; Mancias J. D.; Gray N. S.; Bradner J. E.; Fischer E. S. Plasticity in Binding Confers Selectivity in Ligand-Induced Protein Degradation. Nat. Chem. Biol. 2018, 14, 706–714. 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The Influence of Drug-like Concepts on Decision-Making in Medicinal Chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Mullighan C. G.; Collins-Underwood J. R.; Phillips L. A. A.; Loudin M. G.; Liu W.; Zhang J.; Ma J.; Coustan-Smith E.; Harvey R. C.; Willman C. L.; Mikhail F. M.; Meyer J.; Carroll A. J.; Williams R. T.; Cheng J.; Heerema N. A.; Basso G.; Pession A.; Pui C.-H.; Raimondi S. C.; Hunger S. P.; Downing J. R.; Carroll W. L.; Rabin K. R. Rearrangement of CRLF2 in B-Progenitor– and Down Syndrome–Associated Acute Lymphoblastic Leukemia. Nat. Genet. 2009, 41, 1243–1246. 10.1038/ng.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milde T.; Lodrini M.; Savelyeva L.; Korshunov A.; Kool M.; Brueckner L. M.; Antunes A. S. L. M.; Oehme I.; Pekrun A.; Pfister S. M.; Kulozik A. E.; Witt O.; Deubzer H. E. HD-MB03 Is a Novel Group 3 Medulloblastoma Model Demonstrating Sensitivity to Histone Deacetylase Inhibitor Treatment. J. Neuro-Oncol. 2012, 110, 335–348. 10.1007/s11060-012-0978-1. [DOI] [PubMed] [Google Scholar]
- Bandopadhayay P.; Bergthold G.; Nguyen B.; Schubert S.; Gholamin S.; Tang Y.; Bolin S.; Schumacher S. E.; Zeid R.; Masoud S.; Yu F.; Vue N.; Gibson W. J.; Paolella B. R.; Mitra S. S.; Cheshier S. H.; Qi J.; Liu K.-W.; Wechsler-Reya R.; Weiss W. A.; Swartling F. J.; Kieran M. W.; Bradner J. E.; Beroukhim R.; Cho Y.-J. BET Bromodomain Inhibition of MYC-Amplified Medulloblastoma. Clin. Cancer Res. 2014, 20, 912–925. 10.1158/1078-0432.CCR-13-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel M. F.; Robinson G. W. Role of MYC in Medulloblastoma. Cold Spring Harbor Perspect. Med. 2013, 3, a014308–a014308. 10.1101/cshperspect.a014308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G.; Surka C.; Lu C.-C.; Jang I. S.; Wang K.; Rolfe M. Elucidating the Mechanism of Action of CC-90009, a Novel Cereblon E3 Ligase Modulator, in AML Via Genome-Wide CRISPR Screen. Blood 2019, 134, 405. 10.1182/blood-2019-125492. [DOI] [Google Scholar]
- Lu G.; Lopez-Girona A.; Filvaroff E.. Methods for Treating Cancer and the Use of Biomarkers as a Predictor of Clinical sensitivity to therapies. US20170199193, June 1, 2017.
- Lopez-Girona A.; Lu G.; Rychak E.; Mendy D.; Lu C.-C.; Rappley I.; Fontanillo C.; Cathers B. E.; Daniel T. O.; Hansen J. CC-90009, a Novel Cereblon E3 Ligase Modulator, Targets GSPT1 for Degradation to Induce Potent Tumoricidal Activity Against Acute Myeloid Leukemia (AML). Blood 2019, 134, 2703. 10.1182/blood-2019-127892. [DOI] [Google Scholar]
- Bunnage M. E.; Chekler E. L. P.; Jones L. H. Target Validation Using Chemical Probes. Nat. Chem. Biol. 2013, 9, 195–199. 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- Chauvin C.; Salhi S.; Jean-Jean O. Human Eukaryotic Release Factor 3a Depletion Causes Cell Cycle Arrest at G1 Phase through Inhibition of the MTOR Pathway. MCB 2007, 27, 5619–5629. 10.1128/MCB.00035-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S. D.; Urh M.; Daniels D. L. High-Throughput Cellular Profiling of Targeted Protein Degradation Compounds Using HiBiT CRISPR Cell Lines. JoVE 2020, 61787. 10.3791/61787. [DOI] [PubMed] [Google Scholar]
- Sarojini K.; Krishnan H.; Kanakam C. C.; Muthu S. Synthesis, Structural, Spectroscopic Studies, NBO Analysis, NLO and HOMO–LUMO of 4-Methyl-N-(3-Nitrophenyl)Benzene Sulfonamide with Experimental and Theoretical Approaches. Spectrochim. Acta, Part A 2013, 108, 159–170. 10.1016/j.saa.2013.01.060. [DOI] [PubMed] [Google Scholar]
- Bai B.; Wang X.; Li Y.; Chen P.-C.; Yu K.; Dey K. K.; Yarbro J. M.; Han X.; Lutz B. M.; Rao S.; Jiao Y.; Sifford J. M.; Han J.; Wang M.; Tan H.; Shaw T. I.; Cho J.-H.; Zhou S.; Wang H.; Niu M.; Mancieri A.; Messler K. A.; Sun X.; Wu Z.; Pagala V.; High A. A.; Bi W.; Zhang H.; Chi H.; Haroutunian V.; Zhang B.; Beach T. G.; Yu G.; Peng J. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 106, 700. 10.1016/j.neuron.2020.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino S.; Imai M.; Mizutani M.; Kikuchi Y.; Hanaoka F.; Ui M.; Katada T. Molecular Cloning of a Novel Member of the Eukaryotic Polypeptide Chain-Releasing Factors (ERF): ITS IDENTIFICATION AS ERF3 INTERACTING WITH ERF1. J. Biol. Chem. 1998, 273, 22254–22259. 10.1074/jbc.273.35.22254. [DOI] [PubMed] [Google Scholar]
- Lee J. A.; Park J. E.; Lee D. H.; Park S. G.; Myung P. K.; Park B. C.; Cho S. G1 to S Phase Transition Protein 1 Induces Apoptosis Signal-Regulating Kinase 1 Activation by Dissociating 14-3-3 from ASK1. Oncogene 2008, 27, 1297–1305. 10.1038/sj.onc.1210740. [DOI] [PubMed] [Google Scholar]
- Malta-Vacas J.; Ferreira P.; Monteiro C.; Brito M. Differential Expression of GSPT1 GGCn Alleles in Cancer. Cancer Genet. Cytogenet. 2009, 195, 132–142. 10.1016/j.cancergencyto.2009.08.010. [DOI] [PubMed] [Google Scholar]
- Malta-Vacas J.; Aires C.; Costa P.; Conde A. R.; Ramos S.; Martins A. P.; Monteiro C.; Brito M. Differential Expression of the Eukaryotic Release Factor 3 (ERF3/GSPT1) According to Gastric Cancer Histological Types. J. Clin. Pathol. 2005, 58, 621–625. 10.1136/jcp.2004.021774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R.; Li C.; Chai B. MiRNA-144 Suppresses Proliferation and Migration of Colorectal Cancer Cells through GSPT1. Biomed. Pharmacother. 2015, 74, 138–144. 10.1016/j.biopha.2015.08.006. [DOI] [PubMed] [Google Scholar]
- Nair S.; Bora-Singhal N.; Perumal D.; Chellappan S. Nicotine-Mediated Invasion and Migration of Non-Small Cell Lung Carcinoma Cells by Modulating STMN3 and GSPT1 Genes in an ID1-Dependent Manner. Mol. Cancer 2014, 13, 173. 10.1186/1476-4598-13-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malta-Vacas J.; Chauvin C.; Gonçalves L.; Nazaré A.; Carvalho C.; Monteiro C.; Bagrel D.; Jean-Jean O.; Brito M. ERF3a/GSPT1 12-GGC Allele Increases the Susceptibility for Breast Cancer Development. Oncol. Rep. 2009, 21, 1551–1558. 10.3892/or_00000387. [DOI] [PubMed] [Google Scholar]
- PeCAN Data Portal (Https://Pecan.Stjude.Cloud/Home).
- Zhou X.; Edmonson M. N.; Wilkinson M. R.; Patel A.; Wu G.; Liu Y.; Li Y.; Zhang Z.; Rusch M. C.; Parker M.; Becksfort J.; Downing J. R.; Zhang J. Exploring Genomic Alteration in Pediatric Cancer Using ProteinPaint. Nat. Genet. 2016, 48, 4–6. 10.1038/ng.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John L. B.; Ward A. C. The Ikaros Gene Family: Transcriptional Regulators of Hematopoiesis and Immunity. Mol. Immunol. 2011, 48, 1272–1278. 10.1016/j.molimm.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Olsson L.; Johansson B. Ikaros and Leukaemia. Br. J. Haematol. 2015, 169, 479–491. 10.1111/bjh.13342. [DOI] [PubMed] [Google Scholar]
- Marke R.; van Leeuwen F. N.; Scheijen B. The Many Faces of IKZF1 in B-Cell Precursor Acute Lymphoblastic Leukemia. Haematologica 2018, 103, 565–574. 10.3324/haematol.2017.185603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J. D. E.; Beuling E.; van den Heuvel-Eibrink M. M.; Obulkasim A.; Baruchel A.; Trka J.; Reinhardt D.; Sonneveld E.; Gibson B. E. S.; Pieters R.; Zimmermann M.; Zwaan C. M.; Fornerod M. Recurrent Deletions of IKZF1 in Pediatric Acute Myeloid Leukemia. Haematologica 2015, 100, 1151–1159. 10.3324/haematol.2015.124321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly J. P.; Pruett-Miller S. M. CRIS.Py: A Versatile and High-Throughput Analysis Program for CRISPR-Based Genome Editing. Sci. Rep. 2019, 9, 4194. 10.1038/s41598-019-40896-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauler M.; Roberts J. K.; Wu C.-C.; Fan B.; Ferrara F.; Yip B. H.; Diao S.; Kim Y.-I.; Moore J.; Zhou S.; Wielgosz M. M.; Ryu B.; Throm R. E. Production of Lentiviral Vectors Using Suspension Cells Grown in Serum-Free Media. Mol. Ther.--Methods Clin. Dev. 2020, 17, 58–68. 10.1016/j.omtm.2019.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentmanat M. F.; Peters S. T.; Florian C. P.; Connelly J. P.; Pruett-Miller S. M. A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci. Rep. 2018, 8, 888. 10.1038/s41598-018-19441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E. III; Cruzeiro V. W. D.; Darden T. A.. AMBER 2018 ;University of California: San Francisco, 2018. [Google Scholar]
- Schrödinger Release 2020–2; Maestro, Schrödinger, LLC: New York, NY, 2020. [Google Scholar]
- Bai B.; Tan H.; Pagala V. R.; High A. A.; Ichhaporia V. P.; Hendershot L.; Peng J.. Deep Profiling of Proteome and Phosphoproteome by Isobaric Labeling, Extensive Liquid Chromatography, and Mass Spectrometry. In Methods in Enzymology ;Elsevier: 2017; Vol. 585, pp. 377–395, 10.1016/bs.mie.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.