

Abstract
DNA inter-strand crosslinks (ICLs) are dangerous lesions that can be caused by a variety of endogenous and exogenous bifunctional compounds. Because covalently linking both strands of the double helix locally disrupts DNA replication and transcription, failure to remove even a single ICL can be fatal to the cell. Thus, multiple ICL repair pathways have evolved, with the best studied being the canonical Fanconi anemia (FA) pathway. However, recent research demonstrates that different types of ICLs (e.g., backbone distorting vs. non-distorting) can be discriminated by the cell, which then mounts a specific repair response using the FA pathway or one of a variety of FA-independent ICL repair pathways. This review focuses on the latter, covering current work on the transcription-coupled, base excision, acetaldehyde-induced, and SNM1A/RecQ4 ICL repair pathways and highlighting unanswered questions in the field. Answering these questions will provide mechanistic insight into the various pathways of ICL repair and enable ICL-inducing agents to be more effectively used as chemotherapeutics.
Keywords: Fanconi anemia, DNA inter-Strand crosslink repair, DNA helicase, Nuclease, Hrq1, Pso2
1. Introduction
1.1. DNA inter-strand crosslink repair
The world is a dangerous place for genetic material. Consequently, the susceptibility of DNA to damage has led to the evolution of an arsenal of different DNA repair mechanisms. The inter-strand crosslink (ICL), a covalent linkage between both strands of the double helix (Dronkert and Kanaar, 2001), is among the most dangerous type of DNA lesion. Indeed, a single unrepaired ICL can be lethal to a yeast cell (Grossmann et al., 2001), and a small number of unrepaired ICLs are lethal to mammalian cells (Liu and Wang, 2013). Such lesions are difficult to repair, as simple excision still leaves a potentially mutagenic adduct that must be unhooked before fill-in of the initial incision site by either translesion polymerases or homologous recombination (HR) (Deans and West, 2011) (Fig. 1). The remains of the crosslink on the unrepaired strand must also be removed, potentially by excision repair factors. Therefore, ICL repair involves the cooperation of multiple DNA repair pathways, and deficiency in any one can lead to incomplete ICL removal and mutagenesis.
Fig. 1.
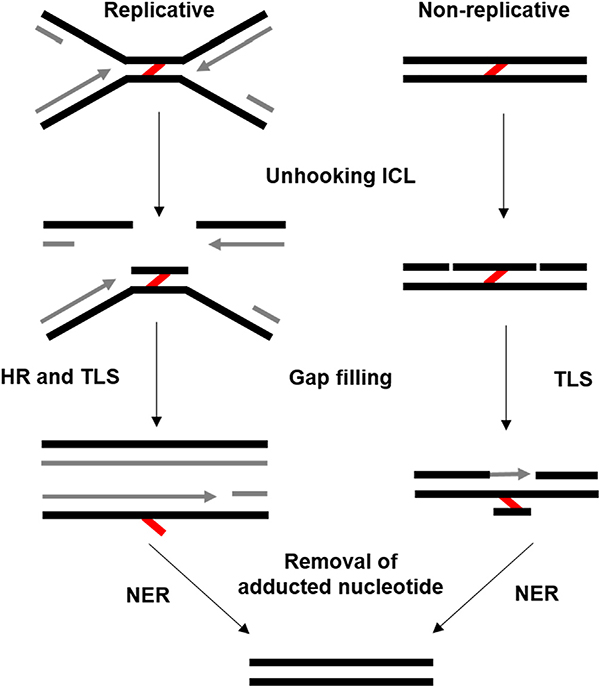
Simplified view of DNA ICL repair. Two DNA structures (replicative and non-replicative) are depicted undergoing the same three stages of ICL repair (unhooking, gap filling, and removal of the adducted nucleotide). ICLs are denoted by the red lines, and newly synthesized DNA is shown in grey. See the text and Fig. 2 for additional details.
1.1.1. ICLs, cancer, and chemotherapy
Failure to properly repair an ICL can lead to cell death either through mitotic catastrophe or p53-dependent apoptosis. The general lethality of ICLs also engenders their strength as chemotherapeutics. As early as 1946, the crosslinking agent nitrogen mustard was used to treat a variety of cancers (Goodman et al., 1946). Today, many ICL-inducing agents are successfully employed in the treatment of a variety of cancers (Huang and Li, 2013). This class of chemotherapeutics creates a large number of ICLs (as well as intra-strand crosslinks, mono-adducts, and/or protein-DNA crosslinks) at any given time. Unable to repair all lesions, the cancer cells are overwhelmed by the DNA damage and are either destroyed through apoptosis or an inability to replicate their DNA, grow, and divide (Rycenga and Long, 2018).
Cancer cells notoriously evolve to become drug-resistant over time, creating unique challenges and dangers for cancer patients and their physicians (Holohan et al., 2013). As described below, the Fanconi anemia (FA) pathway is a major ICL repair pathway in humans. For FA-deficient cancers, the upregulation of compensatory ICL repair pathways is a major source of drug resistance (Niraj et al., 2019). Further, platinum-based chemotherapeutics, which can induce ICLs, are less effective when ICL repair mechanisms become more active following chemotherapy regiments (Wynne et al., 2007) and as patients age (Rudd et al., 1995).
Unfortunately, ICL agents can also yield genomic instability. Many exogenous chemicals (e.g., chemotherapeutics) and endogenous metabolic byproducts (e.g., those formed by lipid peroxidation) can cause numerous types of DNA damage, including ICLs. This damage, when unrepaired, is strongly linked to carcinogenesis (Kozekov et al., 2003; Stone et al., 2008; Muniandy et al., 2010). Blood cells appear to be particularly susceptible to ICLs, including those from chemotherapeutics, because patients can develop acute myeloid leukemia after ICL regiments (Deans and West, 2011; Travis et al., 1999). Cancer predisposition mutations in genes connected to ICL repair, such as those that cause BRCA2 and PALB2 deficiencies, and conditions such as FA, Rothmund-Thomson Syndrome, Baller-Gerold Syndrome, and RAPADILINO Syndrome, may affect a number of pathways resulting in cancer development, several of which are detailed below.
1.2. Endogenous sources of ICLs
As alluded to above, there are currently a wide range of natural and synthetic chemical ICL-inducing agents. However, ICLs are also caused by metabolic by-products within the cell. The structure and source of ICLs can vary dramatically, but in general, ICL-inducing agents are bifunctional to allow for reactivity with both strands of DNA. Although not an exhaustive list of known or hypothesized sources of ICLs, many of the most well-studied causes of crosslinks are discussed below and summarized in Table 1.
Table 1.
Exogenous sources of ICLs, their preferential DNA targets, and representative chemical structures for each.
| ICL Agent | Target Sequence | Structurea |
|---|---|---|
| Mitomycin C | 5′-CG | 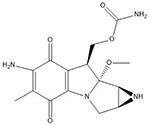 |
| Pyrrolobenzodiazepines | Purine-GATC-pyrimidine | 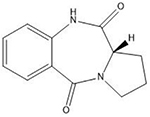 |
| Colibactin | 5′-AATTT and 5′-ATTTT | 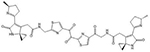 |
| Psoralens | 5′-TA | 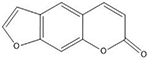 |
| Pyrrolizidine alkaloids | N2 guanine in 5′-CG-3′ | 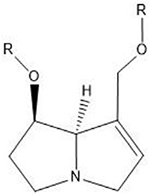 |
| Nitrogen mustards | 5′-GNC | |
| Chloroethylnitrosoureas | O6- and N7 positions of guanine | 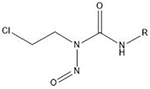 |
| Cisplatin | 5′-GC | 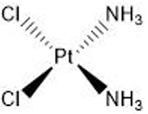 |
| Diepoxybutane | 5′-GNC | 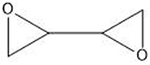 |
| Dianhydrogalactitol | N7 guanine | 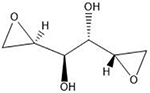 |
Many of these structures can vary by the presence of a number of different possible–R groups. For the sake of brevity, the simplest molecular scaffolds are shown.
1.2.1. Reactive aldehydes
Reactive aldehydes formed via lipid peroxidation and base excision repair (BER) are a known source of DNA damage (reviewed in (Burcham, 1998)). Dietary intake (e.g., fats and alcohol) and environmental exposure to exogenous toxins, such as car exhaust and tobacco smoke, promote the formation of or introduce harmful aldehydes within cells (Burcham, 1998; Nath et al., 1998; Clauson et al., 2013). Reactive aldehydes such as acrolein, crotonaldehyde, and 4-hydroxynonenal interact with guanine bases to form exocyclic 1,N2-dG adducts (Stone et al., 2008), which have demonstrable genotoxicity in humans (Kawanishi et al., 1998; Czerny et al., 1998). Multiple groups have described the ability of the exocyclic 1,N2-dG product to reopen, exposing an aldehyde that, under the correct steric context, can form an ICL in vitro (Stone et al., 2008; Huang et al., 2010). Putative evidence for aldehyde-induced ICL formation in vivo was identified in mice deficient for the FA ICL repair pathway and aldehyde catabolism (Garaycoechea et al., 2012; Langevin et al., 2011). FA in ICL repair is discussed in detail below, but mice deficient in both pathways demonstrate clinical FA phenotypes such as bone marrow failure when exposed to the metabolic acetaldehyde precursor, ethanol (Langevin et al., 2011). In addition, ICL repair-deficient Saccharomyces cerevisiae cells are sensitive to acetaldehyde accumulation (Brendel et al., 2010), further supporting the ability of aldehydes to form ICLs.
1.2.2. Abasic crosslinks
Spontaneous or DNA glycosidic hydrolysis of nucleobases results in the formation of abasic (Ap) sites (Krokan and Bjoras, 2013), and these lesions represent a large percentage of the daily DNA damage burden that cells contend with (Tubbs and Nussenzweig, 2017; Nakamura and Swenberg, 1999; Nakamura et al., 1998). Work from Kent Gates’ lab demonstrates the propensity of Ap sites to form ICLs in vitro (Price et al., 2014; Johnson et al., 2013). Like other ICLs, those formed from Ap DNA only occur under the appropriate sequence context (e.g., 5′-AT-3′), meaning not every Ap is able to form an ICL. However, the high abundance of spontaneous Ap sites in cells suggests that some amount of these lesions become ICLs in vivo. Similar to the mechanism of ICL formation via reactive aldehydes discussed above, the progenitor species in Ap crosslinking is an aldehyde that exists in equilibrium with the cyclic deoxyribose sugar (Price et al., 2014; Dutta et al., 2007). Importantly, these ICLs still act as physical barriers to the replication machinery (Yang et al., 2015; Price et al., 2017), and polymerization activity through the crosslink occurs in an error-prone manner (Price et al., 2017). In Xenopus cell extracts, Ap-ICLs are repaired using a similar mechanism to that employed in the repair of the chemical ICL-inducing agent psoralen (Semlow et al., 2016). Taken together, the cellular response to Ap-ICLs is similar to that of a chemically induced ICL.
1.3. Exogenous ICLs: Therapeutic damage
The classic and better-studied sources of DNA ICLs come from exogenous chemical agents. Although the mechanisms of these bifunctional alkylating agents are generally the same, these compounds have different sequence preferences, topologically constrain the DNA in various ways, and require differential processing within the cell to form the active ICL-inducing species (Guainazzi and Scharer, 2010) (Table 1). Despite their propensity to damage DNA, innovative and insightful uses of these drugs demonstrate the value in understanding the underlying mechanisms of human disease to identify useful therapeutic targets. Some of the most common ICL agents are discussed below in the context of their clinical benefits.
1.3.1. Bacterial sources
1.3.1.1. Mitomycin C.
As a testament to the power and utility of ICLs as therapeutics, mitomycin C (MMC) continues to be a relevant chemotherapeutic agent despite its origin beginning in the 1950s when it was isolated from Streptomyces caespitosus (Tomasz, 1995; Hata et al., 1956; Wakaki et al., 1958). The natural MMC compound is not directly suitable to form ICLs (reviewed in (Tomasz, 1995)). Two reduction steps are required to activate the bifunctional alkylation groups to form the ICL. Reduction of only one group yields the more prominent monoadduct. Indeed, similar to most ICL-inducing agents, ICLs represent a small fraction of the DNA lesions caused by bifunctional compounds. In the case of MMC, this value was found to be between 5 and 13% (Dronkert and Kanaar, 2001; Warren et al., 1998). However, the activation of MMC in reducing conditions has proven to be part of its selectivity as a cancer target. The hypoxic environment found in solid tumors helps to promote the active ICL-inducing product of MMC, whereas the presence of oxygen will render MMC less toxic in normal tissue (Tomasz, 1995). MMC is also sequence specific and requires the dinucleotide 5′-CG-3′ to generate the ICL product, with the crosslink forming between the guanine bases. Only 5% of guanines in mammalian genomes exist within this sequence context, which, combined with the low propensity for ICL formation, lowers the potential efficacy of MMC as a chemotherapeutic. Despite these caveats however, MMC has been successfully used in the treatment of bladder (Tolley et al., 1988) and esophageal (Wolf et al., 2010) cancers.
1.3.1.2. Pyrrolobenzodiazepines.
First discovered in Streptomyces, pyrrolobenzodiazepines (PBDs) are ICL-inducing agents that are generally regarded as much more potent when functionally coupled to a tumor targeting moiety or antibody than systemic chemotherapeutics like MMC (Antonow and Thurston, 2011). PBDs induce ICLs at purine-GATC-pyrimidine sequences in a time-dependent manner, with the greatest effects occurring within the first few hours following exposure (Arnould et al., 2006). PBDs bind to the guanine N2 in the minor groove of DNA and block DNA replication without otherwise distorting the double helix. ICLs that have minor topological effects on DNA are more likely to evade detection and can assist in preventing cancerous cells from developing resistance to PBDs when used as a therapeutic agent (Saunders et al., 2015).
1.3.2. Colibactin
Colibactin is a genotoxic secondary metabolite produced by intestinal microorganisms encoding the pks island gene cluster that has long been associated with colorectal cancer in mammals (reviewed in (Balskus, 2015)). Collaborative work between Seth Herzon’s and Jason Crawford’s groups suggests a structure of colibactin and its associated adducts that reflects previous research on colibactin biosynthesis. In their model, two “precolibactin” structures that each contain a reactive aminocyclopropane group heterodimerize and are each capable of alkylating adenine bases in AT-rich DNA regions, with a second alkylation creating an ICL (Xue et al., 2019; Dziubanska-Kusibab et al., 2020). Gut inflammation is believed to contribute to increased DNA damage by allowing direct contact between microbes and tissue (Arthur et al., 2012). To date, limited progress has been made toward identifying the exact repair mechanisms available for colibactin ICLs, but it is known that exposure to the toxin enhances FA pathway signaling (Bossuet-Greif et al., 2018).
1.3.3. Plant sources
1.3.3.1. Psoralens.
Psoralens are a class of naturally produced furocoumarins from plants and have been used in the treatment of dermatological diseases for thousands of years (reviewed in (Melough and Chun, 2018)). Psoralens require photoactivation to interact with DNA, and the combination of psoralen and ultraviolet A (UVA) radiation is a common treatment of the chronic skin diseases psoriasis (Zhang and Wu, 2018) and vitiligo (McEvoy and Stern, 1987), though the mechanism is unclear. UVA and psoralen are also used in the treatment of cutaneous T-cell lymphoma (Atilla et al., 2017; Oldham et al., 2016). Through appropriate photoactivation, psoralens form ICLs between thymines in 5′-TA-3′ sequences and have a much higher propensity to form ICLs (~40%) when compared to the other crosslinkers discussed (Dronkert and Kanaar, 2001). Psoralen adducts also cause a relatively minor local 25° unwinding of DNA around the lesion (Dronkert and Kanaar, 2001; Spielmann et al., 1995). Although potentially harmful due to their presence in consumed plants, the requirement for UV activation of these DNA damaging agents allows for local targeting in cancer treatments (Melough and Chun, 2018; Melough et al., 2018).
1.3.3.2. Pyrrolizidine Alkaloids.
Pyrrolizidine Alkaloids (PAs) are natural compounds produced in low concentrations by an extensive variety of plant species and have genotoxic effects upon ingestion (Smith and Culvenor, 1981). Oxidation of the pyrrolizidine ring by liver cytochrome P450s activates both C7 and C9 of the PA and results in a combination of DNA monoadducts, DNA-protein crosslinks, DNA intra-strand crosslinks, and ICLs. The propensity for each lesion caused by various PAs is not well understood but has been linked to the saturation and cyclicity of the different ester groups (reviewed in (Chen et al., 2010)). Sequence preference of the alkylation is also a subject of dispute, as there are results claiming preference for the N2 position of guanine in 5′-CG-3′ sequences (Weidner et al.,1990), as well as results that suggest no sequence or base preference at all (Rieben and Coulombe, 2004), even for the same PA compound.
1.3.4. Synthetic sources
1.3.4.1. Nitrogen mustards.
The history of nitrogen mustard in cancer therapies is both tragic in its origin and optimistic in the innovation that was sparked by it. In 1943, an air raid of a ship carrying mustard gas during World War II resulted in mustard gas poisoning of the survivors (Deans and West, 2011). Upon noticing leukopenia in these patients, the first chemotherapy for leukemia using nitrogen mustard was born (Goodman et al., 1946). Since then, nitrogen mustard and its derivatives have been used in the treatment of several cancers, including malignant melanomas (Boone et al., 2018), lymphomas (Vitolo et al., 2017), and others (reviewed in (Singh et al., 2018)). Although they vary in scaffolding, the functional groups in nitrogen mustards are basically the same. Briefly, nitrogen mustards contain two (2-chloroethyl)amines, that undergo cyclization to form an unstable and highly reactive intermediate (Singh et al., 2018). One round of this reaction results in a monoadduct, and when this occurs in the proper 5′-GNC-3′ sequence context, a second reaction can occur to form an ICL (Guainazzi and Scharer, 2010). Similar to MMC, a small percent of nitrogen mustard lesions are ICLs (~5%), in addition to monoadducts and DNA-protein crosslinks (Dronkert and Kanaar, 2001). Compared to non-distorting lesions like those caused by PBDs, nitrogen mustards induce ~15° of DNA bending around the ICL.
1.3.4.2. Chloroethylnitrosoureas.
Chloroethylnitrosoureas (CENUs) are known DNA chloroethylating agents that can react with different positions on nucleotide bases but preferentially target the O6 position of guanine, triggering inter-strand crosslinking between guanine and cystine or thymine (Clauson et al., 2013; Williams and Shaw, 1987). Cellular repair mechanisms for ICLs caused by CENUs and similar chloroethylating agents include O6-methylguanine DNA methyltransferase (Esteller and Herman, 2004) and O6-alkylguanine-DNA alkyltransferase (Yamada et al., 2019). Unsurprisingly, it has been found that other repair mechanisms for CENU-derived ICLs include NER and recombination (Yamada et al., 2019). CENUs are uniquely attractive ICL agents for their ability to cross the blood-brain barrier for treatment of brain cancers. Additionally, as with any DNA alkylation agent, there is a chance for genotoxicity secondary to chemotherapeutic treatment, but there is also evidence that CENU treatment can induce the bystander chemotherapeutic effect, providing protection against secondary tumor growth (Merle et al., 2008).
1.3.5. Cisplatin
Platinum-based DNA damaging agents, such as cisplatin, are among the most common anti-tumor drugs used in medicine and are effective in the treatment of several types of cancers (reviewed in (Dasari and Tchounwou, 2014)). The mechanism by which cisplatin forms DNA adducts is similar to that of nitrogen mustards (Deans and West, 2011), though cisplatin is more properly described as a DNA intra-strand crosslinking agent due to its greater tendency to form crosslinks between bases on the same DNA strand (Dronkert and Kanaar, 2001). Different platinum-based crosslinkers have different propensities to form ICLs (~5% for cisplatin), but the most commonly used platinum drugs all crosslink guanines in the 5′-GC-3′ sequence. Cisplatin significantly distorts the DNA structure by causing 110° unwinding around the lesion and a 47° bend in the DNA backbone (Dronkert and Kanaar, 2001). As a consequence, the cytosine bases across from the crosslinked guanines become bent such that they are extrahelical (Guainazzi and Scharer, 2010). Despite their broad efficacy as chemotherapeutics, cisplatin-treated tumors are subject to drug resistance (reviewed in (Galluzzi et al., 2012)). There are multiple hypothesized mechanisms for chemotherapeutic resistance, including deficient apoptotic cell signaling in the presence of excess damage, increased use of translesion polymerases, and upregulation of DNA repair pathways.
1.3.6. Diepoxybutane
Diepoxybutane (DEB) is a metabolic product of the carcinogen 1,3-butadiene, a compound used in the production of synthetic plastics and rubbers (Millard and White, 1993; Bond and Medinsky, 2001). DEB is not currently used in cancer treatments, and thus, its cytotoxicity has not been as extensively studied as the crosslinkers mentioned above. However, the clinical laboratory diagnosis of FA uses ICL-inducing agents such as DEB or MMC, as elevated sensitivity to ICLs is a hallmark of the disease (Cirkovic et al., 2011; Auerbach, 2009). Recent work from our lab also demonstrates that DEB ICLs are repaired in yeast using a similar suite of proteins as those involved in the repair of more commonly investigated agents such as MMC (Rogers et al., 2020). DEB-derived ICLs share the preferred trinucleotide 5′-GNC-3′ as seen with nitrogen mustard (Millard and White, 1993) and produce a 34 °C bend in DNA (Millard et al., 2012). DEB is noteworthy for its utility in cell biology studies because, relative to other crosslinkers, it is inexpensive, stable, and does not require additional activation (Rogers et al., 2020).
1.3.6.1. Dianhydrogalactitol.
1,2:5,6-dianhydrogalactitol (DAG) is an alkylating agent known for its uses in the treatment of a variety of cancers, most popularly non-small cell and small cell lung cancers. DAG is a synthetic hexitol derivative and is used to target all cell cycle stages (Chiuten et al., 1981a; Chiuten et al., 1981b; Peng et al., 2017). The damage inflicted by DAG is due to ICLs (specifically via N7-guanine alkylation), which ultimately form DNA double-strand breaks (DSBs) that cause cell cycle arrest (Zhai et al., 2018). Cells may attempt to repair DSBs and ICLs created by DAG by HR, though in cases where the HR pathway is inhibited, DAG induces massive damage to the cells. DAG is growing in popularity for the treatment of a variety of cancers, but its mechanism of action remains largely unknown.
1.4. FA and ICL repair
As incredibly dangerous DNA lesions, ICLs require a complex network of repair pathways to recognize and remove the damage to allow normal cell progression. Indeed, components of many DNA repair pathways (e.g., HR, TLS, and NER) briefly mentioned thus far participate in some aspect of ICL repair (Deans and West, 2011). Like most molecular biology processes, ICL repair is extremely complex, and the cellular response to these lesions is largely dependent on the context in which the crosslink is identified (Fig. 2). In mammals, the predominant and most studied mechanism for ICL repair is replication coupled and involves the FA pathway (Zhang and Walter, 2014).
Fig. 2.

Outline of common ICL repair pathways. ICLs are denoted by the red lines, newly synthesized DNA is shown in grey, and RNA is green. In the ICL traverse pathway, the replisome encounters an ICL but can bypass it (dashed grey line). Subsequently, one or more nucleases act on the ICL, generating a substrate for HR and NER. The replication coupled pathway is the canonical FA pathway. In the TCR pathway, RNA polymerase (RNA POL) encounters the ICL. See the main text for additional details.
1.4.1. The FA signaling cascade
The FA complementation group involves 20+ proteins, with new members frequently being identified (Longerich et al., 2014). FA-mediated ICL recognition and repair can broadly be broken down into three steps (Duxin and Walter, 2015) (Fig. 3). First, an eight-member complex of FA proteins, called the FA core complex, is involved in lesion recognition and activation of the FA pathway (reviewed in (Longerich et al., 2014)). FANCM, as part of the core complex, plays an important role in lesion recognition (Kim et al., 2008) and replication fork stabilization (Kim and D’Andrea, 2012). Second, the FA core complex, via the E3 ubiquitin ligase FANCL, monoubiquitinates the FANCI-FANCD2 dimer (Longerich et al., 2014) to prime the lesion for the final components of the FA cascade (Knipscheer et al., 2009). The last step is lesion processing by nucleases and other DNA response elements that participate in unhooking and removal of the ICL (Duxin and Walter, 2015).
Fig. 3.

Basic steps of the general FA repair pathway. Recognition and activation of the FA core complex through FANCM begins FA ICL repair. Ubiquitination of the FANCI-FANCD2 complex is important for recruitment of the subsequent repair factors that remove the lesion (red) and fill-in the gap (newly synthesized DNA is in grey). See the main text for more details.
1.4.2. Mechanistic details of the FA pathway
Over the last decade, Johannes Walter’s laboratory, using the replication-proficient Xenopus cell-free extract system (Walter et al., 1998), has provided beautiful mechanistic insight into the FA repair pathway. The general assay used by the Walter group involves the measurement of replication via incorporation of 32P-α-dATP into a plasmid harboring a single site-specific ICL (Raschle et al., 2008). Although the premise of this assay is relatively simple, the manipulation of various aspects of FA and FA-associated proteins has made this assay a veritable goldmine in breaking down individual components of vertebrate ICL repair.
The initial application of this system in ICL repair observed stalling of the replisome ~20-nt away from the ICL, at which point the replisome either remodels, or the CMG helicase becomes uncoupled from DNA synthesis to physically allow polymerases access up to −1-nt away from the ICL (Raschle et al., 2008). Nucleolytic incision was also observed, essentially creating a DSB that uncouples the sister chromatids (Fig. 2). The strand that contains the newly created monoadduct thus serves as a template for HR of the DSB. Depletion of Rev7, a component of the translesion polymerase DNA pol ζ, results in accumulation of product at the 0 position (i.e., directly across from the lesion), suggesting that the more accommodating pocket of a TLS polymerase, relative to that of a replicative polymerase, is required for complete DNA replication through the ICL. Subsequent analysis determined that inhibiting ubiquitination of the FANCI-FANCD2 complex results in accumulation of the −1 product, suggesting that ubiquitinated FANCI/D2 promotes the incision step of ICL repair (Knipscheer et al., 2009). Indeed, depletion of FANCD2 prevents identification of the DSB-like, post-incision product. The programmed DSB promoted by the FANCI-FANCD2 complex then leads to Rad51 filament formation and subsequent repair via HR (Long et al., 2011).
Several endo- and exonucleases have been implicated in the unhooking step of ICL repair (reviewed in (Zhang and Walter, 2014)). Douwel et al. found that depletion or inactivation of the XPF-ERCC1 endonuclease complex virtually eliminates ICL repair, specifically by abrogating production of the incision product (Klein Douwel et al., 2014). Consistent with the hypotheses discussed above, XPF-ERCC1 recruitment and incision is dependent on FANCI-FANCD2. Other nucleases predicted to participate in unhooking include MUS81-EME1, SLX1-SLX4, and FAN1 (Zhang and Walter, 2014) (Fig. 2). Each enzyme has a different structure preference, suggesting that the nuclease utilized depends on the DNA structure at the ICL.
Recently, Amunugama et al. found that ejection of the CMG helicase is required to promote the incision step (Amunugama et al., 2018). Interestingly, this unloading step is important for pathway choice and commits cells to FA-mediated repair (Wu et al., 2019). Although several questions remain with respect to general ICL repair, the experiments discussed above have provided a wealth of information about the sequence of events and networking that occurs to remove these lesions.
1.4.3. Evidence for replication without repair
Whereas the Walter model for ICL repair requires convergence of two replication forks at an ICL (Zhang et al., 2015) (Fig. 2), single-molecule analysis from Michael Seidman’s group discovered that the majority of replication forks actually progress through ICLs, leaving behind the unrepaired lesion in a “traverse” model (Huang et al., 2013). FA proteins are at least partially involved in this phenomenon, as FANCM translocase activity is required to promote ICL traverse. The ability of the replisome to accommodate and move through an ICL seems contrary to widely accepted mechanisms of how replication works, though a more recent model of ICL repair could explain this phenomenon (discussed below). Follow-up experiments will be required to determine how this pathway operates, and the notion that ICLs are an absolute block to replication obviously needs to be reconsidered.
2. FA-independent ICL repair pathways
Other mechanisms of ICL repair that function independently or synergistically with the FA pathway also play an important role in the repair of these lesions. While FA appears to be the predominant source of ICL repair in S-phase, alternative pathways may be required for repair of ICLs in different cellular contexts (e.g., other cell cycle phases). For example, lesion bypass in the traverse model leaves behind an ICL that still needs to be repaired. Does the FA pathway remove this lesion, or do one of the alternative pathways shown in Fig. 2 fill this role? Further, under what conditions are these FA-independent ICL repair pathways utilized? It is worth noting that there are several factors, such as the unhooking nucleases and translesion polymerases, that are involved in one or more ICL repair pathways. However, we define FA-independent repair as generally functioning outside of the context of the main pathway described above and outlined in Fig. 3. Relative to FA repair, these alternative ICL pathways are poorly understood, but the repair mechanisms as they are currently known are discussed below.
2.1. Transcription-coupled ICL repair
Evidence for NER in replication-independent ICL repair has existed for many years (Sarkar et al., 2006), but this work was conducted in yeast, which does not utilize the FA pathway as the main mechanism for ICL repair (McHugh et al., 2012). More recently, Enoiu et al. identified a novel component of transcription-coupled NER (TC-NER) in ICL repair that operates outside of S-phase (Enoiu et al., 2012). Using a highly transcribed reporter gene containing a site-specific cisplatin ICL, they determined that several NER repair factors are required for removal of the lesion. Interestingly, CSB, a TC-NER-specific ATPase that binds tightly to RNA polymerase upon transcription arrest (Hanawalt and Spivak, 2008), is vital for appropriate repair of the ICL, whereas the global NER recognition protein XPC is not required (Enoiu et al., 2012). CSB-deficient cells that are synchronized in G0/G1 and treated with MMC accumulate in G2/M arrest, providing evidence for TC-NER ICL repair prior to S-phase. Importantly, depletion of FANCD2 in CSB-deficient cells causes a synergistic increase in the cytotoxicity of cisplatin, further supporting the hypothesis that TC-NER functions independently of FA.
The identification of a novel nuclease involved in ICL repair by Andrews et al. suggests additional features of transcription-coupled repair (TCR) of ICLs that are unique to this pathway (Andrews et al., 2018). This nuclease, called SAN1, possesses 5′ → 3′ ssDNA exonuclease activity. Knockout or inactivation of SAN1 sensitizes HeLa cells to MMC and cisplatin, and ICL cytotoxicity is further increased by depletion of FANCD2. The FA pathway appears to function normally in the absence of SAN1, providing additional evidence for a FA-independent repair mechanism. Interestingly, SAN1 was found to interact with SETX, an RNA/DNA helicase involved in R-loop resolution. R-loops form naturally during transcription, and an abundance of them can result in replication blocks and genome instability (reviewed in (Belotserkovskii et al., 2018)). Not only is the SAN1-SETX interaction required for SAN1-dependent ICL repair, but SAN1 knockout also results in accumulation of transcription-dependent R-loops (Andrews et al., 2018). A model has begun to emerge in which the transcription machinery stall at ICLs, resulting in the recruitment of SETX to prevent the build-up of R-loops. SETX then associates with SAN1, which uses its nuclease activity in an unknown mechanism of ICL processing that likely involves the TC-NER factors outlined by Enoiu et al. (Enoiu et al., 2012; Andrews et al., 2018).
2.2. Base excision in ICL repair
Ap sites and small base modifications are one of the more common types of DNA damage (Krokan and Bjoras, 2013). BER is thus one of the most utilized DNA damage response pathways. The general BER scheme involves the removal of the damaged DNA base via a DNA glycosylase to create an Ap site, endonuclease activity to remove a segment of DNA around the lesion, and DNA polymerase and ligase activity to fill the gap. Mammals utilize several different DNA glycosylases to recognize a suite of DNA lesions, and many of these enzymes have been implicated in ICL repair (Kothandapani and Patrick, 2013).
Cells also appear to have a mechanism for inhibiting ICL formation at Ap sites. The DNA glycosylases bind the Ap site tightly and prevent the crosslinking by the reactive intermediate (Admiraal and O’Brien, 2015). Employing the Xenopus extract system, Semlow et al. identified a novel DNA glycosylase-mediated mechanism for ICLs that operates independently of FA (Semlow et al., 2016). Interestingly, where cisplatin and nitrogen mustard ICLs are repaired by the endonuclease incision model discussed above (Fig. 2), psoralen and Ap-ICLs utilize the NEIL3 glycosylase in an incision-independent manner. Inhibition of the DNA glycosylase pathway shifts repair to the FA mechanism, suggesting that psoralen and Ap-ICLs are preferentially repaired by the former pathway. Glycosylase-mediated ICL repair also does not result in a DSB, which prevents the production of this potentially dangerous repair intermediate. Why cells have different repair mechanisms for different ICLs remains an important question, but it could be due to steric obstruction to the N-glycosyl bond in certain crosslinks. This pathway could also help explain the traverse model, as NEIL3-repaired ICLs do not require uncoupling of the replisome. Recently, the Walter group also identified the TRAIP E3 ubiquitin ligase as an important mediator in committing cells to either the glycosylase or FA pathway (Wu et al., 2019).
Interestingly, another method employed by the cell to shield Ap sites from forming ICLs has also recently been reported. In humans, an enzyme called HMCES forms a covalent protein-DNA crosslink at Ap sites, thus preventing spontaneous ICL formation by the Ap site (Mohni et al., 2019). The HMCES crosslink, which is eventually degraded by the proteasome, also prevents the deleterious action of TLS polymerases and endonucleases at the Ap site. Further, this appears to be a conserved mechanism because homologs of HMCES are found in yeast and bacteria.
2.3. Repair of acetaldehyde-induced ICLs
Alcohol metabolism can produce harmful intermediates, one of them being acetaldehyde. When acetaldehyde is generated, it reacts with guanine to produce N2-propanoguanine, which then interacts with the N2-amine of the guanine on the opposite strand to form an ICL. Recently, Hodskinson et al. described two replication-coupled mechanisms for the repair of ICLs derived from alcohol processing metabolites by examining the cellular repair systems for acetaldehyde-induced ICLs (Hodskinson et al., 2020), with one method utilizing the canonical FA pathway.
A new route for ICL repair has also been discovered using the acetaldehyde-induced ICL model. This mechanism works independently of and potentially blocks the FA pathway. This pathway works quickly and does not involve FA-like incision of DNA, but it is dependent upon convergence of two replication forks at the lesion. Interestingly, the ICL is unhooked by cutting between the nucleotide base and the lesion itself and thus can avoid breaking the strands of DNA and creating Ap sites (Hodskinson et al., 2020). This suggests a new repair pathway that may be error prone due to the remaining modified base but can help support genomic stability during the repair of acetaldehyde ICLs. The specific protein(s) responsible for cleaving within the crosslinked lesions has not yet been identified, though it has been speculated that the process may be enzymatically driven, which would suggest this pathway as a potential target for future therapeutic agents (Gallina and Duxin, 2020).
2.4. SNM1 translesion nucleases in ICL repair
The SNM1 family nucleases have long been known to participate in ICL repair, but their mechanism of action is poorly understood (Cattell et al., 2010). Complicating matters is the evolutionary divergent utilization of FA-like and SNM1-dependent ICL repair. For instance, S. cerevisiae possesses a degenerate FA-like pathway that lacks many of the 20+ proteins found in metazoans (McHugh et al., 2012; Ward et al., 2012). ICL repair in this organism operates predominantly via the SNM1 family nuclease Pso2 (Rogers et al., 2020; Henriques and Moustacchi, 1981; Barber et al., 2005; Henriques and Moustacchi, 1980). Indeed, deletion of the FANCM homolog in yeast, Mph 1, does not sensitize cells to nitrogen mustard, but deletion of both mph1 and pso2 results in synergistic sensitivity, suggesting that Pso2 functions dominantly and independently of the proto-FA pathway (Ward et al., 2012).
There are three SNM1 proteins in humans (SNM1A, SNM1B, and SNM1C), with SNM1A being the functional homolog of Pso2 (Hazrati et al., 2008). Both SNM1A and Pso2 possess 5′ → 3′ exonuclease activity and structure-specific endonuclease activity (Li et al., 2005; Buzon et al., 2018; Tiefenbach and Junop, 2012). The model of SNM1/Pso2 in ICL repair largely comes from work by Peter McHugh’s lab. After incision by NER factors, Pso2 uses translesion nuclease activity to further degrade the “unhooked” strand of the lesion to facilitate TLS (McHugh et al., 2012) (Fig. 2). Pso2 has been implicated in lesion processing for several reasons: 1) psoΔ cells are sufficient for NER-mediated incisions flanking the ICL (Grossmann et al., 2000); 2) Pso2 is not required for HR, but DSBs accumulate in the absence of Pso2 (Li and Moses, 2003); and 3) SNM1A can digest through an ICL from an XPF-induced nick in vitro (Wang et al., 2011; Abdullah et al., 2017). SNM1A has also been linked to replication- and transcription-coupled ICL repair (Wang et al., 2011; Iyama et al., 2015), suggesting it has a broad but dedicated role in ICL repair.
2.5. RecQ4 family helicases in ICL repair
The RecQ helicases are known as “guardians of the genome” for their presence in all domains of life and the versatility in which they participate in maintaining genome integrity (Croteau et al., 2014). All members of the RecQ family have a typical Superfamily II (SF2) helicase core domain and translocate along DNA in a 3′ → 5′ direction (Croteau et al., 2014; Beyer et al., 2013). The importance of RecQ helicases in genome maintenance is typified by the presence of five RecQ helicases in humans (Croteau et al., 2014). These helicases, called RECQL1, BLM, WRN, RECQL4, and RECQL5, are involved in divergent and synergistic pathways that all combine to promote genome integrity. RECQL1 is upregulated upon DNA damage (Parvathaneni et al., 2017), and when depleted, renders cells more sensitive to DNA damaging agents (reviewed in (Sharma and Brosh, 2008) and (Sami and Sharma, 2013)). Similarly, RECQL5 has an uncharacterized DNA repair function, along with a unique role in RNA transcription (reviewed in (Popuri et al., 2013)). Despite the importance of RECQL1 and RECQL5 in genome maintenance, deficiencies in either helicase have not yet been linked to any human diseases (Croteau et al., 2014). In contrast, BLM, WRN and RECQL4 cause clinically distinct human diseases when mutated, but they are phenotypically similar in causing various types of cancers and premature aging phenotypes. Of the disease-linked RecQ helicases, RECQL4 is the least well understood and is focused on below.
2.5.1. Human RECQL4
RECQL4 mutations are linked to three distinct diseases, Rothmund-Thomson syndrome (RTS), Baller-Gerold syndrome (BGS), and RAPADILINO, which present a number of overlapping clinical features that are generally summarized by developmental defects and elevated cancer risk (reviewed in (Liu, 2010)). Most of the reported RECQL4 mutations result in partial or complete elimination of the helicase domain (Larizza et al., 2010), and recombinant expression of four different known RECQL4 mutants from RAPADILINO patients revealed a lack of ATPase and helicase activities (Jensen et al., 2012; Croteau et al., 2012). Though little is known about the exact in vivo roles of RECQL4, it is linked to several important aspects of genome integrity, including telomere maintenance (Ghosh et al., 2012; Ferrarelli et al., 2013), DSB repair (Singh et al., 2010; Lu et al., 2016; Shamanna et al., 2014), and DNA ICL repair (Jin et al., 2008). The latter findings are based on the sensitivity of RTS cells to ICL-inducing agents, and mounting evidence also shows that the up-regulation of RECQL4 occurs in a range of drug-resistant cancers. From breast to gastric cancers, increases in RECQL4 expression are strongly linked to poorer prognosis and increased metastatic development (Fang et al., 2013; Li et al., 2018; Mo et al., 2016). Additionally, human prostate cancer cells display elevated rates of RECQL4 transcription, and siRNA suppression of RECQL4 reduces metastasis in prostate cancer cells lines (Su et al., 2010).
2.5.2. RECQL4 homologs in lower eukaryotes
Metazoan RECQL4 proteins present a variety of in vitro and in vivo challenges to investigating their roles in genome maintenance (Rogers et al., 2017). Thus, the search for non-metazoan RECQL4 homologs has been ongoing for many years, and multiple groups have begun work on several of these putative models. Interestingly, RECQL4 homologs have been identified in plants and fungi (Barea et al., 2008), bacteria, and archaea (Yakovleva and Shuman, 2012), making the RecQ4 sub-family helicases the only known RecQ conserved in all three domains of life. Herein, we focus on the S. cerevisiae Hrq1 helicase as a RECQL4 model, but the other identified RECQL4 homologs are briefly discussed to provide perspective into this burgeoning field.
2.5.3. RecQ4 helicases in prokaryotes
The first bacterial RecQ4 sub-family helicase was identified in Mycobacterium smegmatis and is named SftH (Yakovleva and Shuman, 2012). Yakovleva and Shuman biochemically characterized SftH and found it has the typical 3′ → 5′ helicase activity displayed by all RecQ helicases. More importantly, they used phylogenetic analysis to identify SftH homologs in dozens of bacteria and archaea, reinforcing the evolutionary conservation of RecQ4 sub-family helicases. Consistent with its eukaryotic homologs, SftH is also potentially involved in ICL repair in M. smegmatis (Rogers, Strnat, and Bochman, observations).
More recent work with the Bacillus subtilis SftH homolog, called MrfA, suggests a role in ICL repair (Burby and Simmons, 2019). MrfA interacts with the N-terminus of the exonuclease MrfB in this potential pathway. This is reminiscent of the SETX-SAN1 helicase-nuclease pair in TCR of ICLs and is further mirrored by the Hrq1 helicase and Pso2 nuclease in S. cerevisiae (below). Thus, the synergism between a helicase and nuclease could be an ancient and essential ICL repair mechanism. It has been suggested that the MrfAB pathway acts as a second excision repair pathway to repair ICLs (Burby and Simmons, 2019). However, MrfA is not involved in the unhooking step of MMC-induced lesions, so the role of MrfA in ICL repair is currently tenuous.
2.6. The Hrq1 helicase
2.6.1. The discovery of Hrq1
In 2005, a high-throughput screen of ~4700 single gene deletions in S. cerevisiae for sensitivity to a suite of DNA-damaging agents identified cells lacking the gene product of YDR291W as sensitive to the DNA ICL agent nitrogen mustard, noting the sequence similarity of this gene to mammalian RECQL4 (Lee et al., 2005). Three years later, Barea et al. published an in-depth computational search for RECQL4 homologs in plants and fungi and named them Hrq1 (Barea et al., 2008). The discovery of Hrq1 provided new avenues to examine RECQL4 mechanisms with the potential to overcome the practical challenges exhibited by the metazoan RECQL4 enzymes. Over the last decade, research from numerous groups working on a variety of organisms have begun to identify the roles of Hrq1 in several DNA repair pathways, including DNA ICL repair.
2.6.2. Hrq1 in eukaryotes
The first application of the Hrq1 model was performed by Groocock et al. using the Schizosaccharomyces pombe system (Groocock et al., 2012). Deletion of hrq1 resulted in increased cell length, a hallmark of general DNA damage checkpoint activation in fission yeast. While hrq1Δ strains displayed little to no sensitivity to DNA alkylating agents, UV damage, and HU-induced replication stalls, the DNA ICL-inducing agents MMC and cisplatin were highly toxic to hrq1 mutants. A dominant negative phenotype was observed when Hrq1 helicase activity was inactivated, suggesting that helicase activity is required for Hrq1-mediated ICL repair. Other general phenotypes observed in hrq1Δ strains include elevated mutation rates and hyper-recombination.
Since the initial work on S. pombe Hrq1, S. cerevisiae has been the workhorse model for Hrq1 research. Consistent with the role of RECQL4 in telomere maintenance, Hrq1 was found play a significant role in telomere maintenance in budding yeast (Bochman et al., 2014; Nickens et al., 2018). Similar to the S. pombe (Groocock et al., 2012), Arabidopsis (Rohrig et al., 2018), and human (Jin et al., 2008) RecQ4 family helicases, budding yeast also Hrq1 catalytically participates in DNA ICL repair (Rogers et al., 2020; Rogers et al., 2017; Bochman et al., 2014). S. cerevisiae hrq1 mutants are largely insensitive to most DNA lesions, with the exception being those that cause ICLs. In contrast, deletion of SGS1, the gene encoding the other yeast RecQ family helicase, strongly sensitizes cells to most types of DNA damage except ICLs (Bochman et al., 2014), suggesting a dedicated role for Hrq1 in ICL repair.
2.7. Hrq1 and Pso2 in ICL repair
RECQL4 and Hrq1 are known ICL repair factors, but their mechanism of action and the pathway in which they function are unclear (Jin et al., 2008; Bochman et al., 2014). It is recognized that Hrq1 operates with Pso2 in the repair of ICLs in yeast, but several questions remain. In what way does Hrq1 facilitate ICL repair? Do Hrq1 and Pso2 repair all types of ICL lesions? Does Hrq1 also participate in the degenerate FA pathway? To address these and other quandaries, we recently performed a thorough biochemical comparison of Hrq1 and RECQL4 to examine the strengths and limitation of the Hrq1 model (Rogers et al., 2017; Rogers and Bochman, 2017) and began to elucidate the mechanism for Hrq1 and Pso2-dependent ICL repair (Rogers et al., 2020).
Deletion of PSO2 in yeast results in cells that are specifically sensitive to DNA ICLs (Rogers et al., 2017; Bochman et al., 2014). The classic model for Pso2-mediated ICL repair involves translesion nuclease activity across an ICL to provide access for downstream repair processes such as TLS and HR (McHugh et al., 2012). However, previous work with Pso2 did not demonstrate translesion nuclease activity (Tiefenbach and Junop, 2012), suggesting that an accessory factor is required to promote digestion through the lesion. Hrq1 was shown to have a dedicated role in ICL repair, though it appears to be less significant than Pso2 (Rogers et al., 2017; Bochman et al., 2014). Importantly, hrq1Δ is epistatic to pso2Δ, suggesting that they cooperatively participate in ICL repair. Hypothetically, the 5′ → 3′ exonuclease activity of Pso2 could create 3′ → 5′ ssDNA (Li et al., 2005) that acts as a substrate for the Hrq1 helicase (Rogers et al., 2017). Indeed, the combination of Hrq1 and Pso2 results in stimulation of Pso2 nuclease activity in vitro (Rogers et al., 2020). While Pso2 does possess some weak translesion nuclease activity in our hands, digestion through the lesion is greatly increased in the presence of Hrq1. This phenomenon is specific to eukaryotic RecQ4 helicases as the yeast homolog of BLM, Sgs1, is unable to stimulate Pso2, whereas human RECQL4 does. Hrq1-mediated stimulation of Pso2 appears to occur through a direct, albeit transient, protein-protein interaction.
Why did this helicase-nuclease system evolve in which Pso2 needs to be stimulated by Hrq1 for appropriate ICL resistance? It is known that Pso2 is expressed at basal levels, and ICL damage results in only an approximately four-fold increase (Wolter et al., 1996). We found that Pso2 over-expression in yeast is toxic (Rogers et al., 2020), explaining why it is normally not abundant in cells. Thus, Pso2 levels are maintained such that they are sufficient for ICL repair but not cytotoxic. The interaction of Hrq1 with Pso2 during ICL repair likely enables stimulation of Pso2 nuclease activity in a site-specific manner. We hypothesize that Hrq1 and Pso2 are recruited to ICLs, and low levels of Pso2 are sufficient for ICL repair because Hrq1 is present to promote Pso2 translesion nuclease activity (Fig. 4). This scheme allows for cells to maintain a low Pso2 concentration but still have the appropriate amount of nuclease activity at the lesion.
Fig. 4.

Function of Hrq1 and Pso2 in ICL repair and consequences of their deletion. Following NER-induced incisions on either side of the ICL (red line), Hrq1 (blue) and Pso2 (orange) are recruited to digest one strand of DNA between the incisions. In the absence of Hrq1, Pso2 retains minimal translesional nuclease activity, so some lesions persist. In the absence of Pso2, alternative repair machinery can be recruited to repair the lesion, but most ICLs persist and lead to cell death.
Interestingly, six reported phosphorylated residues have been identified between the disordered N-terminus of Pso2 and its nuclease activity-associated MBL domain. These post-translational modifications may also represent another level of regulation, especially considering that the N-terminus of Pso2 is autoinhibitory (Rogers et al., 2020). This phosphorylated cluster could act as a “hinge” that modulates the level of autoinhibition or promotes recruitment to ICLs. Although delineation of the Hrq1-Pso2 mechanism is at an early stage, progress in this pathway and the others discussed above will inevitably lead to new avenues for chemotherapeutic treatments.
3. Outstanding questions in the field
ICL sensitivity is not as severe in hrq1Δ cells relative to pso2Δ, indicating that minimal Pso2 translesion nuclease activity is sufficient for some ICL repair. Alternatively, Pso2 could create a different substrate to facilitate other downstream repair processes such as HR, or other mechanisms (e.g., FA-like repair) become the predominant repair pathway. Thus, an important yet underexplored question in ICL repair is when are these different ICL mechanisms utilized? Work from Johannes Walter’s group suggests most ICL repair is replication-coupled (Raschle et al., 2008), but others have also shown examples of ICL repair pathway outside of S-phase (Enoiu et al., 2012). It is not surprising that essential processes like replication and transcription are important for lesion recognition as they will eventually encounter an ICL when present in the genome, but what about lesions left behind in the traverse model? Are lesions recognized in the absence of processes that require separation of the duplex? While the answer appears to be yes (Williams et al., 2013; Williams et al., 2012), when and why cells utilize replication-and transcription-independent pathways is not clear. The answer likely depends on the type of lesion and the cell cycle in which the ICL is encountered. For example, Xenopus extracts (Semlow et al., 2016) and human cells (Li et al., 2020) can repair ICLs via FA or with DNA glycosylases depending on the lesion. ICLs that strongly distort the DNA topology, like cisplatin, are likely recognized by NER factors in the absence of replication- or transcription-coupled repair, but psoralen adducts could evade a NER response due to more subtle changes in DNA structure (Guainazzi and Scharer, 2010; Smeaton et al., 2008).
There is currently little information with respect to Hrq1 and Pso2 (and other ICL repair factors) in cell cycle utilization, though some work has been performed to observe cell cycle arrest in S. cerevisiae in the presence of different ICL-inducing agents (Grossmann et al., 2000; Meniel et al., 1997; Grossmann et al., 1999). Since these experiments, the role of Pso2 in ICL repair has become more established and the yeast FA-like pathway identified. It would be interesting to determine if the use of these pathways changes in a cell cycle-dependent manner and if hrq1Δ cells cause cells to arrest in a manner similar to pso2Δ. Although the degenerate yeast FA pathway appears to be a back-up ICL repair pathway to Pso2 (Rogers et al., 2017), the real use of the FA-like pathway is likely more complicated. For example, the FA pathway may be specific to S-phase like in metazoans, but in yeast, most lesions may not be repaired until G2 where the Pso2-dependent pathway is predominant (Barber et al., 2005). These types of questions will need to be answered to begin to fully understand the complex network of ICL repair pathways.
Compared to the vast network of 20+ proteins used in FA pathway, our knowledge of the factors used in FA-independent ICL repair is very basic. Identification of additional factors would allow for expansion of the current models and could eventually result in complete in vitro recapitulation of these modes of ICL repair. Mass spectrometry (MS) will likely play an important role in the identification of proteins directly involved in ICL processing, but there are a few technical challenges to overcome. The low abundance of factors such as Hrq1 and Pso2 in the cell makes it difficult to use them as co-immunoprecipitation (co-IP) bait because it is difficult to capture enough protein to potentially identify interacting partners. Furthermore, the timing of the various ICL repair pathways also means it may be difficult capture their discrete interactomes, as the involvement of any one factor may be transient over the course of the several hours it takes for ICL repair (Magana-Schwencke et al., 1982). Taken together, targeting ICL repair proteins for this purpose has a number of technical hurdles to overcome, but the development of methods that allow for IP of the ICL itself could be a novel direction in identifying ICL repair proteins.
4. Parting remarks
ICL repair is as complicated as it is essential. The field is rapidly evolving as understanding of new proteins and pathways continues to improve. Work from Johannes Walter’s lab has set the gold standard for the study of ICL repair with the FA pathway both in the quality of their work and the significance with which this pathway operates. However, their discovery that the alternative replication-coupled NEIL3 pathway is responsible for the repair of psoralen and Ap lesions typifies the complexity of ICL repair and begs the question: Why do cell have so many different ICL repair pathways? An obvious answer is that they are highly toxic lesions that must be repaired for cell viability, and evolution has generated multiple distinct mechanisms to deal with ICLs from a variety of sources. The clinical use of ICL inducing agents to treat a variety of conditions, primarily cancers, demonstrates the value in understanding these mechanisms for application in both the laboratory and the clinic. The work reviewed herein details ICL origins and additional ICL response mechanisms that will certainly continue to be developed and added to the suite of known tools for removal of these dangerous lesions by the cell. The list of putative ICL repair pathways is continually growing, and the field must now begin to define when and why these pathways are activated to gain a full mechanistic understanding of ICL repair.
Funding and acknowledgements
We thank the participants of the Fusion Conferences 4th DNA Repair/Replication Structures and Cancer Conference for stimulating discussions concerning the topics covered in this review. This work was supported by start-up funding from Indiana University and grants from the American Cancer Society (RSG-16–180-01-DMC) and the National Institutes of Health (1R35GM133437).
Abbreviations:
- ICL
inter-strand crosslink
- Ap
abasic
- MMC
mitomycin C
- DEB
Diepoxybutane
- FA
Fanconi anemia
- HR
homologous recombination
- NER
nucleotide excision repair
- TLS
translesion synthesis
- TC-NER
transcription-coupled NER
- TCR
transcription-coupled repair
- BER
base excision repair
- SF2
Superfamily II
- RQC
RecQ C-Terminal Domain
- dsDNA
double-stranded DNA
- HRDC
Helicase and RNase D C-terminal
- RTS
Rothmund-Thomson Syndrome
- BGS
Baller-Gerold Syndrome
- T-loop
telomeric D-loop
- G4
G-quadruplex
- NHEJ
non-homologous end joining
- DSB
double strand break
- CDKs
cyclin-dependent kinases
- RHCD
RecQ4/Hrq1 -conserved domain
- MS
mass spectrometry
- CoIP
co-immunoprecipitation
References
- Abdullah UB, McGouran JF, Brolih S, Ptchelkine D, El-Sagheer AH, Brown T, McHugh PJ, 2017. RPA activates the XPF-ERCC1 endonuclease to initiate processing of DNA interstrand crosslinks. EMBO J. 36, 2047–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Admiraal SJ, O’Brien PJ, 2015. Base excision repair enzymes protect abasic sites in duplex DNA from interstrand cross-links. Biochemistry 54, 1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunugama R, Willcox S, Wu RA, Abdullah UB, El-Sagheer AH, Brown T, McHugh PJ, Griffith JD, Walter JC, 2018. Replication fork reversal during DNA interstrand crosslink repair requires CMG unloading. Cell Rep. 23, 3419–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews AM, McCartney HJ, Errington TM, D’Andrea AD, Macara IG, 2018. A senataxin-associated exonuclease SAN1 is required for resistance to DNA interstrand cross-links. Nat. Commun. 9, 2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonow D, Thurston DE, 2011. Synthesis of DNA-interactive pyrrolo[2,1-c][1,4] benzodiazepines (PBDs). Chem. Rev. 111, 2815–2864. [DOI] [PubMed] [Google Scholar]
- Arnould S, Spanswick VJ, Macpherson JS, Hartley JA, Thurston DE, Jodrell DI, Guichard SM, 2006. Time-dependent cytotoxicity induced by SJG136 (NSC 694501): influence of the rate of interstrand cross-link formation on DNA damage signaling. Mol. Canc. Therapeut. 5, 1602–1609. [DOI] [PubMed] [Google Scholar]
- Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C, 2012. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atilla E, Atilla PA, Bozdag SC, Yuksel MK, Toprak SK, Topcuoglu P, Akay BN, Sanli H, Akan H, Demirer T, Beksac M, Arslan O, Ozcan M, Gurman G, Ilhan O, 2017. Extracorporeal photochemotherapy in mycosis fungoides. Transfus. Clin. Biol. 24, 454–457. [DOI] [PubMed] [Google Scholar]
- Auerbach AD, 2009. Fanconi anemia and its diagnosis. Mutat. Res. 668, 4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balskus EP, 2015. Colibactin: understanding an elusive gut bacterial genotoxin. Nat. Prod. Rep. 32, 1534–1540. [DOI] [PubMed] [Google Scholar]
- Barber LJ, Ward TA, Hartley JA, McHugh PJ, 2005. DNA interstrand cross-link repair in the Saccharomyces cerevisiae cell cycle: overlapping roles for PSO2 (SNM1) with MutS factors and EXO1 during S phase. Mol. Cell Biol. 25, 2297–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barea F, Tessaro S, Bonatto D, 2008. In silico analyses of a new group of fungal and plant RecQ4-homologous proteins. Comput. Biol. Chem. 32, 349–358. [DOI] [PubMed] [Google Scholar]
- Belotserkovskii BP, Tornaletti S, D’Souza AD, Hanawalt PC, 2018. R-loop generation during transcription: formation, processing and cellular outcomes. DNA Repair 71, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer DC, Ghoneim MK, Spies M, 2013. Structure and mechanisms of SF2 DNA helicases. Adv. Exp. Med. Biol. 767, 47–73. [DOI] [PubMed] [Google Scholar]
- Bochman ML, Paeschke K, Chan A, Zakian VA, 2014. Hrq1, a homolog of the human RecQ4 helicase, acts catalytically and structurally to promote genome integrity. Cell Rep. 6, 346–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond JA, Medinsky MA, 2001. Insights into the toxicokinetics and toxicodynamics of 1,3-butadiene. Chem. Biol. Interact. 135–136, 599–614. [DOI] [PubMed] [Google Scholar]
- Boone BA, Perkins S, Bandi R, Santos E, McCluskey K, Bartlett DL, Pingpank JF, 2018. Hepatic artery infusion of melphalan in patients with liver metastases from ocular melanoma. J. Surg. Oncol. 117, 940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossuet-Greif N, Vignard J, Taieb F, Mirey G, Dubois D, Petit C, Oswald E, Nougayrede JP, 2018. The colibactin genotoxin generates DNA interstrand cross-links in infected cells. mBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel M, Marisco G, Ganda I, Wolter R, Pungartnik C, 2010. DNA repair mutant pso2 of Saccharomyces cerevisiae is sensitive to intracellular acetaldehyde accumulated by disulfiram-mediated inhibition of acetaldehyde dehydrogenase. Genet. Mol. Res. 9, 48–57. [DOI] [PubMed] [Google Scholar]
- Burby PE, Simmons LA, 2019. A bacterial DNA repair pathway specific to a natural antibiotic. Mol. Microbiol. 111, 338–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burcham PC, 1998. Genotoxic lipid peroxidation products: their DNA damaging properties and role in formation of endogenous DNA adducts. Mutagenesis 13, 287–305. [DOI] [PubMed] [Google Scholar]
- Buzon B, Grainger R, Huang S, Rzadki C, Junop MS, 2018. Structure-specific endonuclease activity of SNM1A enables processing of a DNA interstrand crosslink. Nucleic Acids Res. 46, 9057–9066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattell E, Sengerova B, McHugh PJ, 2010. The SNM1/Pso2 family of ICL repair nucleases: from yeast to man. Environ. Mol. Mutagen. 51, 635–645. [DOI] [PubMed] [Google Scholar]
- Chen T, Mei N, Fu PP, 2010. Genotoxicity of pyrrolizidine alkaloids. J. Appl. Toxicol. 30, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiuten DF, Rozencweig M, Von Hoff DD, Muggia FM, 1981a. Clinical trials with the hexitol derivatives in the. U.S. Cancer 47, 442–451. [DOI] [PubMed] [Google Scholar]
- Chiuten DF, Vosika GJ, Shaw MT, Boiron M, Gisselbrecht C, Marty M, Higgins G, Muggia FM, 1981b. Clinical trials with diglycoaldehyde (NSC118994): review and reasons for withdrawal from clinical trial. Anticancer Res. 1, 121–124. [PubMed] [Google Scholar]
- Cirkovic S, Guc-Scekic M, Vujic D, Ilic N, Micic D, Skoric D, Jovanovic A, 2011. Diagnosis of fanconi’s anemia by diepoxybutane analysis in children from Serbia. Balkan J. Med. Genet. 14, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauson C, Scharer OD, Niedernhofer L, 2013. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Biol 5, a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau DL, Rossi ML, Ross J, Dawut L, Dunn C, Kulikowicz T, Bohr VA, 2012. RAPADILINO RECQL4 mutant protein lacks helicase and ATPase activity. Biochim. Biophys. Acta 1822, 1727–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croteau DL, Popuri V, Opresko PL, Bohr VA, 2014. Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 83, 519–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerny C, Eder E, Runger TM, 1998. Genotoxicity and mutagenicity of the alpha, beta-unsaturated carbonyl compound crotonaldehyde (butenal) on a plasmid shuttle vector. Mutat. Res. 407, 125–134. [DOI] [PubMed] [Google Scholar]
- Dasari S, Tchounwou PB, 2014. Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol. 740, 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans AJ, West SC, 2011. DNA interstrand crosslink repair and cancer. Nat. Rev. Canc. 11, 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronkert ML, Kanaar R, 2001. Repair of DNA interstrand cross-links. Mutat. Res. 486, 217–247. [DOI] [PubMed] [Google Scholar]
- Dutta S, Chowdhury G, Gates KS, 2007. Interstrand cross-links generated by abasic sites in duplex DNA. J. Am. Chem. Soc. 129, 1852–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, Walter JC, 2015. What is the DNA repair defect underlying Fanconi anemia? Curr. Opin. Cell Biol. 37, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziubanska-Kusibab PJ, Berger H, Battistini F, Bouwman BAM, Iftekhar A, Katainen R, Cajuso T, Crosetto N, Orozco M, Aaltonen LA, Meyer TF, 2020. Colibactin DNA-damage signature indicates mutational impact in colorectal cancer. Nat. Med. [DOI] [PubMed] [Google Scholar]
- Enoiu M, Jiricny J, Scharer OD, 2012. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 40, 8953–8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Herman JG, 2004. Generating mutations but providing chemosensitivity: the role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene 23, 1–8. [DOI] [PubMed] [Google Scholar]
- Fang H, Nie L, Chi Z, Liu J, Guo D, Lu X, Hei TK, Balajee AS, Zhao Y, 2013. RecQL4 helicase amplification is involved in human breast tumorigenesis. PloS One 8, e69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrarelli LK, Popuri V, Ghosh AK, Tadokoro T, Canugovi C, Hsu JK, Croteau DL, Bohr VA, 2013. The RECQL4 protein, deficient in Rothmund-Thomson syndrome is active on telomeric D-loops containing DNA metabolism blocking lesions. DNA Repair 12, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina I, Duxin JP, 2020. A safe fix for alcohol-derived DNA damage. Nature 579, 499–500. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G, 2012. Molecular mechanisms of cisplatin resistance. Oncogene 31, 1869–1883. [DOI] [PubMed] [Google Scholar]
- Garaycoechea JI, Crossan GP, Langevin F, Daly M, Arends MJ, Patel KJ, 2012. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 489, 571–575. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Rossi ML, Singh DK, Dunn C, Ramamoorthy M, Croteau DL, Liu Y, Bohr VA, 2012. RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J. Biol. Chem. 287, 196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman LS, Wintrobe MM, et al. , 1946. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J. Am. Med. Assoc. 132, 126–132. [DOI] [PubMed] [Google Scholar]
- Groocock LM, Prudden J, Perry JJ, Boddy MN, 2012. The RecQ4 orthologue Hrq1 is critical for DNA interstrand cross-link repair and genome stability in fission yeast. Mol. Cell Biol. 32, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann KF, Brown JC, Moses RE, 1999. Cisplatin DNA cross-links do not inhibit S-phase and cause only a G2/M arrest in Saccharomyces cerevisiae. Mutat. Res. 434, 29–39. [DOI] [PubMed] [Google Scholar]
- Grossmann KF, Ward AM, Moses RE, 2000. Saccharomyces cerevisiae lacking Snm1, Rev3 or Rad51 have a normal S-phase but arrest permanently in G2 after cisplatin treatment. Mutat. Res. 461, 1–13. [DOI] [PubMed] [Google Scholar]
- Grossmann KF, Ward AM, Matkovic ME, Folias AE, Moses RE, 2001. S. cerevisiae has three pathways for DNA interstrand crosslink repair. Mutat. Res. 487, 73–83. [DOI] [PubMed] [Google Scholar]
- Guainazzi A, Scharer OD, 2010. Using synthetic DNA interstrand crosslinks to elucidate repair pathways and identify new therapeutic targets for cancer chemotherapy. Cell. Mol. Life Sci. 67, 3683–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G, 2008. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970. [DOI] [PubMed] [Google Scholar]
- Hata T, Hoshi T, Kanamori K, Matsumae A, Sano Y, Shima T, Sugawara R, 1956. Mitomycin, a new antibiotic from Streptomyces. I. J Antibiot (Tokyo) 9, 141–146. [PubMed] [Google Scholar]
- Hazrati A, Ramis-Castelltort M, Sarkar S, Barber LJ, Schofield CJ, Hartley JA, McHugh PJ, 2008. Human SNM1A suppresses the DNA repair defects of yeast pso2 mutants. DNA Repair 7, 230–238. [DOI] [PubMed] [Google Scholar]
- Henriques JA, Moustacchi E, 1980. Isolation and characterization of pso mutants sensitive to photo-addition of psoralen derivatives in Saccharomyces cerevisiae. Genetics 95, 273–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques JA, Moustacchi E, 1981. Interactions between mutations for sensitivity to psoralen photoaddition (pso) and to radiation (rad) in Saccharomyces cerevisiae. J. Bacteriol. 148, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodskinson MR, Bolner A, Sato K, Kamimae-Lanning AN, Rooijers K, Witte M, Mahesh M, Silhan J, Petek M, Williams DM, Kind J, Chin JW, Patel KJ, Knipscheer P, 2020. Alcohol-derived DNA crosslinks are repaired by two distinct mechanisms. Nature 579, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG, 2013. Cancer drug resistance: an evolving paradigm. Nat. Rev. Canc. 13, 714–726. [DOI] [PubMed] [Google Scholar]
- Huang Y, Li L, 2013. DNA crosslinking damage and cancer - a tale of friend and foe. Transl. Cancer Res. 2, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Kozekov ID, Kozekova A, Wang H, Lloyd RS, Rizzo CJ, Stone MP, 2010. DNA cross-link induced by trans-4-hydroxynonenal. Environ. Mol. Mutagen. 51, 625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, Seidman MM, 2013. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol. Cell. 52, 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyama T, Lee SY, Berquist BR, Gileadi O, Bohr VA, Seidman MM, McHugh PJ, Wilson DM 3rd, 2015. CSB interacts with SNM1A and promotes DNA interstrand crosslink processing. Nucleic Acids Res. 43, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MB, Dunn CA, Keijzers G, Kulikowicz T, Rasmussen LJ, Croteau DL, Bohr VA, 2012. The helicase and ATPase activities of RECQL4 are compromised by mutations reported in three human patients. Aging (N Y) 4, 790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Liu H, Zhang Y, Otta SK, Plon SE, Wang LL, 2008. Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Hum. Genet. 123, 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KM, Price NE, Wang J, Fekry MI, Dutta S, Seiner DR, Wang Y, Gates KS, 2013. On the formation and properties of interstrand DNA-DNA cross-links forged by reaction of an abasic site with the opposing guanine residue of 5’-CAp sequences in duplex DNA. J. Am. Chem. Soc. 135, 1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanishi M, Matsuda T, Nakayama A, Takebe H, Matsui S, Yagi T, 1998. Molecular analysis of mutations induced by acrolein in human fibroblast cells using supF shuttle vector plasmids. Mutat. Res. 417, 65–73. [DOI] [PubMed] [Google Scholar]
- Kim H, D’Andrea AD, 2012. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 26, 1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Kee Y, Gurtan A, D’Andrea AD, 2008. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood 111, 5215–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Raschle M, Walter JC, Knipscheer P, 2014. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell. 54, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC, 2009. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326, 1698–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothandapani A, Patrick SM, 2013. Evidence for base excision repair processing of DNA interstrand crosslinks. Mutat. Res. 743–744, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozekov ID, Nechev LV, Moseley MS, Harris CM, Rizzo CJ, Stone MP, Harris TM, 2003. DNA interchain cross-links formed by acrolein and crotonaldehyde. J. Am. Chem. Soc. 125, 50–61. [DOI] [PubMed] [Google Scholar]
- Krokan HE, Bjoras M, 2013. Base excision repair. Cold Spring Harb Perspect Biol 5, a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ, 2011. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58. [DOI] [PubMed] [Google Scholar]
- Larizza L, Roversi G, Volpi L, 2010. Rothmund-Thomson syndrome. Orphanet J. Rare Dis. 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, St Onge RP, Proctor M, Flaherty P, Jordan MI, Arkin AP, Davis RW, Nislow C, Giaever G, 2005. Genome-wide requirements for resistance to functionally distinct DNA-damaging agents. PLoS Genet. 1, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Moses RE, 2003. The beta-lactamase motif in Snm1 is required for repair of DNA double-strand breaks caused by interstrand crosslinks in S. cerevisiae. DNA Repair 2, 121–129. [DOI] [PubMed] [Google Scholar]
- Li X, Hejna J, Moses RE, 2005. The yeast Snm1 protein is a DNA 5’-exonuclease. DNA Repair 4, 163–170. [DOI] [PubMed] [Google Scholar]
- Li J, Jin J, Liao M, Dang W, Chen X, Wu Y, Liao W, 2018. Upregulation of RECQL4 expression predicts poor prognosis in hepatocellular carcinoma. Oncol Lett 15, 4248–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wang J, Wallace SS, Chen J, Zhou J, D’Andrea AD, 2020. Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair. Nucleic Acids Res. 48, 3014–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, 2010. Rothmund-Thomson syndrome helicase, RECQ4: on the crossroad between DNA replication and repair. DNA Repair 9, 325–330. [DOI] [PubMed] [Google Scholar]
- Liu S, Wang Y, 2013. A quantitative mass spectrometry-based approach for assessing the repair of 8-methoxypsoralen-induced DNA interstrand cross-links and monoadducts in mammalian cells. Anal. Chem. 85, 6732–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DT, Raschle M, Joukov V, Walter JC, 2011. Mechanism of RAD51dependent DNA interstrand cross-link repair. Science 333, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longerich S, Li J, Xiong Y, Sung P, Kupfer GM, 2014. Stress and DNA repair biology of the Fanconi anemia pathway. Blood 124, 2812–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Shamanna RA, Keijzers G, Anand R, Rasmussen LJ, Cejka P, Croteau DL, Bohr VA, 2016. RECQL4 promotes DNA end resection in repair of DNA double-strand breaks. Cell Rep. 16, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magana-Schwencke N, Henriques JA, Chanet R, Moustacchi E, 1982. The fate of 8-methoxypsoralen photoinduced crosslinks in nuclear and mitochondrial yeast DNA: comparison of wild-type and repair-deficient strains. Proc. Natl. Acad. Sci. U. S. A. 79, 1722–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy MT, Stern RS, 1987. Psoralens and related compounds in the treatment of psoriasis. Pharmacol. Ther. 34, 75–97. [DOI] [PubMed] [Google Scholar]
- McHugh PJ, Ward TA, Chovanec M, 2012. A prototypical Fanconi anemia pathway in lower eukaryotes? Cell Cycle 11, 3739–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melough MM, Chun OK, 2018. Dietary furocoumarins and skin cancer: a review of current biological evidence. Food Chem. Toxicol. 122, 163–171. [DOI] [PubMed] [Google Scholar]
- Melough MM, Cho E, Chun OK, 2018. Furocoumarins: a review of biochemical activities, dietary sources and intake, and potential health risks. Food Chem. Toxicol. 113, 99–107. [DOI] [PubMed] [Google Scholar]
- Meniel V, Magana-Schwencke N, Averbeck D, Waters R, 1997. Preferential incision of interstrand crosslinks induced by 8-methoxypsoralen plus UVA in yeast during the cell cycle. Mutat. Res. 384, 23–32. [DOI] [PubMed] [Google Scholar]
- Merle P, Morvan D, Caillaud D, Demidem A, 2008. Chemotherapy-induced bystander effect in response to several chloroethylnitrosoureas: an origin independent of DNA damage? Anticancer Res. 28, 21–27. [PubMed] [Google Scholar]
- Millard JT, White MM, 1993. Diepoxybutane cross-links DNA at 5’-GNC sequences. Biochemistry 32, 2120–2124. [DOI] [PubMed] [Google Scholar]
- Millard JT, McGowan EE, Bradley SQ, 2012. Diepoxybutane interstrand crosslinks induce DNA bending. Biochimie 94, 574–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo D, Fang H, Niu K, Liu J, Wu M, Li S, Zhu T, Aleskandarany MA, Arora A, Lobo DN, Madhusudan S, Balajee AS, Chi Z, Zhao Y, 2016. Human helicase RECQL4 drives cisplatin resistance in gastric cancer by activating an AKT-YB1MDR1 signaling pathway. Canc. Res. 76, 3057–3066. [DOI] [PubMed] [Google Scholar]
- Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, Eichman BF, Thompson PS, Mehta KPM, Cortez D, 2019. HMCES maintains genome integrity by shielding abasic sites in single-strand DNA. Cell 176, 144–153 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniandy PA, Liu J, Majumdar A, Liu ST, Seidman MM, 2010. DNA interstrand crosslink repair in mammalian cells: step by step. Crit. Rev. Biochem. Mol. Biol. 45, 23–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura J, Swenberg JA, 1999. Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Canc. Res. 59, 2522–2526. [PubMed] [Google Scholar]
- Nakamura J, Walker VE, Upton PB, Chiang SY, Kow YW, Swenberg JA, 1998. Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Canc. Res. 58, 222–225. [PubMed] [Google Scholar]
- Nath RG, Ocando JE, Guttenplan JB, Chung FL, 1998. 1,N2-propanodeoxyguanosine adducts: potential new biomarkers of smoking-induced DNA damage in human oral tissue. Canc. Res. 58, 581–584. [PubMed] [Google Scholar]
- Nickens DG, Rogers CM, Bochman ML, 2018. The Saccharomyces cerevisiae Hrq1 and Pif1 DNA helicases synergistically modulate telomerase activity in vitro. J. Biol. Chem. 293, 14481–14496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niraj J, Farkkila A, D’Andrea AD, 2019. The Fanconi anemia pathway in cancer. Annu. Rev. Cell Biol. 3, 457–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham M, Yoon P, Fathi Z, Beyer WF, Adamson J, Liu L, Alcorta D, Xia W, Osada T, Liu C, Yang XY, Dodd RD, Herndon JE 2nd, Meng B, Kirsch DG, Lyerly HK, Dewhirst MW, Fecci P, Walder H, Spector NL, 2016. X-ray psoralen activated cancer therapy (X-PACT). PloS One 11, e0162078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvathaneni S, Lu X, Chaudhary R, Lal A, Madhusudan S, Sharma S, 2017. RECQ1 expression is upregulated in response to DNA damage and in a p53-dependent manner. Oncotarget 8, 75924–75942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Qi XM, Miao LL, Ren J, 2017. 1,2:5,6-dianhydrogalactitol inhibits human glioma cell growth in vivo and in vitro by arresting the cell cycle at G2/M phase. Acta Pharmacol. Sin. 38, 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popuri V, Tadokoro T, Croteau DL, Bohr VA, 2013. Human RECQL5: guarding the crossroads of DNA replication and transcription and providing backup capability. Crit. Rev. Biochem. Mol. Biol. 48, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NE, Johnson KM, Wang J, Fekry MI, Wang Y, Gates KS, 2014. Interstrand DNA-DNA cross-link formation between adenine residues and abasic sites in duplex DNA. J. Am. Chem. Soc. 136, 3483–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NE, Li L, Gates KS, Wang Y, 2017. Replication and repair of a reduced 2deoxyguanosine-abasic site interstrand cross-link in human cells. Nucleic Acids Res. 45, 6486–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC, 2008. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 134, 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieben WK Jr., Coulombe RA Jr., 2004. DNA cross-linking by dehydromonocrotaline lacks apparent base sequence preference. Toxicol. Sci. 82, 497–503. [DOI] [PubMed] [Google Scholar]
- Rogers CM, Bochman ML, 2017. Saccharomyces cerevisiae Hrq1 helicase activity is affected by the sequence but not the length of single-stranded DNA. Biochem. Biophys. Res. Commun. 486, 1116–1121. [DOI] [PubMed] [Google Scholar]
- Rogers CM, Wang JC, Noguchi H, Imasaki T, Takagi Y, Bochman ML, 2017. Yeast Hrq1 shares structural and functional homology with the disease-linked human RecQ4 helicase. Nucleic Acids Res. 45, 5217–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers CM, Lee CY, Parkins S, Buehler NJ, Wenzel S, Martinez-Marquez F, Takagi Y, Myong S, Bochman ML, 2020. The yeast Hrq1 helicase stimulates Pso2 translesion nuclease activity and thereby promotes DNA inter-strand cross-link repair. J. Biol. Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrig S, Dorn A, Enderle J, Schindele A, Herrmann NJ, Knoll A, Puchta H, 2018. The RecQ-like helicase HRQ1 is involved in DNA crosslink repair in Arabidopsis in a common pathway with the Fanconi anemia-associated nuclease FAN1 and the postreplicative repair ATPase RAD5A. New Phytol. 218, 1478–1490. [DOI] [PubMed] [Google Scholar]
- Rudd GN, Hartley JA, Souhami RL, 1995. Persistence of cisplatin-induced DNA interstrand crosslinking in peripheral blood mononuclear cells from elderly and young individuals. Canc. Chemother. Pharmacol. 35, 323–326. [DOI] [PubMed] [Google Scholar]
- Rycenga HB, Long DT, 2018. The evolving role of DNA inter-strand crosslinks in chemotherapy. Curr. Opin. Pharmacol. 41, 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sami F, Sharma S, 2013. Probing genome maintenance functions of human RECQ1. Comput. Struct. Biotechnol. J 6, e201303014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Davies AA, Ulrich HD, McHugh PJ, 2006. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J. 25, 1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, Desai R, Escarpe PA, Hampl J, Laysang A, Liu D, Lopez-Molina J, Milton M, Park A, Pysz MA, Shao H, Slingerland B, Torgov M, Williams SA, Foord O, Howard P, Jassem J, Badzio A, Czapiewski P, Harpole DH, Dowlati A, Massion PP, Travis WD, Pietanza MC, Poirier JT, Rudin CM, Stull RA, Dylla SJ, 2015. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 7, 302ra136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semlow DR, Zhang J, Budzowska M, Drohat AC, Walter JC, 2016. Replication-dependent unhooking of DNA interstrand cross-links by the NEIL3 glycosylase. Cell 167, 498–511 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamanna RA, Singh DK, Lu H, Mirey G, Keijzers G, Salles B, Croteau DL, Bohr VA, 2014. RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis 35, 2415–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Brosh RM Jr., 2008. Unique and important consequences of RECQ1 deficiency in mammalian cells. Cell Cycle 7, 989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Karmakar P, Aamann M, Schurman SH, May A, Croteau DL, Burks L, Plon SE, Bohr VA, 2010. The involvement of human RECQL4 in DNA double-strand break repair. Aging Cell 9, 358–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, Kumar S, Prasad DN, Bhardwaj TR, 2018. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: historic to future perspectives. Eur. J. Med. Chem. 151, 401–433. [DOI] [PubMed] [Google Scholar]
- Smeaton MB, Hlavin EM, McGregor Mason T, Noronha AM, Wilds CJ, Miller PS, 2008. Distortion-dependent unhooking of interstrand cross-links in mammalian cell extracts. Biochemistry 47, 9920–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LW, Culvenor CC, 1981. Plant sources of hepatotoxic pyrrolizidine alkaloids. J. Nat. Prod. 44, 129–152. [DOI] [PubMed] [Google Scholar]
- Spielmann HP, Dwyer TJ, Hearst JE, Wemmer DE, 1995. Solution structures of psoralen monoadducted and cross-linked DNA oligomers by NMR spectroscopy and restrained molecular dynamics. Biochemistry 34, 12937–12953. [PubMed] [Google Scholar]
- Stone MP, Cho YJ, Huang H, Kim HY, Kozekov ID, Kozekova A, Wang H, Minko IG, Lloyd RS, Harris TM, Rizzo CJ, 2008. Interstrand DNA cross-links induced by alpha,beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem. Res. 41, 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Meador JA, Calaf GM, Proietti De-Santis L, Zhao Y, Bohr VA, Balajee AS, 2010. Human RecQL4 helicase plays critical roles in prostate carcinogenesis. Canc. Res. 70, 9207–9217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiefenbach T, Junop M, 2012. Pso2 (SNM1) is a DNA structure-specific endonuclease. Nucleic Acids Res. 40, 2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolley DA, Hargreave TB, Smith PH, Williams JL, Grigor KM, Parmar MK, Freedman LS, Uscinska BM, 1988. Effect of intravesical mitomycin C on recurrence of newly diagnosed superficial bladder cancer: interim report from the medical research council subgroup on superficial bladder cancer (urological cancer working party). Br. Med. J. 296, 1759–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasz M, 1995. Mitomycin C: small, fast and deadly (but very selective). Chem. Biol. 2, 575–579. [DOI] [PubMed] [Google Scholar]
- Travis LB, Holowaty EJ, Bergfeldt K, Lynch CF, Kohler BA, Wiklund T, Curtis RE, Hall P, Andersson M, Pukkala E, Sturgeon J, Stovall M, 1999. Risk of leukemia after platinum-based chemotherapy for ovarian cancer. N. Engl. J. Med. 340, 351–357. [DOI] [PubMed] [Google Scholar]
- Tubbs A, Nussenzweig A, 2017. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitolo U, Trneny M, Belada D, Burke JM, Carella AM, Chua N, Abrisqueta P, Demeter J, Flinn I, Hong X, Kim WS, Pinto A, Shi YK, Tatsumi Y, Oestergaard MZ, Wenger M, Fingerle-Rowson G, Catalani O, Nielsen T, Martelli M, Sehn LH, 2017. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J. Clin. Oncol. 35, 3529–3537. [DOI] [PubMed] [Google Scholar]
- Wakaki S, Marumo H, Tomioka K, Shimizu G, Kato E, Kamada H, Kudo S, Fujimoto Y, 1958. Isolation of new fractions of antitumor mitomycins. Antibiot. Chemother. 8, 228–240. [PubMed] [Google Scholar]
- Walter J, Sun L, Newport J, 1998. Regulated chromosomal DNA replication in the absence of a nucleus. Mol. Cell. 1, 519–529. [DOI] [PubMed] [Google Scholar]
- Wang AT, Sengerova B, Cattell E, Inagawa T, Hartley JM, Kiakos K, BurgessBrown NA, Swift LP, Enzlin JH, Schofield CJ, Gileadi O, Hartley JA, McHugh PJ, 2011. Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 25, 1859–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward TA, Dudasova Z, Sarkar S, Bhide MR, Vlasakova D, Chovanec M, McHugh PJ, 2012. Components of a Fanconi-like pathway control Pso2independent DNA interstrand crosslink repair in yeast. PLoS Genet. 8, e1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren AJ, Maccubbin AE, Hamilton JW, 1998. Detection of mitomycin C-DNA adducts in vivo by 32P-postlabeling: time course for formation and removal of adducts and biochemical modulation. Canc. Res. 58, 453–461. [PubMed] [Google Scholar]
- Weidner MF, Sigurdsson ST, Hopkins PB, 1990. Sequence preferences of DNA interstrand cross-linking agents: dG-to-dG cross-linking at 5’-CG by structurally simplified analogues of mitomycin C. Biochemistry 29, 9225–9233. [DOI] [PubMed] [Google Scholar]
- Williams LD, Shaw BR, 1987. Protonated base pairs explain the ambiguous pairing properties of O6-methylguanine. Proc. Natl. Acad. Sci. U. S. A. 84, 1779–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams HL, Gottesman ME, Gautier J, 2012. Replication-independent repair of DNA interstrand crosslinks. Mol. Cell. 47, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams HL, Gottesman ME, Gautier J, 2013. The differences between ICL repair during and outside of S phase. Trends Biochem. Sci. 38, 386–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf M, Zehentmayr F, Niyazi M, Ganswindt U, Haimerl W, Schmidt M, Holzel D, Belka C, 2010. Long-term outcome of mitomycin C- and 5-FU-based primary radiochemotherapy for esophageal cancer. Strahlenther. Onkol. 186, 374–381. [DOI] [PubMed] [Google Scholar]
- Wolter R, Siede W, Brendel M, 1996. Regulation of SNM1, an inducible Saccharomyces cerevisiae gene required for repair of DNA cross-links. Mol. Gen. Genet. 250, 162–168. [DOI] [PubMed] [Google Scholar]
- Wu RA, Semlow DR, Kamimae-Lanning AN, Kochenova OV, Chistol G, Hodskinson MR, Amunugama R, Sparks JL, Wang M, Deng L, Mimoso CA, Low E, Patel KJ, Walter JC, 2019. TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 567, 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne P, Newton C, Ledermann JA, Olaitan A, Mould TA, Hartley JA, 2007. Enhanced repair of DNA interstrand crosslinking in ovarian cancer cells from patients following treatment with platinum-based chemotherapy. Br. J. Canc. 97, 927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Kim CS, Healy AR, Wernke KM, Wang Z, Frischling MC, Shine EE, Wang W, Herzon SB, Crawford JM, 2019. Structure elucidation of colibactin and its DNA cross-links. Science 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovleva L, Shuman S, 2012. Mycobacterium smegmatis SftH exemplifies a distinctive clade of superfamily II DNA-dependent ATPases with 3’ to 5’ translocase and helicase activities. Nucleic Acids Res. 40, 7465–7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Watanabe S, Okamoto K, Arimoto S, Takahashi E, Negishi K, Negishi T, 2019. Chloroethylating anticancer drug-induced mutagenesis and its repair in Escherichia coli, 11 Gene Environ. 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Price NE, Johnson KM, Gates KS, 2015. Characterization of interstrand DNA-DNA cross-links derived from abasic sites using bacteriophage varphi 29 DNA polymerase. Biochemistry 54, 4259–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai B, Steino A, Bacha J, Brown D, Daugaard M, 2018. Dianhydrogalactitol induces replication-dependent DNA damage in tumor cells preferentially resolved by homologous recombination. Cell Death Dis. 9, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Walter JC, 2014. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 19, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wu MX, 2018. A clinical review of phototherapy for psoriasis. Laser Med. Sci. 33, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dewar JM, Budzowska M, Motnenko A, Cohn MA, Walter JC, 2015. DNA interstrand cross-link repair requires replication-fork convergence. Nat. Struct. Mol. Biol. 22, 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]