

Abstract
The epidermal growth factor receptor (EGFR) family is a class of receptor tyrosine kinase playing a central role in carcinogenesis and cancer progression. The members of this family, particularly EGFR and human epidermal growth factor receptor 2 (HER2), are the most extensively studied drug targets for malignancy. Today, numerous tyrosine kinase inhibitors targeting EGFR family have been developed to combat non-small-cell lung cancer and breast cancer. However, severe gastrointestinal (GI) toxicity leading to dose reduction and treatment discontinuation hampers the therapeutic outcome of EGFR inhibitors. Diarrhea is one of the most frequent GI side effects, especially when it comes to second-generation EGFR inhibitors. Enterocytes apoptosis and increased inflammation accompany with many oral EGFR inhibitors. Loperamide and budesonide are the first-line treatment to manage such adverse effects. However, current prophylaxis and management are all empirical interventions to relieve the symptom. They do not specifically target the toxicological mechanism of EGFR inhibitors. Hereby, those anti-diarrhea agents do not work well when used in cancer patients experiencing EGFR inhibitor-induced diarrhea. On the other hand, the toxicological mechanism of EGFR inhibitor-induced diarrhea is poorly understood. Thus, determining the mechanism behind such diarrhea is urgently in need for developing genuinely effective anti-diarrhea agents. This review aims to call attention to EGFR inhibitor-induced diarrhea, a highly occurring and devastating cancer drug toxicity.
Keywords: epidermal growth factor receptor, tyrosine kinase inhibitors, gastrointestinal toxicity, diarrhea
Introduction
Epidermal growth factor receptor (EGFR) is fundamentally important in cell proliferation and differentiation and is one of the most extensively studied drug targets for many cancers. In 2004 and 2009, FDA approved first-generation EGFR tyrosine kinase inhibitors (TKIs), gefitinib and erlotinib, for patients having EGFR mutation-positive non-small-cell lung cancer (NSCLC). However, many responsive patients develop T790M-driven gatekeeper drug resistance after 12 months of treatment [1]. To address this resistance, second-generation EGFR TKIs were developed, like afatinib and dacomtinib, to treat NSCLC patients harboring T790M. Meanwhile, lapatinib and neratinib were approved for human epidermal growth factor receptor 2 (HER2)-positive breast cancer [2]. However, gastrointestinal (GI) toxicities are associated with second-generation EGFR TKIs [3] because of wide-spectrum inhibitory effect on both mutant- and wild-type EGFR. Currently, third-generation EGFR TKIs are emerging to selectively target T790M mutants while sparing wild-type EGFR. Nevertheless, a new mechanism of resistance toward third-generation TKIs was recently discovered, that is, the EGFR C797S mutation [1, 4]. Aiming at targeting C797S mutant, fourth-generation EGFR TKIs are now under development.
To date, many clinical trials have revealed that EGFR TKIs, especially second-generation inhibitors, cause remarkable GI toxicities such as diarrhea, vomiting, and nausea. In a pooled analysis, diarrhea occurred to over half of patients receiving EGFR TKIs-based treatment. Diarrhea happened to 51% of patients having lapatinib monotherapy and to 65% of patients having lapatinib plus capecitabine [5]. Cancer drug-induced diarrhea also causes considerable economic loss. Hogsett et al. [6] suggested that additional costs up to $25 000 (USD) per therapy cycle might happen. Such costs are due to increased risk of mucositis, prolonged hospital stays, and additional supportive care. Thus, the mechanism of EGFR TKIs-induced diarrhea needs to be better understood. Effective assessment, prevention, and management of such GI toxicity should be established.
In this review, we summarize the first three generations of EGFR TKIs. We focus on the differences in the mechanism of action between each generation and its relevance to GI toxicity. We review the primary toxicological mechanisms underlying EGFR TKIs-induced diarrhea. Finally, we discuss contemporary non-medical supportive care and medication for EGFR TKIs-induced diarrhea.
EGFR inhibitors causing diarrhea
A large portion of human carcinoma is caused by the excessive activation of human EGFR family. While staying inactive, all EGFR members remain as monomers. Once one member is activated by ligand, it will couple with another EGFR member to trigger downstream signaling. Without forming dimer with another EGFR member, single EGFR member cannot initiate downstream transduction (Fig. 1). Such dimerization stimulates several oncogenes, including mitogen-activated protein kinase (MAPK), phosphoinositol 3-kinase/protein kinase B (PI3K/PKB), and janus kinase/signal transducer and activator of transcription (JAK/STAT). Eventually, those events lead to increased cellular motility, proliferation, and invasion [7]. Based on this conceptual framework, EGFR family proteins are identified as the first and foremost target in treating many types of carcinoma from NSCLC, breast cancer, head and neck cancers, and pancreatic cancer to renal cancer. Up to date, three generations of EGFR inhibitors have been posted to market for malignant carcinoma (Fig. 2).
Figure 1.
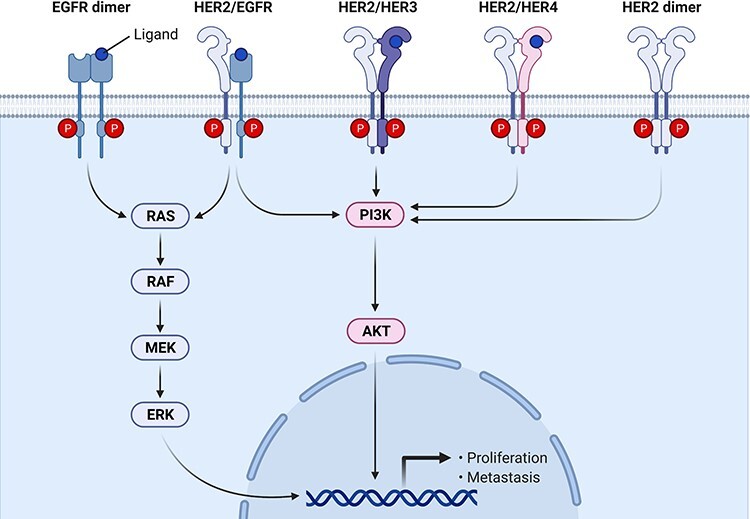
EGFR signaling in cancer proliferation.
Figure 2.
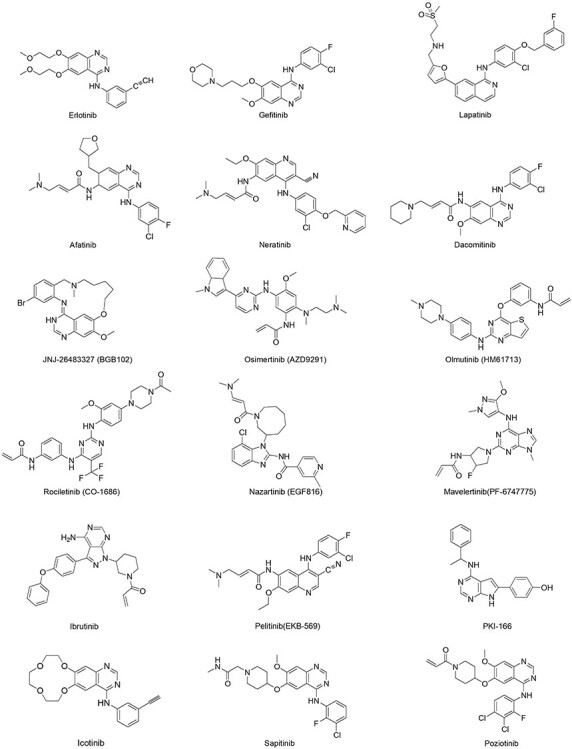
the EGFR TKIs causing severe diarrhea.
First-generation EGFR inhibitors: reversible inhibition
The most frequent mutations in EGFR encoding gene are L858R and Del19 (exon 19 deletions between amino acids 746 and 750), resulting in most of the EGFR-driven carcinogenesis [8]. Patients with these somatic mutants are treated with first-generation EGFR inhibitors, such as gefitinib and erlotinib. First-generation EGFR inhibitors usually have promising response rates for the first 11–14 months [9]. First-generation EGFR inhibitors display potency by blocking the adenosine triphosphate (ATP) binding site of EGFR kinases. Generally, the EGFR inhibitors of this generation are anilinoquinazoline derivatives, a class of ATP homologous [10]. This similarity allows them to compete for the ATP-binding domain of protein kinases to prevent the activation of EGFR downstream signaling, which ultimately suppresses cancer.
However, a big portion of patients develop resistance to such competitive EGFR inhibitors due to a secondary EGFR kinase domain mutation, T790M. T790M restores EGFR-dependent signaling [11]. The mechanism of T790M-driven resistance has not been fully illustrated but is believed to consist of three actions. T790 mutant exerts increased ATP binding affinity. Then, it generates steric clash between Met790 gatekeeper side chain and the aniline moiety of first-generation EGFR inhibitors, hindering the drugs from inserting into kinase back pocket. Besides, such mutation changes the conformational dynamics of EGFR catalytic domain [12, 13].
Second-generation EGFR inhibitors: irreversible inhibition and multiple targeting
The second-generation EGFR inhibitors, irreversible inhibitors, are developed to resolve the resistance to first-generation inhibitors. Irreversible inhibitors have advantages over reversible compounds since they achieve complete and sustained target engagement even with a high concentration of the endogenous ligand, ATP. It requires the physical turnover of targeted protein to restore suppressed signaling [14]. Indeed, irreversible inhibitors of EGFR family protein, like dacomitinib and afatinib, demonstrate increased potency against EGFR oncogenic variants such as EGFR L858R/T790M [13]. The main drawback of second-generation EGFR inhibitors is that they potently block wild-type EGFR too and cause epithelium-based toxicity such as diarrhea [15].
In addition to irreversible inhibition, another signature feature of second-generation EGFR inhibitor is multi-targeting, like lapatinib and neratinib. This class of compound blocks more than one EGFR family member, so it is named as pan-HER inhibitor. It is known that cross-activation between EGFR family members generates complementary cascades. This system is one of the main factors jeopardizing the clinical benefit of first-generation EGFR inhibitors and causing cancer recurring [16, 17]. The strength of multi-targeting is disrupting such crosstalk signaling, but it makes normal epithelium more vulnerable to toxicity as well. One example is that Pifzer suspended the development of canertinib (CI-1033), a pan-HER inhibitor, due to its uncontrollable GI toxicities in Phase I trial [18].
Third-generation EGFR inhibitors: targeting T790M mutant
The third-generation EGFR inhibitors selectively target T790M mutants to tackle T790M-driven resistance. Third-generation inhibitors bind covalently to Cys797 but spare wild-type EGFR. Increasing third-generation EGFR inhibitors have advanced into clinical trials or got approval, such as osimertinib (AZD9291), rociletinib (CO1686), olmutinib (HM617l3), zazartinib (EGF816), and naquotinib (ASP8273) [19]. The Phase II trial of osmertinib in NSCLC patients having T790M showed that the incidence of diarrhea significantly decreased. The Grade 2 diarrhea in this trial is 5%, while Grade 3 diarrhea is less than 1%, which is impressively better than the second-generation inhibitors [20].
Clinical incidence of EGFR inhibitors-induced diarrhea
In cancer patients treated with EGFR inhibitors, diarrhea is the second most common adverse events, affecting up to 95% of them. The occurrence of diarrhea has been suggested to predict tumor response. Diarrhea of Grade 3 or higher occurs in around 30% of patients taking EGFR inhibitors. Diarrhea typically starts as early as 2–3 days after doing EGFR inhibitors. As for most EGFR inhibitors, the severity of diarrhea is dose dependent. Nevertheless, high risk of diarrhea compromises the therapeutic outcome of EGFR inhibitors (Table 1). The current supportive cares are all empirical practices. In the field of cancer supportive care, empirical intervention is distinct from target-oriented intervention. Loperamide and budesonide are used because they alleviate diarrhea in other scenarios, like inflammatory bowel disease. They are not specifically developed to target EGFR-induced diarrhea. Thus, their effectiveness is not always satisfactory when tested in cancer patients taking EFGR inhibitors.
Table 1.
GI adverse effect associated with EGFR inhibitors
| Drug | Regimen | Reviewed trial | Diarrhea | Stomatitis | Nausea | Vomiting | Ref. | ||||
| All grades | ≥3 | All grades | ≥3 | All grades | ≥3 | All grades | ≥3 | ||||
| Erlotinib (OSI-774) | Patients received erlotinib (150 mg/d) or placebo combined with up to six 21-day cycles of chemotherapy (gemcitabine 1250 mg/m2 on Days 1 and 8 and cisplatin 80 mg/m2 on Day 1) | Phase III | NA | 6% | NA | NA | NA | 6% | NA | 7% | [62] |
| Gefitinib | Patients received paclitaxel 225 mg/m2 and carboplatin area under concentration/time curve of 6 mg/min/ml (Day 1 every 3 weeks) plus gefitinib 500 mg/d or placebo | Phase III | 69.3% | 25.4% | NA | NA | 18.7% | 4.1% | 12.9% | 2.9% | [63] |
| Icotinib (BPI-2009H) | Patients were randomly assigned (1:1) to receive icotinib (125 mg, three times per day) until disease progression or unacceptable toxicity | Phase III | 22% | 0% | NA | NA | 4% | <1% | 5% | 0% | [64] |
| Lapatinib (GW572016) | The starting dose of single agent lapatinib was 750 mg twice daily. Dose delays of up to 2 weeks and two dose reductions, first to 1500 mg once daily and second to 1250 mg, were allowed for toxicities | Phase III | <66% | <14% | NA | NA | 27% | 3% | 24% | 4% | [65] |
| Afatinib (BIBW2992) | Patients randomized to afatinib plus paclitaxel received 40 mg daily and 80 mg/m2 weekly, respectively | Phase III | 53.8% | 12.1% | 9.8% | 1.5% | 17.4% | 1.5% | 15.9% | 2.3% | [66] |
| Neratinib | Oral neratinib 240 mg was administered once per day without breaks, and cycle duration was 28 days | Phase III | 41% | 26% | NA | NA | 13% | 5% | 13% | 5% | [22] |
| Sapitinib (AZD8931) | Patients received a single oral dose of AZD8931 on Day 1 (D1), followed by a 4-day observation period to allow sufficient time to determine single dose | Phase III | 75% | 25% | 29% | NA | 14% | NA | 14% | NA | [67] |
| Dacomitinib | A trial of dacomitinib as initial systemic therapy | Phase III | 93% | 15% | 40% | 4% | 26% | 3% | 15% | 1% | [30] |
| Poziotinib | Oral administration | Phase II | 92% | 10% | 59% | 18% | 8% | 0% | 5% | 3% | [68] |
| JNJ-26483327 (BGB102) | Oral solution for doses p1200 mg, or capsules of 50, 100, or 300 mg for doses X1500 mg. Medication was taken in combination with food BID with 12-h intervals. A cycle was defined as 28 days of treatment | Phase I | 63.2% | 0% | NA | NA | 68.4% | 0% | 52.6% | 0% | [69] |
| Osimertinib (AZD9291) | Osimertinib was given 80 mg orally once daily | Phase III | <34% | <1% | 11% | 0% | NA | NA | NA | NA | [70] |
| Olmutinib (HM61713) | 800 mg once daily | Phase III | 59% | 0% | NA | NA | 39% | 0% | NA | NA | [71] |
| Rociletinib (CO-1686) | 1000 mg twice daily | Phase I/II | 33% | NA | NA | NA | 50% | NA | NA | NA | [72] |
| Ibrutinib | Patients received 28-day cycles of once-daily ibrutinib 420 mg together with rituximab (375 mg/m2, intravenously, every week during Cycle 1, then once per cycle until Cycle 6) | Phase II | 26% | NA | NA | NA | 38% | NA | 38% | NA | [73] |
Second-generation inhibitors are reported to cause dose-limiting diarrhea. In NSCLC patients treated with second-generation EGFR inhibitors, the incidence of diarrhea of all grades was 100%, while the incidence of Grade 3 was 23% [21]. In a comparative Phase III study, the incidence of diarrhea in afatinib arm was even higher than the arm of cisplatin plus pemetrexed. About 95.2% of the patients taking afatinib had diarrhea, and diarrhea of Grade 3 was 14%. Meanwhile, the occurrence of diarrhea was 15% in the arm of cisplatin plus pemetrexed and no Grade 3 diarrhea was recorded [3]. Same issue happens to pan-HER inhibitors used for breast cancer. ExteNET, the Phase III trial of neratinib in HER2-positive breast cancer, showed that 96% patients taking neratinib had diarrhea, while the incidence of Grade 3 diarrhea was 41% [22].
Toxicological mechanisms of EGFR inhibitors-induced diarrhea
Although diarrhea is a highly occurring side effect of EGFR inhibitors, the mechanism underlying such toxicity remains unclear. Also, the inter-individual variability in diarrhea incidence is high. It is uncertain whether the enterocytes or enzymes in the GI tract are sensitive to EGFR inhibitors. Typically, drug-associated diarrhea is attributed to three principle reasons: (i) excessive hypertonic substances in lumen, (ii) damage to transporters that control electrolytes flux across enterocyte membrane, and (iii) increased gut motility. Further studies to support the above three hypotheses are expected to be done. Generally, drug-induced diarrhea is driven by multiple mechanisms.
EGFR regulate epithelium regeneration and permeability
It is known that the inhibition of EGFR signaling results in reduced growth and barrier leakage in intestinal epithelium. This leads to mucosal atrophy-associated GI toxicity [23]. GI tract is one of the most rapidly proliferating organs in the body. Berlanga-Acosta et al. [24] proved that, in rodent model, EGF promoted enterocytes growth and altered crypt fission. Crypt fission is to maintain the absorption function of gut barrier. Inhibition of crypt fission will reduce the water absorption in GI tract, leading to diarrhea. In rats’ colon, EGF is a potent stimulus for crypt fission [24]. Besides, it was found that multiple EGFR ligands were induced in Drosophila in response to damage in intestinal epithelium. Increase in those ligands activated the intestinal stem cells. Activation of EGFR signaling promoted epithelium regeneration in Drosophila, which eventually maintained intestine homeostasis. Jiang et al. [25] showed that EGFR-deficit intestinal stem cells could not support intestinal epithelium regeneration after bowel infection.
In addition to enterocyte proliferation, EGFR regulates tight junction and permeability in gut. Raimondi et al. demonstrated that adding cholic acid, deoxycholic acid (DCA), and chenodeoxycholic acid (CDCA) decreased transepithelial electrical resistance and increased dextran flux in Caco-2 cell monolayer. Co-incubation of CDCA or DCA with EGF abolished such effect. Those findings proved that EGFR signaling was essential to support the tight junction of Caco-2 monolayer [26]. Basuroy et al. [27] reported that EGF suppressed H2O2-induced gut leakage by blocking Tyr-phosphorylation, Thr-dephosphorylation and by redistributing zonula occludin-1 (ZO-1). In their study, EGF also protected epithelium from cytoskeleton disruption [27].
EGFR inhibitors induce apoptosis and cytokine secretion
Recently, Hong et al. reported that erlotinib caused barrier dysfunction in rat small intestine epithelial cells (IEC-6) by increasing permeability and by down-regulating E-cadherin. Erlotinib induced endoplasmic reticulum stress (ER stress) in both IEC-6 and human colon epithelial cells in a concentration-dependent manner [28]. Hong et al. showed that knockdown of C/EBP homologous protein protected IEC-6 cells from erlotinib-induced apoptosis and E-cadherin decrease [28]. Such findings implied that ER stress-mediated injury might contribute to erlotinib-induced diarrhea. Another parallel study indicated that gefitinib and icotinib arrested cell cycle at G0/G1 phase by increasing cyclin D1 and p27 in IEC-6 cells [29]. In addition, gefitinib and icotinib reduced cell adhesion molecules while increasing IL-6 and IL-25. Those EGFR inhibitors all triggered ER stress response by activating protein kinase R-like ER kinase (PERK) pathway and by increasing serine/threonine-protein kinase/endoribonuclease inositol-requiring enzyme 1α (IRE1α)-mediated XBP1 splicing [28] (Fig. 3).
Figure 3.
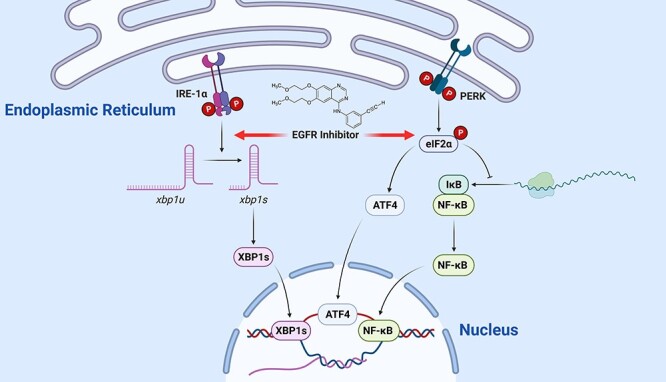
EGFR inhibitor causes inflammation in epithelium by triggering ER stress; following ER stress, PERK activates NF-κB via inhibiting IκB protein translation; this causes the release of NF-κB protein, which then carries out its role as transcription factor to promote inflammation; in addition, ER stress activates IRE-1α to initiate inflammatory response and apoptosis via splicing XBP1 mRNA; the spliced form of XBP1 (XBP1s) translocates into the nucleus and functions as transcription factor; EGFR inhibitors stimulate IRE-1α-mediated XBP1 slicing and PERK-mediated NF-κB activation; abbreviations: eIF2α, eukaryotic initiation factor 2α; IκB protein, inhibitor of kappa B protein; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; IRE-1α, serine/threonine-protein kinase/IRE1α; XBP1, X-box binding protein 1.
Lately, Van Sebille et al. [30] reported a case of dacomitinib GI toxicity. Based on their results, dacomitinib did not affect transepithelial electrical resistance or cell viability in vitro but caused diarrhea and weight loss in vivo. Dacomitinib left serious injury in distal ileum and elevated the level of monocyte chemoattractant protein 1 (MCP1) [30] in rats. In all, those studies demonstrated that EGFR inhibitors-associated diarrhea is owning to multiple mechanisms beyond inhibition of epithelium regeneration.
EGFR activation reduces chloride secretion
Today, the idea that EGFR inhibitors-associated diarrhea is due to excessive chloride secretion is drawing attention. The chloride in lumen builds up the osmotic gradient for water flowing into lumen. Chloride secretion is instrumental for keeping GI tract moist, but excessive secretion will lead to diarrhea. In intestinal epithelial cells, there are two key pathways governing chloride secretion: cAMP-dependent pathway stimulating delayed and prolonged response; and Ca2+-dependent pathway eliciting rapid and transient response [31]. It is known that EGF inhibits Ca2+-dependent chloride secretion via binding with the EGFR on basolateral membrane [32].
Barrett et al. [33] reported several EGFR agonists exerting inhibitory effect on Ca2+-dependent chloride secretion and sodium absorption in intestinal epithelia cells. Another study found that both EGF and carbachol, ligands of M3 muscarinic receptor, reduced chloride secretion in vitro [34]. Follow-up study proved that the activation of M3 muscarinic receptor suppressed chloride secretion via an EGFR-dependent manner [35] (Fig. 4). Furthermore, Barrett et al. [36] demonstrated that activation of EGFR stimulated Ras–Raf–MEK–ERK as well as PI3K–AKT–mTOR pathways to decrease Ca2+-dependent chloride secretion. According to those findings, EGFR inhibitors disrupt the inhibitory mechanism of EGFR toward chloride secretion. This results in excessive chloride resident in lumen and diarrhea. Latest studies by Duan et al. and Kim et al. [37, 38] verified this hypothesis, respectively.
Figure 4.
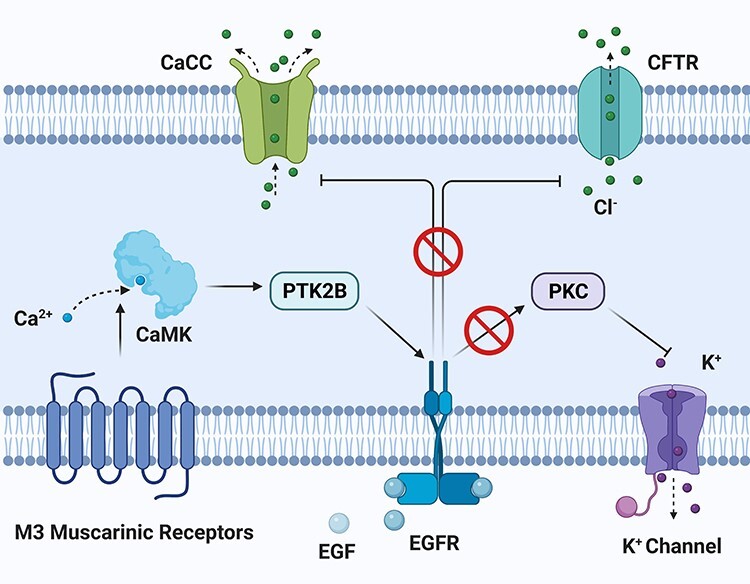
activation of EGFR suppresses Ca2+-dependent chloride secretion in epithelial cells; activation of M3 muscarinic receptor transactivates EGFR and eventually targets CaCC on apical side; the whole signal transduction involves CaMK and PTK2B; direct activation of EGFR by EGF inhibits chloride secretion on lumen side and simultaneously stimulates potassium channel on basal side; EGFR inhibitors are shown to block the inhibitory pathway from EGFR to chloride channel; abbreviations: CaMK, Ca2+/calmodulin-dependent protein kinase; PTK2B, protein tyrosine kinase 2 beta; CaCC, Ca2+-activated chloride channel; CFTR, cystic fibrosis transmembrane conductance regulator; PKC, protein kinase C.
Bowen et al. suggested that dacomitinib-induced diarrhea was mainly due to increased chloride secretion. Bowen et al. showed that crofelemer, a natural product used for diarrhea, suppressed dacomitinib-induced chloride secretion in vitro. However, when tested ex vivo, crofelemer did not inhibit dacomitinib-induced chloride secretion in ileum and colon tissues. Pharmacokinetics study revealed that crofelemer did not change dacomitinib bioavailability. Bowen et al. [39] conducted a large-scale animal experiment to investigate the mechanism of lapatinib-induced diarrhea. Their study showed that lapatinib caused diarrhea symptom but gave no histological damage in the gut. However, the serum level of chloride was decreased in lapatinib group. It suggested that lapatinib-induced diarrhea might be driven chloride loss in GI tract [40].
Saturation of drug transporters
When EGFR inhibitor and other therapeutic agents share the same efflux transporters or enzymes, there will be drug accumulation in the gut, leading to GI toxicity [41]. First-generation EGFR inhibitors, both gefitinib and erlotinib, show affinity with the ATP-binding cassette (ABC) transporter. In vitro experiments showed that gefitinib reversed ABCG2-mediated resistance at high concentration [42]. In clinical setting, patients carrying ABCG2 421C/A polymorphism had higher gefitinib exposure and more diarrhea episodes compared to those carrying wild-type ABCG2 [43]. In mice studies, the absence of ABCB1 and ABCG2 increased the oral bioavailability of erlotinib [44]. It is reported that ABCG2-15622C/T and 1143C/T polymorphisms increased the AUC and Cmax of erlotinib pharmacokinetics profile [45].
Likewise, second-generation EGFR inhibitors, lapatinib and neratinib, exert an inhibitory effect upon ABC transporters. Lapatinib was reported to be both the substrate and inhibitor of ABC transporters. It reversed ABCB1- and ABCG2-driven resistance in cancer cells [46]. Perry et al. [47] reported that lapatinib increased SN-38 intracellular accumulation due to the inhibition of ABCG2. Zhao et al. found that neratinib reversed ABCB1-mediated resistance in vitro and in vivo. Besides, neratinib increased the accumulation of doxorubicin and rhodamine in ABCB1-overexpressing cell lines. It is also known that neratinib suppressed the ATPase activity of ABCB1 [48]. More studies in humans are expected to clarify the role of ABC transporters in the disposition, toxicity of EGFR inhibitors.
Current management
A series of studies have established the causality between EGFR inhibitors-induced diarrhea and excess chloride secretion. In fact, such diarrhea is facilitated by multiple factors from increase in gut motility, damage in epithelium to altered gut microbiome [49]. The complexity of toxicological mechanism is the obstacle for personalized management. Very few clinical studies have been conducted to explore molecular mechanism. Today, very limited options of prophylaxis and management are available in clinical setting. Current guidelines for anti-diarrhea management mainly aim to control dehydration [50], but those managements are not effective in many cancer patients.
Non-medical management
Non-medical supportive care is a critical part to attenuate chemotherapy-induced diarrhea, such as changes in diet and nutrition supplements. Patients should drink three or more liters of clear fluid per day. This quantity should be electrolyte-containing fluids, such as sport drinks, broth, gelatin, decaffeinated tea, and decarbonated soft drinks [51]. After each bowel movement, a barrier cream or ointment, like petroleum jelly, can be applied to the anal area to prevent irritation [51]. The anal area should also be examined for red or broken skin [51]. Grades 3 and 4 diarrhea in some patients require dose reduction or treatment cycle interruption. The dose reduction varies depending on the grade of diarrhea. But it may compromise the drug efficacy. When non-medical intervention cannot alleviate the side effect, anti-diarrhea medication must chime in.
Medication prolonging gut transient time
High-dose loperamide is the first-line treatment for many types of diarrhea. Typically, an initial dose is 4 mg. Follow-up dose of 2 mg every 2 h is given until no loose motions last for 12 h [52]. As an agonist acting on the μ-opioid receptors in colon, loperamide prolongs the bowel transit time of food, decreases feces volume, and diminishes the loss of fluid. Budesonide or derivatives of codeine are suggested for patients who are refractory to loperamide [53]. Budesonide is orally administered, topically active steroid. Its potency to restore mucosa function is because of the inhibition of mucosal prostaglandins [53]. Oral budesonide at a dose of 9 mg once daily for 3–5 days may be effective for the treatment of loperamide-resistant diarrhea [54]. Budesonide at a dose of 3 mg twice daily was found to decrease the number of episodes of diarrhea [54]. Diophenoxylate and atropine are other alternatives to loperamide. Although loperamide and budesonide are the most widely used anti-diarrhea drugs, their effectiveness is not satisfying when it comes to EGFR-induced diarrhea.
Medication reducing intestinal secretion
Octreotide is used to control bowel motility and water flow. Octreotide suppresses the response of gut to gonadotropin-releasing hormone [55]. It decreases splanchnic blood flow as well [55]. It inhibits the release of serotonin, gastrin, vasoactive intestinal peptide, secretin, motilin, and pancreatic polypeptide [55]. Octreotide can be given subcutaneously starting at 100–150 μg. Doses of octreotide ranges from 50 μg twice daily to 2500 μg three times daily based on the diarrhea severity [55]. The currently recommended dose is 100–150 μg three times per day [55]. The dose may be escalated to 500 μg three times daily when diarrhea is persistent [55]. As plenty of evidences suggest that chloride secretion plays an important role in EGFR inhibitors-induced diarrhea, octreotide should be considered as an option to contain this toxicity.
Probiotics
Probiotics, such as Lactobacillus acidophilus and Bifidobacterium spp., competitively block the invasion of pathogens into intestinal mucosa [56]. Probiotics activate immunity by stimulating the release of cytokines [56]. Although they have been used in infectious diarrhea [57], there is very limited study exploring the effect of Lactobacillus plantarum on drug-induced diarrhea [58]. How EGFR inhibitors change the structure of microbiome and how imbalance of microbiome leads to diarrhea need to be answered. It should be noted that cancer patients with weakened immune systems may confront uncontrollable infection when receiving probiotics. Thus, evidence validating the safety of probiotics in cancer patients is needed before we set up the clinical trial to test its anti-diarrhea efficacy.
Case studies of EGFR inhibitors-induced diarrhea
As for afatinib-induced diarrhea, Yang et al. suggested that loperamide treatment should start right after the diarrhea episode until there was no bowel movement. If Grade 2 diarrhea lasts for more than 48 h despite anti-diarrhea treatment, afatinib interruption is recommended. In the event of Grades 3 and 4 diarrhea, patients should be admitted to hospitals and should receive intravenous fluid. In this case, loperamide should continue and antibiotics can be considered if neutropenia is diagnosed [59]. According to previous report, after lapatinib was administered, the onset of diarrhea would occur within 2–5 days [39]. When lapatinib-induced diarrhea persists for more than 24 h, increasing the dose of loperamide is suggested. Reducing lapatinib dose and withholding treatment should be considered for patients experiencing Grade 3 diarrhea [60]. If mild diarrhea does not stop after 24 h of high-dose loperamide, second-line agents like octreotide, budesonide, and laudanum should be taken. If the dehydration is severe, fluoroquinolone needs to be administrated. In Phase III trial of neratinib, Grade 3 diarrhea causing dose reduction was recorded [22]. Latest trial showed that adding colestipol to loperamide prophylaxis reduced diarrhea episodes in cancer patients having neratinib [61]. It is suggested that colestipol may also improve neratinib tolerability by decreasing the rate of other adverse events, including fatigue, headache, and abdominal pain [61]. But further follow-up is still necessary to validate the anti-diarrhea function of colestipol in the neratinib case [61].
Conclusion
Diarrhea is one of the most frequent adverse effects hampering the therapeutic outcome of EGFR inhibitors. The third-generation EGFR inhibitors selectively targeting T790M have been developed to spare wild-type EGFR, in hope to reduce the incidence of diarrhea. However, first- and second-generation inhibitors are still widely prescribed for cancer patients without T790M. The toxicological mechanism of EGFR inhibitors-induced diarrhea is poorly illustrated. Very limited preclinical studies have been done, not to mention the clinical trials. Irinotecan-associated diarrhea is the most extensively studied chemotherapy GI toxicity [41]. Nevertheless, irinotecan is a topoisomerase blocker. The pharmacological mechanism of topoisomerase blocker is distinct from EGFR inhibitors, so what we learn from irinotecan is hardly applicable to EGFR inhibitors (Fig. 5). Diarrhea that resulted from EGFR inhibitors is progressive and requires a prompt and effective management. However, the current guideline for this side effect is not truly effective. The knowledge gap in toxicology is the major barrier for prophylaxis development. Leveraging gut-on-a-chip, humanized mouse model and physiologically based pharmacokinetic (PBPK) modeling, more details behind EGFR-induced diarrhea can be revealed. It will help us to predict the risk of GI toxicity more accurately during new drug development. In terms of reported mechanisms, they mainly fall into three categories: inhibition of epithelium regeneration, excessive chloride secretion, and inhibition of drug transporters. But which one is the main driving force of the EGFR inhibitors-associated diarrhea remains as an open question. We suggest that excessive chloride secretion probably accounts for acute toxicity response, while suppression of epithelium regeneration is responsible for long-term damage. Inhibition of drug transporters partially answers why combinational therapy has a higher diarrhea incidence than the EGFR inhibitor monotherapy.
Figure 5.
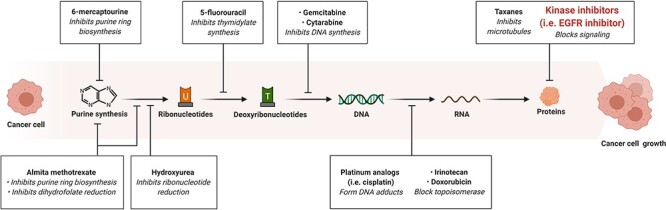
basic methods of chemotherapy.
Acknowledgements
We thank Dr Romi Ghose for offering the critiques and Ms Liye Wang’s assistance during the manuscript preparation. No funding information needs to be disclosed for this work. Figures have been created with Biorender.com.
Contributor Information
Gabriel Tao, Department of Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston, Houston, TX 77204, USA.
Pavan Kumar Chityala, Department of Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston, Houston, TX 77204, USA; Division of Pharmacotherapy and Experimental Therapeutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.
Conflict of interest statement
None declared.
References
- 1. Chen L, Fu W, Zheng L et al. Recent Progress of Small-Molecule Epidermal Growth Factor Receptor (EGFR) Inhibitors against C797S Resistance in Non-Small-Cell Lung Cancer. J Med Chem 2018;61:4290–300. [DOI] [PubMed] [Google Scholar]
- 2. Paranjpe R, Basatneh D, Tao G et al. Neratinib in HER2-Positive Breast Cancer Patients. Ann Pharmacother 2019;53:612–20. [DOI] [PubMed] [Google Scholar]
- 3. Sequist LV, Yang JCH, Yamamoto N et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327–34. [DOI] [PubMed] [Google Scholar]
- 4. Thress KS, Paweletz CP, Felip E et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 2015;21:560–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Geyer CE, Forster J, Lindquist D. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. Adv Breast Cancer 2008;5:45. [DOI] [PubMed] [Google Scholar]
- 6. Carlotto A, Hogsett L, Maiorini EM et al. The economic burden of toxicities associated with cancer treatment: Review of the literature and analysis of nausea and vomiting, diarrhoea, oral mucositis and fatigue. PharmacoEconomics 2013;31:753–66. [DOI] [PubMed] [Google Scholar]
- 7. Richard J, Sainsbury C, Needham GK et al. Epidermal-growth-factor receptor status as predictor of early recurrence of and death from breast cancer. Lancet 1987;329:1398–402. [DOI] [PubMed] [Google Scholar]
- 8. Ercan D, Choi HG, Yun CH et al. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res 2015;21:3913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Su KY, Chen HY, Li KC et al. Pretreatment Epidermal Growth Factor Receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol 2012;30:433–40. [DOI] [PubMed] [Google Scholar]
- 10. Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem 2002;277:46265–72. [DOI] [PubMed] [Google Scholar]
- 11. Kobayashi S, Boggon TJ, Dayaram T et al. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N Engl J Med 2005;352:786–92. [DOI] [PubMed] [Google Scholar]
- 12. Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009;28:S24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yun CH, Mengwasser KE, Toms AV et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li D, Ambrogio L, Shimamura T et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008;27:4702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burstein H, Burstein HJ, Sun Y et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. Artic J Clin Oncol 2010;28:1301–7. [DOI] [PubMed] [Google Scholar]
- 16. Lee CM, Shrieve DC, Zempolich KA et al. Correlation between human epidermal growth factor receptor family (EGFR, HER2, HER3, HER4), phosphorylated Akt (P-Akt), and clinical outcomes after radiation therapy in carcinoma of the cervix. Gynecol Oncol 2005;99:415–21. [DOI] [PubMed] [Google Scholar]
- 17. Zaczek A, Brandt B, Bielawski KP. The diverse signaling network of EGFR, HER2, HER3 and HER4 tyrosine kinase receptors and the consequences for therapeutic approaches. Histol Histopathol 2005;20:1005–15. [DOI] [PubMed] [Google Scholar]
- 18. Garland LL, Hidalgo M, Mendelson DS et al. A phase I clinical and pharmacokinetic study of oral CI-1033 in combination with docetaxel in patients with advanced solid tumors. Clin Cancer Res 2006;12:4274–82. [DOI] [PubMed] [Google Scholar]
- 19. Patel H, Pawara R, Ansari A, Surana S. Recent updates on third generation EGFR inhibitors and emergence of fourth generation EGFR inhibitors to combat C797S resistance. Eur J Med Chem 2017;142:32–47. [DOI] [PubMed] [Google Scholar]
- 20. Yang JCH, Ahn MJ, Kim DW et al. Osimertinib in pretreated T790M-positive advanced non-small-cell lung cancer: AURA study phase II extension component. J Clin Oncol 2017;35:1288–96. [DOI] [PubMed] [Google Scholar]
- 21. Zhang Q, Honko A, Zhou J et al. Cellular Nanosponges Inhibit SARS-CoV-2 Infectivity. Nano Lett 2020;20:5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan A, Delaloge S, Holmes FA et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2016;17:367–77. [DOI] [PubMed] [Google Scholar]
- 23. Bowen JM. Mechanisms of TKI-induced diarrhea in cancer patients. EGFR/Ras/MAPK signaling mediates adult midgut epithelial homeostasis and regeneration in drosophila. Curr Opin Support Palliat Care 2013;7:162–7. [DOI] [PubMed] [Google Scholar]
- 24. Berlanga-Acosta J, Playford RJ, Mandir N et al. Gastrointestinal cell proliferation and crypt fission are separate but complementary means of increasing tissue mass following infusion of epidermal growth factor in rats. Gut 2001;48:803–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang H, Grenley MO, Bravo MJ et al. EGFR/Ras/MAPK signaling mediates adult midgut epithelial homeostasis and regeneration in drosophila. Cell Stem Cell 2011;8:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Raimondi F, Santoro P, Barone MV et al. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol 2007;294:G906–13. doi: 10.1152/ajpgi.00043. [DOI] [PubMed] [Google Scholar]
- 27. Basuroy S, Seth A, Elias B et al. MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem J 2006;393:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fan L, Hu L, Yang B et al. Erlotinib promotes endoplasmic reticulum stress-mediated injury in the intestinal epithelium. Toxicol Appl Pharmacol 2014;278:45–52. [DOI] [PubMed] [Google Scholar]
- 29. Hong S, Gu Y, Gao Z et al. EGFR inhibitor-driven endoplasmic reticulum stress-mediated injury on intestinal epithelial cells. Life Sci 2014;119:28–33. [DOI] [PubMed] [Google Scholar]
- 30. Van Sebille YZA, Gibson RJ, Wardill HR et al. Dacomitinib-induced diarrhoea is associated with altered gastrointestinal permeability and disruption in ileal histology in rats. Int J Cancer 2017;140:2820–9. [DOI] [PubMed] [Google Scholar]
- 31. Verkman AS, Galietta LJV. Chloride channels as drug targets. Nat Rev Drug Discov 2009;8:153–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barrett KE, Keely SJ. Chloride secretion by the intestinal epithelium: Molecular basis and regulatory aspects. Annu Rev Physiol 2000;62:535–72. [DOI] [PubMed] [Google Scholar]
- 33. Keely SJ, Uribe JM, Barrett KE. Carbachol stimulates transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T84 cells: Implications for carbachol-stimulated chloride secretion. J Biol Chem 1998;273:27111–7. [DOI] [PubMed] [Google Scholar]
- 34. Barrett KE, Smitham J, Traynor-Kaplan A et al. Inhibition of Ca2+−dependent Cl−secretion in T84 cells: membrane target(s) of inhibition is agonist specific. Am J Physiol Cell Physiol. 1998;274:C958–65. doi: 10.1152/ajpcell.1998.274.4.c958. [DOI] [PubMed] [Google Scholar]
- 35. Keely SJ, Calandrella SO, Barrett KE. Carbachol-stimulated transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T84 cells is mediated by intracellular Ca2+, PYK-2, and p60(src). J Biol Chem 2000;275:12619–25. [DOI] [PubMed] [Google Scholar]
- 36. Uribe JM, Gelbmann CM, Traynor-Kaplan AE et al. Epidermal growth factor inhibits Ca2+-dependent Cl-transport in T84 human colonic epithelial cells. Am J Physiol Cell Physiol. 1996;271:C914–22. doi: 10.1152/ajpcell.1996.271.3.c914. [DOI] [PubMed] [Google Scholar]
- 37. Duan T, Cil O, Thiagarajah JR et al. Intestinal epithelial potassium channels and CFTR chloride channels activated in ErbB tyrosine kinase inhibitor diarrhea. JCI insight. 2019;4:e126444. doi: 10.1172/jci.insight.126444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim Y, Quach A, Das S et al. Potentiation of calcium-activated chloride secretion and barrier dysfunction may underlie EGF receptor tyrosine kinase inhibitor-induced diarrhea. Physiol Rep. 2020;8:e14490. doi: 10.14814/phy2.14490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bowen JM, Mayo BJ, Plews E et al. Development of a rat model of oral small molecule receptor tyrosine kinase inhibitor-induced diarrhea. Cancer Biol Ther 2012;13:1269–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bowen JM, Mayo BJ, Plews E et al. Determining the mechanisms of lapatinib-induced diarrhoea using a rat model. Cancer Chemother Pharmacol 2014;74:617–27. [DOI] [PubMed] [Google Scholar]
- 41. Tao G, Huang J, Moorthy B et al. Potential role of drug metabolizing enzymes in chemotherapy-induced gastrointestinal toxicity and hepatotoxicity. Expert Opin Drug Metab Toxicol 2020;16:1109–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Özvegy-Laczka C, Hegedus T, Várady G et al. High-affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Mol Pharmacol 2004;65:1485–95. [DOI] [PubMed] [Google Scholar]
- 43. Li J, Cusatis G, Brahmer J et al. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther 2007;6:432–8. [DOI] [PubMed] [Google Scholar]
- 44. Marchetti S, De Vries NA, Buckle T et al. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1-/-/Mdr1a/1b-/- (triple-knockout) and wild-type mice. Mol Cancer Ther 2008;7:2280–7. [DOI] [PubMed] [Google Scholar]
- 45. Rudin CM, Liu W, Desai A et al. Pharmacogenomic and pharmacokinetic determinants of erlotinib toxicity. J Clin Oncol 2008;26:1119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dai CL, Tiwari AK, Wu CP et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res 2008;68:7905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Perry J, Ghazaly E, Kitromilidou C et al. A Synergistic Interaction between Lapatinib and Chemotherapy Agents in a Panel of Cell Lines Is Due to the Inhibition of the Efflux Pump BCRP. Mol Cancer Ther 2010;9:3322–9. [DOI] [PubMed] [Google Scholar]
- 48. Zhao XQ, Xie JD, Chen XG et al. Neratinib reverses ATP-binding cassette B1-mediated chemotherapeutic drug resistance in vitro, in vivo, and ex vivo. Mol Pharmacol 2012;82:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ianiro G, Rossi E, Thomas AM et al. Faecal microbiota transplantation for the treatment of diarrhoea induced by tyrosine-kinase inhibitors in patients with metastatic renal cell carcinoma. Nat Commun 2020;11:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stein A, Voigt W, Jordan K. Review: Chemotherapy-induced diarrhea: Pathophysiology, frequency and guideline-based management. Ther Adv Med Oncol 2010;2:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Joensuu H, Trent JC, Reichardt P. Practical management of tyrosine kinase inhibitor-associated side effects in GIST. Cancer Treat Rev 2011;37:75–88. [DOI] [PubMed] [Google Scholar]
- 52. Wang HH, Shieh MJ, Liao KF. A blind, randomized comparison of racecadotril and loperamide for stopping acute diarrhea in adults. World J Gastroenterol 2005;11:1540–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karthaus M, Ballo H, Abenhardt W et al. Prospective, double-blind, placebo-controlled, multicenter, randomized phase III study with orally administered budesonide for prevention of irinotecan (CPT-11)-induced diarrhea in patients with advanced colorectal cancer. Oncology 2005;68:326–32. [DOI] [PubMed] [Google Scholar]
- 54. Richardson G, Dobish R. Chemotherapy induced diarrhea. J Oncol Pharm Pract 2007;13:181–98. [DOI] [PubMed] [Google Scholar]
- 55. Cascinu S, Fedeli A, Fedeli SL et al. Octreotide versus loperamide in the treatment of fluorouracil-induced diarrhea: A randomized trial. J Clin Oncol 1993;11:148–51. [DOI] [PubMed] [Google Scholar]
- 56. Kailasapathy K, Chin J. Survival and therapeutic potential of probiotic organisms with reference to Lactobacillus acidophilus and Bifidobacterium spp. Immunol Cell Biol 2000;78:80–8. [DOI] [PubMed] [Google Scholar]
- 57. Justino PFC, Melo LFM, Nogueira AF et al. Regulatory role of Lactobacillus acidophilus on inflammation and gastric dysmotility in intestinal mucositis induced by 5-fluorouracil in mice. Cancer Chemother Pharmacol 2015;75:559–67. [DOI] [PubMed] [Google Scholar]
- 58. Mego M, Chovanec J, Vochyanova-Andrezalova I et al. Prevention of irinotecan induced diarrhea by probiotics: A randomized double blind, placebo controlled pilot study. Complement Ther Med 2015;23:356–62. [DOI] [PubMed] [Google Scholar]
- 59. Katakami N, Atagi S, Goto K et al. LUX-Lung 4: a phase II trial of afatinib in patients with advanced non–small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J Clin Oncol 2013;31:3335–42. [DOI] [PubMed] [Google Scholar]
- 60. Frankel C, Palmieri FM. Lapatinib side-effect management. Clin J Oncol Nurs 2010;14:223–33. [DOI] [PubMed] [Google Scholar]
- 61. Ibrahim E, Tripathy D, Wilkinson M et al. Cancer Research, Vol. 77. American Association for Cancer Research (AACR), 2017, CT128–8. [Google Scholar]
- 62. Gatzemeier U, Pluzanska A, Szczesna A et al. Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: The Tarceva lung cancer investigation trial. J Clin Oncol 2007;25:1545–52. [DOI] [PubMed] [Google Scholar]
- 63. Herbst RS, Giaccone G, Schiller JH et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: A phase III trial - INTACT 2. J Clin Oncol 2004;22:785–94. [DOI] [PubMed] [Google Scholar]
- 64. Shi Y, Zhang L, Liu X et al. Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): A randomised, double-blind phase 3 non-inferiority trial. Lancet Oncol 2013;14:953–61. [DOI] [PubMed] [Google Scholar]
- 65. Lin NU, Diéras V, Paul D et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res 2009;15:1452–9. [DOI] [PubMed] [Google Scholar]
- 66. Schuler M, Yang JCH, Park K et al. Afatinib beyond progression in patients with non-small-cell lung cancer following chemotherapy, erlotinib/gefitinib and afatinib: phase III randomized LUX-Lung 5 trial. Ann Oncol 2016;27:417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tjulandin S, Moiseyenko V, Semiglazov V et al. Phase I, dose-finding study of AZD8931, an inhibitor of EGFR (erbB1), HER2 (erbB2) and HER3 (erbB3) signaling, in patients with advanced solid tumors. New Drugs 2014;32:145–53. [DOI] [PubMed] [Google Scholar]
- 68. Han JY, Lee KH, Kim SW et al. A phase II study of poziotinib in patients with epidermal growth factor receptor (EGFR)-mutant lung adenocarcinoma who have acquired resistance to EGFR-tyrosine kinase inhibitors. Cancer Res Treat 2017;49:10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Konings IRHM, De Jonge MJA, Burger H et al. Phase i and pharmacological study of the broad-spectrum tyrosine kinase inhibitor JNJ-26483327 in patients with advanced solid tumours. Br J Cancer 2010;103:987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Goss G, Tsai CM, Shepherd FA et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol 2016;17:1643–52. [DOI] [PubMed] [Google Scholar]
- 71. Park K, Lee J-S, Lee KH et al. BI 1482694 (HM61713), an EGFR mutant-specific inhibitor, in T790M+ NSCLC: Efficacy and safety at the RP2D. J Clin Oncol 2016;34:9055–5. [Google Scholar]
- 72. Sequist LV, Soria JC, Goldman JW et al. Rociletinib in EGFR-mutated non–small-cell lung cancer. New England Journal of Medicine 2015;372:1700–1709. [DOI] [PubMed] [Google Scholar]
- 73. Burger JA, Keating MJ, Wierda WG et al. Safety and activity of ibrutinib plus rituximab for patients with high-risk chronic lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol 2014;15:1090–9. [DOI] [PMC free article] [PubMed] [Google Scholar]