

Abstract
Following our report that A3 adenosine receptor (AR) antagonist 1 exhibited a polypharmacological profile as a dual modulator of peroxisome proliferator-activated receptor (PPAR)γ/δ, we discovered a new template, 1′-homologated adenosine analogues 4a–4t, as dual PPARγ/δ modulators without AR binding. Removal of binding affinity to A3AR was achieved by 1′-homologation, and PPARγ/δ dual modulation was derived from the structural similarity between the target nucleosides and PPAR modulator drug, rosiglitazone. All the final nucleosides were devoid of AR-binding affinity and exhibited high binding affinities to PPARγ/δ but lacked PPARα binding. 2-Cl derivatives exhibited dual receptor-binding affinity to PPARγ/δ, which was absent for the corresponding 2-H derivatives. 2-Propynyl substitution prevented PPARδ-binding affinity but preserved PPARγ affinity, indicating that the C2 position defines a pharmacophore for selective PPARγ ligand designs. PPARγ/δ dual modulators functioning as both PPARγ partial agonists and PPARδ antagonists promoted adiponectin production, suggesting their therapeutic potential against hypoadiponectinemia-associated cancer and metabolic diseases.
Graphical Abstract
INTRODUCTION
Metabolic diseases such as metabolic syndrome, diabetes, and nonalcoholic steatohepatitis are associated with the disruption of the cellular metabolic balance due to genetic or acquired causes.1 Peroxisome proliferator-activated receptors (PPARs) are nuclear transcriptional factors with a pivotal role in the regulation of metabolic homeostasis. PPARs can sense the cellular levels of fatty acids and activate the transcription of various genes associated with the cellular response to changing metabolic needs. Three PPAR isoforms, PPARα, PPARγ, and PPARδ, have been identified in mammalian cells, and PPARs show differentially expressed profiles in some tissues.2 PPARα is highly expressed in liver, and its significant level is also detected in all tissues. PPARγ is the major PPAR isoform in adipose tissue, whereas PPARδ expression is mainly observed in skeletal muscle. The heterodimeric form of PPARs with a retinoid X receptor can bind to a peroxisome proliferator response element, which is essential for the transcription of metabolic target genes. PPARs regulate metabolic homeostasis, and their pharmacological ligands have been developed to treat human metabolic diseases. However, as highly specific PPARγ or PPARδ agonists have raised safety issues of hepatotoxicity and cardiovascular adverse effects,3 PPAR dual- or pan-modulators have instead been suggested with the expectation that the balanced PPAR activation may reduce serious side effects.
Recently, we have reported that truncated 4′-thioadenosine derivatives, acting as potent and selective A3 adenosine receptor (AR) antagonists, also showed potent modulation of PPARs, demonstrating their polypharmacological profiles.4 Among these, compound 1 was discovered as a dual modulator acting as PPARγ partial agonist and PPARδ antagonist, while maintaining its binding affinity at the hA3AR (Figure 1).4 The adiponectin secretion-promoting activity of 1 was found in the phenotypic screening, exploiting the adipogenesis model of human bone marrow mesenchymal stem cells (hBM-MSCs). However, the effect of 1 and its analogues on adiponectin production was not correlated to their A3AR-binding affinities.4 The target identification study revealed that 1 directly bound both PPARγ and PPARδ. Interestingly, compound 1 significantly increased the nuclear receptor corepressor peptide recruitment to the PPARδ ligand-binding domain (LBD), whereas it did not affect the coactivator peptide recruitment, a feature of PPARδ antagonists.4 Thus, if we can design the pure PPAR modulators that lack binding affinity at ARs, it was thought that the undesired effects arising from their AR activity might be avoided, while retaining favorable effects from pure PPAR modulation. To achieve this goal, we turned our attention to the 1′-homologated 4′-thioadenosine analogues 2 because they were totally devoid of binding affinity at any of the ARs.5 Furthermore, the homologated analogues showed a structural similarity to a known thiazolidinedione PPARγ agonist, rosiglitazone (3), which also contains one-carbon homologation linker between the 1,3-thiazolidinedione ring and the aromatic ring. Molecular modeling demonstrated that sugar ring and purine ring of the target compound 2 mimic the binding mode of the 1,3-thiazolidinedione ring and aromatic substituents of 3 against PPARγ, respectively (Figure 1).
Figure 1.
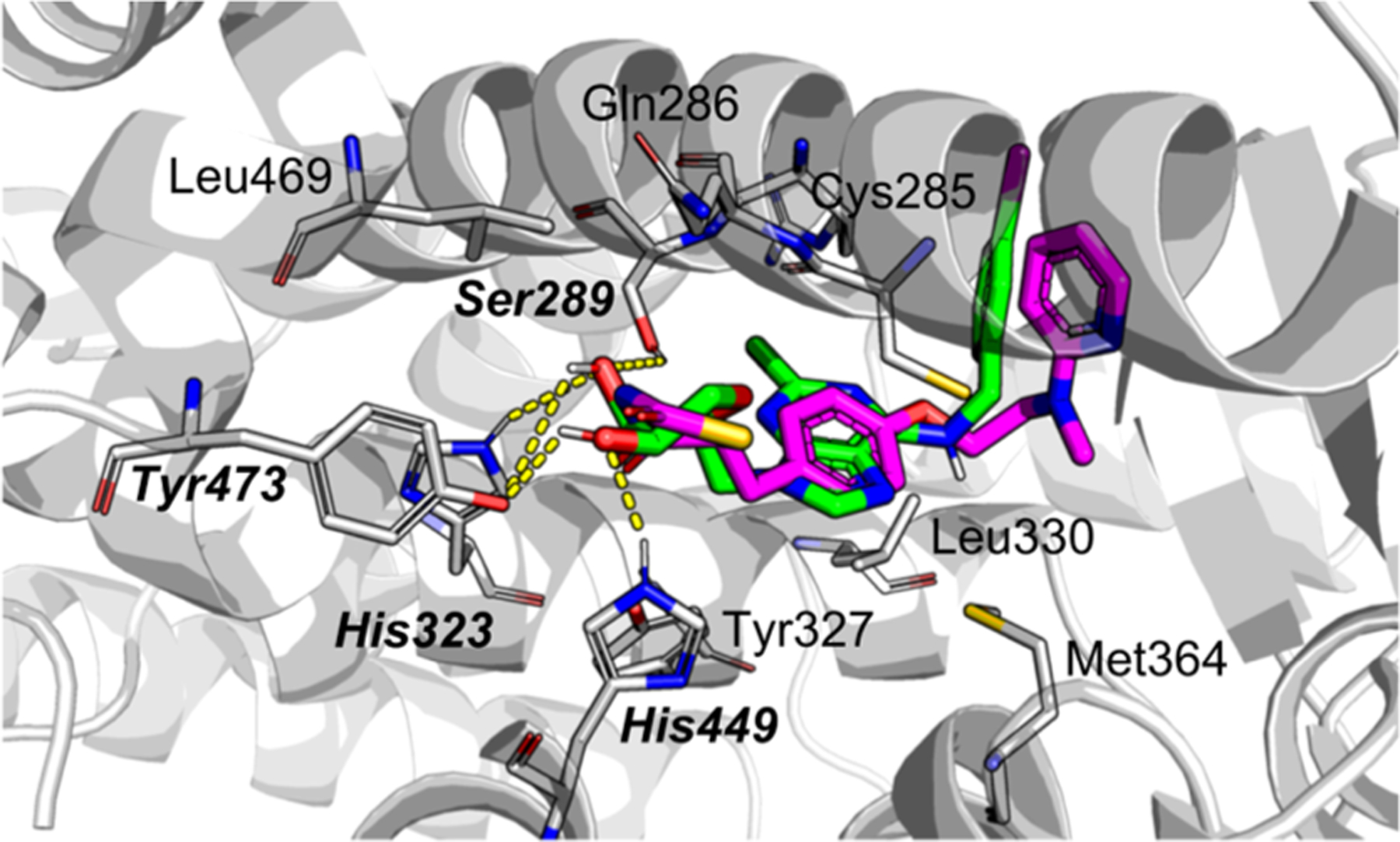
Superimposed structures of rosiglitazone (magenta) and 4p (green) inside binding pocket of PPARγ-LBD (PDB ID: 5YCP). Amino acid residues close-contacting with 4p are presented, and hydrogen-bonding residues are bold-highlighted.
In this study, two hydroxyl groups from 4p were responsible for generating several hydrogen bonds with Ser289, His323, His449, and Tyr473 of PPARγ-LBD. These amino acid residues were also involved in the binding interaction of rosiglitazone against PPARγ-LBD. Unlike the phenyl moiety of rosiglitazone, the nucleobase moiety can help increase the binding affinity by forming the cation–π interaction and a noncovalent bond. A docking score, calculated using AutoDock Vina software, showed that 4p has a better binding affinity (−8.5 kcal/mol) in comparison with that of rosiglitazone (−7.8 kcal/mol). The detailed studies by using Flare are described in the Supporting Information (Figures S28–S30).
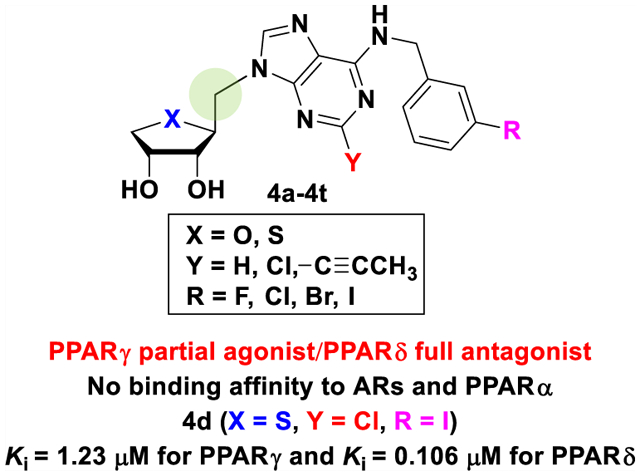
Thus, to design the pure PPAR modulators devoid of the binding affinity at the ARs, we synthesized the one-carbon homologated 4′-oxo- and 4′-thioadenosine derivatives, 4a–4h and 4m–4t with the same substituents with 1 and compared their PPARs and AR binding affinities (Figure 2). As our previous report4 that a hydrophobic group such as 3-halobenzyl at the N6-position of adenine favored the adiponectin secretion-promoting activity or PPAR-binding activity during adipogenesis in hBM-MSCs, we also synthesized 1-propynyl derivatives 4i–4j to introduce a lipophilic substituent at the C2 position of the purine ring to increase adiponectin secretion-promoting activity during adipogenesis in hBM-MSCs. Another reason to introduce a 1-propynyl group is to determine if steric effects are important in binding to PPARγ and PPARδ. These target compounds are expected to overcome the intrinsic toxicity associated with 1,3-thiazolidinedione antidiabetic drugs such as troglitazone and rosiglitazone, while maintaining the same high binding affinity as 1,3-thiazolidinediones.
Figure 2.
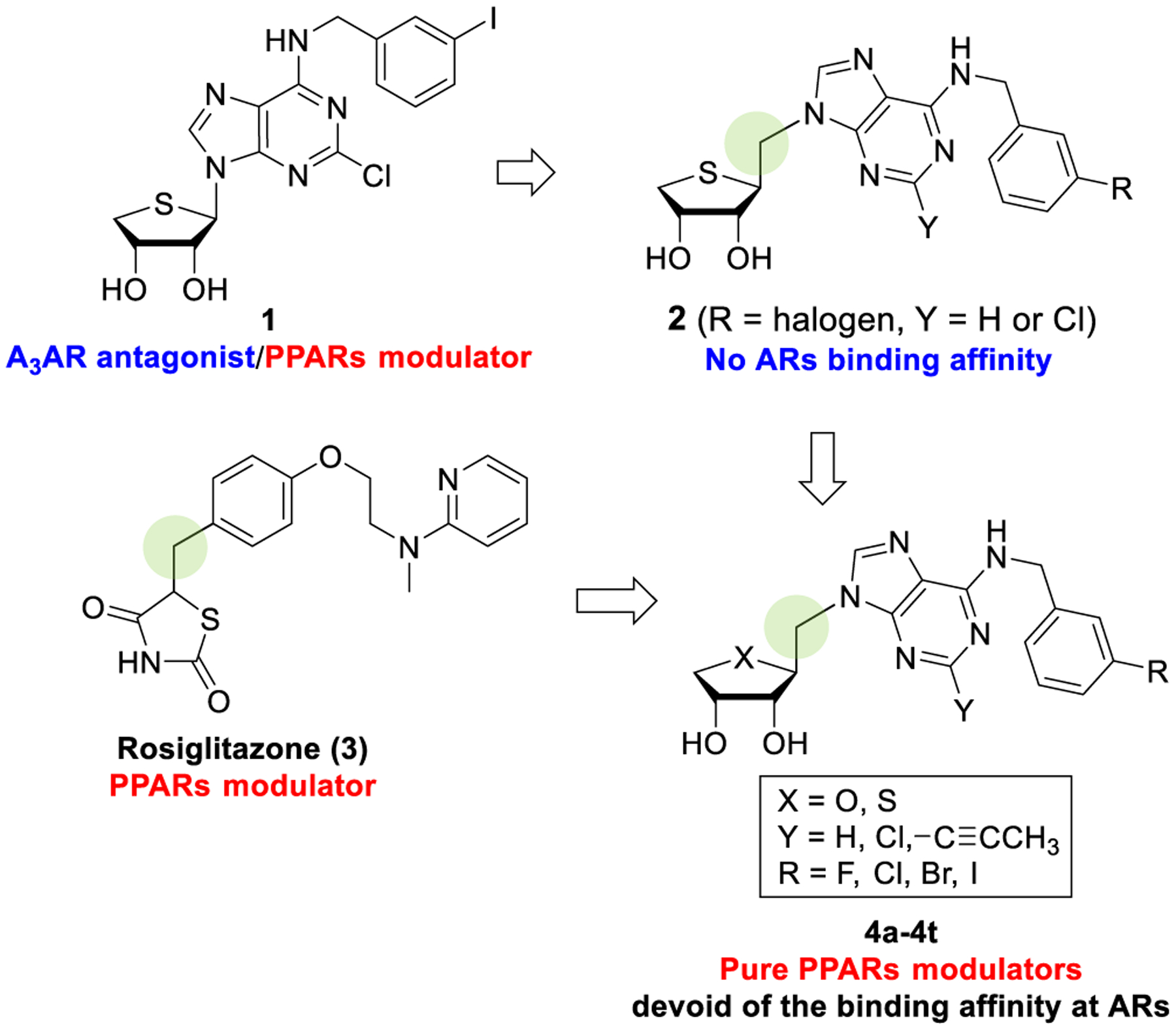
Design of novel PPAR modulators lacking AR-binding affinity by the homologation strategy.
As expected, our target compounds 4a–4t exhibited good binding affinities to both PPARγ and PPARδ without binding affinity to AR and a significant adiponectin secretion-promoting activity during adipogenesis in hBM-MSCs through phenotypic screening. Herein, we report the discovery and structure–activity relationships (SARs) of novel template, truncated 1′-homologated adenosine analogues 4a–4t as dual PPARγ/δ modulators, and their potent adiponectin secretion-promoting activity.
RESULTS AND DISCUSSION
Chemistry.
The synthetic strategy for the synthesis of the final nucleosides was first to synthesize the glycosyl donor and then to condense it with the purine base. Truncated-homologated 4′-thioadenosine derivatives, 4a–4h, were synthesized according to our previously published procedures.5,6
As shown in Scheme 1, the glycosyl donor 105 was synthesized, starting from d-mannose. d-Mannose was converted to the diacetonide 5 by treating with 2,2-dimethoxypropane and acetone under acidic conditions. Reduction of 5 with sodium borohydride, followed by the treatment of the resulting diol with mesyl chloride afforded the dimesylate 6. Heating of 6 with sodium sulfide in dimethylformamide (DMF) produced the 4-thiosugar 7. Regioselective hydrolysis of 5,6-acetonide of 7 with 60% AcOH yielded 5,6-diol 8. Oxidative cleavage of 8 with Pb(OAc)4 at 0 °C produced the aldehyde, which without purification, was reduced with sodium borohydride to give the primary alcohol 9. It is interesting to note that oxidative cleavage using excess amounts of Pb(OAc)4 at room temperature (rt) directly produced the 1-acetate, which was formed via further oxidation of 8 to the acid followed by successive oxidative decarboxylation. Treatment of 9 with phosphorus oxychloride yielded the glycosyl donor 10.
Scheme 1. Synthesis of Truncated-Homologated Glycosyl Donor 10a.
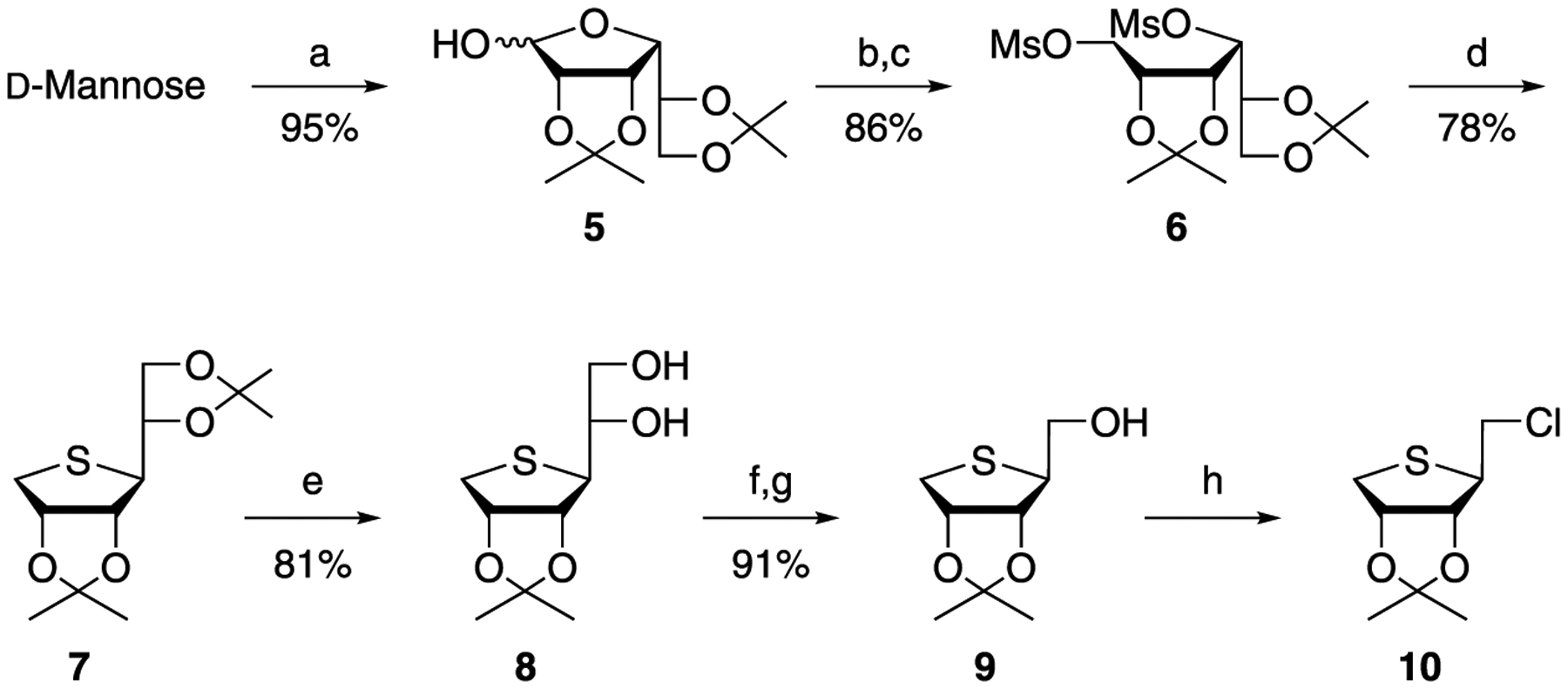
aReagents and conditions: (a) 2,2-dimethoxypropane, CSA, acetone, rt; (b) NaBH4, EtOH, rt; (c) MsCl, Et3N, DMAP, CH2Cl2, rt; (d) Na2S, DMF, 80 °C; (e) 60% AcOH, rt; (f) Pb(OAc)4, EtOAc, 0 °C; (g) NaBH4, EtOH, 0 °C; and (h) POCl3, CH3CN, rt.
The glycosyl donor 10 was condensed with 2,6-dichloropurine and 6-chloropurine anion in DMF to give 11 and 12, respectively, as shown in Scheme 2. Removals of the acetonide of 11 and 12 using 2 N HCl, followed by the treatment of the resulting diol with 3-halobenzylamines yielded the final nucleosides, 4a–4d and 4e–4h, respectively.5
Scheme 2. Synthesis of Truncated 1′-Homologated 4′-Thioadenosine Derivatives, 4a–4ha.
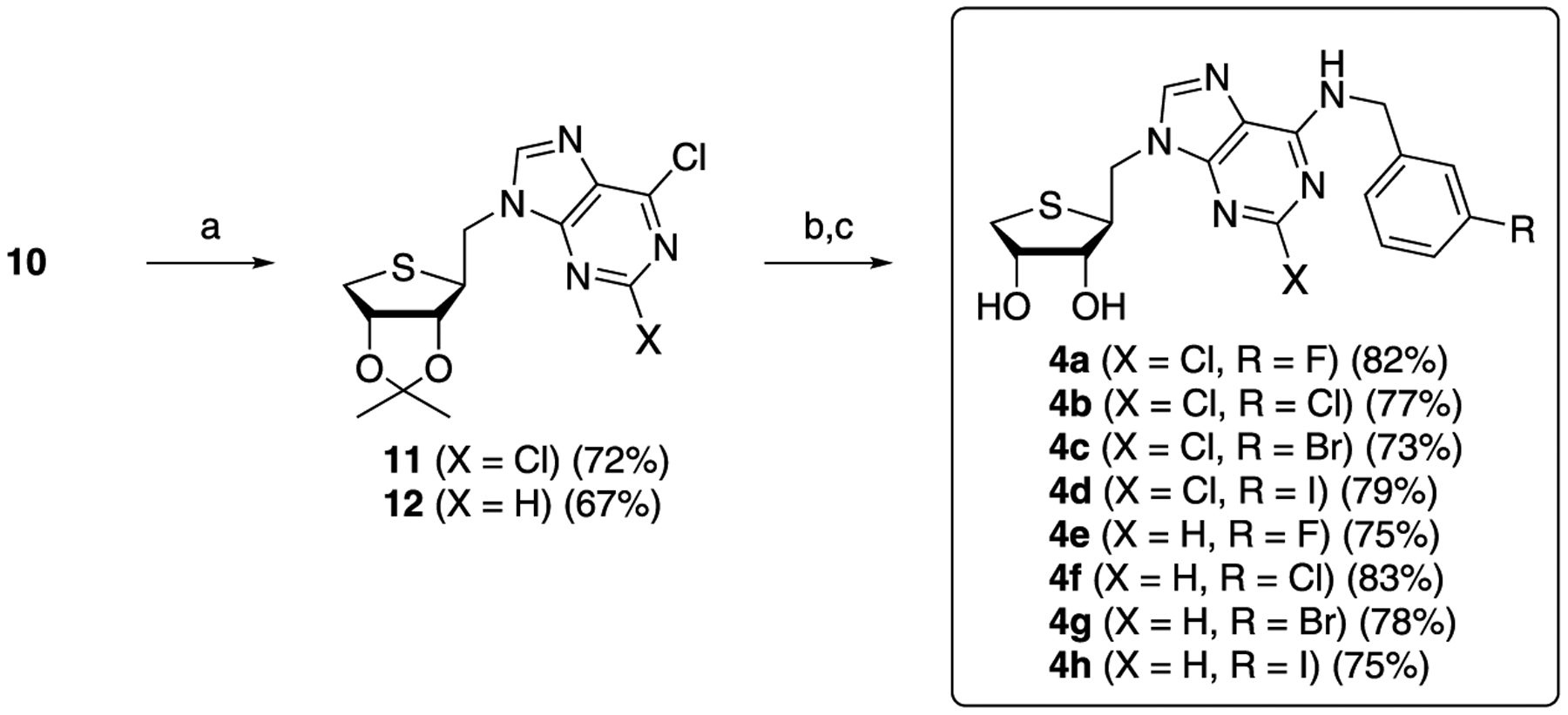
aReagents and conditions: (a) NaH, 2,6-dichloropurine or 6-chloropurine, DMF, rt; (b) 2 N HCl, THF, rt; and (c) 3-halobenzylamines, Et3N, EtOH, rt.
Next, we synthesized the truncated C2-propynyl-4′-thioadenosine derivatives 4i–4l to determine if bulky and lipophilic 1-propynyl substituent might affect the adiponectin secretion-promoting activity, PPAR-binding activity, or binding to PPARγ and PPARδ. To functionalize the C2 position with the propynyl group, using Sonogashira coupling, we synthesized the 6-chloro-2-iodopurine 17 as the coupling substrate, as shown in Scheme 3.7 The N9 position of 6-chloropurine (13) was protected with the tetrahydropyran group by treating with 2,3-dihydropyran under acidic conditions to afford 14, which was subjected to stannylation by treating with n-Bu3SnCl in the presence of LiTMP to give the tin derivative 15. Treatment of 15 with iodine at rt yielded 2-iodo derivative 16, which was treated with 10 mol % CuCl2 to afford 17.
Scheme 3. Synthesis of 6-Chloro-2-iodopurine 17a.
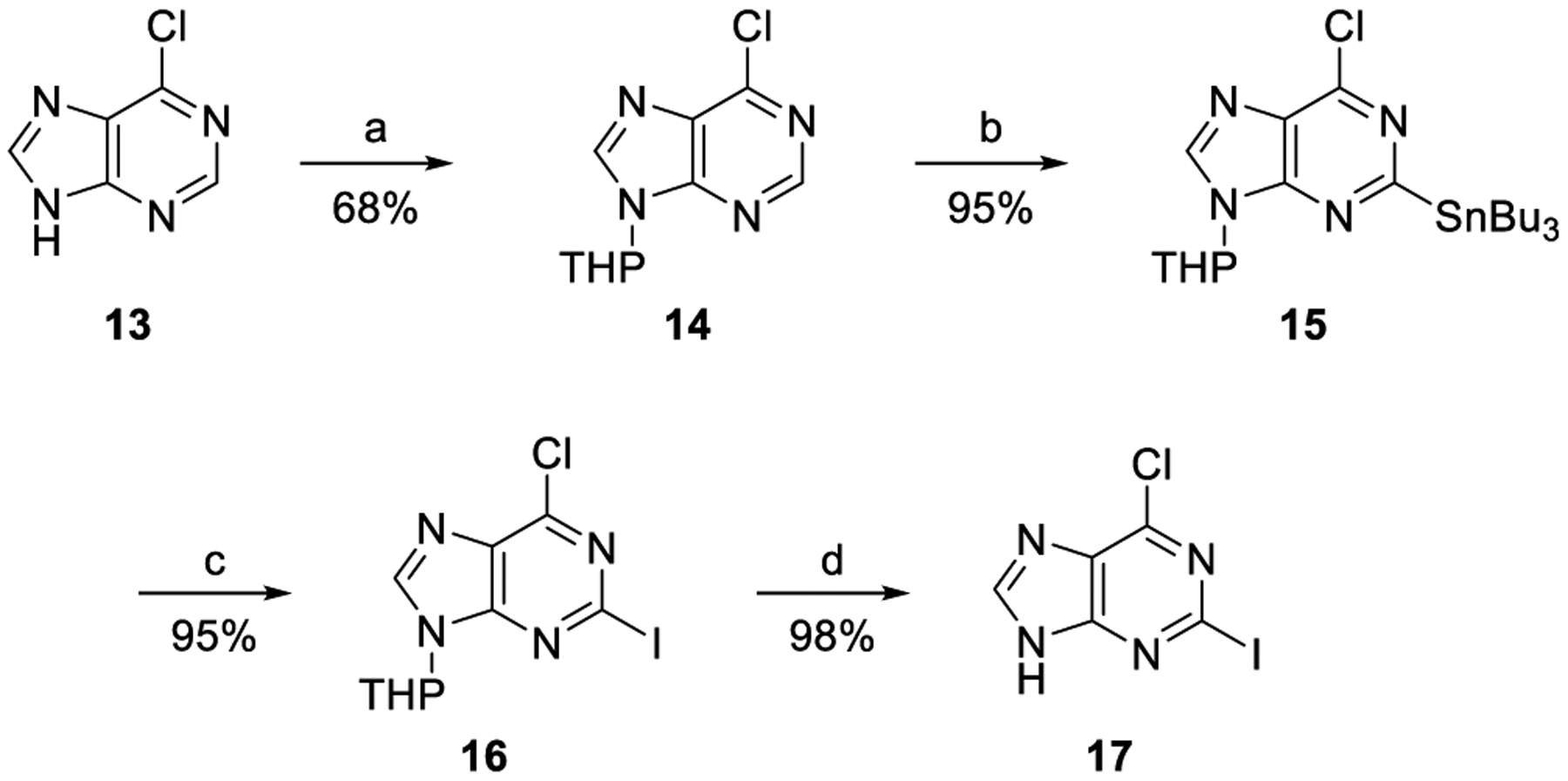
aReagents and conditions: (a) 2,3-dihydropyran, PTSA, THF, 80 °C; (b) n-Bu3SnCl, LiTMP, THF/n-hexane (2:1), −78 °C; (c) I2, THF, rt; and (d) 10 mol % CuCl2, EtOH/H2O (19:1), rt.
Condensation of 9 with 6-chloro-2-iodopurine (17) under the standard Mitsunobu conditions yielded the protected nucleoside 18 (Scheme 4). Sonogashira coupling of 18 with propyne in the presence of CuI, Pd(PPh3)4, and Cs2CO3 produced C2-propynyl derivative 19 in 75% yield. Removal of the acetonide of 19 with 2 N HCl, followed by treating the resulting diol with 3-halobenzylamines yielded the truncated 1′-homologated 2-propynyl-N6-(3-halobenzyl)adenosines 4i–4l.
Scheme 4. Synthesis of Truncated 1′-Homologated 2-Propynyl-4′-thioadenosine Derivatives, 4i–4la.
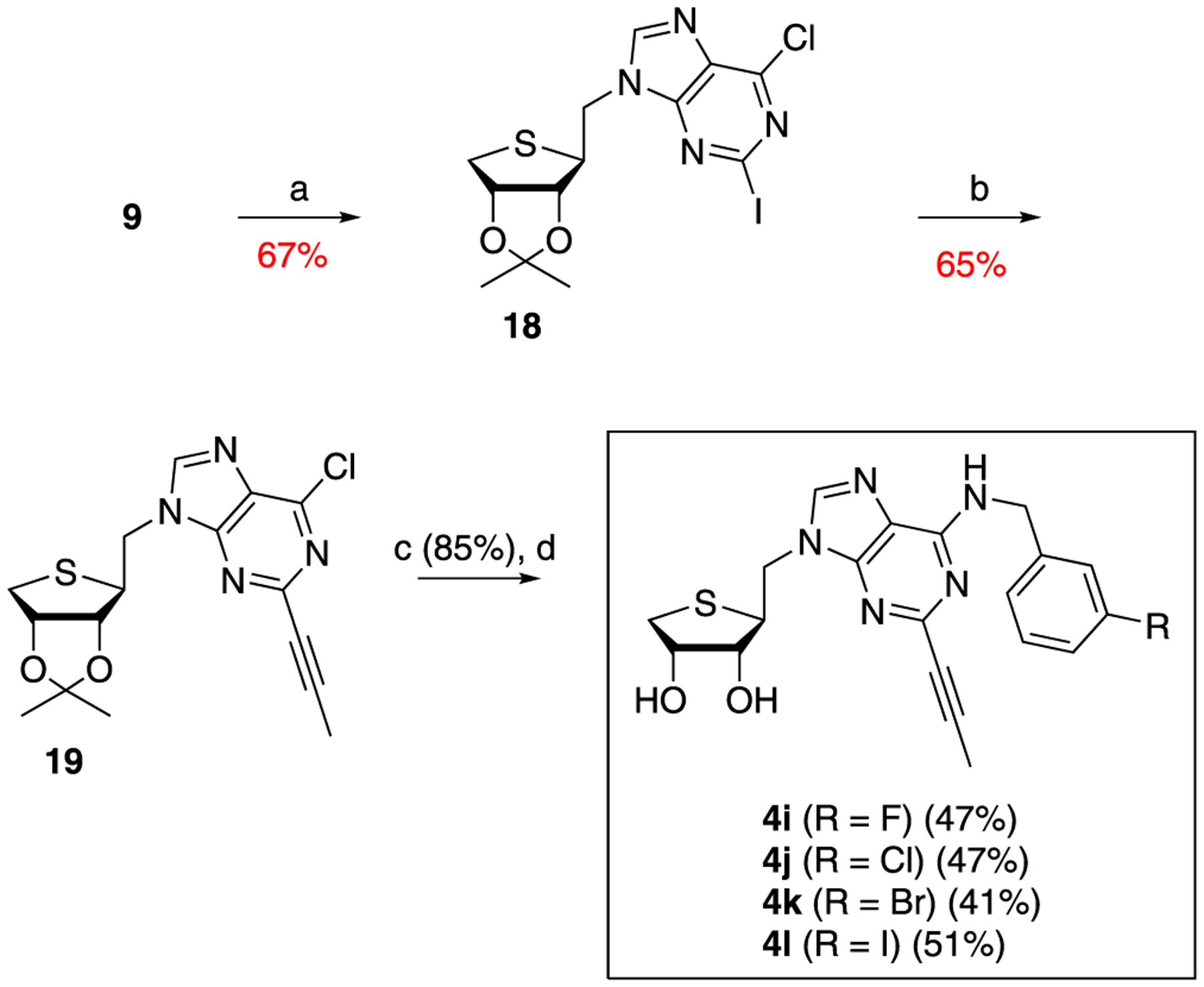
aReagents and conditions: (a) PPh3, DIAD, THF, 0 °C, 1 h; 6-chloro-2-iodopurine (17); (b) propyne, CuI, Pd(PPh3)4, Cs2CO3, DMF, rt; (c) 2 N HCl, THF, rt; and (d) 3-halobenzylamines, Et3N, EtOH, rt.
Based on a bioisosteric rationale, we designed and synthesized the corresponding 4′-oxo derivatives, 4m–4t from l-ribose, as illustrated in Scheme 5. l-Ribose was converted to the 2,3-acetonide 20 by treating with acetone under acidic conditions. Protection of the primary hydroxyl group of 20 with trityl (Tr) followed by reduction with NaBH4 afforded the diol 21. Regioselective tosylation of the primary alcohol of 21 smoothly converted the mono tosylate into 4-oxo sugar 22.8 Removal of the Tr group of 22 was achieved by treating with Et2AlCl in CH2Cl2 at −40 °C to produce the glycosyl donor 23. The Mitsunobu condensation of 23 with 2,6-dichloropurine and 6-chloropurine afforded the 2,6-dichloropurine nucleoside 24 and the 6-chloropurine nucleoside 25, respectively. Treatment of 24 and 25 with 3-halobenzylamines followed by acetonide deprotection yielded the truncated 1′-homologated 4′-oxoadenosine derivatives, 4m–4t.
Scheme 5. Synthesis of Truncated 1′-Homologated 4′-Oxoadenosine Derivatives, 4m–4ta.
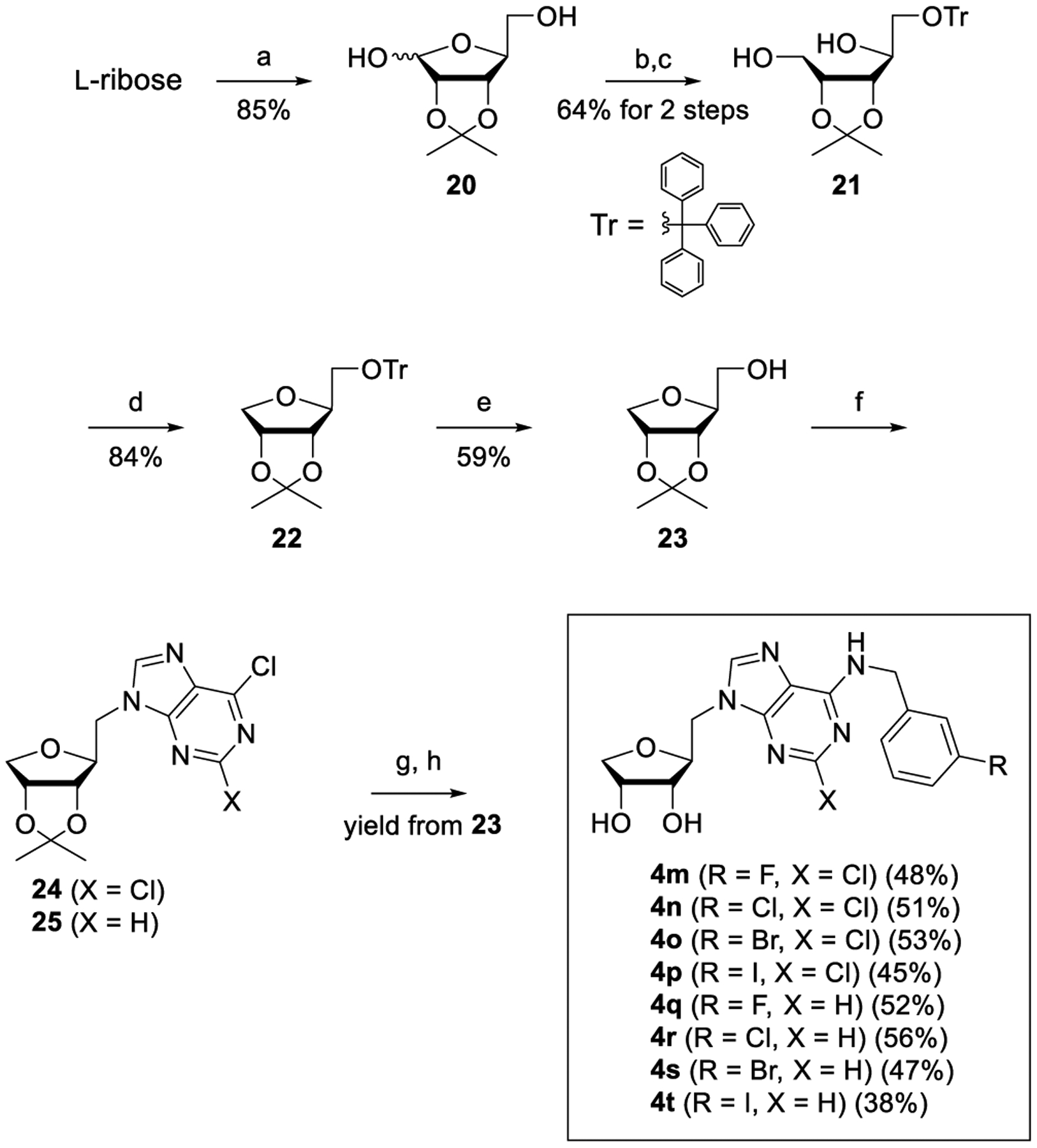
aReagents and conditions: (a) cH2SO4, acetone, rt, 3 h; (b) trityl chloride, DMAP, pyridine, 80 °C, 3 h; (c) NaBH4, MeOH, rt, 1 h; (d) TsCl, pyridine, rt, 12 h; (e) Et2AlCl, CH2Cl2, −40 °C, 3 h; (f) PPh3, DIAD, THF, rt, 12 h; 2,6-dichloropurine or 6-chloropurine; (g) 3-halobenzylamines, Et3N, THF, rt; and (h) 1 N HCl, MeOH, rt.
Biological Evaluation.
PPAR-Binding Profiles of Compounds 4a–4t.
Compound 1 has a polypharmacological receptor-binding profile recognizing A3AR, PPARγ, and PPARδ.4 Therefore, we investigated binding profiles of compounds 4a–4t, which are structurally similar to 1, for A3AR and PPARs (Table 1). First, their A3AR-binding affinity was measured using the human A3AR expressed in the Chinese hamster ovary cells. Although 1 showed high binding affinity (Ki = 4.16 nM for A3AR), compounds 4a–4t did not significantly bind A3AR up to 10 μM. Next, we determined whether compounds 4a–4t directly bind PPARs by the time-resolved fluorescence resonance energy transfer (TR-FRET)-based nuclear receptor-binding assay (Table 1). In the TR-FRET-based assays, compounds 4a–4t did not bind to PPARα as compound 1 did, whereas several compounds showed high receptor-binding activity for PPARγ and/or PPARδ. Compounds 4a, 4c, 4d, and 4n–4p with 2-Cl exhibited a significant dual receptor-binding activity against both PPARγ and PPARδ, although their efficacy to PPARγ or PPARδ was not as potent as positive control drugs, a PPARγ agonist pioglitazone (26), a PPARδ agonist 2-[2-methyl-4-[[[4-methyl-2-[4-(trifluoromethyl)phenyl]-5-thiazolyl]methyl]thio]phenoxy]-acetic acid (27, GW501516), and a PPARδ antagonist 3-(((2-methoxy-4-(phenylamino)phenyl)amino)sulfonyl)-2-thiophenecarboxylic acid methyl ester (28, GSK0660). Notably, compounds 4i–4l with 1-propynyl substituent at the C2 position lost their binding activity to PPARδ, whereas they retained their PPARγ-binding activity except 4j (Table 1), indicating that the PPARδ ligand-binding pocket (LBP) was unable to accommodate the relatively bulky substituent at the C2 position. This result matches the previous report that the LBP of PPARδ is smaller and narrower than that of PPARγ.9 Interestingly, there was no significant PPARγ- or PPARδ-binding activity up to 30 μM for compounds 4e–4h and 4q–4t with no substituent at the C2 position, indicating that proper size of the lipophilic substituent is essential for binding activity to PPARγ and PPARδ. In summary, 1′-homologation of 1 resulted in a dramatic loss of the A3AR-binding affinity, while retaining the PPARγ/δ dual modulator potential (Table 1).
Table 1.
Receptor-Binding Profiles of Truncated 1′-Homologated Adenosine Derivatives 4a–4t
 | |||||||
|---|---|---|---|---|---|---|---|
| cpd | X | Y | R | hA3AR Ki (nM) or % replacementa | PPARα % replacementb at 30 μM (%) | PPARγ Ki (μM) or % replacementb | PPARδ Ki (μM) or % replacementb |
| 1 | 4.16 | 35.6 | 3.33 ± 1.67 | 0.0106 ± 0.015 | |||
| 4a | S | Cl | F | 17% at 10 μM | 15.5 | 3.89 ± 0.47 | 1.91 ± 0.39 |
| 4b | S | Cl | Cl | 13% at 10 μM | 12.8 | 34.1% at 30 μM | 7.8% at 30 μM |
| 4c | S | Cl | Br | 21% at 10 μM | 3.0 | 4.07 ± 0.86 | 0.592 ± 0.26 |
| 4d | S | Cl | I | 10% at 10 μM | 20.4 | 1.23 ± 0.25 | 0.106 ± 0.031 |
| 4e | S | H | F | 9% at 10 μM | 25.6 | 24.4% at 30 μM | 0.8% at 30 μM |
| 4f | S | H | Cl | 9% at 10 μM | 7.7 | 24.9% at 30 μM | 10.0% at 30 μM |
| 4g | S | H | Br | 6% at 10 μM | 5.4 | 33.5% at 30 μM | 9.9% at 30 μM |
| 4h | S | H | I | 14% at 10 μM | 9.4 | 39.2% at 30 μM | 5.2% at 30 μM |
| 4i | S | −C≡CCH3 | F | 2% at 10 μM | 15.5 | 7.66 ± 0.65 | 13.8% at 30 μM |
| 4j | S | −C≡CCH3 | Cl | 1% at 10 μM | 12.6 | 41.7% at 30 μM | 3.8% at 30 μM |
| 4k | S | −C≡CCH3 | Br | 12% at 10 μM | 12.8 | 7.04 ± 0.40 | 15.3% at 30 μM |
| 4l | S | −C≡CCH3 | I | 2% at 10 μM | 24.0 | 7.30 ± 1.01 | 15.9% at 30 μM |
| 4m | O | Cl | F | 2% at 10 μM | 5.0 | 9.0% at 30 μM | 7.3% at 30 μM |
| 4n | O | Cl | Cl | 1% at 10 μM | 3.6 | 7.66 ± 2.45 | 0.504 ± 0.29 |
| 4o | O | Cl | Br | 14% at 10 μM | 23.2 | 9.55 ± 6.61 | 1.43 ± 0.67 |
| 4p | O | Cl | I | 3% at 10 μM | 0.3 | 3.56 ± 0.75 | 0.374 ± 0.036 |
| 4q | O | H | F | 1% at 10 μM | 1.4 | 13.3% at 30 μM | 14.6% at 30 μM |
| 4r | O | H | Cl | 1% at 10 μM | 8.3 | 0.8% at 30 μM | 10.6% at 30 μM |
| 4s | O | H | Br | 14% at 10 μM | 5.6 | 4.1% at 30 μM | 16.5% at 30 μM |
| 4t | O | H | I | 3% at 10 μM | 1.7 | 16.0% at 30 μM | 6.5% at 30 μM |
| 26 | ND | 3.5 | 0.096 ± 0.049 | 14.7% at 30 μM | |||
| 27 | ND | 44.9 | 38.5% at 30 μM | 0.00608 ± 0.0055 | |||
| 28 | ND | 12.4 | 59.9% at 30 μM | 0.00465 ± 0.0023 | |||
Effects of Compounds 4a–4t on Adiponectin Biosynthesis.
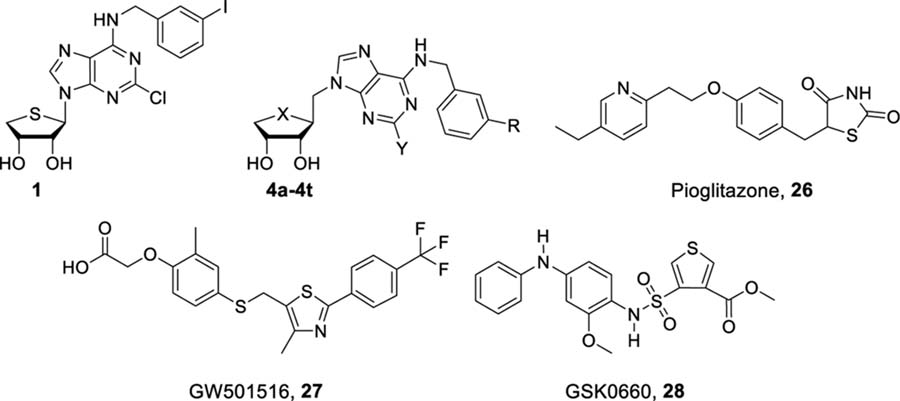
Next, the effect of compounds 4a–4t on adiponectin production was evaluated with the adipogenesis model of hBM-MSCs (Figure 3). hBM-MSCs have been used as a pharmacological tool for screening adiponectin secretion-inducing compounds by the phenotype.4,10 PPARs are known to contribute to insulin sensitivity, in part through the regulation of synthesis and secretion of adiponectin, an adipocyte-derived cytokine that suppresses gluconeogenesis in the liver.11 Adiponectin has received substantial attention because of its important role as an integrative modulator for communication between adipose tissue and other metabolism-related organs.11b,12 Adiponectin has an antiatherogenic and anti-inflammatory activity as well as an insulin-sensitizing activity.13 Thus, it has a potential in the treatment of various diseases, including type 2 diabetes, atherosclerosis, and cardiovascular disease.13 When 10 μM of each compound was cotreated with the adipogenesis-inducing medium consisting of insulin, dexamethasone, and 3-isobutyl-1-methylxanthine (IDX medium) for 5 d, compounds 4a, 4c, 4d, 4i, 4k, 4l, 4n, 4o, and 4p significantly promoted adiponectin production compared to the IDX vehicle control during adipogenesis in hBM-MSCs, indicating the correlation with PPAR-binding affinity (Figure 3A). As expected, compounds 4e–4h and 4q–4t with no substituent at the C2 position, which are also inactive at PPARs, could not significantly promote adiponectin production (Figure 3A). In phenotypic analysis with potent adiponectin secretion-promoting PPARγ/δ dual modulators, compounds 4d and 4p significantly promoted lipid droplet formation during adipogenesis in hBM-MSCs compared to the IDX control (Figure 3B). In concentration-effect analysis for the adiponectin production phenotypic assay, the half-maximal effective concentration values (EC50) of compounds 4d and 4p were 9.20 and 16.14 μM, respectively (Figure 3C).
Figure 3.
Effects of truncated 1′-homologated adenosine derivatives 4a–4t on adiponectin biosynthesis. (A) hBM-MSCs were grown under IDX conditions and cotreated with 26 or compounds 4a–4t. On the 5th day of adipogenesis induction, supernatants were harvested and adiponectin ELISA was performed. (B) ORO staining was performed to visualize lipid droplets after the 5-day adipogenesis induction. (C) Concentration-effect analysis was performed for compounds 4d and 4p. Results are the mean ± SD of three measurements using hBM-MSCs from three independent cultures (n = 3, three independent experiments); (*) p ≤ 0.05 and (**) p ≤ 0.01.
Evaluation of a Pharmacological Profile of PPARγ/δ Dual Modulators.
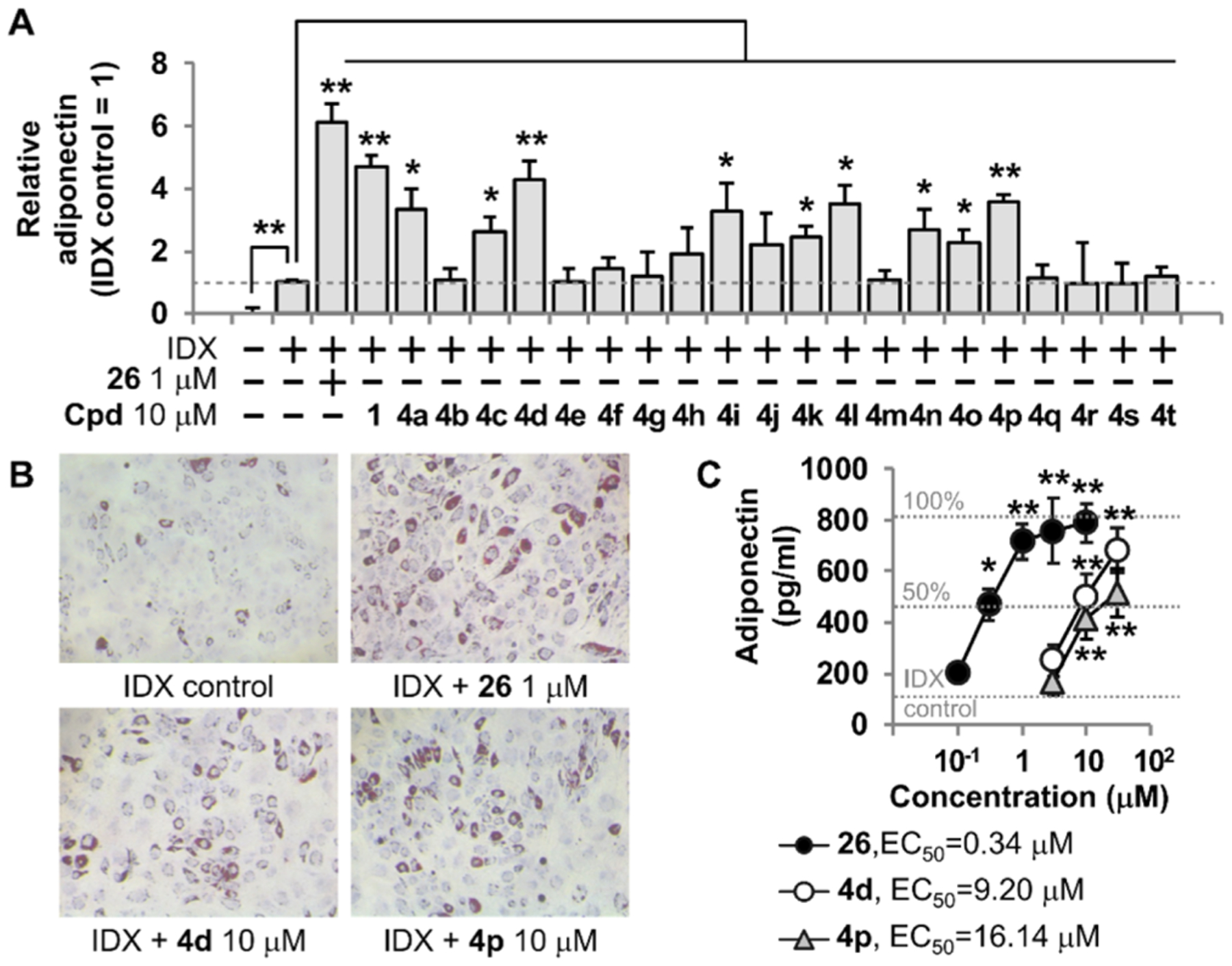
One of the polypharmacological features of 1 is a PPARγ partial agonist.4 To investigate how compounds 4a–4t functionally affected PPARγ activation, a TR-FRET-based PPARγ coactivator assay was performed for compounds 4a–4d, 4i–4l, and 4m–4p, which showed a significant PPARγ-binding activity (Figure 4). The level of interaction between the PPARγ-LBD and a fluorescein-labeled coactivator peptide thyroid hormone receptor-associated protein 220/vitamin D receptor interacting protein (TRAP220/DRIP) derived from MED1, a nuclear receptor coactivator encoded by MED1 gene, was measured.14 MED1, a component of the mediator complex, ligand dependently interacts with PPARγ and other nuclear receptors and regulates transcription of RNA polymerase II-dependent genes, by directly facilitating promoter recruitment and function of RNA polymerase II.15 MED1 is also known to be essential for PPARγ-mediated adipogenesis in mouse embryonic fibroblasts.16 Compounds 4a, 4c, 4d, 4i–4l, 4n, 4o, and 4p significantly increased the coactivator peptide recruitment. However, the effects were less potent compared to those of PPARγ agonist controls, N-(2-benzoylphenyl)-O-[2-(methyl-2-pyridinylamino)ethyl]-l-tyrosine hydrochloride (29, GW1929) and troglitazone (30) (Figure 4A). It is interesting to note that the binding affinity of 4j was weaker than that of 4i, 4k, and 4l, but 4j can still function as a PPARγ partial agonist. Because the maximum binding efficacy of 4j was around 40% of GW1929 (PPARγ full agonist), this binding level was enough for 4j to modulate the PPARγ function. It should be mentioned that compounds showing weak activity tend to show more biological variations, and 4j with weak activity had no statistical significance in coactivation analyses (Figure 4A). In the Pearson correlation analysis, the levels of the coactivator peptide recruitment on PPARγ-LBD by compounds 4a–4d, 4i–4l, and 4m–4p were significantly correlated to their adiponectin secretion-promoting activity (R2 = 0.71, p < 0.001) (Figure 4B). When the maximal activity for the coactivator peptide recruitment by a PPARγ full agonist 29 was taken as the 100% effect, EC50 values of compounds 4d and 4p were calculated as 6.85 and 9.95 μM, respectively (Figure 4C). Next, we evaluated whether compound 4d had characteristics of a PPARγ partial agonist because 1 exhibited a PPARγ partial agonism among its polypharmacological properties (Figure 4D). By pharmacological definition, a receptor partial agonist functions as a competitive antagonist against its full agonist.17 When compound 4d was cotreated with a PPARγ full agonist 30 in the TR-FRET-based PPAR coactivator assay, compound 4d competitively inhibited PPARγ coactivation of 30 (Figure 4D). Therefore, compound 4d and other PPARγ-binding compounds function as a PPARγ partial agonist.
Figure 4.
Effects of truncated 1′-homologated adenosine derivatives on PPARγ coactivation. (A) TR-FRET-based PPARγ coactivation assay was performed using fluorescein-TRAP220/DRIP peptide. PPARγ agonists 29 and 30 were used as positive controls. The degree of the recruiting coactivator peptide to PPARγ-LBD was normalized to % coactivation. (B) Coefficient of determination (R2) between the PPARγ coactivation activity and the level of relative adiponectin secretion-promoting activity was calculated using the Pearson correlation analysis. (C) Concentration-dependent effects of 4d and 4p on the TRAP220/DRIP peptide-based PPARγ coactivation were investigated. (D) Evaluation of the PPARγ partial agonism of 4d. Results are the mean ± SD of three measurements (n = 3); (*) p ≤ 0.05 and (**) p ≤ 0.01.
As the second polypharmacological feature of 1 is a PPARδ antagonist,4 we next examined whether the PPARδ binding compounds function as antagonists (Figure 5). To determine a functional outcome for the PPARδ antagonism, a TR-FRET-based PPARδ coactivator assay was performed. A fluorescein-labeled peptide, the interaction domain 2 (ID2) of silencing mediator of retinoid and thyroid hormone receptors (SMRT), was used as a corepressor and peptide C33 was used as a coactivator.18 SMRT, also known as nuclear receptor corepressor 2, can control various adipogenic set points and whole-body metabolic homeostasis by repressing nuclear receptors.19 Compounds 4a, 4c, 4d, 4n, 4o, and 4p significantly increased the interaction between PPARδ-LBD and the corepressor peptide, SMRT-ID2 (Figure 5A), whereas these compounds had no effect on the recruitment of coactivator peptide (Figure 5B). Compounds 4d and 4p showed concentration-dependent effects on the corepressor peptide binding to PPARδ-LBD, and their EC50 values were 12.53 and 20.20 μM, respectively (Figure 5C). Importantly, the levels of the corepressor peptide recruitment to PPARδ-LBD by compounds 4a–4d and 4m–4p showed a significant correlation to the adiponectin secretion-promoting activity (R2 = 0.80, p < 0.001) (Figure 5D).
Figure 5.

Effects of truncated 1′-homologated adenosine derivatives on PPARδ coregulation. (A) TR-FRET-based PPARδ corepression assay was performed using fluorescein-SMRT corepressor peptide. A PPARδ agonist 27 and a PPARδ antagonist 28 were used as positive or negative controls. The degree of the recruiting corepressor to PPARγ-LBD was normalized to % corepression. (B) TR-FRET-based PPARδ coactivation assay was performed using coactivator C33. (C) Concentration-dependent effects of 4d and 4p on the PPARδ corepression were investigated. (D) Pearson’s correlation coefficient (R2) between the PPARδ corepression activity and the level of relative adiponectin secretion-promoting activity was calculated using the Pearson correlation analysis. (E) Effects of PPARδ agonist 27 (1 μM) on PPARδ antagonism of 4d (10 μM) and 28 (1 μM). Results are the mean ± SD of three measurements (n = 3); (*) p ≤ 0.05 and (**) p ≤ 0.01.
Next, we investigated whether the PPARδ antagonism of 4d was affected by a specific PPARδ agonist 27 (Figure 5E). The recruitment of corepressor peptides by a PPARδ antagonist 28 was competitively inhibited by 27. The effect of 4d on the PPARδ corepressor recruitment was also significantly inhibited by 27 (Figure 5E). This result supported that 4d was a PPARδ antagonist. In conclusion, compound 4d and other PPARγ/δ compounds were dual modulators functioning as PPARγ partial agonists and PPARδ antagonists.
In Vivo Study of 4p in Noninsulin-Dependent Type 2 Diabetes Mellitus.
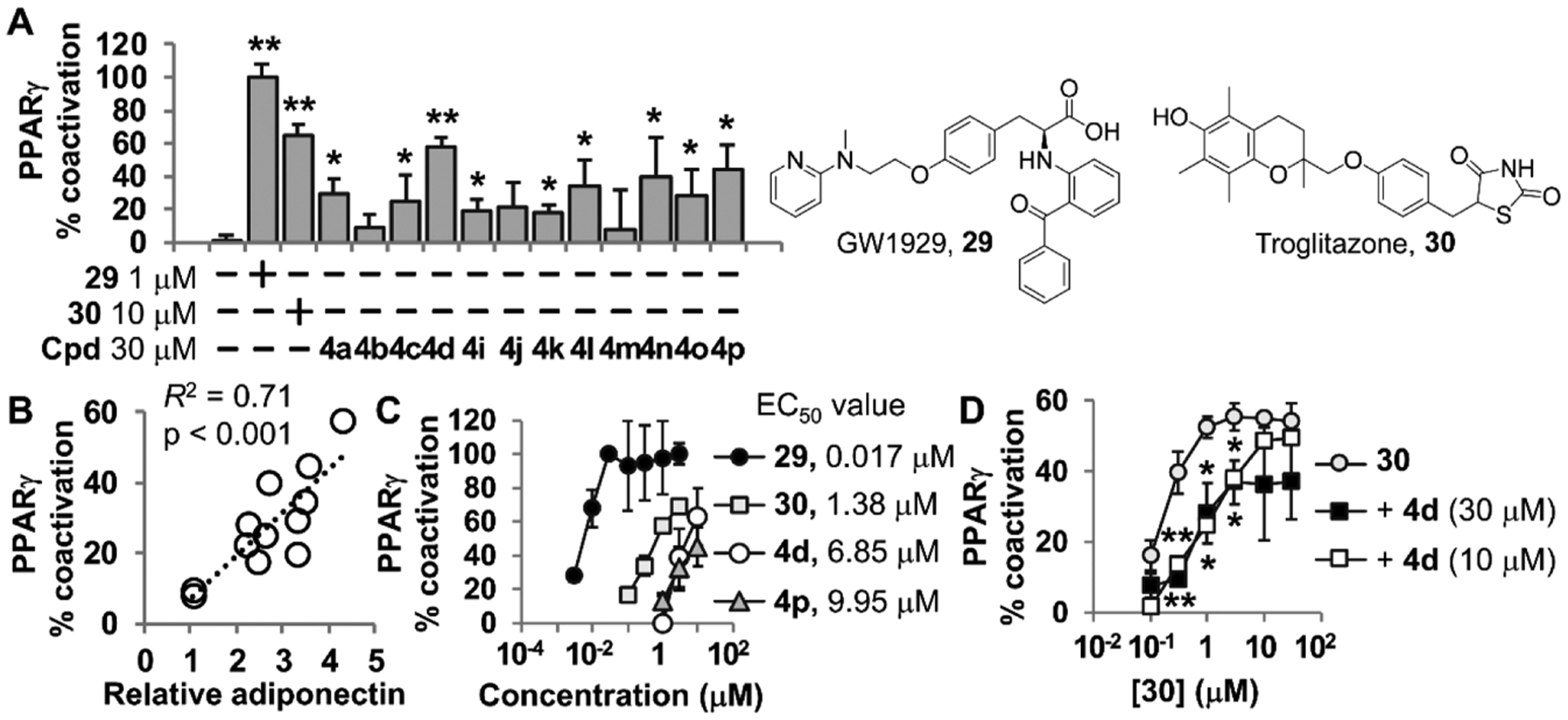
A leptin-deficient ob/ob mouse model is commonly used for in vivo study of noninsulin-dependent type 2 diabetes mellitus (T2D).20 We evaluated efficacy of 4p on glucose tolerance and insulin sensitivity of obese–diabetic ob/ob mice. 7 week old ob/ob mice were administered 30 mg/kg of 4p six times per week. Mice orally administered with pioglitazone were used as positive control. Glucose response curves during an oral glucose tolerance test (OGTT) were plotted after 3 and 6 weeks. The area under the curve of OGTT showed no difference between ob/ob and control mice, and ob/ob mice received 4p. Unfortunately, 4p administration for 6 weeks did not result in improving fasting glucose levels of ob/ob mice (see the Supporting Information; Figure S1). To examine the effects of 4p on T2D in more detail, the serum insulin level was measured through enzyme-linked immunosorbent assay (ELISA). Although the blood glucose levels were not significantly different, serum insulin levels of fed conditioned ob/ob mice were decreased after administration of 4p (Figure 6A). In addition, no weight gain was observed in the 4p administered group (Figure 6B). Although 4p administration could not normalize blood glucose levels of ob/ob mice, decreased serum insulin levels demonstrate potential use of 4p as a molecular entity for treatment of T2D.
Figure 6.
Effects of 4p on insulin sensitivity in ob/ob mice. (A) Fed serum insulin levels were analyzed through the mouse insulin ELISA kit. (B) Weight gain was calculated using body weights of mice prior to the experiment and after 6 weeks of administration. **p < 0.01 compared to lean mice; #p < 0.05 compared to vehicle-administered ob/ob mice.
Docking Analysis on the SAR of PPARγ/δ Dual Modulators.
The 1′-homologation of 1 resulted in the loss of the A3AR-binding affinity. To investigate the structural basis of A3AR activity of the target compounds, a homology model of the A3AR was constructed based on the human A2AAR structure in an active conformation (PDB ID: 5G53), and the optimum binding mode of 1 was determined by protein–ligand docking analysis (Figure 7B).21 A2AAR and A3AR showed 43.45% of sequence identity, indicating that the structure of A3AR can be predicted from the known A2AAR structure with an accuracy equivalent to a low-resolution X-ray structure.22 In the optimum binding mode, the adenine ring of 1 formed the π–π stacking interaction with Phe168 of extracellular loop 2 and interacted with amino acid residues Thr94, Ser271, and His272 through 4′-thiosugar (Figure 7B,C). Such interaction patterns of ligands in the binding pocket were observed in many cases including A2AAR-ligand co-crystallized structures and conserved in other AR subtypes.23,24 Notably, it was reported that the site-directed mutagenesis on Thr94 or His272 residue can significantly decrease the binding affinity of A3AR ligands regardless of their agonistic or antagonistic features.24 In contrast to the case of 1, it was difficult to determine the optimum docking model of compound 4d against the homology-modeled A3AR because of the lack of the A3AR-binding affinity. Among the various docking modes of 4d to A3AR, we selected the docking model in which the adenine ring of compound 4d formed a π–π stacking with Phe168 for comparing with the docking model of 1 (Figure 7A,B). The binding stabilization of 1 and 4d with the A3AR, calculated using AutoDock Vina, was −9.0 kcal/mol and −8.0 kcal/mol, respectively. The introduction of the methylene group in 1 made it harder to achieve a close contact with Thr94 and His272 (Figure 7C,D). In addition, the distance between the 3′-OH of 4d and the donor hydrogen of His272 was 6.5 Å, too far to form a hydrogen bonding interaction (Figure 7D), whereas the distance in the model of 1 was 2.9 Å (Figure 7C). Therefore, the lack of interaction with residues important for activity was responsible for the decrease of A3AR-binding affinity of 4d. One-carbon homologation exhibited significant outcomes to induce the loss of the A3AR-binding activity.
Figure 7.
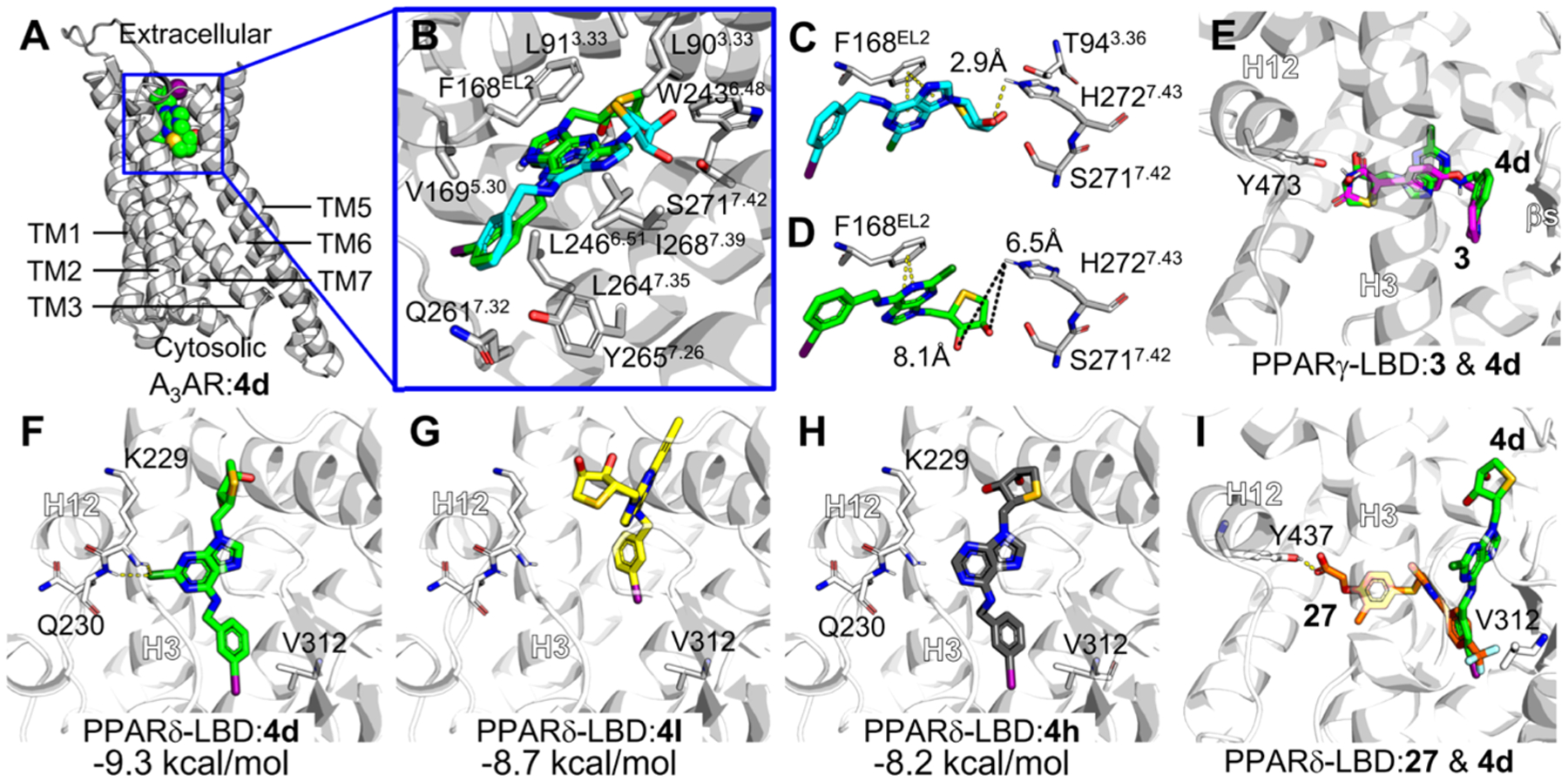
Molecular docking modes of compound 4d against A3AR, PPARγ, and PPARδ. (A) Optimum binding mode of compound 4d was generated in the A3AR homology model constructed based on the A2AAR structure (PDB ID: 5G53). TM, transmembrane. (B) Selected docking mode of compound 4d (green) for the comparison with A3AR antagonist 1 (cyan), forming π–π stacking with Phe168 with the adenine ring. Major amino acid residues interacting with compound 4d in the optimum docking mode against the A3AR homology model were labeled. The amino acid residues closely contacting with ligand (<van der Waals radii) or forming hydrogen bond with ligand are labeled. TM2 is shown transparently for clarity. The residue numbers were assigned following Ballesteros–Weinstein notation.27 (C,D) Comparison of binding models of 1 (cyan, C) and 4d (green, D) in the homology-modeled A3AR. Representative residues important for A3AR binding are shown in sticks. Distances between the hydroxyl group of thiosugar of ligands and hydrogen of His272 were measured, given in Ångstrom. (E) Putative binding modes suggested by docking simulation of 3 (magenta) and 4d against PPARγ-LBD (PDB ID: 5YCP). Binding models of 4d and 3 were overlaid. (F–I) Putative binding modes suggested by docking simulation of 4d (F), 4l (G), 4h (H) against PPARδ-LBD (PDB ID: 5U46). (I) Comparison of binding models of 27 (orange) and 4d in PPARδ-LBP. H, helix. Possible hydrogen bonds are shown in yellow dashes. Binding free energies calculated by AutoDock Vina are presented in kcal/ mol. All molecular graphics works were conducted using PyMOL software.
Next, to understand the molecular basis for the SAR of PPARγ/δ dual modulators, the binding modes for compounds 4d, 4h, and 4l against PPAR-LBDs were compared (Figure 7). The docking simulations of these compounds were performed with receptor 3-dimensional coordinates prepared from the PPARγ-LBD/3 complex structure (PDB ID: 5YCP).25 Notably, one-carbon homologation of the truncated adenosine derivatives was designed to improve PPARγ activity by structurally mimicking PPARγ full agonists like pioglitazone and rosiglitazone. In the energy-minimized docking model for the PPARγ-LBP, compound 3 or 4d interacted with amino acid residues in both the hydrophilic and hydrophobic pockets interacting with Tyr473 of helix (H) 12 (Figures 1 and 7E).
Similarly, PPARδ agonist 27-bound PPARδ-LBD was processed for docking analysis (PDB ID: 5U46). In the docking models against PPARδ-LBD, the 3-iodobenzyl group of 4d interacted hydrophobically with Val312, the essential amino acid residue significantly affecting the PPARδ ligand binding (Figure 7F).26 In addition, when compared with those of well-known PPARδ agonist 27, compound 4d in the optimum binding mode for the PPARδ binding was preferentially located between H3 and β-sheets, lacking an interaction with Tyr437, the amino acid residue important for the potency of PPARδ agonists through stabilizing H12 (Figure 7I).26 Meanwhile, 2-Cl of 4d was used as an anchor, forming a halogen bond with backbone nitrogen atoms of Lys229 and Gln230 on H2′–H3 loop of PPARδ-LBD. Compound 4d and its derivatives may have acted as antagonists by tightly occupying the space between H3 and the β-sheet, preventing PPARδ ligands from interacting with Val312 and Tyr437. On the contrary, compound 4l with bulky 2-propyne and compound 4h without 2-Cl could not form or maintain such binding models, resulting in weaker binding affinities compared to 4d (Figures 7G,H). Therefore, the docking simulation study further supported that compound 4d and its congeners were PPARγ/δ dual modulators, agonists for PPARγ and antagonists against PPARδ.
Therapeutic Potential of Adiponectin Secretion-Promoting PPARγ/δ Dual Modulators.
Compound 4d and its related compounds exhibited the unique PPAR dual modulator profile, which functioned as both PPARγ partial agonists and PPARδ antagonists. Most of PPAR dual- or pan-modulators have been developed as co-agonists with the expectation that the balanced PPAR activation may reduce serious side effects caused by highly specific PPARγ or PPARδ agonists. In fact, many selective PPARγ agonists and even PPARα/γ dual agonists were discontinued from their therapeutic use or clinical development because of their cardiovascular risks or other safety issues.3,28 In addition, PPARδ agonists have raised the controversial issues regarding to their increase in cancer risk.29 Compound 4d and its related compounds with the dual pharmacological profile of a partial agonist for PPARγ and an antagonist against PPARδ may have therapeutic benefits which are different from those of other dual- or pan-PPAR modulators.
Adiponectin secretion-promoting compound 4d may have the potential to treat various diseases associated with hypoadiponectinemia such as metabolic syndrome and cancer. It has been reported that the exogenous treatment of adiponectin improved the pathogenic phenotypes in animal models for atherosclerosis and nonalcoholic fatty liver diseases.30 Thus, the upregulation of adiponectin levels by adiponectin secretion-promoting PPARγ/δ dual modulators may provide therapeutic benefits to these metabolic conditions. Interestingly, a PPARγ partial agonist and a PPARδ antagonist may synergistically contribute to treat cancer. It has been reported that the activation of PPARγ induced apoptosis of cancer cells.31 PPARδ antagonists have been suggested as new drugs for antiproliferative therapeutics32 because a specific PPARδ agonist GW501516 significantly induced tumors in animal models.2a,29a Currently, the therapeutic potential of PPARδ antagonists has not been yet fully investigated compared with those of other PPAR modulators. Recently, it was reported that PPARδ antagonists were effective to attenuate the psoriasis-like pathogenic outcomes in a transgenic animal model.33 Psoriasis is a hyperproliferative skin disease, so the antipsoriatic activity of PPARδ antagonists may be reasonably expected. In this regard, the anticancer potential of a PPARγ/δ dual modulator 4d should be further investigated.
CONCLUSIONS
In this study, novel PPARγ/δ dual modulators functioning as both PPARγ partial agonists and PPARδ antagonists were designed and synthesized. These pure PPARγ/δ dual modulators were discovered from the polypharmacology of 1 that can simultaneously affect A3AR, PPARγ, and PPARδ, as well as the structural similarity between the target compounds 4a–4t and 1,3-thiazolidinedione antidiabetic drugs. Compounds 4d and 4p were discovered as the most promising compounds in 4′-thioand 4′-oxonucleosides, respectively, whose adiponectin secretion-promoting PPARγ/δ dual modulators may have therapeutic potential against cancer and metabolic diseases, and pharmacological studies to demonstrate their therapeutic benefits may be required in the near future.
EXPERIMENTAL SECTION
Chemical Synthesis.
General Methods.
Proton (1H) and carbon (13C) NMR spectra were obtained on a Bruker AV 400 (400/100 MHz), Bruker AMX 500 (500/125 MHz), Jeol JNM-ECA600 (600/150 MHz), or Bruker AVANCE III 800 (800/200 MHz) spectrometer. Chemical shifts are reported as parts per million (δ) relative to the solvent peak. Coupling constants (J) are reported in hertz (Hz). Mass spectra were recorded on a Thermo LCQ XP instrument. Optical rotations were determined on Jasco III in appropriate solvent. UV spectra were recorded on U-3000 made by Hitachi in methanol or water. Infrared spectra were recorded on Fourier transform infrared (FTS-135) made by Bio-Rad. Melting points were determined on a Buchan B-540 instrument and are uncorrected. The crude compounds were purified by column chromatography on a silica gel (Kieselgel 60, 70–230 mesh, Merck). Elemental analyses (C, H, and N) were used to determine the purity of all synthesized compounds, and the results were within ±0.4% of the calculated values, confirming ≥95% purity. High-performance liquid chromatography analysis was also performed to determine the purity of the most promising compounds 4d and 4p, confirming ≥95% purity. All animal experiments were performed in compliance with by Institutional Animal Care and Use Committee (IACUC, SNU-200605-5-1) in the Seoul National University.
(3aS,6R,6aS)-6-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-ol (5).
To a stirred suspension of d-mannose (1.74 g, 6.52 mmol) and 2,2-dimethoxypropane (2.45 mL, 19.55 mmol) in acetone (50 mL) was added portionwise camphosulfonic acid (0.45 g, 1.96 mmol) at 0 °C under a N2 atmosphere. After being stirred for 24 h at rt, the mixture was neutralized with Et3N and evaporated under reduced pressure. The resulting residue was purified by flash silica gel column chromatography (n-hexanes/EtOAc = 1:1) to give 5 (1.61 g, 95%) as a white solid: mp 120.3–120.5 °C; 1H NMR (CDCl3, 400 MHz): δ 5.34 (s, 1H), 4.76–4.79 (m, 1H), 4.58 (d, 1H, J = 6.0 Hz), 4.34–4.39 (m, 1H), 4.15 (dd, 1H, J = 3.6, 7.2 Hz), 4.00–4.08 (m, 2H); +11.71 (c 0.11, CH2Cl2); FAB-MS m/z: 261 (M + H+); Anal. Calcd for C12H20O6: C, 55.37; H, 7.74. Found: C, 55.30; H, 7.75.
((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)((4R,5R)-2,2-dimethyl-5-(((methylsulfonyl)oxy)methyl)-1,3-dioxolan-4-yl)methyl Methane-sulfonate (6). Reduction.
To a stirred solution of diacetonide 5 (1.50 g, 5.76 mmol) in EtOH (25 mL) was added, cautiously in several portions, sodium borohydride (440 mg, 11.53 mmol) at 0 °C under a N2 atmosphere, and the mixture was stirred at rt for 2 h. The mixture was neutralized with acetic acid and evaporated to remove solvent. The mixture was partitioned between EtOAc and water, and the organic layer was dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by flash silica gel column chromatography (n-hexanes/EtOAc = 1:1) to give the diol 11 (1.38 g, 92%) as a syrup: 1H NMR (CDCl3, 400 MHz): δ 4.33 (dd, J = 1.6, 7.2 Hz, 1H), 4.24–4.28 (m, 1H), 4.06–4.13 (m, 2H), 3.92–3.97 (m, 1H), 3.76–3.85 (m, 2H), 3.59–3.61 (m, 1H), 1.48 (s, 3H), 1.38 (s, 3H), 1.36 (s, 3H), 1.33 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 110.5, 109.5, 79.2, 77.6, 77.5, 71.4, 68.4, 62.0, 27.1, 25.7, 25.4; −3.88 (c 0.44, CH2Cl2); FAB-MS m/z: 263 (M + H+); Anal. Calcd for C12H22O6: C, 54.95; H, 8.45. Found: C, 54.90; H, 8.35.
Mesylation.
To a stirred solution of diol X (38.52 g, 146.85 mmol) and 4-DMAP (5.38 mg, 44.06 mmol) in a mixture of CH2Cl2 (300 mL) and Et3N (163.75 mL, 1.17 mol) was added cautiously dropwise methanesulfonyl chloride (47.59 mL, 587.42 mmol) at 0 °C under a N2 atmosphere. After being stirred for 1 h, at rt, the mixture was diluted with CH2Cl2 and washed with saturated NaHCO3 solution. The organic layer was dried with anhydrous MgSO4, filtered, and evaporated to give the dimesylate compounds as brownish syrup. It was purified by flash silica gel column chromatography (n-hexanes/EtOAc = 5:1) to give a dimesylate 6 (57.83 g, 94%) as a syrup; 1H NMR (CDCl3, 400 MHz): δ 4.75 (pseudo t, J = 7.4 Hz, 1H), 4.33–4.45 (m, 4 H), 4.06–4.20 (m, 3H), 3.12 (s, 3H), 3.07 (s, 3H), 1.51 (s, 3H), 1.43 (s, 3H), 1.37 (s, 3H), 1.33 (s, 3H); +38.32 (c 0.29, CH2Cl2); FAB-MS m/z: 419 (M + H+); Anal. Calcd for C14H26O10S2: C, 40.18; H, 6.26; S, 15.32. Found: C, 39.99; H, 6.65.
(3aR,4S,6aS)-4-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxole (7).
To a stirred solution of dimesylate (933.80 mg, 2.23 mmol) in DMF (50 mL) was added sodium sulfide (348.30 g, 4.46 mmol), and the mixture was stirred with heating at 80 °C for overnight under a N2 atmosphere. After the solvent was removed under reduced pressure, the residue was partitioned between EtOAc and water. The organic layer was dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by flash silica gel column chromatography (n-hexanes/EtOAc = 8:1) to give 7 (453.0 mg, 78%) as a syrup: 1H NMR (CDCl3, 400 MHz): δ 4.92 (dt, J = 1.8, 5.6 Hz, 1H), 4.72 (dd, J = 2.0, 6.0 Hz, 1H), 4.26–4.30 (m, 1H), 4.04 (s, 1H), 3.79 (t, J = 3.8 Hz, 1H), 3.31–3.32 (m, 1H), 3.19 (dd, J = 5.4, 12.0 Hz, 1H), 2.84 (dd, J = 1.6, 12.0 Hz, 1H), 1.51 (s, 3H), 1.43 (s, 3H), 1.32 (dd, J = 8.4 Hz, 6 H); 13C NMR (CDCl3): δ 111.5, 109.8, 87.3, 84.0, 78.0, 68.3, 56.3, 56.2, 38.8, 26.8, 26.3, 25.8, 24.9; −96.04 (c 0.20, CH2Cl2); FAB-MS m/z: 261 (M + H+); Anal. Calcd for C12H20O4S: C, 55.36; H, 7.74; S, 12.32. Found: C, 55.29; H, 7.85; S, 12.20.
(R)-1-((3aR,4S,6aS)-2,2-Dimethyltetrahydrothieno[3,4-d][1,3]-dioxol-4-yl)ethane-1,2-diol (8).
A solution of 7 (21.78 g, 83.66 mmol) in 60% aqueous AcOH (250 mL) was stirred at rt for 2 h. The reaction mixture was evaporated under reduced pressure, and the resulting residue was purified by flash silica gel column chromatography (n-hexanes/EtOAc = 1:2) to give the diol 8 (14.85 g, 81%) as a white solid, and 6.15 g of the starting material 13 was recovered after being repeated for three times: mp 100.7–101.8 °C; 1H NMR (CDCl3, 400 MHz): δ 4.93 (dt, J = 1.8, 5.6 Hz, 1H), 4.76 (dd, J = 2.0, 5.6 Hz, 1H), 3.76–3.80 (m, 1H), 3.70–3.74 (m, 1H), 3.64–3.68 (m, 1H), 3.40 (dd, J = 1.6, 5.2 Hz, 1H), 3.16 (dd, J = 5.0, 12.8 Hz, 1H), 2.90 (dd, J = 1.6, 12.8 Hz, 1H), 1.52 (s, 3H), 1.33 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 111.6, 87.3, 84.0, 72.3, 65.3, 57.7, 32.0, 26.9, 24.9; −52.38 (c 0.13, CH2Cl2); FAB-MS m/z: 221 (M + H+); Anal. Calcd for C9H16O4S: C, 49.07; H, 7.32; S, 14.56. Found: C, 49.47; H, 7.72; S, 14.15.
((3aR,4S,6aS)-2,2-Dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)methanol (9).
To a stirred solution of 8 (20 g, 90.8 mmol) in ethyl acetate (500 mL) was added Pb(OAc)4 (48.3 g, 109 mmol) at 0 °C under a N2 atmosphere, and the reaction mixture was stirred for 10 min at which time thin-layer chromatography (TLC) indicated the absence of the starting material. The reaction mixture was filtered, and the filtrate was diluted with EtOAc. The organic layer was washed with saturated aqueous NaHCO3 solution, dried over anhydrous MgSO4, and evaporated to give the aldehyde intermediate, which was used for the next step without further purification. To a stirred solution of aldehyde intermediate (5.6 g, 30.0 mmol) in EtOH (70 mL) was carefully added sodium borohydride (1.3 g, 33.6 mmol) in several portions at 0 °C, and the reaction mixture was stirred for 30 min at the same temperature and neutralized with glacial AcOH. After the removal of the solvent, the mixture was partitioned between EtOAc (150 mL) and brine (100 mL). The organic layer was dried over anhydrous MgSO4, filtered, and evaporated. The resulting residue was purified by silica gel column chromatography (n-hexanes/EtOAc = 2:1) to give 9 (5.2 g, 91%) as a colorless syrup: 1H NMR (CDCl3, 400 MHz): δ 1.27 (s, 3H), 1.47 (s, 3H), 2.68 (br s, 1H), 2.84 (dd, J = 2.0, 12.6 Hz, 1H), 3.06 (dd, J = 5.2, 13.0 Hz, 1H), 3.38 (m, 1H), 3.56 (dd, J = 2.8, 8.8 Hz, 2H), 4.66 (dd, J = 1.6, 5.6 Hz, 1H), 4.86 (td, J = 1.6, 4.8 Hz, 1H); 13C NMR (CDCl3): δ 111.2, 85.9, 83.7, 63.3, 56.6, 26.6, 24.7; FAB-MS m/z: 191 (M + H+).
(3aR,4R,6aS)-4-(Chloromethyl)-2,2-dimethyltetrahydrothieno-[3,4-d][1,3]dioxole (10).
To a stirred solution of 8 (8.5 g, 40.7 mmol) in anhydrous acetonitrile (100 mL) was added POCl3 (4.47 mL, 48.9 mmol) at 0 °C under a N2 atmosphere, and the reaction mixture was stirred for 30 min at rt. The reaction mixture was dried in vacuo and coevaporated with toluene twice to give 10, which was directly used for the next step without further purification: 1H NMR (CDCl3, 400 MHz): δ 4.89 (m, 1H), 4.81 (dd, J = 0.8, 5.6 Hz, 1H), 2.88 (m, 2H), 3.65 (dd, J = 4.0, 8.2 Hz, 1H), 3.44 (m, 1H), 3.08 (dd, J = 4.8, 13.2 Hz, 1H), 1.46 (s, 3H), 1.27 (s, 3H); 13C NMR (CDCl3): δ 111.3, 86.1, 83.5, 55.3, 45.7, 37.8, 26.5, 24.7.
General Procedure for the Condensation.
To a solution of 6-chloropurine (15.18 mmol) and 2,6-dichloropurine (15.18 mmol) in a solution of anhydrous DMF (50 mL) was added NaH (15.18 mmol), and the mixture was stirred at rt until the solution became clear. A solution of 10 (12.64 mmol) in anhydrous DMF (7 mL) was added to the resulting solution at rt and stirred overnight at rt. The reaction mixture was diluted with EtOAc (70 mL) and washed with water several times, dried with anhydrous MgSO4, and evaporated. The residue was purified by flash silica gel column chromatography (n-hexanes/EtOAc = 2:1) to give the condensed products 11 and 12, respectively.
2,6-Dichloro-9-(((3aR,4S,6aS)-2,2-dimethyltetrahydrothieno[3,4-d][1,3]dioxol-4-yl)methyl)-9H-purine (11).
Yield = 72%; white foam; UV (MeOH) λmax: 275 nm (pH 7); +173.3 (c 0.30, MeOH); FAB-MS m/z: 361 (M + H+); 1H NMR (CDCl3, 400 MHz): δ 8.23 (s, 1H), 4.89 (m, 1H), 4.69 (dd, J = 2.0, 5.6 Hz, 1H), 4.47 (dd, J = 6.8, 14.2 Hz, 1H), 4.21 (dd, J = 8.8, 14.2 Hz, 1H), 3.74 (td, J = 2.0, 6.2 Hz, 1H), 2.98 (m, 2H), 1.47 (s, 3H), 1.27 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 153.37, 153.32, 152.1, 146.1, 145.5, 112.2, 86.2, 83.3, 54.1, 46.1, 37.0, 26.6, 24.8; Anal. Calcd for C13H14Cl2N4O2S: C, 43.22; H, 3.91; N, 15.51; S, 8.88. Found: C, 43.29; H, 3.96; N, 15.63; S, 8.82.
6-Chloro-9-(((3aR,4S,6aS)-2,2-dimethyltetrahydrothieno[3,4-d]-[1,3]dioxol-4-yl)methyl)-9H-purine (12).
Yield = 67%; white foam; UV (MeOH) λmax: 265 nm (pH 7); +128.0 (c 0.25, MeOH); FAB-MS m/z: 327 (M + H+); 1H NMR (CDCl3, 400 MHz): δ 8.67 (s, 1H), 8.28 (s, 1H), 4.79 (m, 1H), 4.66 (dd, J = 2.0, 5.0 Hz, 1H), 4.59 (dd, J = 6.8, 14.0 Hz, 1H), 4.26 (dd, J = 8.0, 16.0 Hz, 1H), 3.72 (td, J = 1.2, 2.4 Hz, 1H), 2.88 (m, 2H), 1.38 (s, 3H), 1.19 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 152.1, 151.8, 151.0, 145.5, 131.1, 111.8, 86.0, 83.2, 54.1, 46.0, 36.9, 26.4, 24.7; Anal. Calcd for C13H15ClN4O2S: C, 47.78; H, 4.63; N, 17.14; S, 9.81. Found: C, 47.82; H, 4.65; N, 17.20; S, 9.85.
General Procedure for the N6-Substitution Reaction.
To a stirred solution of 11 or 12 in THF was added dropwise 2 N HCl. The acetonide group was deprotected by using HCl under a N2 atmosphere. After being stirred at rt for 5 h, the reaction mixture was evaporated under reduced pressure. The residue was purified by flash column chromatography (reverse phase silica gel, H2O/MeOH, 10/1). To a stirred solution of acetonide deprotected 6-chloropurine derivatives (0.45 mmol) or 2,6-dichloropurine derivatives (0.45 mmol) and an appropriate amine hydrochloride salts or free amines (0.90 mmol) in EtOH (10 mL) was added Et3N (1.35 mmol), and the solution was stirred overnight at rt. After removing the solvent under reduced pressure, the residue was purified by flash silica gel column chromatography (CH2Cl2/EtOAc/MeOH = 10:10:1) to give the N6-substituted amine derivatives 4a–h.
(2S,3R,4S)-2-((2-Chloro-6-((3-fluorobenzyl)amino)-9H-purin-9-yl)methyl)tetrahydrothiophene-3,4-diol (4a).
Yield = 82%; white solid; mp 164–167 °C; UV (MeOH) λmax: 275 nm (pH 7); +48.0 (c 0.50, MeOH); 1H NMR (DMSO-d6, 400 MHz): δ 8.83 (br s, 1H), 8.17 (s, 1H), 3.65 (m, 1H), 7.74 (s, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.36 (d, J = 7.6 Hz, 1H), 7.12 (t, J = 7.6 Hz, 1H), 5.09 (d, J = 6.0 Hz, 1H), 5.03 (d, J = 4.8 Hz, 1H), 4.60 (br d, J = 5.2 Hz, 2H), 4.43 (dd, J = 6.4, 14.0 Hz, 1H), 4.16 (m, 2H), 3.78 (m, 1H), 2.89 (dd, J = 4.8, 10.4 Hz, 1H), 2.61 (dd, J = 4.8, 10.8 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz): δ 154.7, 152.9, 149.9, 141.9, 141.6, 136.0, 135.5, 130.5, 126.8, 118.0, 94.7, 77.1, 73.5, 48.3, 47.0, 42.5, 32.9; FAB-MS m/z: 410 (M + H+); Anal. Calcd for C17H17ClFN5O2S: C, 49.82; H, 4.18; N, 17.09; S, 7.82. Found: C, 49.94; H, 4.25; N, 17.12; S, 7.85.
(2S,3R,4S)-2-((2-Chloro-6-((3-chlorobenzyl)amino)-9H-purin-9-yl)methyl)tetrahydrothiophene-3,4-diol (4b).
Yield = 77%; white solid; mp 154–158 °C; UV (MeOH) λmax: 275 nm (pH 7); +77.5 (c 0.40, MeOH); 1H NMR (DMSO-d6, 400 MHz): δ 8.84 (br s, 1H), 8.17 (s, 1H), 7.55 (s, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.35 (d, J = 7.6 Hz, 1H), 7.27 (t, J = 7.6 Hz, 1H), 5.09 (d, J = 5.6 Hz, 1H), 5.04 (d, J = 4.8 Hz, 1H), 4.63 (br d, J = 6.5 Hz, 2H), 4.44 (dd, J = 6.0, 14.0 Hz, 1H), 4.11–4.20 (m, 2H), 3.78 (m, 1H), 3.65 (m, 1H), 2.90 (dd, J = 4.8, 10.8 Hz, 1H), 2.62 (dd, J = 4.8, 10.6 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz): δ 154.7, 152.9, 149.9, 142.0, 141.6, 130.5, 130.1, 129.7, 126.4, 121.5, 118.0, 77.1, 73.5, 48.3, 47.0, 42.6, 32.9; FAB-MS m/z: 426 (M+); Anal. Calcd for C17H17Cl2N5O2S: C, 47.89; H, 4.02; N, 16.43; S, 7.52. Found: C, 47.92; H, 4.05; N, 16.50; S, 7.55.
(2S,3R,4S)-2-((6-((3-Bromobenzyl)amino)-2-chloro-9H-purin-9-yl)methyl)tetrahydrothiophene-3,4-diol (4c).
Yield = 73%; white solid; mp 165–169 °C; UV (MeOH) λmax: 274 nm (pH 7); +70.0 (c 0.50, MeOH); 1H NMR (DMSO-d6, 400 MHz): δ 8.84 (br s, 1H), 8.17 (s, 1H), 7.02–7.30 (m, 4 H), 5.10 (d, J = 5.6 Hz, 1H), 5.04 (d, J = 4.8 Hz, 1H), 4.65 (br d, J = 5.2 Hz, 2H), 4.44 (dd, J = 6.4, 13.8 Hz, 1H), 4.13–4.21 (m, 2H), 3.79 (m, 1H), 3.66 (m, 1H), 2.89 (dd, J = 5.2, 10.6 Hz, 1H), 2.62 (dd, J = 4.4, 10.6 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz): δ 163.3, 160.9, 154.8, 152.9, 149.9, 142.3, 141.6, 130.2, 130.2, 123.3, 118.0, 77.1, 73.6, 48.3, 47.0, 42.7, 32.9; FAB-MS m/z: 472 (M + H+); Anal. Calcd for C17H17BrClN5O2S: C, 43.37; H, 3.64; N, 14.88; S, 6.81. Found: C, 43.40; H, 3.70; N, 14.90; S, 6.85.
(2S,3R,4S)-2-((2-Chloro-6-((3-iodobenzyl)amino)-9H-purin-9-yl)-methyl)tetrahydrothiophene-3,4-diol (4d).
Yield = 79%; white solid; mp 174–180 °C; UV (MeOH) λmax: 274 nm (pH 7); +136.4 (c 0.33, MeOH); 1H NMR (DMSO-d6, 400 MHz): δ 8.84 (br s, 1H), 8.17 (s, 1H), 7.27–7.40 (m, 4 H), 5.09 (d, J = 5.6 Hz, 1H), 5.04 (d, J = 4.8 Hz, 1H), 4.64 (br d, J = 4.8 Hz, 2H), 4.44 (dd, J = 6.0, 14.0 Hz, 1H), 4.13–4.20 (m, 2H), 3.78 (m, 1H), 3.65 (m, 1H), 2.89 (dd, J = 4.8, 10.4 Hz, 1H), 2.62 (dd, J = 4.8, 10.6 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz): δ 154.7, 152.9, 149.9, 141.8, 141.6, 132.9, 130.1, 127.2, 126.8, 126.0, 118.0, 77.1, 73.6, 48.3, 47.0, 42.7, 32.9; FAB-MS m/z: 518 (M + H+); Anal. Calcd for C17H17ClIN5O2S: C, 39.43; H, 3.31; N, 13.53; S, 6.19. Found: C, 39.52; H, 3.35; N, 13.58; S, 6.22.
(2S,3R,4S)-2-((6-((3-Fluorobenzyl)amino)-9H-purin-9-yl)methyl)-tetrahydrothiophene-3,4-diol (4e).
Yield = 75%; white solid; mp 117–120 °C; UV (MeOH) λmax: 274 nm (pH 7); +53.3 (c 0.30, MeOH); 1H NMR (DMSO-d6, 400 MHz): δ 8.36 (br s, 1H), 8.21 (s, 1H), 8.16 (s, 1H), 7.73 (s, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.36 (d, J = 7.6 Hz, 1H), 7.10 (t, J = 8.0 Hz, 1H), 5.09 (d, J = 5.6 Hz, 1H), 5.02 (d, J = 4.8 Hz, 1H), 4.66 (br s, 2H), 4.47 (dd, J = 6.0, 14.2 Hz, 1H), 4.19 (dd, J = 7.6, 14.0 Hz, 1H), 4.14 (m, 1H), 3.78 (m, 1H), 3.68 (m, 1H), 2.88 (dd, J = 5.2, 10.4 Hz, 1H), 2.61 (dd, J = 5.2, 10.8 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz): δ 154.1, 152.3, 148.9, 142.9, 141.0, 135.7, 135.3, 130.4, 126.6, 118.9, 94.7, 76.8, 73.5, 48.7, 46.8, 42.2, 32.8; FAB-MS m/z: 376 (M + H+); Anal. Calcd for C17H18FN5O2S: C, 54.39; H, 4.83; N, 18.65; S, 8.54. Found: C, 54.32; H, 4.96; N, 18.77; S, 8.62.
(2S,3R,4S)-2-((6-((3-Chlorobenzyl)amino)-9H-purin-9-yl)methyl)-tetrahydrothiophene-3,4-diol (4f).
Yield = 83%; white solid; mp 143–147 °C; UV (MeOH) λmax: 275 nm (pH 7); +71.8 (c 0.32, MeOH); 1H NMR (DMSO-d6, 400 MHz): δ 8.38 (br s, 1H), 8.22 (s, 1H), 8.16 (s, 1H), 7.54 (s, 1H), 7.39 (d, J = 7.6 Hz, 1H), 7.35 (d, J = 7.6 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 5.08 (d, J = 5.6 Hz, 1H), 5.01 (d, J = 4.8 Hz, 1H), 4.69 (br s, 2H), 4.46 (dd, J = 8.0, 14.0 Hz, 1H), 4.19 (dd, J = 7.6, 14.0 Hz, 1H), 4.16 (m, 1H), 3.78 (m, 1H), 3.68 (m, 1H), 2.87 (dd, J = 5.2, 10.4 Hz, 1H), 2.61 (dd, J = 5.2, 10.4 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz): δ 154.1, 152.3, 148.9, 143.0, 141.0, 130.4, 129.8, 129.4, 126.2, 121.5, 118.9, 76.8, 73.5, 48.7, 46.8, 42.4, 32.8; FAB-MS m/z: 392 (M + H+); Anal. Calcd for C17H18ClN5O2S: C, 52.10; H, 4.63; N, 17.87; S, 8.18. Found: C, 52.33; H, 4.64; N, 17.90; S, 8.16.
(2S,3R,4S)-2-((6-((3-Bromobenzyl)amino)-9H-purin-9-yl)methyl)-tetrahydrothiophene-3,4-diol (4g).
Yield = 78%; white solid; mp 134–139 °C; UV (MeOH) λmax: 270 nm (pH 7); +72.0 (c 0.25, MeOH); 1H NMR (DMSO-d6, 400 MHz): δ 8.37 (br s, 1H), 8.21 (s, 1H), 8.16 (s, 1H), 7.39 (s, 1H), 7.31 (m, 3H), 5.08 (d, J = 5.6 Hz, 1H), 5.02 (d, J = 5.2 Hz, 1H), 4.70 (br s, 2H), 4.47 (dd, J = 6.4, 14.0 Hz, 1H), 4.19 (dd, J = 7.6, 14.2 Hz, 1H), 4.14 (m, 1H), 3.78 (m, 1H), 3.67 (m, 1H), 2.87 (dd, J = 4.8, 10.2 Hz, 1H), 2.62 (dd, J = 5.2, 10.6 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz): δ 154.2, 152.3, 148.9, 142.8, 141.0, 132.8, 130.1, 126.9, 126.5, 125.8, 118.9, 76.8, 73.5, 48.7, 46.8, 42.4, 32.1; FAB-MS m/z: 436 (M + H+); Anal. Calcd for C17H18BrN5O2S: C, 46.80; H, 4.16; N, 16.05; S, 7.35. Found: C, 46.89; H, 4.21; N, 16.07; S, 7.41.
(2S,3R,4S)-2-((6-((3-Iodobenzyl)amino)-9H-purin-9-yl)methyl)-tetrahydrothiophene-3,4-diol (4h).
Yield = 75%; white solid; mp 172–175 °C; UV (MeOH) λmax: 272 nm (pH 7); +68.5 (c 0.35, MeOH); 1H NMR (DMSO-d6, 400 MHz): δ 8.36 (br s, 1H), 8.21 (s, 1H), 8.16 (s, 1H), 7.32 (m, 1H), 7.00–7.19 (m, 3H), 5.08 (d, J = 5.6 Hz, 1H), 5.02 (d, J = 4.8 Hz, 1H), 4.71 (br s, 2H), 4.46 (dd, J = 6.4, 14.0 Hz, 1H), 4.19 (dd, J = 7.6, 18.0 Hz, 1H), 4.14 (m, 1H), 3.78 (m, 1H), 3.67 (m, 1H), 2.87 (dd, J = 5.2, 10.6 Hz, 1H), 2.61 (dd, J = 5.2, 10.4 Hz, 1H); 13C NMR (DMSO-d6, 100 MHz): δ 163.3, 160.9, 154.2, 152.3, 148.9, 143.2, 141.0, 130.1, 130.0, 123.1, 118.9, 76.8, 73.5, 48.7, 46.8, 42.5, 32.8; FAB-MS m/z: 484 (M + H+); Anal. Calcd for C17H18IN5O2S: C, 42.25; H, 3.75; N, 14.49; S, 6.63. Found: C, 42.34; H, 3.78; N, 14.51; S, 6.65.
6-Chloro-9-(((3aR,4S,6aS)-2,2-Dimethyltetrahydrothieno[3,4-d]-[1,3]dioxol-4-yl)methyl)-2-iodo-9H-purine (18).
To a stirred solution of 2-iodo-6-chloropurine (1.23 g, 4.4 mmol) and triphenylphosphine (Ph3P) (1.90 g, 4.4 mmol) in anhydrous THF (20 mL) was added diisopropyl azodicarboxylate (DIAD) (1.44 mL, 9.16 mmol) in THF (10 mL) at 0 °C under a N2 atmosphere, and the mixture was stirred at the same temperature for 15 min. To this solution was added a solution of compound 9 (0.5 g, 2.62 mmol) in THF (10 mL) at 0 °C, and the reaction mixture was stirred at rt for 1 h. The reaction mixture was concentrated under reduced pressure, and the crude residue was purified by flash silica gel column chromatography (n-hexanes/EtOAc = 5:1) to give 18 (0.79 g, 67%) as a white solid: 1H NMR (CDCl3, 300 MHz): δ 8.10 (s, 1H), 4.86 (m, 1H), 4.71 (dd, J = 1.6, 5.6 Hz, 1H), 4.44 (dd, J = 6.6, 14.4 Hz, 1H), 4.22 (dd, J = 8.2, 14.0 Hz, 1H), 3.72 (t, J = 6.6 Hz, 1H), 2.94 (s, 1H), 2.94 (s, 1H), 1.47 (s, 3H), 1.26 (s, 3H); Anal. Calcd for C13H14ClIN4O2S: C, 34.49; H, 3.12; N, 12.38. Found: C, 34.29; H, 3.22; N, 12.49.
6-Chloro-9-(((3aR,4S,6aS)-2,2-dimethyltetrahydrothieno[3,4-d]-[1,3]dioxol-4-yl)methyl)-2-(prop-1-yn-1-yl)-9H-purine (19).
To a stirred solution of 18 (315 mg, 0.70 mmol) in DMF (5 mL) were added cesium carbonate (274 mg, 0.84 mmol, 1.2 equiv), tetrakis-(triphenylphosphine)palladium(0) (81 mg, 0.07 mmol, 0.1 equiv), and copper(I) iodide (16 mg, 0.084 mmol, 0.12 equiv) at rt under a N2 atmosphere. After being stirred at the same temperature for 3 h, the reaction mixture was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (n-hexanes/EtOAc = 1:1) to give 19 as colorless oil (165 mg, 0.45 mmol, 65%): 1H NMR (CDCl3, 500 MHz): δ 8.20 (s, 1H), 4.85 (quin, J = 3.2 Hz, 1H), 4.67 (dd, J = 1.6, 5.6 Hz, 1H), 4.45 (dd, J = 6.8, 14.4 Hz, 1H), 4.22 (dd, J = 8.2, 14.4 Hz, 1H), 3.74 (t, J = 8.2 Hz, 1H), 2.95 (s, 1H), 2.95 (s, 1H), 2.13 (s, 3H), 1.46 (s, 3H), 1.26 (s, 3H); Anal. Calcd for C16H17ClN4O2S: C, 52.67; H, 4.70; N, 15.36. Found: C, 52.69; H, 4.77; N, 15.16.
General Procedure for the N6-Substitution Reaction.
To a stirred solution of 6-chloropurine derivative and an appropriate free amines (3 equiv) in EtOH (0.2 M) was added Et3N (3 equiv), and the solution was stirred overnight at rt under a N2 atmosphere. After removing the solvent under reduced pressure, the residue was purified by flash silica gel column chromatography (CH2Cl2/MeOH) to give the N6-substituted amine derivatives 4i–4l.
(2S,3R,4S)-2-((6-((3-Fluorobenzyl)amino)-2-(prop-1-yn-1-yl)-9H-purin-9-yl)methyl)tetrahydrothiophene-3,4-diol (4i).
Yield = 47%; white solid; mp 118–121 °C; 1H NMR (CD3OD, 500 MHz): δ 8.10 (s, 1H), 7.31 (dd, J = 7.6, 13.9 Hz, 1H), 7.19 (d, J = 7.6 Hz, 1H), 7.11 (d, J = 9.9 Hz, 1H), 6.96 (ddd, J = 2.2, 8.5, 10.5 Hz, 1H), 4.80 (br s, 2H), 4.53 (dd, J = 5.6, 14.2 Hz, 1H), 4.32 (dd, J = 7.5, 14.2 Hz, 1H), 4.23 (dd, J = 4.1, 7.9 Hz, 1H), 3.87 (dd, J = 3.4, 6.6 Hz, 1H), 3.75 (dd, J = 6.6, 12.9 Hz, 1H), 2.98 (dd, J = 4.8, 11.0 Hz, 1H), 2.73 (dd, J = 4.1, 11.0 Hz, 1H), 2.05 (s, 3H); 13C NMR (CD3OD, 125 MHz): δ 166.1, 164.2, 156.5, 148.7, 144.2, 143.9, 132.0 (d, J = 8.0 Hz), 125.1, 120.3, 116.0 (d, J = 21.7 Hz), 115.6 (d, J = 21.3 Hz), 84.6, 81.3, 79.9, 76.3, 50.7, 48.8, 45.4, 34.7, 4.4; −61.7 (c 0.08, MeOH); ESI-MS m/z: 414.1382 (M + H+); Anal. Calcd for C20H20FN5O2S: C, 58.10; H, 4.88; N, 16.94. Found: C, 58.40; H, 4.58; N, 16.64.
(2S,3R,4S)-2-((6-((3-Chlorobenzyl)amino)-2-(prop-1-yn-1-yl)-9H-purin-9-yl)methyl)tetrahydrothiophene-3,4-diol (4j).
Yield = 47%; white solid; mp 125–128 °C; 1H NMR (CD3OD, 500 MHz): δ 8.10 (s, 1H), 7.39 (s, 1H), 7.27–7.34 (m, 2H), 7.21–7.26 (m, 1H), 4.78 (br s, 2H), 4.53 (dd, J = 5.7, 14.2 Hz, 1H), 4.32 (dd, J = 7.5, 14.2 Hz, 1H), 4.23 (dd, J = 4.1, 8.0 Hz, 1H), 3.87 (dd, J = 3.4, 6.6 Hz, 1H), 3.75 (dd, J = 6.7, 13.0 Hz, 1H), 2.98 (dd, J = 4.8, 11.1 Hz, 1H), 2.73 (dd, J = 4.1, 11.0 Hz, 1H), 2.05 (s, 3H); 13C NMR (CD3OD, 125 MHz): δ 156.5, 148.7, 143.9, 143.7, 136.1, 131.8, 129.8, 129.4, 129.0, 127.8, 120.3, 84.6, 81.3, 79.9, 76.3, 50.4, 48.8, 45.3, 34.7, 4.3; −125.0 (c 0.05, MeOH); ESI-MS m/z: 430.1087 (M + H+); Anal. Calcd for C20H20ClN5O2S: C, 55.87; H, 4.69; N, 16.29. Found: C, 55.57; H, 4.89; N, 16.59.
(2S,3R,4S)-2-((6-((3-Bromobenzyl)amino)-2-(prop-1-yn-1-yl)-9H-purin-9-yl)methyl)tetrahydrothiophene-3,4-diol (4k).
Yield = 41%; white solid; mp 124–126 °C; 1H NMR (CD3OD, 500 MHz): δ 8.10 (s, 1H), 7.56 (s, 1H), 7.39 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 7.7 Hz, 1H), 7.23 (t, J = 7.8 Hz, 1H), 4.77 (br s, 2H), 4.53 (dd, J = 5.6, 14.2 Hz, 1H), 4.32 (dd, J = 7.5, 14.2 Hz, 1H), 4.23 (dd, J = 4.1, 8.0 Hz, 1H), 3.87 (dd, J = 3.4, 6.6 Hz, 1H), 3.75 (dd, J = 6.6, 13.0 Hz, 1H), 2.98 (dd, J = 4.8, 11.0 Hz, 1H), 2.73 (dd, J = 4.1, 11.1 Hz, 1H), 2.06 (s, 3H); 13C NMR (CD3OD, 125 MHz): δ 156.4, 151.0, 148.7, 143.9, 142.4, 132.4, 132.1, 132.0, 128.2, 124.2, 120.3, 84.6, 81.3, 79.9, 76.3, 50.4, 48.7, 45.1, 34.7, 4.4; −75.4 (c 0.05, MeOH); ESI-MS m/z: 476.0559 (M + H+); Anal. Calcd for C20H20BrN5O2S: C, 50.64; H, 4.25; N, 14.76. Found: C, 50.84; H, 4.45; N, 14.96.
(2S,3R,4S)-2-((6-((3-Iodobenzyl)amino)-2-(prop-1-yn-1-yl)-9H-purin-9-yl)methyl)tetrahydrothiophene-3,4-diol (4l).
Yield = 51%; white solid; mp 116–118 °C; 1H NMR (CD3OD, 500 MHz): δ 8.10 (s, 1H), 7.77 (s, 1H), 7.59 (d, J = 7.7 Hz, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.08 (t, J = 7.8 Hz, 1H), 4.74 (br s, 2H), 4.53 (dd, J = 5.7, 14.2 Hz, 1H), 4.32 (dd, J = 7.5, 14.2 Hz, 1H), 3.87 (dd, J = 3.4, 6.6 Hz, 1H), 3.75 (dd, J = 6.7, 13.0 Hz, 1H), 2.98 (dd, J = 4.8, 11.0 Hz, 1H), 2.73 (dd, J = 4.1, 11.0 Hz, 1H), 2.06 (s, 3H); 13C NMR (CD3OD, 125 MHz): δ 156.4, 151.0, 148.7, 143.9, 143.8, 138.6, 138.1, 132.1, 128.8, 127.1, 120.3, 95.7, 84.6, 81.3, 79.9, 76.3, 50.4, 48.8, 34.7, 4.4; −99.6 (c 0.05, MeOH); ESI-MS m/z: 522.0441 (M + H+); Anal. Calcd for C20H20IN5O2S: C, 46.07; H, 3.87; N, 13.43. Found: C, 46.27; H, 3.97; N, 13.63.
(3aS,6S,6aS)-6-(Hydroxymethyl)-2,2-dimethyltetrahydrofuro-[3,4-d][1,3]dioxol-4-ol (20).
To a stirred suspension of d-ribose (4 g, 26.64 mmol) in acetone (50 mL) was added dropwise conc. H2SO4 (0.12 mL) at rt, and the reaction mixture was stirred at rt for 3 h. The mixture was neutralized with solid NaHCO3, filtered, and evaporated under reduced pressure to give a colorless syrup. The residue was purified by silica gel column chromatography using hexane and ethyl acetate (1:2) as the eluent to afford 20 as a colorless syrup (4.3 g, 85%): +25.3 (c 1.05, CHCl3), lit.34 +21.3 (c 1.02, CHCl3); 1H NMR (CD3OD, 400 MHz), δ 5.26 (s, 1H), 4.77 (d, J = 6.0 Hz, 1H), 4.52 (d, J = 6.0 Hz, 1H), 4.19 (irregular t, J = 4.4, 5.2 Hz, 1H), 3.63 (dd, J = 4.8, 12.0 Hz, 1H), 3.59 (dd, J = 5.6, 12.0, 1H), 1.44 (s, 3H), 1.31 (s, 3H); Anal. Calcd for C8H14O5: C, 50.52; H, 7.42. Found: C, 50.48; H, 7.36.
(S)-1-((4S,5R)-5-(Hydroxymethyl)-2,2-dimethyl-1,3-dioxolan-4-yl)-2-(trityloxy)ethanol (21).
To a stirred solution of 20 (2.01 g, 10.56 mmol) in pyridine (36 mL, 0.3 M) were added trityl chloride (3.24 g, 11.62 mmol) and 4-dimethylaminopyridine (258 mg, 2.11 mmol) at 0 °C, and the mixture was stirred with heating at 80 °C under a N2 atmosphere. After being stirred for 3 h, the mixture was cooled, diluted with EtOAc, and washed with saturated NH4Cl solution and brine and evaporated to give the hemiacetal intermediate, which was used for the next step without further purification. To a stirred solution of the intermediate in methanol was added portionwise sodium borohydride (1.19 g, 31.68 mmol) at 0 °C under a N2 atmosphere. After being stirred for 1 h, the mixture was evaporated and the resulting white suspension was diluted with EtOAc and washed with saturated NH4Cl solution and brine. The separated organic layer was evaporated, and the residue was purified by silica gel column chromatography (n-hexanes/EtOAc = 3:1) to give diol 21 (2.93 g, 64% for 2 step) as a syrup: 1H NMR (CDCl3, 400 MHz): δ 7.40–7.46 (m, 6 H), 7.28–7.34 (m, 6 H), 7.22–7.28 (m, 3H), 4.29–4.36 (m, 1H), 4.10 (dd, J = 5.8, 9.6 Hz, 1H), 3.70–3.90 (m, 3H), 3.47 (dd, J = 3.0, 9.8 Hz, 1H), 3.31 (dd, J = 6.8, 9.8 Hz, 1H), 3.01 (dd, J = 5.0, 8.7 Hz, 1H), 2.89 (d, J = 3.9 Hz, 1H), 1.309 (s, 3H), 1.30 (s, 3H); 13C NMR (CDCl3, 100 MHz): δ 149.8, 147.6, 143.6, 135.9, 128.6, 127.9, 127.2, 123.7, 108.5, 87.1, 69.0, 65.0, 60.7, 27.8, 25.3; Anal. Calcd for C27H30O5: C, 74.63; H, 6.96. Found: C, 74.53; H, 6.66.
(3aS,4S,6aR)-2,2-Dimethyl-4-((trityloxy)methyl)tetrahydrofuro-[3,4-d][1,3]dioxole (22).
To a cooled (0 °C) solution of 21 (14.18 g, 32.64 mmol) in anhydrous pyridine (110 mL, 0.3 M) were added 4-toluenesulfonyl chloride (12.44 g, 65.28 mmol, 2 equiv) and 4-dimethylaminopyridine (0.39 g, 3.26 mmol, 0.1 equiv) under a N2 atmosphere. After the reaction mixture was being stirred with heating (80 °C) for 3 h, the reaction mixture was cooled and quenched with saturated ammonium chloride solution. The organic layer was washed with brine, dried with anhydrous MgSO4, and evaporated. The residue was purified by silica gel column chromatography (n-hexanes/EtOAc = 30:1) to give 22 as a colorless syrup (11.41 g, 84%): 1H NMR (CDCl3, 500 MHz): δ 7.40 (d, J = 7.5 Hz, 6 H), 7.29 (dd, J = 7.3, 15.2 Hz, 6 H), 7.20–7.25 (m, 3H), 4.87 (t, J = 5.1 Hz, 1H), 4.64 (d, J = 6.1 Hz, 1H), 4.20 (dd, J = 3.8, 7.7 Hz, 1H), 4.15 (dd, J = 4.3, 10.2 Hz, 1H), 4.04 (d, J = 10.2 Hz, 1H), 3.25 (dd, J = 4.0, 10.0 Hz, 1H), 3.09 (dd, J = 4.3, 10.0 Hz, 1H), 1.50 (s, 3H), 1.32 (s, 3H); 13C NMR (CDCl3, 125 MHz): δ 143.6, 128.6, 127.8, 127.0, 112.3, 87.0, 84.0, 83.0, 81.6, 74.3, 64.5, 26.6, 25.0; Anal. Calcd for C27H28O4: C, 77.86; H, 6.78. Found: C, 77.65; H, 6.59.
((3aS,4S,6aR)-2,2-Dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methanol (23).
To a cooled (−40 °C) solution of 22 (1.04 g, 2.50 mmol) in anhydrous CH2Cl2 (12.5 mL, 0.2 M) was added dropwise diethylaluminium chloride 1.0 M solution in hexanes (7.50 mL, 7.50 mmol, 3 equiv) under a N2 atmosphere. After the reaction mixture was stirred at the same temperature for 3 h, the mixture was quenched with saturated potassium sodium tartrate solution and stirred for additional 1 h. The organic layer was washed with brine, dried with anhydrous MgSO4, and evaporated at 5 °C. The residue was purified by silica gel column chromatography (n-hexanes/EtOAc = 4:1) to give 23 as a colorless syrup (0.256 g, 59%): 1H NMR (CDCl3, 500 MHz): δ 4.79 (m, 1H), 4.58 (dd, J = 1.9, 6.3 Hz, 1H), 4.11 (ddd, J = 2.1, 3.9, 6.2 Hz, 1H), 3.95 (m, 2H), 3.65 (dd, J = 3.7, 11.6 Hz, 1H), 3.58 (dd, J = 6.7, 11.5 Hz, 1H), 1.50 (s, 3H), 1.32 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 112.9, 84.8, 81.8, 81.0, 72.8, 61.7, 26.6, 24.9; Anal. Calcd for C8H14O4: C, 55.16; H, 8.10. Found: C, 55.36; H, 8.30.
General Procedure for the Synthesis of 4m–4t.
To a stirred solution of 6-chloropurine or 2,6-dichloropurine (1.7 equiv) and triphenylphosphine (Ph3P) (1.7 equiv) in anhydrous THF (0.15 M) was added DIAD (3.5 equiv) in anhydrous THF (0.3 M) at 0 °C under a N2 atmosphere, and the mixture was stirred at the same temperature for 15 min. To this solution was added a solution of compound 23 (1 equiv) in anhydrous THF (0.3 M) at 0 °C, and the reaction mixture was stirred at rt for 1 h. The reaction mixture was concentrated under reduced pressure, and the crude residue was purified by flash silica gel column chromatography to give 24 and 25 contaminated with triphenylphosphine oxide, respectively. To a stirred solution of 24 or 25 and an appropriate amine hydrochloride salts or free amines (3 equiv) in anhydrous THF (0.3 M) was added Et3N (3 equiv), and the solution was stirred overnight at rt. After removing the solvent under reduced pressure, the residue was purified by flash silica gel column chromatography to give the triphenylphosphine oxide contaminated with the N6-substituted amine intermediates. To a stirred solution of N6-substituted amine intermediates in methanol (0.1 M) was added dropwise 1 N HCl (0.1 equiv) at rt. After being stirred at rt at which time TLC indicated the absence of the starting material, the reaction mixture was evaporated under reduced pressure. The residue was purified by flash column chromatography (reverse phase silica gel, H2O/MeOH, 10/1) to give 4m–4t.
2,6-Dichloro-9-(((3aS,4S,6aR)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl)-9H-purine (24).
1H NMR (CD3OD, 400 MHz): δ 8.51 (s, 1H), 4.83–4.90 (m, 1H), 4.31–4.39 (m, 3H), 4.06 (dd, J = 3.6, 10.8 Hz, 1H), 3.90 (d, J = 10.8 Hz, 1H), 1.41 (s, 3H), 1.30 (s, 3H).
6-Chloro-9-(((3aS,4S,6aR)-2,2-dimethyltetrahydrofuro[3,4-d]-[1,3]dioxol-4-yl)methyl)-9H-purine (25).
1H NMR (CDCl3, 400 MHz): δ 8.77 (s, 1H), 8.22 (s, 1H), 4.78–4.81 (m, 1H), 4.55 (dd, J = 2.5, 6.2 Hz, 1H), 4.47 (dd, J = 2.4, 12.8 Hz, 1H), 4.28–4.37 (m, 2H), 3.97–4.05 (m, 2H), 1.50 (s, 3H), 1.33 (s, 3H).
(2S,3R,4R)-2-((2-Chloro-6-((3-fluorobenzyl)amino)-9H-purin-9-yl)methyl)tetrahydrofuran-3,4-diol (4m).
Yield = 48%; white solid; mp 185–186 °C; 1H NMR (CD3OD, 400 MHz): δ 8.00 (s, 1H), 7.30 (ddd, J = 6.0, 7.6, 13.6 Hz, 1H), 7.18 (d, J = 7.6 Hz, 1H), 7.10 (d, J = 10.0 Hz, 1H), 6.95 (ddd, J = 2.4, 8.4, 11.2 Hz, 1H), 4.41 (dd, J = 3.6, 14.8 Hz, 1H), 4.27 (dd, J = 6.8, 14.4 Hz, 1H), 3.94–4.04 (m, 3H), 3.78 (dd, J = 4.4, 7.2 Hz, 1H), 3.68 (dd, J = 2.0, 9.2 Hz, 1H); 13C NMR (CD3OD, 100 MHz): δ 165.7, 164.5, 157.1, 156.3, 152.4, 144.1, 143.8, 132.0 (d, J = 8.1 Hz), 125.3, 119.7, 116.2 (d, J = 21.7 Hz), 115.7 (d, J = 21.2 Hz), 81.8, 75.6, 74.8, 73.1, 47.3; −239.4 (c 0.05, MeOH); ESI-MS m/z: 394.1065 (M + H+); Anal. Calcd for C17H17ClFN5O3: C, 51.85; H, 4.35; N, 17.78. Found: C, 51.75; H, 4.75; N, 17.68.
(2S,3R,4R)-2-((2-Chloro-6-((3-chlorobenzyl)amino)-9H-purin-9-yl)methyl)tetrahydrofuran-3,4-diol (4n).
Yield = 51%; white solid; mp 139–144 °C; 1H NMR (CD3OD, 400 MHz): δ 7.99 (s, 1H), 7.75 (s, 1H), 7.57 (d, J = 7.6 Hz, 1H), 7.37 (d, J = 7.2 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 4.68 (br s, 2H), 4.41 (dd, J = 3.6, 14.4 Hz, 1H), 4.26 (dd, J = 6.4, 14.4 Hz, 1H), 4.02 (dd, J = 3.6, 7.6 Hz, 1H), 3.93–4.00 (m, 2H), 3.78 (dd, J = 5.2, 7.6 Hz, 1H), 3.67 (dd, J = 2.0, 8.8 Hz, 1H); 13C NMR (CD3OD, 100 MHz): δ 157.1, 156.3, 152.4, 144.1, 143.5, 138.7, 138.2, 132.1, 129.0, 119.7, 95.7, 81.7, 75.6, 74.8, 73.1, 47.3, 45.2; −203.8 (c 0.05, MeOH); FAB-MS m/z: 410.0729 (M + H+); Anal. Calcd for C17H17Cl2N5O3: C, 49.77; H, 4.18; N, 17.07. Found: C, 49.97; H, 4.38; N, 17.37.
(2S,3R,4R)-2-((6-((3-Bromobenzyl)amino)-2-chloro-9H-purin-9-yl)methyl)tetrahydrofuran-3,4-diol (4o).
Yield = 53%; white solid; mp 130–132 °C; 1H NMR (CD3OD, 800 MHz): δ 8.01 (s, 1H), 7.55 (s, 1H), 7.36 (dd, J = 7.9, 25.9 Hz, 2H), 7.21 (t, J = 7.8 Hz, 1H), 4.71 (br s, 2H), 4.42 (dd, J = 3.5, 14.4 Hz, 1H), 4.27 (dd, J = 6.8, 14.4 Hz, 1H), 4.03 (ddd, J = 3.4, 6.9, 10.4 Hz, 1H), 3.97 (dd, J = 4.5, 9.6 Hz, 1H), 3.80 (dd, J = 5.0, 7.2 Hz, 1H), 3.69 (dd, J = 2.8, 9.6 Hz, 1H); 13C NMR (CD3OD, 200 MHz): δ 157.0, 156.3, 152.4, 144.1, 143.5, 142.7, 132.5, 132.1, 128.4, 124.1, 119.6, 81.7, 75.6, 74.8, 73.1, 47.3, 45.2; −150.7 (c 0.1, MeOH); FAB-MS m/z: 454.0287 (M + H+); Anal. Calcd for C17H17BrClN5O3: C, 44.90; H, 3.77; N, 15.40. Found: C, 44.90; H, 3.87; N, 15.60.
(2S,3R,4R)-2-((2-Chloro-6-((3-iodobenzyl)amino)-9H-purin-9-yl)-methyl)tetrahydrofuran-3,4-diol (4p).
Yield = 45%; white solid; mp 146–148 °C; 1H NMR (CD3OD, 400 MHz): δ 8.00 (s, 1H), 7.76 (s, 1H), 7.58 (d, J = 7.2 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.07 (t, J = 8.4 Hz, 1H), 4.68 (br s, 2H), 4.41 (dd, J = 3.6, 14.4 Hz, 1H), 4.27 (dd, J = 7.2, 14.8 Hz, 1H), 3.94–4.03 (m, 3H), 3.78 (dd, J = 5.2, 7.2 Hz, 1H), 3.68 (dd, J = 2.4, 9.2 Hz, 1H); 13C NMR (CD3OD, 100 MHz): δ 163.8, 157.1, 144.2, 143.5, 138.7, 138.2, 132.1, 130.3, 129.1, 129.0, 119.7, 108.8, 81.8, 75.6, 74.8, 73.1, 47.3; −131.4 (c 0.1, MeOH); FAB-MS m/z: 502.0151 (M + H+); Anal. Calcd for C17H17ClIN5O3: C, 40.70; H, 3.42; N, 13.96. Found: C, 40.90; H, 3.62; N, 13.96.
(2S,3R,4R)-2-((6-((3-Fluorobenzyl)amino)-9H-purin-9-yl)methyl)-tetrahydrofuran-3,4-diol (4q).
Yield = 52%; white solid; mp 140–141 °C; 1H NMR (CD3OD, 400 MHz): δ 8.24 (s, 1H), 8.05 (s, 1H), 7.29 (ddd, J = 6.0, 8.0, 13.6 Hz, 1H), 7.17 (d, J = 7.6 Hz, 1H), 7.07–7.12 (m, 1H), 6.94 (ddd, J = 2.8, 8.8, 11.6 Hz, 1H), 4.54 (br s, 2H), 4.46 (dd, J = 3.2, 14.4 Hz, 1H), 4.33 (dd, J = 6.4, 14.4 Hz, 1H), 4.04 (ddd, J = 3.6, 6.8, 10.4 Hz, 1H), 3.93–4.01 (m, 2H), 3.78 (dd, J = 4.4, 7.2 Hz, 1H), 3.68 (dd, J = 2.0, 8.8 Hz, 1H); 13C NMR (CD3OD, 100 MHz): δ 156.2, 153.9, 143.6, 143.3, 135.2, 131.4, 131.3, 124.6, 115.3, 115.1, 115.0, 114.8, 81.2, 74.9, 74.2, 72.4, 46.5; −391.6 (c 0.05, MeOH); ESI-MS m/z: 360.1461 (M + H+); Anal. Calcd for C17H18FN5O3: C, 56.82; H, 5.05; N, 19.49. Found: C, 56.92; H, 5.15; N, 19.59.
(2S,3R,4R)-2-((6-((3-Chlorobenzyl)amino)-9H-purin-9-yl)methyl)-tetrahydrofuran-3,4-diol (4r).
Yield = 56%; white solid; mp 150–153 °C; 1H NMR (CD3OD, 400 MHz): δ 8.24 (s, 1H), 8.06 (s, 1H), 7.37–7.41 (m, 2H), 7.19–7.25 (m, 2H), 4.88 (br s, 2H), 4.47 (dd, J = 3.2, 14.0 Hz, 1H), 4.33 (dd, J = 6.4, 14.4 Hz, 1H), 4.04 (ddd, J = 3.6, 6.8, 10.4 Hz, 1H), 3.93–4.00 (m, 2H), 3.78 (dd, J = 4.8, 7.2 Hz, 1H), 3.68 (dd, J = 2.4, 9.2 Hz, 1H); 13C NMR (CD3OD, 100 MHz): δ 156.7, 154.6, 151.1, 143.9, 138.2, 135.2, 131.3, 130.9, 130.5, 128.9, 120.8, 108.8, 81.8, 75.6, 74.8, 73.1, 47.2; +193.5 (c 0.04, MeOH); ESI-MS m/z: 376.1182 (M + H+); Anal. Calcd for C17H18ClN5O3: C, 54.33; H, 4.83; N, 18.64. Found: C, 54.53; H, 4.93; N, 18.84.
(2S,3R,4R)-2-((6-((3-Bromobenzyl)amino)-9H-purin-9-yl)methyl)-tetrahydrofuran-3,4-diol (4s).
Yield = 47%; white solid; mp 148–151 °C; 1H NMR (CD3OD, 500 MHz): δ 8.25 (s, 1H), 8.07 (s, 1H), 7.53 (s, 1H), 7.35 (dd, J = 7.8, 16.8 Hz, 2H), 7.20 (t, J = 7.8 Hz, 1H), 4.78 (br s, 2H), 4.47 (dd, J = 3.4, 14.4 Hz, 1H), 4.33 (dd, J = 6.6, 14.4 Hz, 1H), 4.05 (ddd, J = 3.4, 6.9, 10.4 Hz, 1H), 3.98–4.02 (m, 1H), 3.94–3.99 (m, 1H), 3.79 (dd, J = 5.0, 7.2 Hz, 1H), 3.69 (dd, J = 2.5, 9.6 Hz, 1H), 3.29–3.30 (m, 1H); 13C NMR (CD3OD, 125 MHz): δ 156.1, 153.9, 143.39, 143.33, 131.5 (d, J = 11.4 Hz), 131.3, 127.4, 123.5, 120.1, 108.5, 81.2, 74.9, 74.2, 72.4, 46.5; +56.5 (c 0.04, MeOH); ESI-MS m/z: 422.0642 (M + H+); Anal. Calcd for C17H18BrN5O3: C, 48.58; H, 4.32; N, 16.66. Found: C, 48.78; H, 4.52; N, 16.86.
(2S,3R,4R)-2-((6-((3-Iodobenzyl)amino)-9H-purin-9-yl)methyl)-tetrahydrofuran-3,4-diol (4t).
Yield = 38%; white solid; mp 177–179 °C; 1H NMR (DMSO-d6, 800 MHz): δ 8.35 (br s, 1H), 8.20 (br s, 1H), 8.07 (s, 1H), 7.72 (s, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 7.6 Hz, 1H), 7.10 (t, J = 7.7 Hz, 1H), 4.96 (d, J = 6.2 Hz, 1H), 4.83 (d, J = 4.6 Hz, 1H), 4.64 (br s, 2H), 4.34 (dd, J = 3.8, 14.2 Hz, 1H), 4.20 (dd, J = 7.3, 14.2 Hz, 1H), 3.95 (ddd, J = 3.9, 6.9, 10.7 Hz, 1H), 3.88–3.92 (m, 1H), 3.86 (dd, J = 4.8, 9.2 Hz, 1H), 3.71 (dd, J = 6.4, 11.5 Hz, 1H), 3.50 (dd, J = 3.3, 9.2 Hz, 1H), 3.29 (br s, 1H); 13C NMR (DMSO-d6, 200 MHz): δ 154.1, 152.2, 149.0, 142.9, 141.3, 135.7, 135.2, 130.4, 126.6, 94.6, 79.6, 73.2, 72.4, 70.2, 64.8, 45.2; +29.8 (c 0.1, MeOH); FAB-MS m/z: 468.0529 (M + H+); Anal. Calcd for C17H18IN5O3: C, 43.70; H, 3.88; N, 14.99. Found: C, 43.90; H, 3.98; 14.99.
Cell Culture and Differentiation.
hBM-MSCs (Lonza, Walkers-ville, MD, USA) were maintained in low glucose (1 g/L) Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum and supplemented with penicillin–streptomycin and Glutamax (Invitrogen, Carlsbad, CA, USA). Cell culture media were exchanged every 3 d. hBM-MSC were differentiated for 7 d under IDX conditions (10 μg/mL insulin, 1 μM dexamethasone, 0.5 mM IDX) with DMEM containing a high concentration of glucose (4.5 g/L) for cell growth medium. 1, truncated 1′-homologated adenosine analogues (4a–4t) or 26 were cotreated with IDX conditions, and cell culture supernatants were harvested on the 7th day in culture and 24 h after medium exchange. 26 was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Oil Red O and Hematoxylin Staining.
Oil red O (ORO) staining was performed to evaluate effects of compounds on lipid accumulation during differentiation. After the differentiation of hBM-MSC into adipocytes, cells were cleansed twice with phosphate-buffered saline (PBS) and fixed with 10% formalin in PBS (pH 7.4) for 1 h. Then, cells were washed with 60% isopropanol and then dried at 25 °C. hBM-MSCs were stained with 0.2% ORO solution (Sigma-Aldrich) for 20 min at 25 °C and then washed with tap water three times. After staining lipid droplets, cells were further incubated for 1 min in hematoxylin solution to stain the nucleus. Stained cells were photographed using an Eclipse TS100 inverted microscope (Nikon Co., Tokyo, Japan).
Enzyme-Linked Immunosorbent Assay.
ELISA was conducted to quantitatively evaluate the effects of the compounds on adiponectin secretion. The Quantikine immunoassay kit (R&D Systems, Minneapolis, MN, USA) was used to measure adiponectin amount in cell culture supernatants, according to the manufacturer instruction.
TR-FRET Assays.
The TR-FRET-based receptor-binding assay was performed using LanthaScreen competitive binding assay kits (Invitrogen). LanthaScreen coactivator assay kits (Invitrogen) were used to determine the receptor activation of PPARγ and PPARδ. The PPARγ coactivator assay was performed using fluorescein-TRAP220/DRIP coactivator peptide (sequence KVSQNPILTSLLQITGNGG).35 PPARδ coactivator and corepressor assays were performed using fluorescein-C33 coactivator peptide (sequence HVEMHPLLMGLLMESQWGA) and SMRT-ID2 peptide (sequence HASTNMGLEAHRKALMGKYDQW), respectively.36 Assay plates were read at wavelengths of 520 and 495 nm after 1 to 2 h of incubation, using a CLARIOstar plate reader (BMG LABTECH, Ortenberg, Germany). All procedures and instrument settings were in agreement with the manufacturer instruction. PPAR ligands, 27, 28, and 29 were purchased from Tocris Bioscience (Bristol, U.K.), and 30 was purchased from Sigma-Aldrich.
Animal Study.
Animals.
6 week old male C57BL/6J ob/ob mice were obtained from SLC Inc. (Kotoh-cho, Japan). Mice were acclimatized for 1 week prior to use and were housed in a 12 h light/dark cycle at a temperature of 21 ± 2 °C and 50 ± 5% relative humidity. Mice had free access to food except the time when they were fasted before the measurement of blood glucose. The experimental procedures were approved by IACUC (SNU-200605-5-1) in the Seoul National University.
Drug Preparation and Administration.
Pioglitazone and the test drug (4p) were dissolved in 60% polyethylene glycol (PEG) containing 5% DMSO for an in vivo experiment. Pioglitazone (20 mg/kg) or 4p at a dose of 30 mg/kg was orally administered 6 times per week for 3 weeks followed by intraperitoneal administration for another 3 weeks. Lean mice group and ob/ob mice control group were administered with an equivalent volume of 60% PEG containing 5% DMSO. No adverse effects were observed.
OGTT and Measurement of Fasting Blood Glucose Level.
OGTT and fasting blood glucose levels were measured after mice were fasted for 10 h (starting from 11:00 pm, day before the experiment). Blood glucose levels were determined in blood samples from the tail vein using a G Care glucose analyzer (Green Cross, Inc., Seoul, South Korea). For OGTT, the mice were administered with glucose (0.2 g/kg) and blood samples were collected at t = 0, 15, 30, 60, 90, and 120 min.
Measurement of Serum Insulin Level.
Fed serum insulin levels were measured in blood samples obtained from cardiac puncture at final sacrifice. Blood samples were centrifuged, and the serum part was used to measure insulin levels through a LBIS mouse insulin ELISA kit (Shibayagi, Gunma, Japan).
Statistical Analysis.
The data are shown as the mean ± standard deviation (SD). Comparisons of multiple groups were assessed by oneway analysis of variance (ANOVA), followed by Tukey’s test. p value < 0.05 was considered to be statistically significant. GraphPad PRISM software version 7.0 (GraphPad Software, San Diego, CA, USA) was used for statistical analysis.
Molecular Modeling Study.
Homology Modeling.
An A3AR homology model for molecular docking was built on the SWISS-MODEL server.37 The crystal structure of the adenosine A2A receptor bound to an engineered G protein in complex with AR agonist NECA was downloaded from RCSB PDB (PDB ID: 5G53) and used as the template for homology modeling.21,38 Human adenosine A3 receptor target sequence for homology modeling was fetched from UniProtKB (entry: P0DMS8).39
Computational Docking Simulation.
Ligand-binding site for docking was defined as a 30 Å3 size grid box centered on the centroid of co-crystallized native ligands. The crystal structures of PPARγ and PPARδ LBD were also downloaded from RCSB PDB (PDB ID: 5YCP, 5U46),25,26 and computational docking was performed using AutoDock Vina version 1.1.2 (The Scripps Research Institute, La Jolla, CA, USA).40 For each macromolecule–ligand pair, binding models of ligands with the lowest binding free energy (kcal/mol) were used for further analysis. All figures to show the molecular modeling results were visualized using PyMOL (Schrödinger, LLC, New York, NY, USA).41
Statistical Analysis.
Experimental values were presented in the mean ± SD from three independent experiments. Statistical analyses were conducted using R (R Foundation for Statistical Computing, Vienna, Austria).42 Comparisons were performed using one-way ANOVA followed by the posthoc Welch two sample t-test. The threshold of statistical significance was set at (*) p ≤ 0.05 and (**) p ≤ 0.01.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation (NRF) grants (NRF - 2016R1A2B3010164, 2018R1A5A2024425, and 2019R1F1A1063905) of Korea.
ABBREVIATIONS
- NASH
nonalcoholic steatohepatitis
- PPAR
peroxisome proliferator-activated receptor
- RXR
retinoid X receptor
- PPRE
peroxisome proliferator response element
- AR
adenosine receptor
- hBM-MSCs
human bone marrow mesenchymal stem cells
- LBD
ligand-binding domain
- SAR
structure–activity relationship
- TR-FRET
time-resolved fluorescence resonance energy transfer
- LBP
ligand binding pocket
- EC50
half-maximal effective concentration values
- TRAP220/DRIP-1
thyroid hormone receptor-associated protein 220/vitamin D receptor interacting protein-1
- ELISA
enzyme-linked immunosorbent assay
- EL
extracellular loop
- NCoR2
nuclear receptor corepressor 2
- SMRT
silencing mediator of retinoid and thyroid hormone receptor
- SMRT-ID2
SMRT-interaction domain 2
- TM
transmembrane
- DMEM
Dulbecco modified Eagle’s medium
- FBS
fetal bovine serum
- ORO
oil red O
- PBS
phosphate buffered saline
- ANOVA
one-way analysis of variance
- T2D
type 2 diabetes
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01874.
HPLC data of 4d and 4p; 1H and 13C NMR copies; computational docking study; and animal study (PDF)
Molecular formula strings (CSV)
A3AR/4d (PDB); A3AR/1 (PDB); PPARγ-LBD/4d (PDB); PPARδ-LBD/4d (PDB); PPARδ-LBD/4h (PDB); PPARδ-LBD/4l (PDB) (ZIP)
Accession Codes
The crystal structures of A2AAR (PDB ID: 5G53), PPARγ-LBD (PDB ID: 5YCP), and PPARδ-LBD (PDB ID: 5U46) were obtained from the Protein Data Bank. Authors will release the atomic coordinates and experimental data upon article publication.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c01874
The authors declare no competing financial interest.
Contributor Information
Seungchan An, Research Institute of Pharmaceutical Sciences, College of Pharmacy and Natural Products Research Institute, Seoul National University, Seoul 08826, Korea;.
Gyudong Kim, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Korea; College of Pharmacy and Research Institute of Drug Development, Chonnam National University, Gwangju 61186, Korea.
Hyun Jin Kim, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Korea.
Sungjin Ahn, Research Institute of Pharmaceutical Sciences, College of Pharmacy and Natural Products Research Institute, Seoul National University, Seoul 08826, Korea.
Hyun Young Kim, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Korea.
Hyejin Ko, Research Institute of Pharmaceutical Sciences, College of Pharmacy and Natural Products Research Institute, Seoul National University, Seoul 08826, Korea.
Young Eum Hyun, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Korea.
Mai Nguyen, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Korea; College of Pharmacy and Research Institute of Drug Development, Chonnam National University, Gwangju 61186, Korea.
Juri Jeong, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Korea; College of Pharmacy and Research Institute of Drug Development, Chonnam National University, Gwangju 61186, Korea.
Zijing Liu, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Korea; College of Pharmacy and Research Institute of Drug Development, Chonnam National University, Gwangju 61186, Korea.
Jinhe Han, College of Pharmacy and Research Institute of Drug Development, Chonnam National University, Gwangju 61186, Korea.
Hyuk Woo Lee, Future Medicine Company Ltd., Seongnam, Gyeonggi-do 13449, Korea.
Kenneth A. Jacobson, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes, and Digestive and Kidney Disease, National Institutes of Health, Bethesda, Maryland 20892, United States.
Won Jea Cho, College of Pharmacy and Research Institute of Drug Development, Chonnam National University, Gwangju 61186, Korea.
Young-Mi Kim, Department of Pharmacy, College of Pharmacy and Institute of Pharmaceutical Science and Technology, Hanyang University, Ansan, Gyeonggi-do 15588, Korea.
Keon Wook Kang, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Korea.
Minsoo Noh, Research Institute of Pharmaceutical Sciences, College of Pharmacy and Natural Products Research Institute, Seoul National University, Seoul 08826, Korea.
Lak Shin Jeong, Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, Seoul 08826, Korea.
REFERENCES
- (1).Wahli W; Michalik L PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab 2012, 23, 351–363. [DOI] [PubMed] [Google Scholar]
- (2).(a) Sahebkar A; Chew GT; Watts GF New peroxisome proliferator-activated receptor agonists: potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin. Pharmacother 2014, 15, 493–503. [DOI] [PubMed] [Google Scholar]; (b) Cave MC; Clair HB; Hardesty JE; Falkner KC; Feng W; Clark BJ; Sidey J; Shi H; Aqel BA; McClain CJ; Prough RA Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2016, 1859, 1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Chojkier M Troglitazone and liver injury: in search of answers. Hepatology 2005, 41, 237–246. [DOI] [PubMed] [Google Scholar]; (b) Nissen SE; Wolski K Rosiglitazone revisited: an updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch. Intern. Med 2010, 170, 1191–1201. [DOI] [PubMed] [Google Scholar]; (c) Loke YK; Kwok CS; Singh S Comparative cardiovascular effects of thiazolidinediones: systematic review and meta-analysis of observational studies. BMJ 2011, 342, d1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Yu J; Ahn S; Kim HJ; Lee M; Ahn S; Kim J; Jin SH; Lee E; Kim G; Cheong JH; Jacobson KA; Jeong LS; Noh M Polypharmacology of N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (IB-MECA) and related A3 adenosine receptor ligands: peroxisome proliferator activated receptor (PPAR) γ partial agonist and PPARδ antagonist activity suggests their antidiabetic potential. J. Med. Chem 2017, 60, 7459–7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lee HW; Kim HO; Choi WJ; Choi S; Lee JH; Park S -g.; Yoo, L.; Jacobson, K. A.; Jeong, L. S. Design, synthesis, and binding of homologated truncated 4′-thioadenosine derivatives at the human A3 adenosine receptors. Bioorg. Med. Chem 2010, 18, 7015–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Jeong LS; Choe SA; Gunaga P; Kim HO; Lee HW; Lee SK; Tosh DK; Patel A; Palaniappan KK; Gao Z-G; Jacobson KA; Moon HR Discovery of a new nucleoside template for human A3 adenosine receptor ligands: D-4′-thioadenosine derivatives without 4′-hydroxymethyl group as highly potent and selective antagonists. J. Med. Chem 2007, 50, 3159–3162. [DOI] [PubMed] [Google Scholar]; (b) Jeong LS; Pal S; Choe SA; Choi WJ; Jacobson KA; Gao Z-G; Klutz AM; Hou X; Kim HO; Lee HW; Lee SK; Tosh DK; Moon HR Structure-activity relationships of truncated D- and L-4′-thioadenosine derivatives as species-independent A3 adenosine receptor antagonists. J. Med. Chem 2008, 51, 6609–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Taddei D; Kilian P; Slawin AMZ; Derek Woollins J Synthesis and full characterisation of 6-chloro-2-iodopurine, a template for the functionalisation of purines. Org. Biomol. Chem 2004, 2, 665–670. [DOI] [PubMed] [Google Scholar]
- (8).Bennis K; Calinaud P; Gelas J; Ghobsi M A new route to some enantiomerically pure substituted morpholines from D-ribono- and D-gulono-1,4-lactones. Carbohydr. Res 1994, 264, 33–44. [Google Scholar]
- (9).Xu HE; Lambert MH; Montana VG; Plunket KD; Moore LB; Collins JL; Oplinger JA; Kliewer SA; Gampe RT Jr.; McKee DD; Moore JT; Willson TM Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. U.S.A 2001, 98, 13919–13924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ahn S; Ma CT; Choi JM; An S; Lee M; Le THV; Pyo JJ; Lee J; Choi MS; Kwon SW; Park JH; Noh M Adiponectin-secretion-promoting phenylethylchromones from the agarwood of aquilaria malaccensis. J. Nat. Prod 2019, 82, 259–264. [DOI] [PubMed] [Google Scholar]
- (11).(a) Harmon GS; Lam MT; Glass CK PPARs and lipid ligands in inflammation and metabolism. Chem. Rev 2011, 111, 6321–6340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang X; Wang G; Shi Y; Sun L; Gorczynski R; Li Y-J; Xu Z; Spaner DE PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis 2016, 5, No. e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Romacho T; Elsen M; Röhrborn D; Eckel J Adipose tissue and its role in organ crosstalk. Acta Physiol. 2014, 210, 733–753. [DOI] [PubMed] [Google Scholar]
- (13).(a) Matsuzawa Y; Funahashi T; Kihara S; Shimomura I Adiponectin and metabolic syndrome. Arterioscler., Thromb., Vasc. Biol 2004, 24, 29–33. [DOI] [PubMed] [Google Scholar]; (b) Menzaghi C; Trischitta V; Doria A Genetic influences of adiponectin on insulin resistance, type 2 diabetes, and cardiovascular disease. Diabetes 2007, 56, 1198–1209. [DOI] [PubMed] [Google Scholar]
- (14).Zhu Y; Qi C; Jain S; Rao MS; Reddy JK Isolation and characterization of PBP, a protein that interacts with peroxisome proliferator-activated receptor. J. Biol. Chem 1997, 272, 25500–25506. [DOI] [PubMed] [Google Scholar]
- (15).Baek HJ; Kang YK; Roeder RG Human Mediator enhances basal transcription by facilitating recruitment of transcription factor IIB during preinitiation complex assembly. J. Biol. Chem 2006, 281, 15172–15181. [DOI] [PubMed] [Google Scholar]
- (16).Ge K; Guermah M; Yuan C-X; Ito M; Wallberg AE; Spiegelman BM; Roeder RG Transcription coactivator TRAP220 is required for PPARγ2-stimulated adipogenesis. Nature 2002, 417, 563–567. [DOI] [PubMed] [Google Scholar]
- (17).(a) Gronemeyer H; Gustafsson J-Å; Laudet V Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discovery 2004, 3, 950–964. [DOI] [PubMed] [Google Scholar]; (b) Germain P; Staels B; Dacquet C; Spedding M; Laudet V Overview of nomenclature of nuclear receptors. Pharmacol. Rev 2006, 58, 685–704. [DOI] [PubMed] [Google Scholar]
- (18).Yu S; Reddy J Transcription coactivators for peroxisome proliferator-activated receptors. Biochim. Biophys. Acta 2007, 1771, 936–951. [DOI] [PubMed] [Google Scholar]
- (19).Nofsinger RR; Li P; Hong S-H; Jonker JW; Barish GD; Ying H; Cheng S.-y.; Leblanc M; Xu W; Pei L; Kang Y-J; Nelson M; Downes M; Yu RT; Olefsky JM; Lee C-H; Evans RM SMRT repression of nuclear receptors controls the adipogenic set point and metabolic homeostasis. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 20021–20026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lindström P The Physiology of Obese-Hyperglycemic Mice [ob/ob Mice]. Sci. World J 2007, 7, 666–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Carpenter B; Nehmé R; Warne T; Leslie AGW; Tate CG Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xiang Z Advances in homology protein structure modeling. Curr. Protein Pept. Sci 2006, 7, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).(a) Lebon G; Warne T; Edwards PC; Bennett K; Langmead CJ; Leslie AGW; Tate CG Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu F; Wu H; Katritch V; Han GW; Jacobson KA; Gao Z-G; Cherezov V; Stevens RC Structure of an agonist-bound human A2A adenosine receptor. Science 2011, 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lebon G; Edwards PC; Leslie AGW; Tate CG; Stevens RC Molecular determinants of CGS21680 binding to the human adenosine A2A receptor. Mol. Pharmacol 2015, 87, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ciancetta A; Jacobson K Structural probing and molecular modeling of the A3 adenosine receptor: a focus on agonist binding. Molecules 2017, 22, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jang JY; Bae H; Lee YJ; Choi YI; Kim HJ; Park SB; Suh SW; Kim SW; Han BW Structural basis for the enhanced anti-diabetic efficacy of lobeglitazone on PPARγ. Sci. Rep 2018, 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wu C-C; Baiga TJ; Downes M; La Clair JJ; Atkins AR; Richard SB; Fan W; Stockley-Noel TA; Bowman ME; Noel JP; Evans RM Structural basis for specific ligation of the peroxisome proliferator-activated receptor δ. Proc. Natl. Acad. Sci. U.S.A 2017, 114, E2563–E2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ballesteros JA; Weinstein H Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- (28).Friedland SN; Leong A; Filion KB; Genest J; Lega IC; Mottillo S; Poirier P; Reoch J; Eisenberg MJ The cardiovascular effects of peroxisome proliferator-activated receptor agonists. Am. J. Med 2012, 125, 126–133. [DOI] [PubMed] [Google Scholar]
- (29).(a) Peters JM; Gonzalez FJ; Müller R Establishing the Role of PPARβ/δ in Carcinogenesis. Trends Endocrinol. Metab 2015, 26, 595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang ZV; Scherer PE Adiponectin, the past two decades. J. Mol. Cell Biol 2016, 8, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li Y-J; Sun L; Shi Y; Wang G; Wang X; Dunn SE; Iorio C; Screaton RA; Spaner DE PPAR-delta promotes survival of chronic lymphocytic leukemia cells in energetically unfavorable conditions. Leukemia 2017, 31, 1905–1914. [DOI] [PubMed] [Google Scholar]
- (30).(a) Luo N; Liu J; Chung BH; Yang Q; Klein RL; Garvey WT; Fu Y Macrophage adiponectin expression improves insulin sensitivity and protects against inflammation and atherosclerosis. Diabetes 2010, 59, 791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu A; Wang Y; Keshaw H; Xu LY; Lam KSL; Cooper GJS The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Invest 2003, 112, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).(a) Youssef J; Badr M Peroxisome proliferator-activated receptors and cancer: challenges and opportunities. Br. J. Pharmacol 2011, 164, 68–82. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Piemontese L; Cerchia C; Laghezza A; Ziccardi P; Sblano S; Tortorella P; Iacobazzi V; Infantino V; Convertini P; Dal Piaz F; Lupo A; Colantuoni V; Lavecchia A; Loiodice F New diphenylmethane derivatives as peroxisome proliferator-activated receptor alpha/gamma dual agonists endowed with anti-proliferative effects and mitochondrial activity. Eur. J. Med. Chem 2017, 127, 379–397. [DOI] [PubMed] [Google Scholar]
- (32).Elikkottil J; Kohli DR; Gupta K Antitumor activity of a PPARδ antagonist. Cancer Biol. Ther 2009, 8, 1262–1264. [DOI] [PubMed] [Google Scholar]
- (33).Hack K; Reilly L; Palmer C; Read KD; Norval S; Kime R; Booth K; Foerster J Skin-Targeted Inhibition of PPARβ/δ by Selective Antagonists to Treat PPAR β/δ—Mediated Psoriasis-Like Skin Disease In Vivo. PLoS One 2012, 7, No. e37097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Forsman JJ; Leino R Acetonation of L-pentoses and 6-deoxy-L-hexoses under kinetic control using heterogeneous acid catalysts. Carbohydr. Res 2010, 345, 1548–1554. [DOI] [PubMed] [Google Scholar]
- (35).Rachez C; Gamble M; Chang C-P; Atkins GB; Lazar MA; Freedman FP The DRIP complex and SRC-1/p160 coactivators share similar nuclear receptor binding determinants but constitute functionally distinct complexes. Mol. Cell. Biol 2000, 20, 2718–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).(a) Chen JD; Evans RM A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 1995, 377, 454–457. [DOI] [PubMed] [Google Scholar]; (b) Chang C.-y.; Norris JD; Grøn H; Paige LA; Hamilton PT; Kenan DJ; Fowlkes D; McDonnell DP Dissection of the LXXLL Nuclear Receptor-Coactivator Interaction Motif Using Combinatorial Peptide Libraries: Discovery of Peptide Antagonists of Estrogen Receptors α and β. Mol. Cell. Biol 1999, 19, 8226–8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Waterhouse A; Bertoni M; Bienert S; Studer G; Tauriello G; Gumienny R; Heer FT; de Beer TAP; Rempfer C; Bordoli L; Lepore R; Schwede T SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Burley SK; Berman HM; Bhikadiya C; Bi C; Chen L; Di Costanzo L; Christie C; Dalenberg K; Duarte JM; Dutta S; Feng Z; Ghosh S; Goodsell DS; Green RK; Guranović V; Guzenko D; Hudson BP; Kalro T; Liang Y; Lowe R; Namkoong H; Peisach E; Periskova I; Prlić A; Randle C; Rose A; Rose P; Sala R; Sekharan M; Shao C; Tan L; Tao Y-P; Valasatava Y; Voigt M; Westbrook J; Woo J; Yang H; Young J; Zhuravleva M; Zardecki C RCSB protein data bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2018, 47, D464–D474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Trott O; Olson AJ AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Schrödinger LLC. The PyMOL Molecular Graphics System, version 2.4.0, 2002. [Google Scholar]
- (42).R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria. 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.