

Summary
The natural product cepafungin I was recently reported to be one of the most potent covalent inhibitors of the 20S proteasome core particle through a series of in vitro activity assays. Here, we report a short chemoenzymatic total synthesis of cepafungin I featuring the use of a regioselective enzymatic oxidation to prepare a key hydroxylated amino acid building block in a scalable fashion. The strategy developed herein enabled access to a chemoproteomic probe, which in turn revealed the exceptional selectivity and potency of cepafungin I towards the β2 and β5 subunits of the proteasome. Further structure-activity relationship studies suggest the key role of the hydroxyl group in the macrocycle and the identity of the lipid tail in modulating the potency of this natural product family. This study lays the groundwork for further medicinal chemistry exploration to fully realize the anticancer potential of cepafungin I.
Introduction
Cepafungin I and glidobactin A are two macrolactams belonging to a larger family of natural products called the syrbactins (Fig. 1). Members of this family share a common 12-membered macrolactam core consisting of a vinylogous amino acid and a lysine residue that may exist in various oxidation states. Further diversity can be found within the tail portion of the natural products: while the cepafungins and the glidobactins possess unsaturated fatty acid tails, all syrbactins contain a Val-Val unit that is linked via a unique ureido moiety (Krahn et al., 2017). Initial biological evaluation suggested that the glidobactins and the cepafungins exhibit moderate antifungal activity and potent antitumor activity against P388 leukemia in mice (Oka et al., 1988a; Oka et al., 1988b; Oka et al., 1988c; Shoji et al., 1990). More recently, it has been determined that the latter activity arises from inhibition of the 20S proteasome core particle (CP) via covalent engagement of two distinct catalytically-active Thr1Oγ residues by the unsaturated lactam motif of the macrocycle (Groll et al., 2008). Cepafungin I, in particular, exhibits remarkably strong inhibitory activity relative to all known proteasome inhibitors to date (IC50 of 4 nM against the β5 subunit of yeast CP, (Stein et al., 2012)). The proteasome is a multiprotein complex that plays a critical role in protein degradation. Given the centrality of the proteasome in the regulation of cell cycle and apoptosis, its inhibition constitutes a promising modality for cancer therapy. In support of this notion, three proteasome inhibitors, bortezomib, carfilzomib and ixazomib, have been approved by the U.S. Food and Drug Administration for the treatment of multiple myeloma. However, both bortezomib and carfilzomib have been reported to exhibit several side effects such as thrombocytopenia and high occurrence of relapse and chemoresistance have also been reported (Guerrero-Garcia et al., 2018). Thus, there is still an urgent need to advance new proteasome inhibitors as drug candidates that will address these issues.
Figure 1. Select syrbactin structures and biological activity.
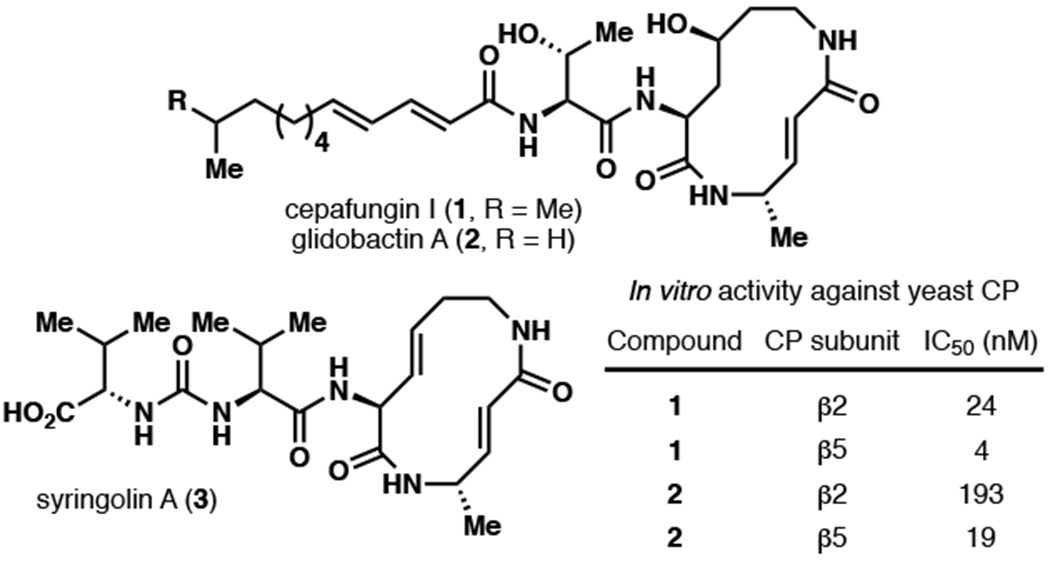
Molecular structures of cepafungin I and related natural products. In vitro yeast CP IC50 values from Stein et al. (2012).
Similar to bortezomib, the syrbactins trigger apoptosis by eliciting p53 accumulation and inhibiting NF-κB activity. Pioneering studies by Bachmann and co-workers using a syrbactin derivative additionally showed that while this compound exhibited cytotoxic effects on cancer cell lines, the effect was most pronounced in multiple myeloma (Archer et al., 2012). Recent research has begun to elucidate certain resistance mechanisms in multiple myeloma. While current proteasome inhibitors primarily inhibit the β5 subunit, it has been shown that co-inhibition of the β2 subunit prevents recovery of proteasome activity by causing aggregation/inactivation of the proteasomal transcription factor Nrf1. Notably, the cytotoxicities of bortezomib and carfilzomib towards solid tumor cells (e.g., triple negative breast cancer) are significantly enhanced in this manner, whereas either drug had otherwise not shown clinical efficacy on its own. Moreover, co-inhibition of β5 with β2 provides a stronger antineoplastic effect than with β1, highlighting the importance of advancing new proteasome inhibitors with equipotent activity for these subunits (Weyburne et al., 2017; Oerlemans et al., 2008). Cepafungin I, with its low nanomolar activity towards β5 and β2 (Fig. 1) (IC50 = 4 nM and 24 nM, respectively), stands as an attractive candidate for such purpose.
A seminal work by Schmidt and coworkers in 1992 established the first total synthesis of glidobactin A (Schmidt et al., 1992). Starting from malic acid, the synthesis of the natural product was achieved in 21 steps. While being a landmark achievement, this approach suffers from high step count due to inefficient functional group interconversions and extraneous protecting group manipulations. For example, the synthesis of the key 4-hydroxylysine moiety took place in 12 steps from malic acid, featuring HWE homologation, asymmetric hydrogenation and displacement of the terminal alcohol with NaN3. Efforts by Ichikawa (Chiba et al., 2014; Kitahata et al., 2016; Kitahata et al., 2017), Pirrung (Pirrung et al., 2010), Stephenson (Dai and Stephenson, 2010) and others (Clerc et al., 2009; Clerc et al., 2010b) have established viable synthetic routes to the syrbactins. However, these routes would not be amenable to the introduction of the key 2° alcohol within the embedded lysine unit.
Results and Discussion
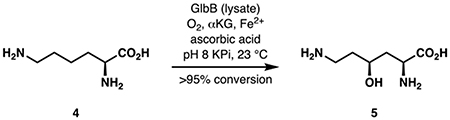
We recognized that an efficient route to 4-hydroxylysine or its synthetic equivalent would hold the key to the development of a practical synthesis of cepafungin I. If such a route could be identified, iterative condensations with alanine and an acetate equivalent would rapidly generate the macrocyclic core of the natural product. Prior approaches to either diastereomers of protected 4-hydroxylysine suffer from high step count arising from protecting group and functional group manipulations (Izumiya et al., 1965; Schmidt et al., 1992; Marin et al., 2004). Among these three approaches, only that by Witkop (Izumiya et al., 1965) described the production of protected 5 while the other two produced only its C4 diastereomer. Witkop’s route to protected 5, however, proceeded through the intermediacy of its C4 diastereomer, required the use of harsh photolysis in the presence of Cl2 gas and involved an inefficient stereoinversion process at the C4 stereocenter. We have previously established the biosynthetic origin of the 4-hydroxylysine moiety of glidobactin A (Amatuni and Renata, 2019). Within the pathway, a nonheme dioxygenase, GlbB, catalyzes the direct C–H hydroxylation of free-standing lysine at the C4 position prior to loading of the product onto the nonribosomal peptide synthetase assembly line. Substrate scope examination of GlbB revealed that while the enzyme exhibits a narrow substrate specificity, it is able to hydroxylate lysine with remarkably high catalytic efficiency. Large-scale biocatalytic hydroxylation with purified GlbB, however, was hampered by poor scalability as the reaction gave poor conversion when conducted at > 100 mg scale. While this issue could be solved by conducting the reaction with clarified lysate of cells expressing GlbB, the poor soluble expression of GlbB led to sub-optimal titer (Fig. 2). Prior reports from our laboratory and others have shown the benefits of co-expressing molecular chaperones, especially GroES/EL, in improving heterologous production of enzymes from secondary metabolite pathways (Payne et al., 2013; Zhang et al., 2018; Li et al., 2019). Indeed, co-expression of chaperones GroES/EL was found to increase soluble expression of GlbB, which translates to ca. 5-fold improvement in reaction conversion and yield on small scale. For large scale reactions, ca. 6–7 g of lysine could be fully converted to its C4-hydroxylated counterpart in a single pass with 1 L of clarified cell lysate (final OD600 = 12.5), corresponding to a titer of ca. 6–7 g/L based on the original volume of the expression culture (Fig. 2).
Figure 2. Optimization of preparative scale hydroxylation of 4 with GlbB.
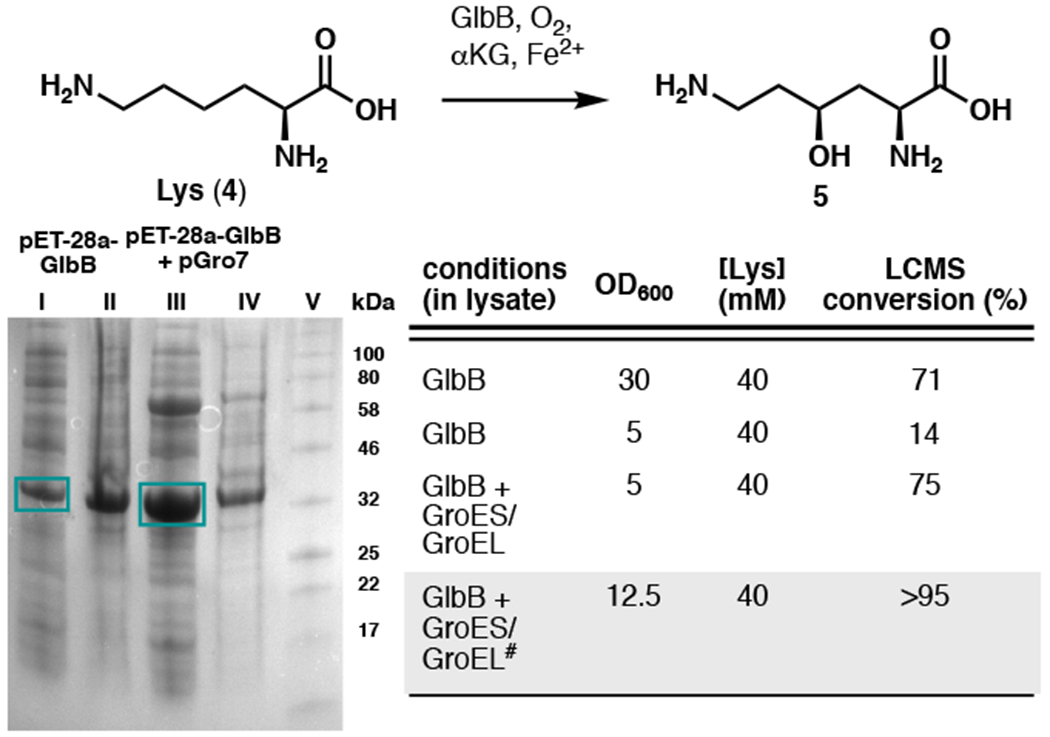
Co-expression of chaperones GroEs/GroEL was found to improve the soluble expression of GlbB, resulting in ca. 5-fold improvement in reaction conversion. Lanes (I) and (III): soluble fraction, lanes (II) and (IV): insoluble fraction, lane (V): protein ladder. #Reaction conducted at 6-gram scale.
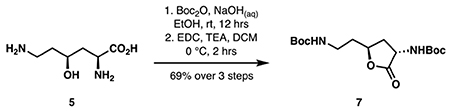
Having solved the material throughput issue for the production of 5, we turned our attention to its conversion to the macrocyclic core of 1. We have previously described the conversion of 5 to intermediate 6 by way of coupling with alaninol (Amatuni and Renata, 2019). However, attempts to selectively oxidize the 1° alcohol of 6 were beset by low yields and poor selectivity (Fig. 3A). Efforts to effect differential protections of the α- and μ-amine also proved problematic due to the presence of the C4-OH. Finally, aminolysis (Sund et al., 2011; Sabot et al., 2007; Foley and Jamison, 2010) of lactone 7 with protected (4S)-amino-2(E)-pentenoic acid derivatives (8) led to only non-productive decomposition of 8. All of these setbacks led to the non-obvious tactical development involving the use of Weinreb amide 9 in an AlMe3-assisted coupling with lactone 7 (Fig. 3B). This protocol reliably afforded dipeptide 10 in 90% yield on multi-gram scale without any observable side reactions (e.g., polymerization of 9, epimerization or re-lactonization of 10). To the best of our knowledge, this is the first report of generating an aluminium amide reagent in situ that itself contains a Weinreb amide moiety. Treatment of 10 with LiAlH4 effected a clean reduction of the Weinreb amide moiety to the corresponding aldehyde. This transformation was regarded as a risky endeavor at the outset due to the presence of several carbonyl groups in 10. Nevertheless, we observed remarkable chemoselectivity as no over-reduction or side reactivity with any of the carbonyl groups in the molecule could be detected. Olefination with Wittig reagent 11 furnished enoate 12, which constitutes the protected linear form of the target macrocyclic core. As a testament to the robustness of the route, more than 1.5 g of 12 could be prepared in a single pass.
Figure 3. Chemoenzymatic synthesis of cepafungin I.
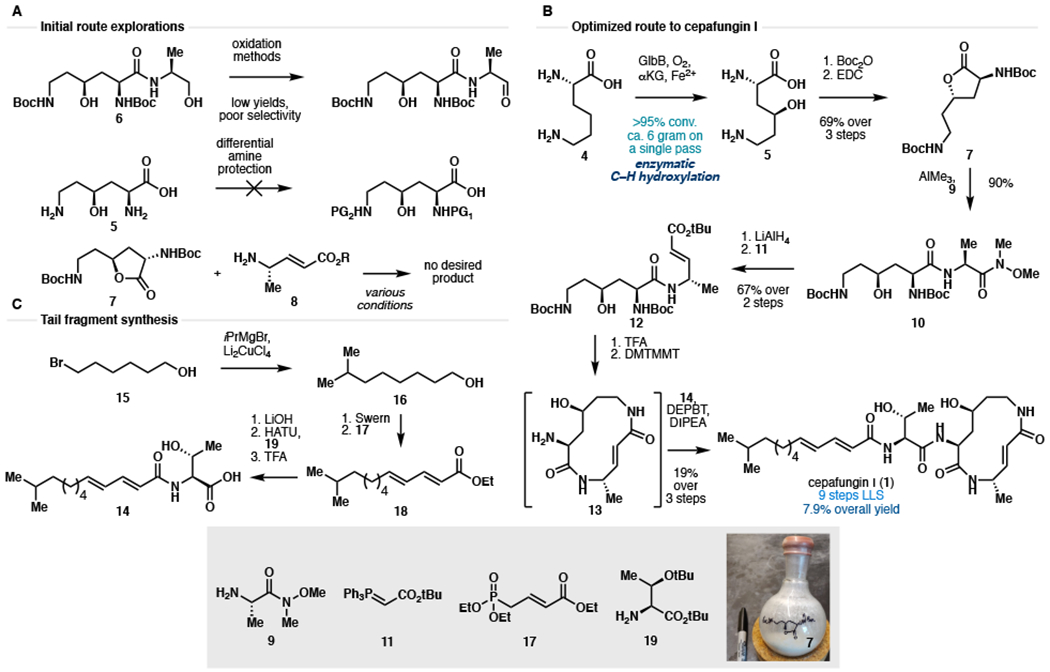
(A) Initial strategies for elaborating 5 to the macrocyclic core of 1. (B) Optimized chemoenzymatic synthesis of cepafungin I. (C) Modular synthesis of tail fragment 14. See STAR Methods for detailed reaction conditions and full characterization. Boc2O: di-tert-butyl carbonate; EDC: 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; TFA: trifluoroacetic acid; HATU: 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate; DMTMMT: 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium tetrafluoroborate; DEPBT: 3-(Diethoxyphosphoryloxy)-1,2,3- benzotriazin-4(3H)-one; DIPEA: N,N-diisopropylethylamine.
While global deprotection of 12 could be achieved cleanly under acidic conditions, the subsequent macrolactamization step required extensive optimization (See Table S1 for optimization). This observation was in line with previous efforts in the total synthesis of glidobactin A and the syrbactins where late-stage macrolactamization typically proceeded in less than 30% yield. Eventually, DMTMMT (Kaminski et al., 2005) was identified as the optimal coupling reagent, providing ~60% NMR yield of the desired macrolactam. In contrast to other approaches to the syrbactins, we conducted this macrocyclization step in the absence of any protecting groups to maximize step economy and synthetic ideality (Gaich and Baran, 2010). Several side reactions including oligomerization, lactonization by the 2° alcohol, or alternative macrolactamization by the lysyl α-amino group could have derailed this approach. Successful execution of our coupling strategy was made possible likely due to a combination of substrate conformational bias and the intrinsic difference in rate of ring formation among medium ring sizes (Illuminati and Mandolini, 1981). A synthetic route to the fully elaborated tail fragment 14 was pursued in parallel (Fig 3C). Given the lack of commercial availability of the 7-methyloctan-1-ol precursor (16), we developed a modular route to access it via a Kochi coupling (Tamura and Kochi, 1971) of bromoalcohol 15 and iPrMgBr. Subsequent oxidation, Horner-Wadsworth-Emmons homologation, peptide coupling and deprotection completed the synthesis of 14. Due to the high polarity of 12, we elected to bypass any purification step and used the compound in a crude form for subsequent coupling with 14. Extensive screening of peptide coupling conditions (Table S2) eventually identified DEPBT as an optimal coupling reagent for this step, leading to the formation of the target natural product in 19% yield from 11. Overall, our chemoenzymatic synthesis of cepafungin I proceeded in 9 steps (longest linear sequence) and 7.9% overall yield from lysine. By virtue of this chemoenzymatic strategy, our route provides exceptionally rapid access to the key hydroxylysine residue and overcomes longstanding challenges associated with synthetic access to syrbactins bearing 2° alcohol at the L-lysine fragment. This route also highlights DMTMMT as a superior macrolactamization reagent and the use of substrate conformational bias to preferentially construct the strained 12-membered ring from a “protection-free” linear precursor. Finally, the modularity of this route enables the rapid construction of synthetic analogs and chemoproteomic probes for further biological evaluation (vide infra).
Multiple myeloma (MM) has remained the main indication for clinical trials of novel proteasome inhibitor drug candidates since the FDA approval of the first-in-class drug Velcade (bortezomib) in 2003 (Manasanch et al., 2017). Accordingly, we started our biological investigations with cytotoxicity measurements of 1 in the commonly used MM cell lines RPMI 8226 and MM1.R to evaluate its potential as a cancer drug candidate. Cells were treated with various concentrations of 1 for 24 h or 48 h and the percentage of viable cells was determined via the WST-1 assay (Fig. 4A). Indeed, 1 proved potently toxic and the measured cytotoxicity after 48 h treatment (EC50s of 31 nM for RPMI 8226 and 23 nM for MM1.R) aligned well with the previously reported data for clinical proteasome inhibitor drugs bortezomib (30 nM for RPMI 8226; 3 nM for MM1.R, Hideshima et al., 2001) and carfilzomib (5 nM for RPMI 8226, Kuhn et al., 2007). Although 1 and structurally related natural products have previously been shown to inhibit proteasomal subunits PSMB2 (β2) and PSMB5 (β5) in purified yeast (Stein et al., 2012) and mammalian (Pawar et al., 2020) proteasomes, direct target engagement of proteasomes by the syrbactins in mammalian cells has not yet been shown. Accordingly, we decided to perform a deep profiling of cellular targets of 1 using a classical chemoproteomics approach. For this purpose, we synthesized covalent probe 20, an alkyne-tagged derivative of 1 (Fig. 4). RPMI 8226 lysates were treated with increasing concentrations of 20 for 1 h at r.t. and the proteins labeled by the covalent probe were then conjugated to TAMRA azide using copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC, Speers and Cravatt, 2004), separated by SDS-PAGE and visualized by in-gel fluorescence scanning (Fig. S1A). The gel profile revealed only a few bands, thus suggesting low proteomic promiscuity of 20. To examine whether 20 can be used as a clickable analog of 1, we performed a gel-based competitive profiling experiment. RPMI 8226 cells were treated in situ with various concentrations of 1 for 6 h, lysed, and then treated with 10 μM 20 for 1 h, followed by TAMRA conjugation and protein visualization, as described above (Figs. 4B, S1B). Excitingly, only a few bands were successfully competed at nanomolar concentrations, suggesting high potency and selectivity of 1 towards its targets. To identify the competed protein targets, we performed an in situ competitive LC-MS/MS-based experiment. Briefly, RPMI 8226 cells were treated with 100 nM 1 for 6 hours, lysed, then treated with 10 μM 20 followed by CuAAC-mediated conjugation of biotin azide, enrichment with streptavidin beads, trypsin digestion, and LC-MS/MS analysis. Out of 764 proteins enriched by 20, only 5 were >50% competed by 1 (Figs. 4C, 4D; Table S5). Strikingly, all 5 identified targets were 20S proteasome subunits: PSMB5, PSMB10, PSMA5, PSMB1, and PSMB2. Such high selectivity is remarkable, especially considering that the structure of 1 contains two electrophilic α, β-unsaturated amides that could potentially engage hundreds of reactive cysteines (Abegg et al., 2015).
Figure 4. Cytotoxicity and cellular target identification of cepafungin I.
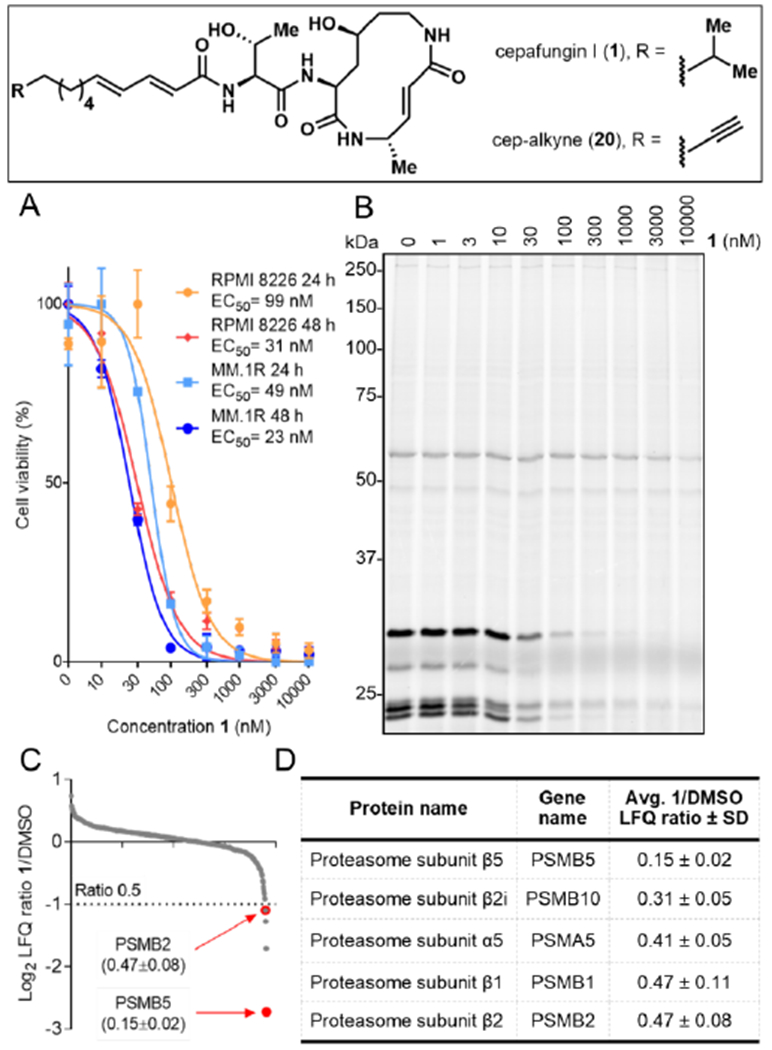
(A) Cytotoxicity curves of RPMI 8226 and MM1.R cells treated with indicated concentrations of 1 for 24 or 48 hours. Quantification was performed using the WST-1 assay (relative values ± SD and EC50s; n = 3). (B) Gel-based competitive in situ profiling of targets of 1. RPMI 8226 cells were treated with various concentrations of 1 for 6 h, lysed, and treated with 10 μM 20. (C) Log2 LFQ ratios of proteins identified from the in situ competitive pull-down experiment with 100 nM 1 or DMSO and with 10 μM 20 (n = 6, three biologicals and two technicals each). (D) Cellular targets of 1 (>50% competition) in RPMI 8226 cells. Quantification was performed using the label-free quantification (LFQ) method (n = 6).
We chose to further validate the engagement of PSMB2 (Proteasome 20S Subunit β2) and PSMB5 (Proteasome 20S Subunit β5) by overexpression and labeling by 1, because their chemical co-inhibition has been shown to be particularly cytotoxic in proteasome inhibitor-resistant MM cells and has therefore emerged as a promising therapeutic strategy (Besse et al., 2019). While single isolated proteasome subunits do not display enzymatic activity and can thus not be labeled by 1, successful incorporation of various overexpressed tagged and untagged proteasome subunits into functional proteasome has previously been documented in yeast and mammalian cells (Howell et al., 2005; Chondrogianni et al., 2005). We therefore hypothesized that overexpression of β2 and β5 subunits would result in their partial incorporation into the proteolytic core (CP) where they can be labeled by our probe. Both proteins were cloned as FLAG-tagged versions and overexpressed in HEK293T cells. PSMB2 and PSMB5-expressing cells were lysed and the lysates labeled with 30 μM 20 for 1 h, followed by conjugation to TAMRA azide, separation by SDS-PAGE, and visualization of probe-bound proteins by in-gel fluorescence scanning (Figs. 5A, S1C). Indeed, a new labeled band was clearly seen in the overexpressed lane of both gels, indicating successful labeling of both overexpressed proteins, PSMB2-F and PSMB5-F.
Figure 5. Validation of target engagement, structure-activity relationship, and downstream effects of 1.
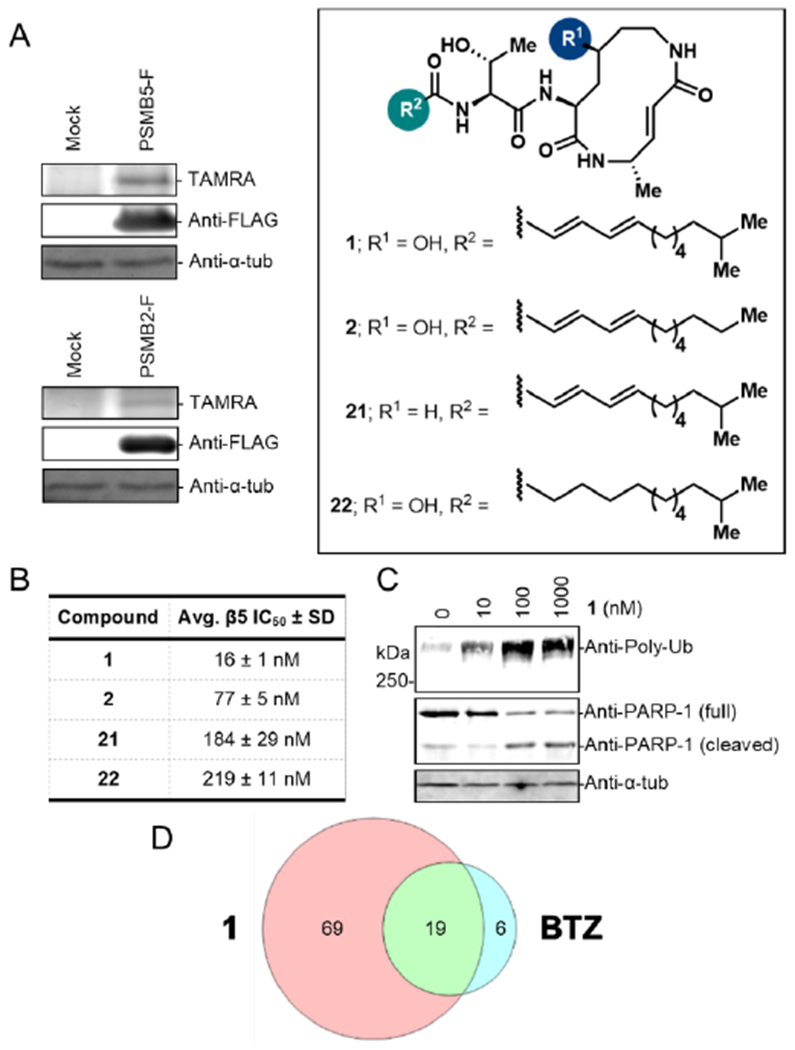
(A) Labeling of mock and overexpressed PSMB2-FLAG and PSMB5-FLAG in HEK293T lysate with 30 μM 20. Shown are the TAMRA fluorescence profile of 20 (top), Western blot membrane probed for FLAG (middle), and α-tubulin as a loading control (bottom). See also Fig. S1C. (B) In situ inhibition of chymotrypsin-like (β5) proteasome activity by 1 and its derivatives. RPMI 8226 cells were treated with various concentrations of compounds for 6 h, lysed, treated with 100 μM fluorogenic substrate Suc-LLVY-AMC, and incubated for 2 hours at 37 °C. IC50 values were quantified based on A360ex/A460em readouts (n = 3). (C) Western blotting of lysates obtained from RPMI 8226 cells treated with various concentrations of 1 for 6 h. Membranes were probed for mono- and poly-ubiquitin, PARP-1, and α-tubulin as a loading control. (D) Venn diagram representing the overlap in significantly upregulated proteins (FDR 0.05; S0 0.01) upon 14 h treatment of RPMI 8226 cells with 100 nM 1 or 2.5 nM BTZ versus vehicle (see Fig. 4A, S2). Quantification was performed using label-free quantification (LFQ) method (n = 6).
Having confirmed both proteins as targets of cepafungin I, we successfully assigned the fluorescence bands in the gel profile of 20 to respective proteasome subunits as previously described (Chondrogianni et al., 2005; Altun et al., 2005; Britton et al., 2009) and quantified the in situ IC50 values of 1 for the endogenously expressed proteasome subunits, yielding IC50s of 7.5 nM for PSMB5 and 35 nM for PSMB2 (Figs. S1D, E). Interestingly, these values only slightly deviate from previously reported IC50s for inhibition of purified yeast proteasome subunits by 1 (PSMB5: 4 nM; PSMB2: 24 nM, Stein et al., 2012). In order to gain more insight into the structure-activity relationship between 1 and PSMB5, we synthesized several derivatives of 1 (Fig. 5) and compared their ability to inhibit β5 activity in RPMI 8226 cells. These derivatives were designed to assess the contribution of various portions of the scaffold to its inhibitory activity, namely the macrocyclic secondary alcohol, the 2,4-dienamide portion of the tail and the terminal methyl branching. Prior medicinal chemistry explorations of the natural product family have focused on the syringolin series, which contain an additional degree of unsaturation in the lysine unit instead of C4 hydroxylation. Within this series, independent studies by Pirrung (Ibarra-Rivera et al., 2011) and by Ichikawa (Chiba et al., 2014; Kitahata et al., 2016; Kitahata et al., 2017) have shed light on the importance of the macrocyclic ring size and the location of the double bond, as well as the need for a hydrophobic tail for high inhibitory activity. In contrast, the cepafungin/glidobactin series have been less explored for analog synthesis and structure-activity relationship studies. Oka et al. had semi-synthetically prepared various derivatives of glidobactin with modifications in the tail portion of the molecule. These compounds were tested for antifungal activity and in vivo cytotoxicity against B16 melanoma and P388 leukemia cells in mice but were not studied in the context of proteasome inhibition.
To assess the inhibitory activity of the synthesized derivatives, cells were treated with various concentrations of compounds for 6 hours, then lysed and treated with the β5 subunit-selective fluorogenic substrate Suc-LLVY-AMC (Maher, 2014). Release of the 7-Amino-4-methylcoumarin (AMC) fluorophore via proteolytic cleavage by β5 was followed over time and quantified to obtain IC50 values (Fig. 5B). From all compounds tested, 1 most potently inhibited the β5 subunit activity. In agreement with prior work (Stein et al., 2012), terminal methyl branching in 1 leads to a nearly fivefold increase in β5 inhibitory activity compared to 2 (glidobactin A). Based on prior structural data (Stein et al. 2012), the lipid tail occupies a hydrophobic patch formed by the adjacent β6 subunit in the active site. Thus, it is likely that the improved activity observed with additional terminal branching arises from increased van der Waals interaction in this region. The difference in potency between 21 (desoxycepafungin) and 1, over tenfold loss, is in agreement with a recently reported potency difference between 2 and its corresponding desoxy-macrocycle variant luminmycin A (Pawar et al., 2020). While the origin of this difference cannot be readily explained by the available structural data, it is well-established that the β5 subunit prefers hydrophilic residues at its S3 site (Clerc et al., 2010a). Thus, it is possible that the 4-hydroxylysine residue increases the hydrophilicity of the macrocycle for more potent binding. Ichikawa has previously reported the bioactivity of several syringolin derivatives bearing alternative alkyl tails. However, no direct head-to-head comparison has been done to investigate the relative importance of the unsaturation in the lipid tail for proteasomal inhibition. While the lipid tail occupies a hydrophobic patch in the binding pocket, it is unclear at the outset whether its rigidity will have any contribution to activity. Indeed, 22 (saturated cepafungin) demonstrated a fourteen-fold loss in potency compared to 1, suggesting that the structural rigidity conferred by the lipid tail unsaturation is important for potent proteasome engagement. Taken together, these results suggest that the secondary alcohol on the macrocycle and the 2,4-dienamide motif on the tail region of 1 are critical for the β5 inhibitory activity.
Next, we sought to investigate if 1 mediates the expected biological downstream response similar to other proteasome inhibitors. One of the key cellular responses of proteasome inhibition is the accumulation of polyubiquitinated proteins, gradually leading to dysfunction and apoptosis (Adams, 2004). Indeed, Western blot of lysates from 1-treated RPMI 8226 cells using a mono-/poly-ubiquitin-conjugate specific antibody showed a concentration-dependent increase in ubiquitinated proteins (Fig. 5C). Next, we probed the same lysates by Western blot for Poly(ADP-Ribose) Polymerase (PARP-1) and observed PARP-1 cleavage in 1-treated samples, indicating that 1 induced apoptotic cell death (Fig. 5C, Kaufmann et al., 1993). Finally, we performed a global proteomics experiment to identify proteins that accumulate or are upregulated in RPMI 8226 cells upon 1 treatment in comparison to the treatment with the clinical multiple myeloma drug bortezomib (BTZ), which is known to covalently inhibit the 20S proteasome (Groll et al., 2006). Previous gel-based ABPP study with vinyl sulfone-based proteasome probes has shown that BTZ engages the proteasome subunits β5, β1, and β2 (Berkers et al., 2005). We therefore anticipated that 1 and BTZ treated myeloma cells may share many upregulated protein targets. Cells were treated for 14 h with either 1 (100 nM) or BTZ (2.5 nM), at concentrations matching their corresponding EC50 values at the 24 h timepoint to avoid protein expression changes linked to cell death (Figs. 4A, S2). Cells were lysed, proteins digested, and peptides analyzed by LC-MS/MS. Out of 3700 quantified proteins, 88 proteins were significantly upregulated (FDR 0.05; S0 0.01) in 1-treated samples and 25 in BTZ-treated samples compared to DMSO control samples (Figs. 5D, S3; Table S6). Indeed, 19 of 25 proteins upregulated in BTZ-treated samples, were also upregulated in proteomic samples from 1-treated cells, thus confirming highly similar mode of action for these two compounds. We believe that even better overlap can be achieved if factors such as differential cell permeability, binding kinetics, and metabolic stability are taken into account. Notably, 11 out of these 19 common targets have previously been reported to be upregulated (Hong et al., 2007a; Hong et al., 2007b; Fabre et al., 2019; Blanchard et al., 2002; Matondo et al., 2017; Mertins et al., 2013; Tatsumi et al., 2003; Yew et al., 2005; Kwan et al., 2015; Chen et al., 2014) upon proteasome inhibition in general (HERPUD1, KIAA0101, RRM2, CDC6, CCNB1, NUSAP1, ORC1, CYBA, CKAP2, KRT18, TPX2) and 5 more targets (BAG3, SRXN1, HMOX1, SQSMT1, HSPA1A/B) specifically upon bortezomib treatment (Liu et al., 2009; Starheim et al., 2016; Shah et al., 2016). Altogether, our data show that 1 is a potent and highly selective covalent inhibitor of the 20S proteasome that mediates downstream effects similar to the clinical drug BTZ in multiple myeloma cells.
Significance
By harnessing the ability of GlbB to hydroxylate l-lysine in a selective and efficient fashion, we have developed a concise chemoenzymatic synthesis of cepafungin I. Despite challenges in the late-stage macrocyclization and amide coupling steps, cepafungin I could be obtained in ca. 8% yield over 9 steps (longest linear sequence). The modularity of this route allowed the development of a chemoproteomic probe to interrogate the cellular targets of cepafungin I. Our chemoproteomics studies revealed that cepafungin I is able to covalently engage 20S proteasome subunits PSMB2 and PSMB5 with exceptional selectivity and that cepafungin I elicits many similar downstream biological responses to the clinically-approved proteasome inhibitor drug bortezomib. Contemporaneous to our efforts, Böttcher and co-workers reported the development of an activity-based probe by derivatization of syringolin A that was obtained from its native producer and showed the ability of the syrbactins to coinhibit PSMB2 and PSMB5 with high potency. Complementary to Böttcher’s findings, our work provides the first proteome-wide identification of syrbactin protein targets in cancer cells and validation of their downstream biological effects via in-depth quantitative proteomics analyses. Moreover, the synthetic strategy developed herein enabled access to both natural and unnatural cepafungin analogs for initial structure-activity relationship studies. Especially notable in this regard is the surprising observation that the macrocyclic secondary alcohol, as well as the degree of unsaturation and the terminal branching of the lipid tail, are critical for high inhibitory potency. In combination with prior structural data of the 20S proteasome:cepafungin complex, our synthetic strategy facilitates structure-guided design of further analogs featuring alternative macrocycle and lipid functionalities. Such studies are ongoing in our laboratory.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hans Renata.
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The published article includes all datasets generated and analyzed during this study. This study did not generate any code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and lysate preparation
RPMI 8226 and MM1.R cells were maintained in RPMI-1640 media. HEK293T cells were maintained in DMEM. All media were supplemented with 10% fetal bovine serum (FBS), non-essential amino acids and penicillin/streptomycin. Cells were grown at 37 °C under 5% CO2 atmosphere. Cells were allowed to grow to confluence, harvested by pipeting, then washed 2 times in PBS by brief centrifugation at 300 x g at 4 °C and resuspended in PBS. Cells were lysed by needle sonication to obtain cell lysates and protein concentration was determined using the Bradford assay.
METHOD DETAILS
Chemistry materials
Unless otherwise noted, all chemicals and reagents for chemical reactions were purchased at the highest commercial quality and used without further purification. Reactions were monitored by thin layer chromatography (TLC) and liquid chromatography/mass spectrometry (LC/MS). TLC was performed with 0.25 mm E. Merck silica plates (60F-254) using short-wave UV light as the visualizing agent, and ninhydrin or KMnO4 and heat as developing agents. Sonication was performed using a Qsonica Q500 sonicator. LC/MS was performed with an Agilent 1260 Infinity System equipped with Poroshell 120 EC-C18 column (3.0 x 50 mm, 2.7 μm). HRMS(ESI-TOF) was performed with an Agilent 1260 Infinity System equipped with Poroshell 120 EC-C18 column (4.6 x 5.0 mm, 2.7 μm) and an Agilent G6230B TOF LC/MS. Preparative HPLC was performed on an Agilent 1260 Infinity II system equipped with a Dr. Maisch Reprosil 100 C18 column (10 x 250 mm, 5 μm), or on an Agilent 1260 Infinity system equipped with a Varian Dynamax C18 column (21.4 x 250 mm, 10 μm). NMR spectra were recorded on a Bruker AVANCE AV400 (400 MHz and 100 MHz) or Bruker AVANCE AV600 (600 MHz and 151 MHz) at 23 °C and calibrated using residual non-deuterated solvent. NMR spectra can be found under Supplemental Data S1. Specific rotations were measured on Autopol IV polarimeter (Rudolph Research Analytical).
Bacterial strain construction
pET-28a(+)-GlbB expression vector was obtained from Genscript as described previously (Amatuni and Renata, 2019). E. coli BL21(DE3) harboring pET-28a(+)-GlbB and pGro7 (Takara Bio, Cat#3340) was constructed by transforming electrocompetent E. coli BL21(DE3) (Lucigen, Cat#60300-2) with the appropriate plasmids.
Chemical synthesis and characterization
Chemoenzymatic synthesis of cepafungin I
(2S,4S)-2,6-diamino-4-hydroxyhexanoic acid (5):
200 mL of TB media containing 25 μg/mL chloramphenicol and 50 μg/mL kanamycin was inoculated with 1 mL of an overnight culture of E. coli BL21(DE3) cells harboring pET-28a(+)-GlbB and pGro7. The culture was shaken at 250 rpm at 36 °C for approximately 2.5 hours or until an optical density of 0.6 was reached. The culture was then cooled on ice (20 minutes) and induced by adding IPTG and arabinose to a final concentration of 0.025 mM and 1 mg/mL, respectively. The culture was allowed to continue for another 20 hours at 23 °C and shaking at 250 rpm. Cells were harvested by centrifugation at 4200 rpm at 4 °C for 15 minutes, then resuspended in pH = 8 kPi buffer (150 mL) to an optical density of 25. The cells were lysed by sonication for 3 minutes (1 second on, 4 seconds off) at 0 °C and 50% amplitude. Cell debris was pelleted by centrifugation and the supernatant was diluted to 250 mL with pH = 8 kPi buffer into a 1L Erlenmeyer flask containing L-lysine (4) (1.46 g, 10 mmol, 1 eq, 40 mM final concentration), α-ketoglutaric acid (2.83 g, 12.5 mmol, 1.25 eq, 50 mM final concentration), ascorbic acid (440 mg, 2.5 mmol, 0.25 eq, 10 mM final concentration) and FeSO4•7H2O (139 mg, 0.5 mmol, 0.05 eq, 2 mM final concentration). The mixture was shaken at 200 rpm at 23 °C overnight or until completion as judged by LC-MS.
The reaction mixture was then acidified to pH = 2 with concentrated HCl and centrifugated at 4200 rpm at 4 °C for 15 minutes. The supernatant containing crude 4-hydroxylysine 5 was carried forward to the next step without further purification. For characterization purposes, the crude product was purified by ion exchange chromatography with Dowex 50WX8 resin.
1H NMR (600 MHz, D2O with 1% TFA):
δ 4.09 – 3.99 (m, 1H), 3.89 – 3.78 (m, 1H), 3.21 – 3.08 (m, 2H), 2.18 – 2.09 (m, 1H), 1.98 – 1.91 (m, 1H), 1.91 – 1.80 (m, 2H).
13C NMR (151 MHz, D2O with 1% TFA):
δ 174.4, 67.7, 53.5, 37.1, 36.6, 34.0.
= 3.7 (c = 0.5, H2O)
HRMS (ESI-TOF):
calculated for C6H16N2O3+ ([M+H]+) 163.1083, found 163.1078
tert-butyl (2-((2S,4S)-4-((tert-butoxycarbonyl)amino)-5-oxotetrahydrofuran-2-yl)ethyl)carbamate (7):
The above supernatant was concentrated to ~33 mL then basified to pH 11 with 2 M NaOH. Boc2O (8.73 g, 40 mmol, 4 eq) in 17 mL ethanol was added and the mixture was allowed to stir at room temperature. The mixture was re-basified to pH 11 after 3.5 hours and allowed to continue stirring overnight or until completion as indicated by LC-MS. Ethanol was removed in vacuo and the mixture was diluted with 50 mL ethyl acetate. The pH of the mixture was adjusted to 2 with 6 M HCl and the aqueous layer was separated and further extracted with ethyl acetate (3 x 50 mL). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated in vacuo to provide 4.29 g crude di-Boc-4-hydroxylysine that was used without further purification.
The crude Boc-protected hydroxylysine (4.29 g) was dissolved in 50 mL anhydrous DCM, cooled to 0 °C, then treated with EDC•HCl (1.92 g, 10 mmol, 1 eq) followed by triethylamine (1.39 mL, 10 mmol, 1 eq). The reaction was stirred under argon at 0 °C for 2 hours or until completion as indicated by TLC. The reaction was concentrated to dryness. The resulting residue was dissolved in 100 mL ethyl acetate and washed with water (3 x 20 mL) and brine, then dried over Na2SO4. Purification by silica flash chromatography (step gradient 35-40-50% ethyl acetate in hexanes) provided pure lactone 7 (2.39 g, 6.93 mmol, 69% over 3 steps).
Note: the above 3-step sequence can be scaled up to 6.90 grams of L-lysine (47.2 mmol) to provide 9.53 grams of lactone 7 (59% yield).
1H NMR (400 MHz, (CD3)2SO):
δ 7.45 (d, J = 8.3 Hz, 1H), 6.89 (t, J = 5.7 Hz, 1H), 4.62 – 4.48 (m, 1H), 4.28 (q, J = 9.1 Hz, 1H), 3.07 – 2.90 (m, 2H), 2.32 – 2.09 (m, 2H), 1.82 – 1.60 (m, 2H), 1.38 (s, 9H), 1.37 (s, 9H).
13C NMR (101 MHz, (CD3)2SO):
δ 175.2, 155.6, 155.1, 78.6, 77.6, 75.4, 48.5, 36.4, 35.1, 32.5, 28.2, 28.1.
= 26.7 (c = 0.5, CHCl3)
HRMS (ESI-TOF):
calculated for C16H29N2O6+ ([M+H]+) 345.2026, found 345.2026
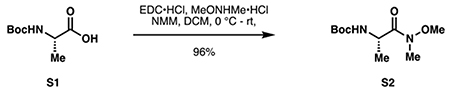
tert-butyl (S)-(1-(methoxy(methyl)amino)-1-oxopropan-2-yl)carbamate (S2):
Boc-L-alanine (S1, 9.10 g, 48.1 mmol, 1.0 eq), N,O-dimethylhydroxylamine hydrochloride (5.16 g, 52.9 mmol, 1.1 eq) and N-methylmorpholine (5.82 mL, 52.9 mmol, 1.1 eq) were dissolved in 120 mL anhydrous DCM. The mixture was cooled to 0 °C, and EDC•HCl (9.22 g, 48.1 mmol, 1.0 eq) was added portionwise. The reaction was stirred at 0 °C for two hours, and then at room temperature overnight. Upon complete consumption of S1 by TLC, the reaction was diluted with 75 mL water and the pH of the mixture was adjusted to 2 with 1 M HCl. The layers were separated, and the aqueous phase extracted twice with 20 mL DCM. The combined organic layers were washed with 30 mL water, saturated NaHCO3, brine, then dried over Na2SO4 and concentrated in vacuo to provide pure S2 as a crystalline white+ solid (10.7 g, 46.1 mmol, 96%).
1H NMR (600 MHz, CD3OD):
δ 4.60 – 4.53 (m, 1H), 3.81 (s, 3H), 3.20 (s, 3H), 1.44 (s, 9H), 1.26 (d, J = 7.1 Hz, 3H).
13C NMR (151 MHz, CD3OD):
δ 175.7, 157.6, 80.4, 62.0, 48.0, 32.4, 28.7, 17.4.
= −26.7 (c 2.45, MeOH)
HRMS (ESI-TOF):
calculated for C10H20N2O4Na+ ([M+Na]+) 255.1315, found 255.1315
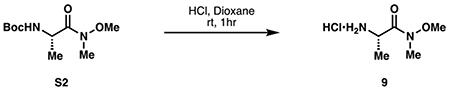
(S)-2-amino-N-methoxy-N-methylpropanamide hydrochloride (9):
Boc-L-alanine amide S2 (7.00 g, 29.4 mmol, 1 eq) was dissolved in 49 mL of 4 M HCl in dioxane at room temperature. Upon complete consumption of S2 by TLC, the reaction was concentrated to dryness, then evaporated twice from 50 mL 1:1 methanol:toluene to provide 9 as a white solid (5.72 g, quantitative).
1H NMR (600 MHz, CD3OD):
δ 4.35 (q, J = 7.0 Hz, 1H), 3.81 (s, 3H), 3.25 (s, 3H), 1.50 (d, J = 7.0 Hz, 3H).
13C NMR (151 MHz, CD3OD):
δ 170.9, 62.4, 48.7, 32.5, 16.3.
= −27.7 (c 2.23, MeOH)
HRMS (ESI-TOF):
calculated for C5H13N2O2+ ([M+H]+) 133.0972, found 133.0974

di-tert-butyl ((3S,5S)-3-hydroxy-6-(((S)-1-(methoxy(methyl)amino)-1-oxopropan-2-yl)amino)-6-oxohexane-1,5-diyl)dicarbamate (10):
Amine hydrochloride 9 (5.71g, 33.9 mmol, 5 eq) was set stirring (undissolved) in 85 mL anhydrous THF. The mixture was cooled to −20 °C, then a 2 M solution of trimethylaluminum in anhydrous toluene was added dropwise (16.8 mL, 4.95 eq). The reaction was stirred at −20 °C for 30 minutes under argon to provide a clear, light yellow solution. To this mixture was added lactone 7 in 17 mL anhydrous THF (2.33 g, 6.77 mmol, 1 eq) dropwise at −20 °C. The reaction was stirred to room temperature overnight, until completion by TLC. After 28 hours, the reaction was cooled to 0 °C and quenched by the careful addition of 20 mL 2 M NaHSO4 solution dropwise. The pH of the mixture was adjusted to 1 with 50 mL 1 M HCl, then concentrated in vacuo to remove most of the organic solvent. The remaining aqueous mixture was extracted with ethyl acetate (5 x 50 mL). The combined organic layers were then washed with water, saturated NaHCO3, water, brine and finally dried over Na2SO4. Concentration in vacuo provided 3.15 g of crude dipeptide 10 that was further purified by silica flash chromatography (60:30:10 hexanes:ethyl acetate:methanol) to provide pure 10 as a white solid (2.90 g, 6.09 mmol, 90 %).
1H NMR (400 MHz, CD3OD):
δ 6.52 (s, 1H), 4.86 – 4.77 (m, 1H), 4.17 (t, J = 7.3 Hz, 1H), 3.82 (s, 3H), 3.80 – 3.73 (m, 1H), 3.21 (s, 3H), 3.19 – 3.13 (m, 2H), 1.90 – 1.75 (m, 1H), 1.72 – 1.61 (m, 2H), 1.60 – 1.49 (m, 1H), 1.44 (s, 9H), 1.43 (s, 9H), 1.34 (d, J = 7.1 Hz, 3H).
13C NMR (151 MHz, CD3OD):
δ 174.6, 174.5, 158.6, 157.6, 80.6, 80.0, 67.2, 62.1, 53.5, 47.1, 40.8, 38.4, 38.1, 32.5, 28.8, 28.7, 17.1.
= −28.8 (c 1.01, MeOH)
HRMS (ESI-TOF):
calculated for C21H41N4O8+ ([M+H]+) 477.2919, found 477.2917
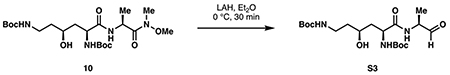
di-tert-butyl ((3S,5S)-3-hydroxy-6-oxo-6-(((S)-1-oxopropan-2-yl)amino)hexane-1,5-diyl)dicarbamate (S3):
In a procedure adapted from Fehrentz et al. (1985), dipeptide 10 (100 mg, 0.210 mmol, 1 eq) was dissolved in 2.1 mL anhydrous diethyl ether and cooled to 0 °C. A solution of lithium aluminum hydride (1.05 M in Et2O, 0.50 mL, 2.5 eq) was then added dropwise at 0 °C. Upon complete addition, the reaction was stirred for 30 minutes at 0 °C then quenched with 2 mL 2M NaHSO4. The layers were separated and the aqueous phase was extracted with Et2O (2 x 2 mL). The combined organic layers were washed with saturated NaHCO3 and brine, then dried over Na2SO4. Concentration in vacuo provided aldehyde S3 as a white foam, which was used without further purification. A sample of the material was purified by silica flash chromatography (9:0.9:0.1 ethyl acetate:dichloromethane:triethylamine) to aid characterization by 1H NMR.
1H NMR (400 MHz, CDCl3):
δ 9.53 (d, J = 2.4 Hz, 1H), 7.23 – 6.99 (m, 1H), 5.82 – 5.55 (m, 1H), 4.91 – 4.73 (m, 1H), 4.50 – 4.36 (m, 1H), 4.35 – 4.18 (m, 2H), 3.86 – 3.68 (m, 1H), 3.55 – 3.36 (m, 1H), 3.18 – 3.00 (m, 1H), 2.03 – 1.91 (m, 1H), 1.85 – 1.71 (m, 1H), 1.63 – 1.53 (m, 2H), 1.45 – 1.41 (m, 18H), 1.35 (d, J = 7.3 Hz, 2H).
HRMS (ESI-TOF):
calculated for C19H36N3O7+ ([M+H]+) 418.2548, found 418.2548
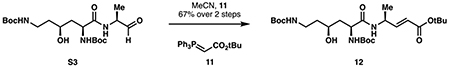
tert-butyl (S,E)-4-((2S,4S)-2,6-bis((tert-butoxycarbonyl)amino)-4-hydroxyhexanamido)pent-2-enoate (12):
Ylide 11 (106 mg, 0.281 mmol, 1.25 eq) was dissolved in 0.5 mL anhydrous acetonitrile, then a solution of aldehyde S3 (94 mg) in 3 mL acetonitrile was added dropwise at room temperature. The reaction was refluxed for 2 hours at 60 °C, then concentrated to dryness in vacuo. Purification by silica flash chromatography (step gradient 70:29.5:0.5 to 70:27:3 dichloromethane:ethyl acetate:methanol) provided 12 as an off-white solid (97 mg, 0.188 mmol, 67% over 2 steps).
1H NMR (600 MHz, CD3OD):
δ 6.78 (dd, J = 15.7, 5.0 Hz, 1H), 5.84 (dd, J = 15.7, 1.8 Hz, 1H), 4.59 (qdd, J = 7.0, 4.9, 1.7 Hz, 1H), 4.16 – 4.03 (m, 1H), 3.73 – 3.62 (m, 1H), 3.16 (t, J = 6.9 Hz, 2H), 1.89 – 1.77 (m, 1H), 1.75 – 1.64 (m, 2H), 1.58 – 1.51 (m, 1H), 1.47 (s, 9H), 1.45 (s, 9H), 1.43 (s, 9H), 1.28 (d, J = 7.0 Hz, 3H).
13C NMR (151 MHz, CD3OD):
δ 174.4, 167.4, 158.7, 157.7, 149.1, 122.9, 81.7, 80.7, 80.0, 67.7, 54.4, 47.0, 40.7, 38.4, 38.1, 28.8, 28.8, 28.4, 19.9.
= −23.5 (c 0.72)
HRMS (ESI-TOF):
calculated for C25H46N3O8+ ([M+H]+) 516.3279, found 516.3299
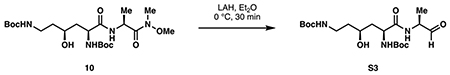
di-tert-butyl ((3S,5S)-3-hydroxy-6-oxo-6-(((S)-1-oxopropan-2-yl)amino)hexane-1,5-diyl)dicarbamate (S3, gram scale):
Dipeptide 10 (2.84 g, 5.96 mmol, 1 eq) was dissolved in 60 mL anhydrous diethyl ether and cooled to 0 °C. A solution of lithium aluminum hydride (1 M in Et2O, 14.9 mL, 2.5 eq) was then added dropwise at 0 °C. Upon complete addition, the reaction was stirred for 30 minutes at 0 °C then quenched with 30 mL 2 M NaHSO4. The layers were separated and the aqueous phase was extracted with Et2O (4 x 25 mL). The combined organic layers were washed with water, saturated NaHCO3 and brine, then dried over Na2SO4. Concentration in vacuo provided aldehyde S3 as a white foam that was used for the next step without further purification (2.15 g).

tert-butyl (S,E)-4-((2S,4S)-2,6-bis((tert-butoxycarbonyl)amino)-4-hydroxyhexanamido)pent-2-enoate (12, gram scale):
Ylide 11 (2.42 g, 6.42 mmol, 1.25 eq) was dissolved in 63 mL anhydrous acetonitrile, then a solution of aldehyde S3 (2.15 g) in 10 mL acetonitrile was added dropwise at room temperature. The reaction was stirred for 30 minutes or until completion by 1H NMR or TLC, and then concentrated to dryness. Purification by silica flash chromatography (100% diethyl ether) provided 12 as an off-white solid (1.59 g, 3.08 mmol, 52% over 2 steps).

(S,E)-4-((2S,4S)-2,6-diamino-4-hydroxyhexanamido)pent-2-enoic acid--2,2,2-trifluoroacetic acid (1/2) (S4):
Dipeptide 12 (500 mg, 0.970 mmol, 1 eq) was dissolved in 24.2 mL of a freshly prepared solution of “Reagent B” (88:5:5:2 trifluoroacetic acid:phenol:water:triisopropylsilane). After stirring for 1 hour at room temperature, the reaction was diluted with 30 mL toluene and concentrated in vacuo to dryness, followed by an additional evaporation from 30 mL toluene. The residue was then dissolved in 2 mL methanol and added dropwise to 100 mL diethyl ether at 0 °C. The solids were collected by centrifugation at 4200 RPM for 10 minutes at 4 °C, then precipitated again into 50 mL diethyl ether at 0 °C from 1.5 mL methanol. The solids were collected by centrifugation as above, then dried from methanol to provide S4 as an off-white solid (442 mg, 0.907 mmol, 94 %).
1H NMR (600 MHz, CD3OD):
δ 6.82 (dd, J = 15.7, 5.4 Hz, 1H), 5.89 (dd, J = 15.7, 1.6 Hz, 1H), 4.67 – 4.61 (m, 1H), 4.04 – 3.97 (m, 1H), 3.96 – 3.87 (m, 1H), 3.13 – 3.01 (m, 2H), 2.06 – 2.00 (m, 1H), 1.94 – 1.84 (m, 2H), 1.84 – 1.74 (m, 1H), 1.33 (d, J = 7.0 Hz, 3H).
13C NMR (151 MHz, CD3OD):
δ 170.3, 169.4, 148.9, 122.9, 68.3, 53.6, 47.4, 39.2, 37.8, 36.0, 19.7.
= −1.8 (c 4.03, MeOH)
HRMS (ESI-TOF):
calculated for C11H22N3O4+ ([M+H]+) 260.1605, found 260.1605
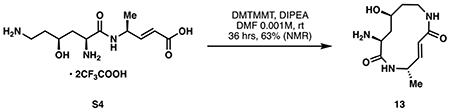
(5S,8S,10S,E)-8-amino-10-hydroxy-5-methyl-1,6-diazacyclododec-3-ene-2,7-dione (13):
Linear macrocycle precursor S4 (366 mg, 0.751 mmol, 1 eq) was dissolved in 750 mL anhydrous DMF. The solution was treated with diisopropylethylamine (0.523 mL, 3.00 mmol, 4 eq), followed by solid 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium tetrafluoroborate (DMTMMT) (369 mg, 1.13 mmol, 1.50 eq) at room temperature. The reaction was stirred under argon for 36 hours, then quenched with 20 mL water and concentrated in vacuo to dryness. The residue was dissolved in 3 mL methanol and precipitated into 50 mL diethyl ether with rapid stirring. The solids were collected by centrifugation at 4200 RPM for 15 minutes at 4 °C to yield crude 13 as a tan solid (540 mg, 0.473 mmol, 63% from S4, 59% from 12). Yield was determined by 1H NMR analysis of a 7.4 mg crude sample with 4-toluenesulfonamide (7.8 mg, 0.0465 mmol) added as internal standard. See Table S1 for macrolactamization optimization.
1H NMR (400 MHz, CD3OD):
4-toluenesulfonamide: δ 7.82 – 7.72 (m, 2H), 7.40 – 7.27 (m, 2H), 2.42 (s, 3H). Compound 13: 6.63 (dd, J = 16.0, 6.1 Hz, 1H), 6.38 (dd, J = 16.0, 1.1 Hz, 1H), 4.73 – 4.64 (m, 1H), 3.83 – 3.76 (m, 1H), 2.27 – 2.10 (m, 1H), 2.01 – 1.92 (m, 1H), 1.92 – 1.79 (m, 0H), 1.75 – 1.64 (m, 2H).
HRMS (ESI-TOF):
calculated for C11H20N3O3+ ([M+H]+) 242.1499, found 242.1495
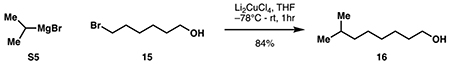
7-methyloctan-1-ol (16):
Following a procedure adapted from Melaugh et al. (2014), lithium chloride (6.66 g, 157 mmol, 2.05 eq) was flame-dried in a 250 mL round-bottom flask under high vacuum. Solid copper(II) chloride (10.3 g, 76.6 mmol, 1 eq) was added under argon. The combined reagents were then dissolved in 105 mL anhydrous THF to provide a dark red solution of Li2CuCl4.
A flame-dried 3-neck 1L flask fitted with a reflux condenser was charged with magnesium turnings (12.9 g, 531 mmol, 1.18 eq with respect to 2-bromopropane) and 200 mL anhydrous THF. With rapid stirring, 2-bromopropane (42.3 mL, 451 mmol, 6.5 eq with respect to 15) was added dropwise until refluxing began, at which point the flask was lowered into a water bath. Upon complete addition, the water bath was removed and the reaction was allowed to stir under argon for 30 minutes to produce a dark gray suspension of S5. The mixture was cooled to −78 °C, and 6-bromohexan-1-ol 15 (12.6 g, 69.6 mmol, 1 eq) was added in 10 mL dry THF, followed by the above solution of Li2CuCl4 (~0.7 M in THF, 105 mL, 1.1 eq with respect to 15). Upon complete addition, the reaction was removed from its cooling bath and allowed to warm to room temperature. Reaction progress was monitored by TLC until completion at 1 hour. The reaction was then carefully quenched at 0 °C with 50 mL saturated NH4Cl solution, filtered through a sintered glass funnel, and the filtrate was concentrated in vacuo to remove most of the THF. The mixture was diluted with 100 mL ethyl acetate and 100 mL water. The layers were separated, and the aqueous phase was extracted with ethyl acetate (3 x 200 mL). The combined organic layers were washed with 50 mL each of saturated aqueous NaHCO3 and brine, then dried over Na2SO4. Purification by silica flash chromatography (90:10 hexanes:ethyl acetate) provided pure 16 as a colorless liquid (8.38 g, 58.1 mmol, 84%).
1H NMR (600 MHz, CDCl3):
δ 3.63 (t, J = 6.7 Hz, 2H), 1.60 – 1.45 (m, 3H), 1.38 – 1.31 (m, 2H), 1.31 – 1.22 (m, 4H), 1.20 – 1.10 (m, 2H), 0.85 (d, J = 6.7 Hz, 6H).
13C NMR (151 MHz, CDCl3):
δ 63.2, 39.1, 32.9, 29.8, 28.1, 27.5, 25.9, 22.8.
HRMS (ESI-TOF):
calculated for C11H24NO+ ([M+MeCN+H]+) 186.1852, found 186.1852

7-methyloctanal (S6):
Oxalyl chloride (893 μL, 10.4 mmol, 1.5 eq) was dissolved in 8 mL anhydrous DCM at −78 °C. An anhydrous solution of DMSO (1.48 mL, 20.8 mmol, 3 eq) in 3 mL DCM was added dropwise and the resulting mixture was stirred at −78 °C for 15 minutes. A solution of branched alcohol 16 (1.00 g, 6.93 mmol, 1 eq) in 6 mL anhydrous DCM was then added dropwise over 10 minutes. The mixture was stirred at −78 °C for 1 hour, and treated with triethylamine (4.83 mL, 34.7 mmol, 5 eq) dropwise. The resulting thick slurry was diluted with 10 mL DCM to aid stirring, and reaction progress was monitored by TLC until completion at 1 hour. The reaction was quenched at −78 °C with 20 mL water and allowed to warm to room temperature. The layers were separated, and the aqueous layer was extracted with DCM (3 x 10 mL). The combined organic layers were washed with water (twice) and brine, then dried over Na2SO4. Concentration in vacuo provided S6 as a pale yellow oil, which was used without further purification (1.19 g).
1H NMR (400 MHz, CDCl3):
δ 9.75 (t, J = 1.9 Hz, 1H), 2.41 (td, J = 7.3, 1.9 Hz, 2H), 1.66 – 1.58 (m, 2H), 1.56 – 1.44 (m, 1H), 1.34 – 1.24 (m, 4H), 1.20 – 1.10 (m, 2H), 0.85 (d, J = 6.6 Hz, 6H).
HRMS (ESI-TOF):
calculated for C9H19O+ ([M+H]+) 143.1430, found 143.1431
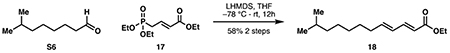
Ethyl (2E,4E)-11-methyldodeca-2,4-dienoate (18):
In a procedure adapted from Imker et al. (2010), ethyl (E)-4-(diethoxyphosphoryl)but-2-enoate 17 (2.60 g, 10.4 mmol, 1.5 eq) in 20 mL anhydrous THF was cooled to −78 °C. A solution of lithium hexamethyldisilazide (1 M THF, 9.91 mL, 1.4 eq) was then added dropwise and allowed to stir for an additional 1 hour at −78 °C. Crude aldehyde S6 (1.19 g) in 5 mL THF was added dropwise at −78 °C, and the mixture was allowed to stir to room temperature overnight. Upon completion by TLC, the reaction was cooled to 0 °C and quenched with 20 mL saturated NH4Cl. The layers were separated, and the aqueous layer was extracted with ethyl acetate (3 x 30 mL). The combined organic layers were washed with 20 mL brine and dried over Na2SO4. Purification by silica flash chromatography (97:3 hexanes:ethyl acetate) provided 18 as a yellow oil (957 mg, 4.01 mmol, 58% from 16).
1H NMR (600 MHz, CDCl3):
δ 7.26 (dd, J = 18.1, 15.4 Hz, 1H), 6.20 – 6.07 (m, 2H), 5.78 (d, J = 15.4 Hz, 1H), 4.19 (q, J = 7.2 Hz, 2H), 2.16 (td, J = 7.5, 6.1 Hz, 2H), 1.55 – 1.48 (m, 1H), 1.46 – 1.38 (m, 2H), 1.29 (t, J = 7.1 Hz, 3H), 1.30 – 1.24 (m, 4H), 1.18 – 1.12 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H).
13C NMR (151 MHz, CDCl3):
δ 167.5, 145.3, 144.9, 128.5, 119.3, 60.3, 39.0, 33.1, 29.6, 28.9, 28.1, 27.3, 22.8, 14.5.
HRMS (ESI-TOF):
Calculated for C15H27O2+ ([M+H]+) 239.2006, found 239.2004
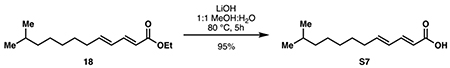
(2E,4E)-11-methyldodeca-2,4-dienoic acid (S7):
Ester 18 (450 mg, 1.89 mmol, 1 eq) was dissolved in 9.5 mL methanol. With rapid stirring, a solution of lithium hydroxide (226 mg, 9.44 mmol, 5 eq) in 9.5 mL water was added. The reaction was refluxed at 80 °C for 5 hours or until completion by TLC. Methanol was evaporated in vacuo, then the mixture was adjusted to pH 1 with 1 M HCl and diluted with 20 mL DCM. The layers were separated, and the aqueous layer was extracted with DCM (3 x 20 mL). The combined organic layers were washed with 10 mL of brine then dried over Na2SO4. Concentration in vacuo provided pure S7 as a waxy white solid (379 mg, 1.80 mmol, 95%).
1H NMR (600 MHz, CDCl3):
δ 7.38 – 7.31 (m, 1H), 6.24 – 6.14 (m, 2H), 5.78 (d, J = 15.3 Hz, 1H), 2.18 (td, J = 7.4, 5.5 Hz, 2H), 1.56 – 1.48 (m, 1H), 1.46 – 1.40 (m, 2H), 1.32 – 1.26 (m, 4H), 1.19 – 1.12 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H).
13C NMR (151 MHz, CDCl3):
δ 172.8, 147.7, 146.5, 128.3, 118.3, 39.0, 33.2, 29.6, 28.8, 28.1, 27.3, 22.8.
HRMS (ESI-TOF):
calculated for C13H23O2+ ([M+H]+) 211.1693, found 211.1695

tert-butyl O-(tert-butyl)-N-((2E,4E)-11-methyldodeca-2,4-dienoyl)-L-threoninate (S8):
Fatty acid S7 (125 mg, 0.594 mmol, 1 eq) was combined with tert-butyl O-(tert-butyl)-L-threoninate 19 (206 mg, 0.892 mmol, 1.5 eq) in 3 mL anhydrous DMF. Diisopropylethylamine was added (0.311 mL, 1.78 mmol, 3 eq), followed by HATU (237 mg, 0.624 mmol, 1.05 eq). The reaction was stirred at room temperature under argon for 4 hours or until completion by LCMS. The reaction was then quenched with 0.3 mL water and concentrated in vacuo. The residue was dissolved in 50 mL ethyl acetate and washed with 1M HCl (twice), water, NaHCO3 and brine, then dried over Na2SO4. Concentration in vacuo followed by silica flash chromatography (90:10 hexanes:ethyl acetate) provided S8 as a colorless oil (203 mg, 0.479 mmol, 81%).
1H NMR (600 MHz, CDCl3):
δ 7.21 (dd, J = 15.0, 10.6 Hz, 1H), 6.19 – 6.11 (m, 2H), 6.06 (dt, J = 15.1, 6.8 Hz, 1H), 5.87 (d, J = 15.1 Hz, 1H), 4.49 (dd, J = 9.2, 2.2 Hz, 1H), 4.21 (qd, J = 6.3, 2.2 Hz, 1H), 2.17 – 2.11 (m, 2H), 1.55 – 1.47 (m, 1H), 1.45 (s, 9H), 1.43 – 1.38 (m, 2H), 1.30 – 1.23 (m, 4H), 1.17 (s, 9H), 1.16 (d, J = 6.3 Hz, 3H), 1.15 – 1.12 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H).
13C NMR (151 MHz, CDCl3):
δ 170.2, 166.6, 143.6, 141.9, 128.4, 121.6, 82.0, 73.9, 67.6, 58.4, 39.0, 33.1, 29.5, 29.0, 28.9, 28.3, 28.1, 27.3, 22.8, 21.0.
= 2.8 (c 2.01, MeOH)
HRMS (ESI-TOF):
calculated for C25H46NO4+ ([M+H]+) 424.3421, found 424.3423
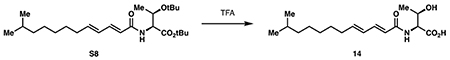
((2E,4E)-11-methyldodeca-2,4-dienoyl)-L-threonine (14):
Amide S8 (170 mg, 0.401 mmol, 1 eq) was dissolved in 4 mL trifluoroacetic acid at 0 °C, then stirred at room temperature for 1 hour. The reaction was then diluted with 10 mL toluene and concentrated in vacuo to dryness. The residue was evaporated twice more from 10 mL toluene to provide 14 as a colorless resin (138 mg, quantitative).
1H NMR (600 MHz, CDCl3):
δ 7.22 (dd, J = 15.0, 9.5 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.18 – 6.07 (m, 2H), 5.93 (d, J = 15.1 Hz, 1H), 4.67 – 4.59 (m, 1H), 4.53 – 4.43 (m, 1H), 2.21 – 2.09 (m, 2H), 1.55 – 1.47 (m, 1H), 1.46 – 1.37 (m, 2H), 1.30 – 1.24 (m, 4H), 1.21 (d, J = 6.3 Hz, 3H), 1.18 – 1.12 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H).
13C NMR (151 MHz, CDCl3):
δ 173.6, 168.6, 145.3, 143.7, 128.2, 120.3, 67.8, 57.8, 39.1, 33.3, 29.7, 29.0, 28.1, 27.3, 22.8, 19.3.
= 6.2 (c 2.66, MeOH)
HRMS (ESI-TOF):
calculated for C17H30NO4+ ([M+H]+) 312.2169, found 312.2170
(2E,4E)-N-((2S,3R)-3-hydroxy-1-(((5S,8S,10S,E)-10-hydroxy-5-methyl-2,7-dioxo-1,6-diazacyclododec-3-en-8-yl)amino)-1-oxobutan-2-yl)-11-methyldodeca-2,4-dienamide (1):
Acid 14 (38 mg, 0.122 mmol, 2 eq) and crude glidobamine 13 (75 mg, ~0.0610 mmol as judged by 1H NMR analysis, 1 eq) were dissolved in 1.5 mL anhydrous DMF and cooled to 0 °C. To the mixture was added diisopropylethylamine (64 μL, 0.366 mmol, 6 eq), followed by solid DEPBT (37 mg, 0.122 mmol, 2 eq). The reaction was warmed to room temperature and stirred overnight, and the reaction progress was monitored by LCMS. After 12 hours, the reaction was cooled to 0 °C and quenched with 0.5 mL water, then concentrated in vacuo to dryness. The residue was precipitated into 50 mL diethyl ether at 0 °C from 0.9 mL methanol, and the resulting solids were collected by centrifugation at 4200 RPM at 4 °C for 15 minutes. The solids were dissolved in methanol, filtered through a 0.2 μm PTFE membrane filter, then purified by reversed-phase HPLC (gradient 30–60% acetonitrile in water over 35 minutes, 0.1% formic acid added) to provide cepafungin I (1) as a fluffy white powder after lyophilization (10.9 mg, 0.0204 mmol, 33%, 20% from 12). See Table S2 for coupling optimization. See Table S3 for comparison of literature NMR data with synthetic cepafungin I.
1H NMR (600 MHz, (CD3)2SO):
δ 8.79 – 8.53 (m, 1H), 7.99 – 7.91 (m, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.46 – 7.36 (m, 1H), 7.00 (dd, J = 15.1, 10.8 Hz, 1H), 6.40 (d, J = 14.4 Hz, 1H), 6.22 – 6.04 (m, 4H), 5.03 – 4.80 (m, 1H), 4.80 – 4.56 (m, 1H), 4.46 – 4.30 (m, 2H), 4.28 (dd, J = 8.9, 4.1 Hz, 1H), 4.01 – 3.90 (m, 1H), 3.63 – 3.51 (m, 1H), 3.09 – 2.91 (m, 2H), 2.16 – 2.09 (m, 2H), 1.93 – 1.73 (m, 1H), 1.58 (d, J = 11.1 Hz, 1H), 1.53 – 1.46 (m, 1H), 1.46 – 1.41 (m, 1H), 1.41 – 1.34 (m, 3H), 1.29 – 1.18 (m, 7H), 1.16 – 1.11 (m, 2H), 1.00 (d, J = 6.3 Hz, 3H), 0.84 (d, J = 6.6 Hz, 6H).
13C NMR (151 MHz, (CD3)2SO):
δ 171.1, 169.4, 167.6, 165.4, 143.1, 142.1, 139.7, 128.6, 123.3, 123.1, 66.9, 66.7, 58.2, 51.2, 44.7, 42.5, 38.4, 32.3, 28.9, 28.4, 27.4, 26.6, 22.5, 19.9, 18.5.
= −106.5° (c 0.20, MeOH)
HRMS (ESI-TOF):
calculated for C28H47N4O6+ ([M+H]+) 535.3490, found 535.3491
Synthesis of cepafungin analogs
Synthesis of cepafungin alkyne (20):
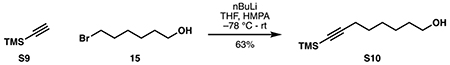
8-(trimethylsilyl)oct-7-yn-1-ol (S10):
In a procedure adapted from Ye et al. (2018), ethynyltrimethylsilane S9 (2.95 g, 30.0 mmol, 2.72 eq) was dissolved in 21 mL anhydrous THF, cooled to −78 °C and treated with n-butyllithium (2.5 M in hexanes, 12.5 mL, 2.82 eq) dropwise. The mixture was stirred at −78 °C for 1 hour, then treated dropwise with a solution of 6-bromohexan-1-ol 15 (2.00 g, 11.0 mmol, 1 eq) in 17 mL anhydrous THF, followed by 8.4 mL HMPA. The reaction was allowed to stir under argon at −78 °C to room temperature overnight or until completion by TLC analysis. The reaction was quenched at –20 °C with 20 mL saturated NH4Cl, diluted with 20 mL ethyl acetate and the layers were separated. The aqueous layer was extracted with ethyl acetate (3 x 25 mL). The combined organic layers were washed with 20 mL water, brine and dried over Na2SO4. Concentration in vacuo followed by silica flash chromatography (85:15 hexanes:ethyl acetate) provided S10 as a colorless oil (1.37 g, 6.91 mmol, 63%).
1H NMR (600 MHz, CDCl3):
δ 3.63 (t, J = 6.6 Hz, 2H), 2.21 (t, J = 7.1 Hz, 2H), 1.60 – 1.48 (m, 4H), 1.43 – 1.32 (m, 4H), 0.13 (s, 9H).
13C NMR (151 MHz, CDCl3):
δ 107.7, 84.5, 63.0, 32.7, 28.6, 28.6, 25.3, 19.9, 0.3.
HRMS (ESI-TOF):
calculated for C11H23OSi+ ([M+H]+) 198.1513, found 199.1517

8-(trimethylsilyl)oct-7-ynal (S11):
Oxalyl chloride (439 μL, 5.12 mmol, 1.5 eq) was dissolved in 4 mL anhydrous DCM at −78 °C. An anhydrous solution of DMSO (727 μL, 10.2 mmol, 3 eq) in 1 mL DCM was added dropwise and the resulting mixture was stirred at −78 °C for 15 minutes. A solution of alkynyl alcohol S10 (677 mg, 3.41 mmol, 1 eq) in 4 mL anhydrous DCM was then added dropwise. The mixture was stirred at −78 °C for 30 minutes, then treated with triethylamine (2.38 mL, 17.1 mmol, 5 eq) dropwise. Reaction progress was monitored by TLC until completion at 1.5 hours. The reaction was quenched at −78 °C with 20 mL water and allowed to warm to room temperature. The layers were separated, and the aqueous layer was extracted with DCM (3 x 10 mL). The combined organic layers were washed with water (twice) and brine, then dried over Na2SO4. Concentration in vacuo provided S11 as a pale yellow oil which was used without further purification (975 mg).
1H NMR (600 MHz, CDCl3):
δ 9.76 (t, J = 1.8 Hz, 1H), 2.43 (td, J = 7.4, 1.8 Hz, 2H), 2.22 (t, J = 7.1 Hz, 2H), 1.64 (p, J = 7.4 Hz, 2H), 1.57 – 1.48 (m, 2H), 1.46 – 1.40 (m, 2H), 1.27 – 1.17 (m, 1H), 0.13 (s, 9H).
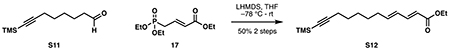
Ethyl (2E,4E)-12-(trimethylsilyl)dodeca-2,4-dien-11-ynoate (S12):
Ethyl (E)-4-(diethoxyphosphoryl)but-2-enoate 17 (1.28 g, 5.12 mmol, 1.5 eq) in 17 mL anhydrous THF was cooled to −78 °C. A solution of lithium hexamethyldisilazide (1 M THF, 4.78 mL, 1.4 eq) was then added dropwise and allowed to stir for 30 minutes at −78 °C. Crude aldehyde S11 (975 mg) in 2 mL THF was added dropwise at −78 °C, and the mixture was allowed to stir to room temperature overnight. Upon its completion as judged by TLC, the reaction was quenched with 20 mL saturated NH4Cl at room temperature. After evaporation of THF in vacuo, the mixture was diluted with 30 mL water, 30 mL ethyl acetate and minimal brine to aid separation. The aqueous layer was extracted with ethyl acetate (3 x 40 mL). The combined organic layers were washed with 20 mL water and brine, then dried over Na2SO4. Purification by silica flash chromatography (97:3 hexanes:ethyl acetate) provided S12 as a yellow oil (498 mg, 1.70 mmol, 50% from S10).
1H NMR (600 MHz, CDCl3):
δ 7.24 (dd, J = 15.4, 10.5 Hz, 1H), 6.21 – 6.04 (m, 2H), 5.77 (d, J = 15.4 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 2.21 (t, J = 7.1 Hz, 2H), 2.17 (q, J = 7.4, 7.0 Hz, 2H), 1.51 (p, J = 7.3 Hz, 2H), 1.47 – 1.36 (m, 4H), 1.28 (t, J = 7.1 Hz, 3H), 0.13 (s, 9H).
13C NMR (151 MHz, CDCl3):
δ 167.4, 145.1, 144.4, 128.6, 119.4, 107.5, 84.6, 60.3, 32.9, 28.5, 28.4, 28.3, 19.9, 14.4, 0.3.
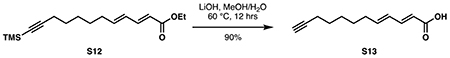
(2E,4E)-dodeca-2,4-dien-11-ynoic acid (S13):
Ester S12 (374 mg, 1.28 mmol, 1 eq) was dissolved in 6.4 mL methanol. With rapid stirring, a solution of lithium hydroxide (153 mg, 6.40 mmol, 5 eq) in 6.4 mL water was added. The reaction was heated at 60 °C overnight or until completion as judged by TLC. Methanol was evaporated in vacuo. The mixture was diluted with 10 mL water and the mixture was adjusted to pH 1 with 1 M HCl and diluted with 10 mL DCM. The layers were separated, and the aqueous layer was extracted with DCM (3 x 10 mL). The combined organic layers were washed with 10 mL brine then dried over Na2SO4. Concentration in vacuo provided pure S13 as a white solid (221 mg, 1.15 mmol, 90%).
1H NMR (400 MHz, CDCl3):
δ 7.34 (dd, J = 15.2, 9.5 Hz, 1H), 6.27 – 6.10 (m, 2H), 5.79 (d, J = 15.3 Hz, 1H), 2.23 – 2.16 (m, 4H), 1.95 (t, J = 2.6 Hz, 1H), 1.54 (p, J = 7.0 Hz, 2H), 1.49 – 1.39 (m, 4H).
13C NMR (101 MHz, CDCl3):
δ 172.5, 147.6, 146.0, 128.5, 118.4, 84.6, 68.5, 33.0, 28.4, 28.3, 28.2, 18.4.
HRMS (ESI-TOF):
calculated for C12H15O2− ([M-H]−) 191.1077, found 191.1078
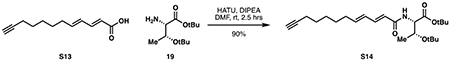
tert-butyl O-(tert-butyl)-N-((2E,4E)-dodeca-2,4-dien-11-ynoyl)-L-threoninate (S14):
Alkynyl fatty acid S13 (216 mg, 1.12 mmol, 1 eq) was combined with diisopropylethylamine (880 μL, 5.05 mmol, 4.5 eq) and HATU (427 mg, 1.12 mmol, 1 eq) in 5 mL anhydrous DMF and allowed to stir for 5 minutes. tert-butyl O-(tert-butyl)-L-threoninate 19 (390 mg, 1.69 mmol, 1.5 eq) dissolved in 0.6 mL DMF was then added and the reaction was stirred under argon for 2.5 hours or until completion by TLC or LCMS. The reaction was then quenched at 0 °C with 20 mL saturated NH4Cl, diluted with 15 mL ethyl acetate and the resulting layers were separated. The aqueous layer was extracted with ethyl acetate (4 x 15 mL). The combined organic layers were washed with water (2 x 10 mL) and 10 mL brine, then dried over Na2SO4. Concentration in vacuo followed by silica flash chromatography (85:15 hexanes:ethyl acetate) provided S14 as a yellow oil (410 mg, 1.01 mmol, 90%).
1H NMR (600 MHz, CDCl3):
δ 7.19 (dd, J = 15.0, 10.8 Hz, 1H), 6.21 – 6.10 (m, 2H), 6.08 – 6.01 (m, 1H), 5.87 (d, J = 15.0 Hz, 1H), 4.48 (dd, J = 9.2, 2.1 Hz, 1H), 4.20 (qd, J = 6.3, 2.2 Hz, 1H), 2.16 (dtd, J = 14.1, 7.0, 1.9 Hz, 4H), 1.93 (t, J = 2.7 Hz, 1H), 1.55 – 1.49 (m, 2H), 1.44 (s, 9H), 1.43 – 1.38 (m, 4H), 1.16 (s, 9H), 1.15 (d, J = 6.3 Hz, 3H).
13C NMR (151 MHz, CDCl3):
δ 170.2, 166.6, 143.0, 141.8, 128.6, 121.8, 84.6, 82.0, 73.9, 68.4, 67.6, 58.4, 32.9, 28.8, 28.4, 28.4, 28.3, 28.2, 21.0, 18.4.
= 5.0 (c 0.680, MeOH)
HRMS (ESI-TOF):
calculated for C24H39NO4Cl− ([M+Cl]−) 440.2573, found 440.2573

((2E,4E)-dodeca-2,4-dien-11-ynoyl)-L-threonine (S15):
Alkynyl fatty acid amide S14 (50 mg, 0.123 mmol, 1 eq) was dissolved in 1.2 mL trifluoroacetic acid at 0 °C, then stirred at room temperature for 1 hour. The reaction was then diluted with 10 mL toluene and concentrated in vacuo to dryness to provide S15 as a pale yellow resin (50 mg, quant.).
1H NMR (600 MHz, CDCl3):
δ 7.21 (dd, J = 15.0, 10.1 Hz, 1H), 7.05 (d, J = 8.1 Hz, 1H), 6.12 (dtd, J = 21.8, 15.1, 8.4 Hz, 2H), 5.94 (d, J = 15.0 Hz, 1H), 4.63 (d, J = 8.5 Hz, 1H), 4.46 (s, 1H), 2.21 – 2.13 (m, 4H), 1.95 (t, J = 2.6 Hz, 1H), 1.56 – 1.49 (m, 2H), 1.46 – 1.37 (m, 4H), 1.20 (d, J = 6.3 Hz, 3H).
13C NMR (151 MHz, CDCl3):
δ 173.7, 168.3, 144.6, 143.4, 128.5, 120.6, 84.6, 68.5, 67.8, 57.8, 33.0, 28.4, 28.4, 28.4, 19.4, 18.5.
= 8.3 (c 2.14, MeOH)
HRMS (ESI-TOF):
calculated for C32H45N2O8− ([2M-H]−) 585.3181, found 585.3192

(2E,4E)-N-((2S,3R)-3-hydroxy-1-(((5S,8S,10S,E)-10-hydroxy-5-methyl-2,7-dioxo-1,6-diazacyclododec-3-en-8-yl)amino)-1-oxobutan-2-yl)dodeca-2,4-dien-11-ynamide (20):
Acid S15 (38 mg, 0.128 mmol, 2 eq) and crude glidobamine 13 (82 mg, ~0.0641 mmol as judged by 1H NMR analysis, 1 eq) were dissolved in 3.2 mL anhydrous DMF and cooled to 0 °C. To the mixture was added diisopropylethylamine (34 μL, 0.192 mmol, 3 eq), followed by solid DEPBT (38 mg, 0.128 mmol, 2 eq). The reaction was stirred to room temperature overnight, and reaction progress was monitored by LCMS. At 12 hours, the reaction was cooled to 0 °C and quenched with 0.5 mL water, then concentrated in vacuo to dryness. The residue was precipitated into 50 mL diethyl ether at 0 °C from 1.2 mL methanol, and the resulting solids were collected by centrifugation at 4200 RPM at 4 °C for 15 minutes. The solids were dissolved in methanol, filtered through a 0.2 μm PTFE membrane filter, then purified by reversed-phase HPLC (gradient 30–60% acetonitrile in water over 35 minutes, 0.1% formic acid added) to provide cepafungin alkyne (20) as a fluffy white powder after lyophilization (8.1 mg, 0.0157 mmol, 14% from 12).
1H NMR (600 MHz, (CD3)2SO):
δ 8.77 – 8.46 (m, 1H), 7.91 (d, J = 9.1 Hz, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.41 (t, J = 6.3 Hz, 1H), 7.00 (dd, J = 15.2, 10.8 Hz, 1H), 6.40 (d, J = 13.2 Hz, 1H), 6.24 – 6.03 (m, 4H), 4.87 (s, 1H), 4.67 (s, 1H), 4.50 – 4.30 (m, 2H), 4.28 (dd, J = 8.9, 4.1 Hz, 1H), 4.03 – 3.88 (m, 1H), 3.65 – 3.49 (m, 1H), 3.08 – 2.92 (m, 2H), 2.74 (t, J = 2.6 Hz, 1H), 2.19 – 2.06 (m, 4H), 1.93 – 1.74 (m, 1H), 1.58 (d, J = 13.5 Hz, 1H), 1.48 – 1.41 (m, 3H), 1.41 – 1.32 (m, 5H), 1.29 – 1.11 (m, 3H), 1.00 (d, J = 6.3 Hz, 3H).
13C NMR (151 MHz, (CD3)2SO):
δ 171.0, 169.3, 167.6, 165.4, 143.1, 141.9, 139.7, 128.6, 123.3, 123.1, 84.5, 71.2, 67.0, 66.7, 58.1, 51.2, 44.7, 42.5, 32.1, 27.8, 27.8, 27.7, 19.9, 18.5, 17.6.
= −74.2 (c 0.260, MeOH)
HRMS (ESI-TOF):
calculated for C28H41N4O8− ([M+HCOOH–H]− 561.2930, found 561.2936.

Synthesis of desoxycepafungin (21)
di-tert-butyl ((S)-6-(((S)-1-(methoxy(methyl)amino)-1-oxopropan-2-yl)amino)-6-oxohexane-1,5-diyl)dicarbamate (S17):
N2,N6-bis(tert-butoxycarbonyl)-L-lysine S16 (2.38 g, 6.86 mmol, 1 eq) and amine hydrochloride 9 (1.45 g, 8.57 mmol, 1.25 eq) were combined in 34 mL anhydrous DMF. The mixture was treated with diisopropylethylamine (5.37 mL, 30.9 mmol, 4.5 eq) followed by HATU (2.61 g, 6.86 mmol, 1 eq) and allowed to stir at room temperature under argon for 1.5 hours or until completion by TLC. The reaction was then quenched at 0 °C with 5 mL saturated aqueous NH4Cl and concentrated to dryness. The residue was diluted with 200 mL ethyl acetate and washed with 30 mL 1 M HCl, water, NaHCO3 and brine. The combined aqueous layers were back-extracted twice with 30 mL ethyl acetate. The combined organic layers were then dried over Na2SO4, concentrated in vacuo, and purified by flash silica chromatography (55:40:5 hexanes:ethyl acetate:methanol) to provide S17 as a colorless foam (3.10 g, 6.73 mmol, 98%).
1H NMR (600 MHz, CD3OD):
δ 4.85 – 4.78 (m, 1H), 4.05 – 3.91 (m, 1H), 3.82 (s, 3H), 3.20 (s, 3H), 3.04 (h, J = 6.8 Hz, 2H), 1.80 – 1.66 (m, 1H), 1.63 – 1.52 (m, 1H), 1.52 – 1.45 (m, 3H), 1.44 (s, 9H), 1.43 (s, 9H), 1.42 – 1.36 (m, 2H), 1.32 (d, J = 7.0 Hz, 3H).
13C NMR (151 MHz, CD3OD):
δ 174.8, 174.6, 158.6, 157.9, 80.6, 79.8, 62.1, 55.7, 46.9, 41.0, 33.0, 32.4, 30.6, 28.8, 28.7, 24.0, 17.3.
= −20.0 (c 1.45, MeOH)
HRMS (ESI-TOF):
calculated for C22H41N4O9− ([M+HCOOH–H]−) 505.2879, found 505.2880
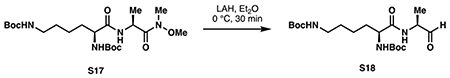
di-tert-butyl ((S)-6-oxo-6-(((S)-1-oxopropan-2-yl)amino)hexane-1,5-diyl)dicarbamate (S18):
Dipeptide S17 (1.00 g, 2.17 mmol, 1 eq) was dissolved in 22 mL anhydrous diethyl ether and cooled to 0 °C. A solution of lithium aluminum hydride (1 M in Et2O, 8.14 mL, 3.75 eq) was then added dropwise at 0 °C. Upon complete addition, the reaction was stirred for 30 minutes at 0 °C then quenched with 16 mL 2 M NaHSO4. The layers were separated and the aqueous phase was extracted with Et2O (3 x 25 mL). The combined organic layers were washed with water, saturated NaHCO3 and brine, then dried over Na2SO4. Concentration in vacuo provided aldehyde S18 as a white foam that was used for the next step without further purification (0.892 g).
1H NMR (600 MHz, CDCl3):
δ 9.44 (s, 1H), 6.84 (d, J = 6.3 Hz, 1H), 5.17 (d, J = 7.5 Hz, 1H), 4.65 – 4.53 (m, 1H), 4.41 – 4.32 (m, 1H), 4.07 – 3.98 (m, 1H), 3.06 – 2.94 (m, 2H), 1.81 – 1.67 (m, 1H), 1.63 – 1.48 (m, 1H), 1.47 – 1.37 (m, 4H), 1.34 (s, 9H), 1.34 (s, 9H), 1.26 (d, J = 7.4 Hz, 3H).
HRMS (ESI-TOF):
calculated for C19H34N3O6− ([M–H]−) 400.2453, found 400.2457
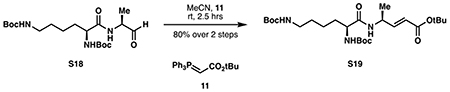
tert-butyl (S,E)-4-((S)-2,6-bis((tert-butoxycarbonyl)amino)hexanamido)pent-2-enoate (S19):
tert-Butyl(triphenylphosphoranylidene)acetate 11 (1.02 g, 2.71 mmol, 1.25 eq) was dissolved in 21 mL anhydrous acetonitrile, then a solution of crude aldehyde S18 (0.892 g) in 10 mL acetonitrile was added dropwise at room temperature. The reaction was stirred for 30 minutes or until completion by 1H NMR or TLC, and then concentrated to dryness. Purification by silica flash chromatography (60:40 hexanes:ethyl acetate) provided S19 as a white foam (0.863 g, 1.73 mmol, 80% from S17).
1H NMR (600 MHz, CD3OD):
δ 6.77 (dd, J = 15.7, 5.0 Hz, 1H), 5.80 (dd, J = 15.7, 1.8 Hz, 1H), 4.59 (qdd, J = 6.9, 4.9, 1.7 Hz, 1H), 4.03 – 3.84 (m, 1H), 3.03 (td, J = 6.8, 2.8 Hz, 2H), 1.78 – 1.66 (m, 1H), 1.66 – 1.56 (m, 1H), 1.54 – 1.48 (m, 2H), 1.47 (s, 9H), 1.45 (s, 9H), 1.43 (s, 9H), 1.40 – 1.31 (m, 2H), 1.28 (d, J = 7.0 Hz, 3H).
13C NMR (151 MHz, CD3OD):
δ 174.6, 167.3, 158.6, 157.8, 149.2, 122.8, 81.7, 80.6, 79.8, 56.3, 46.9, 41.0, 33.1, 30.6, 28.8, 28.8, 28.4, 24.2, 19.9.
= −17.6 (c 1.46, MeOH)
HRMS (ESI-TOF):
calculated for C26H46N3O9− ([M+HCOOH–H]−) 544.3240, found 544.3246
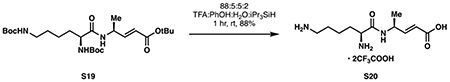
(S,E)-4-((S)-2,6-diaminohexanamido)pent-2-enoic acid−-2,2,2-trifluoroacetic acid (1/2) (S20):
Dipeptide S19 (431 mg, 0.863 mmol, 1 eq) was dissolved in 22 mL of a freshly prepared solution of “Reagent B” (88:5:5:2 trifluoroacetic acid:phenol:water:triisopropylsilane). After stirring for 1 hour at room temperature, the reaction was diluted with 30 mL toluene and concentrated in vacuo to dryness, followed by an additional evaporation from 30 mL toluene. The residue was then dissolved in 2 mL methanol and added dropwise to 100 mL diethyl ether at 0 °C. The solids were collected by centrifugation at 4200 RPM for 10 minutes at 4 °C, then precipitated again into 50 mL diethyl ether at 0 °C from 1.5 mL methanol. The solids were collected by centrifugation as above, then dried from methanol to provide S20 as an off-white solid (356 mg, 0.755 mmol, 88%).
1H NMR (600 MHz, CD3OD):
δ 6.83 (dd, J = 15.7, 5.4 Hz, 1H), 5.86 (dd, J = 15.7, 1.6 Hz, 1H), 4.69 – 4.59 (m, 1H), 3.87 (t, J = 6.4 Hz, 1H), 2.98 – 2.89 (m, 2H), 1.99 – 1.82 (m, 2H), 1.76 – 1.66 (m, 2H), 1.54 – 1.42 (m, 2H), 1.33 (d, J = 7.0 Hz, 3H).
13C NMR (151 MHz, CD3OD):
δ 170.1, 169.3, 149.0, 122.7, 54.2, 47.4, 40.3, 32.1, 28.2, 22.9, 19.7.
= −13.6 (c 0.530, MeOH)
HRMS (ESI-TOF):
calculated for free base C11H20N3O3− ([M–H]−) 242.2521, found 242.1511

(5S,8S,E)-8-amino-5-methyl-1,6-diazacyclododec-3-ene-2,7-dione (S21):
Macrocycle precursor S20 (200 mg, 0.424 mmol, 1 eq) was dissolved in 424 mL anhydrous DMF. The solution was treated with diisopropylethylamine (296 μL, 1.70 mmol, 4 eq), followed by solid 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium tetrafluoroborate (DMTMMT) (209 mg, 0.637 mmol, 1.5 eq) at room temperature. The reaction was stirred under argon for 27 hours, then quenched with 10 mL water and concentrated in vacuo to dryness. The residue was dissolved in 2 mL methanol and precipitated into 50 mL diethyl ether with rapid stirring. The solids were collected by centrifugation at 4200 RPM for 15 minutes at 4 °C to yield crude S21 as a tan solid (276 mg, 0.187 mmol, 44% from S20, 39% from S19). Yield was determined by 1H NMR analysis of a 6.2 mg crude sample with 4-toluenesulfonamide (7.7 mg, 0.0450 mmol) added as internal standard.
1H NMR (400 MHz, CD3OD):
4-toluenesulfonamide: δ 7.82 – 7.72 (m, 2H), 7.40 – 7.30 (m, 2H), 2.42 (s, 3H). Compound S21: 7.03 (dd, J = 15.4, 4.9 Hz, 1H), 6.35 (dd, J = 15.5, 1.4 Hz, 1H), 4.66 – 4.58 (m, 1H), 4.16 (t, J = 3.7 Hz, 1H), 3.58 – 3.44 (m, 2H), 2.36 – 2.24 (m, 1H), 1.98 – 1.77 (m, 2H), 1.77 – 1.62 (m, 2H).
HRMS (ESI-TOF):
calculated for C11H18N3O2− ([M–H]−) 224.1404, found 224.1406

(2E,4E)-N-((2S,3R)-3-hydroxy-1-(((5S,8S,E)-5-methyl-2,7-dioxo-1,6-diazacyclododec-3-en-8-yl)amino)-1-oxobutan-2-yl)-11-methyldodeca-2,4-dienamide (21):
Acid 14 (42 mg, 0.134 mmol, 2 eq) and crude desoxyglidobamine S21 (100 mg, ~0.0670 mmol as judged by 1H NMR analysis, 1 eq) were dissolved in 3.4 mL anhydrous DMF and cooled to 0 °C. To the mixture was added diisopropylethylamine (35 μL, 0.201 mmol, 3 eq), followed by solid DEPBT (40 mg, 0.134 mmol, 2 eq). The reaction was warmed to room temperature and stirred overnight, and the reaction progress was monitored by LCMS. At 24 hours, the reaction was cooled to 0 °C and quenched with 2 mL saturated NH4Cl. The mixture was extracted with ethyl acetate (4 x 4 mL), then n-butanol (3 x 4 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was filtered through a 0.2 μm PTFE membrane filter in methanol, then purified by reversed-phase HPLC (gradient 30–60% acetonitrile in water over 35 minutes, 0.1% formic acid added) to provide desoxycepafungin I (21) as a fluffy white powder after lyophilization (7.9 mg, 0.0152 mmol, 23%, 9% from S24).
1H NMR (600 MHz, (CD3)2SO):
δ 8.44 (d, J = 7.9 Hz, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 7.2 Hz, 1H), 7.35 (dd, J = 8.0, 6.1 Hz, 1H), 7.00 (dd, J = 15.1, 10.8 Hz, 1H), 6.78 (dd, J = 15.3, 4.7 Hz, 1H), 6.23 (dd, J = 15.4, 1.4 Hz, 1H), 6.21 – 6.15 (m, 2H), 6.09 (dt, J = 14.8, 6.9 Hz, 1H), 4.54 – 4.48 (m, 1H), 4.44 – 4.36 (m, 1H), 4.32 (dd, J = 8.5, 3.9 Hz, 1H), 4.02 – 3.96 (m, 1H), 3.80 (s, 1H), 3.32 – 3.23 (m, 1H), 2.98 – 2.91 (m, 1H), 2.13 (q, J = 7.2 Hz, 2H), 2.10 – 2.03 (m, 1H), 1.74 – 1.64 (m, 1H), 1.50 (hept, J = 6.7 Hz, 1H), 1.46 – 1.41 (m, 1H), 1.41 – 1.35 (m, 2H), 1.35 – 1.29 (m, 1H), 1.29 – 1.22 (m, 5H), 1.20 (d, J = 7.0 Hz, 3H), 1.17 – 1.11 (m, 2H), 1.02 (d, J = 6.3 Hz, 3H), 0.98 – 0.90 (m, 1H), 0.85 (d, J = 6.6 Hz, 6H).
13C NMR (151 MHz, (CD3)2SO):
δ 170.5, 169.4, 165.8, 165.5, 146.7, 142.1, 139.8, 128.6, 122.9, 118.4, 66.6, 58.1, 51.5, 45.6, 38.4, 37.9, 32.2, 30.2, 29.6, 28.8, 28.4, 27.4, 26.6, 22.5, 19.6, 18.3, 17.1.
= −14.4 (c = 0.160, MeOH)
HRMS (ESI-TOF):
calculated for C28H47N4O5+ ([M+H]+) 519.3541, found 519.3564
Synthesis of cepafungin analog with saturated lipid tail (22)

11-methyldodecan-1-ol (S23):
Following a procedure adapted from Melaugh et al. (2014), lithium chloride (1.61 g, 42.4 mmol, 2.05 eq) was flame-dried in a 250 mL round-bottom flask under high vacuum. Solid copper(II) chloride (2.50 g, 18.6 mmol, 1 eq) was added under argon. The combined reagents were then dissolved in 25 mL anhydrous THF to provide a dark red solution of Li2CuCl4.
A flame-dried 3-neck 500 mL flask fitted with a reflux condenser was charged with magnesium turnings (3.14 g, 129 mmol, 1.18 eq with respect to 2-bromopropane) and 50 mL anhydrous THF. With rapid stirring, 2-bromopropane (10.3 mL, 110 mmol, 6.5 eq with respect to S22) was added dropwise until refluxing began, at which point the flask was lowered into a water bath. Upon complete addition, the water bath was removed and the reaction was allowed to stir under argon for 30 minutes to produce a dark gray suspension of S5. The mixture was cooled to −78 °C, and 10-bromodecan-1-ol S22 (4.00 g, 16.9 mmol, 1 eq) was added in 4.2 mL dry THF, followed by the above solution of Li2CuCl4 (~0.7 M in THF, 25 mL, 1.1 eq with respect to S22). Upon complete addition, the reaction was removed from its cooling bath and allowed to warm to room temperature. Reaction progress was monitored by TLC until completion at 1 hour. The reaction was then carefully quenched at 0 °C with 50 mL saturated NH4Cl solution, filtered through a sintered glass funnel, and the filtrate concentrated in vacuo to remove most of the THF. The mixture was diluted with 100 mL ethyl acetate and 100 mL 1M HCl. The layers were separated, and the aqueous phase was extracted with ethyl acetate (3 x 50 mL). The combined organic layers were washed with 50 mL each of water, saturated aqueous NaHCO3 and brine, then dried over Na2SO4. Purification by silica flash chromatography (90:10 hexanes:ethyl acetate) provided pure S23 as a colorless oil (3.22 g, 16.1 mmol, 95%).
1H NMR (600 MHz, CDCl3):
δ 3.63 (t, J = 6.7 Hz, 2H), 1.59 – 1.54 (m, 2H), 1.53 – 1.47 (m, 1H), 1.36 – 1.22 (m, 14H), 1.19 – 1.10 (m, 2H), 0.85 (d, J = 6.7 Hz, 6H).
13C NMR (151 MHz, CDCl3):
δ 63.2, 39.2, 32.9, 30.1, 29.8, 29.8, 29.7, 29.6, 28.1, 27.6, 25.9, 22.8.
HRMS (ESI-TOF):
calculated for C14H32O1N1+ ([M+CH3CN+H]+) 242.2478, found 242.2475

11-methyldodecanoic acid (S24):
Alcohol S23 (1.60 g, 7.96 mmol, 1 eq) was dissolved in 50 mL acetone and cooled to 0 °C. A solution of Jones’ reagent (CrO3/H2SO4/H2O) (6.52 mL, 19.9 mmol, 2.5 eq) was then added dropwise and allowed to stir at 0 °C. Upon completion by TLC in five minutes, the reaction was quenched with 3 mL isopropanol, filtered through a plug of celite and concentrated in vacuo to remove the organic solvent. The remaining aqueous mixture was diluted with 25 mL 1 M HCl and extracted with ethyl acetate (4 x 25 mL). The combined organic layers were washed with 25 mL water and brine, then dried over Na2SO4 and concentrated in vacuo to provide pure S24 (1.58 g, 7.37 mmol, 93%).
1H NMR (600 MHz, CDCl3):
δ 2.34 (t, J = 7.5 Hz, 2H), 1.67 – 1.59 (m, 2H), 1.56 – 1.46 (m, 1H), 1.36 – 1.21 (m, 12H), 1.19 – 1.11 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H).
13C NMR (151 MHz, CDCl3):
δ 180.5, 39.3, 34.4, 30.1, 29.9, 29.7, 29.5, 29.3, 28.2, 27.6, 24.9, 22.9.
HRMS (ESI-TOF):
calculated for C13H25O2− ([M–H]−) 213.1860, found 213.1861

tert-butyl O-(tert-butyl)-N-(11-methyldodecanoyl)-L-threoninate (S25):
Fatty acid S24 (636 mg, 2.97 mmol, 1 eq) was dissolved in 14 mL anhydrous DMF at 0 °C. Diisopropylethylamine was added (2.33 mL, 13.4 mmol, 4.5 eq), followed by HATU (1.13 g, 2.97 mmol, 1 eq). The mixture was stirred at 0 °C under argon for five minutes before adding tert-butyl O-(tert-butyl)-L-threoninate 19 (1.03 g, 4.45 mmol, 1.5 eq) in 1 mL DMF. The reaction was stirred to room temperature under argon for 4 hours or until completion by LCMS. The reaction was then diluted at 0 °C with 50 mL saturated NH4Cl and ethyl acetate. The pH of the mixture was adjusted to 7 with 1 M HCl, and the aqueous layer was extracted with ethyl acetate (3 x 50 mL). The combined organic layers were washed with 30 mL 1M HCl, water, NaHCO3 and brine, then dried over Na2SO4. Concentration in vacuo followed by silica flash chromatography (85:15 hexanes:ethyl acetate) provided S25 as a colorless oil (955 mg, 2.23 mmol, 75%).
1H NMR (600 MHz, CDCl3):
δ 6.12 (d, J = 9.2 Hz, 1H), 4.39 (dd, J = 9.2, 2.1 Hz, 1H), 4.19 (qd, J = 6.2, 2.1 Hz, 1H), 2.26 (d, J = 7.5 Hz, 2H), 1.69 – 1.61 (m, 2H), 1.51 (dq, J = 13.3, 6.6 Hz, 1H), 1.45 (s, 9H), 1.38 – 1.21 (m, 14H), 1.16 (s, 9H), 1.15 (d, J = 6.3 Hz, 3H), 0.85 (d, J = 6.6 Hz, 6H).
13C NMR (151 MHz, CDCl3):
δ 173.5, 170.3, 81.9, 73.9, 67.4, 58.3, 39.2, 36.9, 30.0, 29.8, 29.6, 29.5, 29.4, 28.9, 28.3, 28.1, 27.5, 25.8, 22.8, 21.1.
= −4.4 (c 2.09, MeOH)
HRMS (ESI-TOF):
calculated for C25H50NO4+ ([M+H]+) 428.3735, found 428.3748
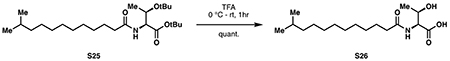
(11-methyldodecanoyl)-L-threonine (S26):
Fatty acid amide S25 (100 mg, 0.234 mmol, 1 eq) was dissolved in 2.3 mL trifluoroacetic acid at 0 °C, then stirred at room temperature for 1 hour. The solution was then diluted with 10 mL toluene and concentrated in vacuo to dryness, followed by an additional evaporation from 10 mL toluene to provide S26 as a colorless oil (91 mg, quantitative).
1H NMR (600 MHz, CDCl3):
δ 6.99 (s, 1H), 4.76 – 4.10 (m, 2H), 2.28 (s, 2H), 1.62 (s, 2H), 1.53 – 1.46 (m, 1H), 1.35 – 1.22 (m, 14H), 1.19 (s, 1H), 1.17 – 1.11 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H).
13C NMR (151 MHz, CDCl3):
δ 175.5, 39.2, 36.6, 32.0, 30.1, 29.9, 29.8, 29.6, 29.5, 28.1, 27.6, 26.3, 26.0, 22.8, 19.5, 14.3.
= 4.6 (c 1.40, MeOH)
HRMS (ESI-TOF):
calculated for C17H32NO4− ([M–H]−) 314.2337, found 314.2339
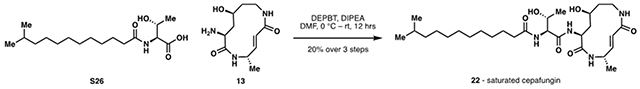
N-((2S,3R)-3-hydroxy-1-(((5S,8S,10S,E)-10-hydroxy-5-methyl-2,7-dioxo-1,6-diazacyclododec-3-en-8-yl)amino)-1-oxobutan-2-yl)-11-methyldodecanamide (22):
Acid S26 (33 mg, 0.104 mmol, 2 eq) and crude glidobamine 13 (62 mg, ~0.0521 mmol as judged by 1H NMR analysis, 1 eq) were dissolved in 2.6 mL anhydrous DMF and cooled to 0 °C. To the mixture was added diisopropylethylamine (27 μL, 0.156 mmol, 3 eq), followed by solid DEPBT (31 mg, 0.104 mmol, 2 eq). The reaction was stirred to room temperature overnight, and reaction progress was monitored by LCMS. At 12 hours, the reaction was cooled to 0 °C and quenched with 0.5 mL water, then concentrated in vacuo to dryness. The residue was precipitated into 50 mL diethyl ether at 0 °C from 0.75 mL methanol, and the resulting solids were collected by centrifugation at 4200 RPM at 4 °C for 15 minutes. The solids were dissolved in methanol, filtered through a 0.2 μm PTFE membrane filter, then purified by reversed-phase HPLC (gradient 30–100% acetonitrile in water over 20 minutes, 0.1% formic acid added) to provide saturated cepafungin (22) as a fluffy white powder after lyophilization (9.6 mg, 0.0178 mmol, 34%, 20% from 12).
1H NMR (600 MHz, (CD3)2SO):
δ 8.66 (s, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.64 (d, J = 7.9 Hz, 1H), 7.41 (t, J = 6.3 Hz, 1H), 6.40 (d, J = 15.9 Hz, 1H), 6.18 (dd, J = 15.8, 1.2 Hz, 1H), 4.85 – 4.74 (m, 1H), 4.73 – 4.60 (m, 1H), 4.48 – 4.25 (m, 2H), 4.18 (dd, J = 8.8, 3.9 Hz, 1H), 3.99 – 3.88 (m, 1H), 3.63 – 3.50 (m, 1H), 3.09 – 2.91 (m, 2H), 2.24 – 2.09 (m, 2H), 1.92 – 1.74 (m, 1H), 1.64 – 1.54 (m, 1H), 1.54 – 1.41 (m, 4H), 1.41 – 1.33 (m, 1H), 1.27 – 1.18 (m, 15H), 1.17 – 1.10 (m, 2H), 0.99 (d, J = 6.3 Hz, 3H), 0.84 (d, J = 6.6 Hz, 6H).
13C NMR (151 MHz, (CD3)2SO):
δ 172.5, 171.0, 169.4, 167.6, 142.7, 123.5, 67.0, 66.6, 57.9, 51.2, 44.7, 42.5, 38.5, 35.2, 29.3, 29.1, 29.0, 28.8, 28.6, 27.4, 26.8, 25.4, 22.5, 19.9, 18.5.
= −76.8 (c 0.190, MeOH)
HRMS (ESI-TOF):
calculated for C28H49N4O6− ([M–H]−) 537.3657, found 537.3657
Synthesis of glidobactin A (2)
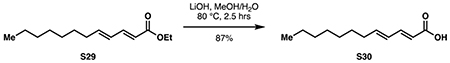
(2E,4E)-dodeca-2,4-dienoic acid (S30):
Ethyl (2E,4E)-dodeca-2,4-dienoate S29 (Imker et al., 2010) (1.00 g, 4.46 mmol, 1 eq) was dissolved in 22.5 mL methanol. With rapid stirring, a solution of lithium hydroxide (534 mg, 22.3 mmol, 5 eq) in 22.5 mL water was added. The reaction was heated at 80 °C for 2.5 hours or until completion as judged by TLC. Methanol was evaporated in vacuo. The mixture was adjusted to pH 1 with 1 M HCl and diluted with 50 mL DCM. The layers were separated, and the aqueous layer was extracted with DCM (3 x 20 mL). The combined organic layers were washed with 20 mL brine then dried over Na2SO4. Concentration in vacuo provided pure S30 as a waxy white solid (764 mg, 3.89 mmol, 87%).
1H NMR (600 MHz, CDCl3):
δ 7.39 – 7.29 (m, 1H), 6.25 – 6.11 (m, 2H), 5.78 (d, J = 15.3 Hz, 1H), 2.24 – 2.10 (m, 2H), 1.47 – 1.39 (m, 2H), 1.33 – 1.22 (m, 8H), 0.88 (t, J = 7.0 Hz, 3H).
13C NMR (151 MHz, CDCl3):
δ 173.0, 147.7, 146.4, 128.4, 118.4, 33.2, 31.9, 29.3, 29.2, 28.8, 22.8, 14.2.
HRMS (ESI-TOF):
calculated for C12H19O2− ([M–H]−) 195.1391, found 195.1392

((2E,4E)-dodeca-2,4-dienoyl)-L-threonine (S32):
tert-butyl O-(tert-butyl)-N-((2E,4E)-dodeca-2,4-dienoyl)-L-threoninate S31 was prepared in a similar fashion to S8. S31 (100 mg, 0.244 mmol, 1 eq) was dissolved in 2.4 mL trifluoroacetic acid at 0 °C, then stirred at room temperature for 30 minutes. The solution was then diluted with 8 mL toluene and concentrated in vacuo to dryness, followed by two additional evaporations from 8 mL toluene to provide pure S32 as a colorless resin (92 mg, quantitative).
1H NMR (400 MHz, CDCl3):
δ 7.25 – 7.15 (m, 1H), 7.08 (d, J = 8.4 Hz, 1H), 6.21 – 6.03 (m, 2H), 5.92 (d, J = 15.0 Hz, 1H), 4.65 (d, J = 8.0 Hz, 1H), 4.55 – 4.37 (m, 1H), 2.20 – 2.07 (m, 2H), 1.45 – 1.35 (m, 2H), 1.33 – 1.24 (m, 8H), 1.21 (d, J = 6.3 Hz, 3H), 0.88 (t, J = 6.9 Hz, 3H).
13C NMR (101 MHz, CDCl3):
δ 173.8, 168.4, 145.1, 143.5, 128.3, 120.4, 67.9, 57.8, 33.3, 31.9, 29.4, 29.3, 29.0, 22.8, 19.4, 14.2.
= 13.5 (c 0.720, MeOH)
HRMS (ESI-TOF):
calculated for C16H26NO4− ([M–H]−) 296.1867, found 296.1870

(2E,4E)-N-((2S,3R)-3-hydroxy-1-(((5S,8S,10S,E)-10-hydroxy-5-methyl-2,7-dioxo-1,6-diazacyclododec-3-en-8-yl)amino)-1-oxobutan-2-yl)dodeca-2,4-dienamide (2):
Acid S32 (60 mg, 0.223 mmol, 2 eq) and crude glidobamine 13 (129 mg, ~0.111 mmol as judged by 1H NMR analysis, 1 eq) were dissolved in 5.6 mL anhydrous DMF and cooled to 0 °C. To the mixture was added diisopropylethylamine (116 μL, 0.668 mmol, 6 eq), followed by solid DEPBT (67 mg, 0.223 mmol, 2 eq). The reaction was warmed to room temperature and stirred overnight, and reaction progress was monitored by LCMS. At 12 hours, the reaction was cooled to 0 °C and quenched with 1 mL water, then concentrated in vacuo to dryness. The residue was precipitated into 50 mL diethyl ether at 0 °C from 0.75 mL methanol, and the resulting solids were collected by centrifugation at 4200 RPM at 4 °C for 15 minutes. The solids were dissolved in methanol, filtered through a 0.2 μm PTFE membrane filter, then purified by reversed-phase HPLC (32% acetonitrile in water) to provide glidobactin A (2) as a fluffy white powder after lyophilization (25 mg, 0.0480 mmol, 43%, 25% from 12). See Table S4 for comparison of literature NMR data with synthetic glidobactin A.
1H NMR (600 MHz, (CD3)2SO):
δ 8.79 – 8.51 (m, 1H), 7.91 (d, J = 8.8 Hz, 1H), 7.74 (d, J = 7.9 Hz, 1H), 7.41 (t, J = 6.5 Hz, 1H), 7.00 (dd, J = 15.1, 10.8 Hz, 1H), 6.40 (d, J = 13.9 Hz, 1H), 6.23 – 6.05 (m, 4H), 4.88 (s, 1H), 4.67 (s, 1H), 4.46 – 4.30 (m, 2H), 4.28 (dd, J = 8.8, 4.1 Hz, 1H), 4.02 – 3.88 (m, 1H), 3.64 – 3.46 (m, 1H), 3.10 – 2.90 (m, 2H), 2.18 – 2.07 (m, 2H), 1.92 – 1.77 (m, 1H), 1.58 (d, J = 13.3 Hz, 1H), 1.50 – 1.41 (m, 1H), 1.41 – 1.33 (m, 3H), 1.30 – 1.22 (m, 8H), 1.22 – 1.16 (m, 2H), 1.00 (d, J = 6.3 Hz, 3H), 0.86 (t, J = 6.8 Hz, 3H).
13C NMR (151 MHz, (CD3)2SO):
δ 171.0, 169.4, 167.6, 165.4, 143.1, 142.1, 139.7, 128.6, 123.3, 123.1, 66.9, 66.7, 58.1, 51.2, 44.7, 42.5, 32.2, 31.2, 28.5, 28.5, 28.4, 22.1, 19.9, 18.5, 14.0.
= −92.0 (c = 0.460, MeOH)
HRMS (ESI-TOF):
calculated for C27H43N4O6− ([M–H]−) 519.3188, found 519.3180
Cell toxicity assays
RPMI 8226 (20,000 cells/well) or MM1.R (20,000 cells/well) cells were seeded in a 96-well plate in cell culture media supplemented with indicated concentrations of either 1 or bortezomib (BTZ) (1,000x stock in DMSO) and cultured for either 24 or 48 hours. Toxicity was determined using the WST-1 assay (Roche) according to the protocol of the manufacturer. EC50 values were calculated with GraphPad Prism (V7.02).
Sample preparation for SDS-PAGE experiments
Proteins in full cell lysate (2 mg/mL, 25 μL) were treated with indicated concentrations of 20 (1 μL of 25x stock in DMSO) or DMSO for one hour at r.t. Click chemistry was then initiated by the addition of TAMRA azide (Click Chemistry Tools, 25x stock in DMSO), tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Alfa Aesar, 1 mM, fresh 50x stock in water), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, Sigma-Aldrich, 100 μM, 16x stock in DMSO:tBuOH 1:4), and copper(II) sulfate (1 mM, 50x stock in water) to the lysate and incubated in the dark for one hour at r.t. SDS-PAGE reducing loading buffer (4x) was added and proteins were separated using an 11% SDS-PAGE gel. Gels were visualized using a Sapphire Biomolecular Imager (Azure Biosystems), then stained using Coomassie Brilliant Blue (CBB). Images were quantified with ImageJ.
Gel-based in situ competitive experiments
RPMI 8226 cells were seeded in a 6-well plate (1,000,000 cells/well) in cell culture media supplemented with indicated concentrations of 1 (1,000x stock in DMSO) or DMSO and cultured for 6 hours. Cells were then collected and lysed as described above. Lysates (2 mg/mL, 25 μL) were treated with 20 (10 μM; 1 μL of 25x stock in DMSO) for one hour at r.t. Click chemistry, reducing SDS-PAGE and visualization were performed as described above.
In situ 20 competitive experiment for MS
RPMI 8226 cells were seeded in 14 cm Petri dishes and cultured as previously described until they reached confluence. Cell culture media was then removed and the cells were incubated in fresh media with either 1 (100 nM; 25 μL of 1000x stock in DMSO) or DMSO for 6 hours. Cells were washed two times with PBS and harvested by centrifugation, resuspended in PBS and lysed as described above. The protein concentration of the lysate was determined using the Bradford assay. Lysates (2.32 mg/mL, 645 μL) were treated with 20 (10 μM; 6.51 μL of 100x stock in DMSO) for one hour at r.t. and subjected to click chemistry using biotin azide (30 μM, 50x stock in DMSO), tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (1 mM, 50x fresh stock in water), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (100 μM, 16x stock in DMSO:tBuOH 1:4), and copper(II) sulfate (1 mM, 50x stock in water) for one hour at r.t. Protein was precipitated by adding methanol/chloroform/water (4:1:3 v/v) to the reaction mixture and the turbid mixture was centrifuged for five minutes at 14,000 x g at 4 °C yielding a protein layer between the aqueous and organic layers. The protein layer was isolated, dried and solubilized in 2 % SDS in PBS via sonication. The proteome was centrifuged at 4,700 x g for five minutes and the supernatant was transferred to a new tube. PBS was added to give a final SDS concentration of 0.2 %. Streptavidin agarose beads (150 μL, ProteoChem) were added and the mixture was rotated overnight at r.t. Beads were washed with 1 % SDS in PBS (1x 10 mL), PBS (3x 10 mL), and water (3x 10 mL). Beads were resuspended in 6 M urea in PBS (500 μL), reduced with 10 mM neutralized TCEP (200 mM fresh stock in water) for 30 minutes at r.t. and alkylated with 25 mM iodoacetamide (400 mM fresh stock in water) for 30 minutes at r.t. in the dark. Beads were pelleted by centrifugation (1’400 x g, 2 minutes) and resuspended in 150 μL of 2 M urea in 50 mM NH4HCO3, 1 mM CaCl2 (100x stock in water) and trypsin (Thermo Scientific, 1.5 μL of 0.5 μg/μL). The digestion was performed for 12 hours at 37 °C. Samples were acidified to a final concentration of 5 % acetic acid, desalted over a self-packed C18 spin column and dried. Samples were analyzed by LC-MS/MS (described below) and the MS data was processed with MaxQuant (described below).
LC-MS/MS analysis
Peptides were resuspended in water with 0.1 % formic acid (FA) and analyzed using EASY-nLC 1200 nano-UHPLC coupled to a Q Exactive HF-X Quadrupole-Orbitrap mass spectrometer (Thermo Scientific). The chromatography column consisted of a 50 cm long, 75 μm i.d. microcapillary capped by a 5 μm tip and packed with ReproSil-Pur 120 C18-AQ 2.4 μm beads (Dr. Maisch GmbH). LC solvents were 0.1 % FA in H2O (Buffer A) and 0.1 % FA in 90 % MeCN: 10 % H2O (Buffer B). Peptides were eluted into the MS at a flow rate of 300 nL/min. over a 240 min. linear gradient (5-35 % Buffer B) at 65 °C. Data was acquired in data-dependent mode (top-20, NCE 28, R = 7,500) after full MS scan (R = 60,000, m/z 400-1,300). Dynamic exclusion was set to 10 s, peptide match to prefer and isotope exclusion was enabled.
MaxQuant analysis
The MS data was analyzed with MaxQuant (V1.6.1.0) (Cox and Mann, 2008) and searched against the human proteome (Uniprot) and a common list of contaminants (included in MaxQuant). The first peptide search tolerance was set at 20 ppm, 10 ppm was used for the main peptide search and fragment mass tolerance was set to 0.02 Da. The false discovery rate for peptides, proteins and sites identification was set to 1%. The minimum peptide length was set to 6 amino acids and peptide re-quantification and label-free quantification (MaxLFQ) were enabled. The ‘match between runs’ function was activated for the global changes in protein expression experiment. The minimal number of peptides per protein was set to two. Methionine oxidation was searched as a variable modification and carbamidomethylation of cysteines was searched as a fixed modification.
Transfection and western blot
HEK293T cells were seeded (6,000,000 cells in 20 mL DMEM 10% FCS without Pen/Strep) in a 10 cm Petri dish and left overnight to attach. Cells were transfected using a mixture of 96 μg PEI MAX 40000 (Polysciences Inc.) and 24 μg DNA in 2 mL opti-MEM. After 24 hours, cells were harvested by scraping, washed two times with PBS and lysates were prepared and treated with 30 μM 20 as described above. Click chemistry, reducing SDS-PAGE and visualization were performed as described above. Proteins were transferred to a PVDF membrane using a Trans-Blot Turbo transfer system (Bio-Rad). Membrane was blocked with 5% milk in Tris-Buffered Saline with 0.1 % Tween 20 (TBST) for one hour. Rabbit polyclonal antibody against DYKDDDDK (FLAG) (CST, #2368S, 1/1,000 in 5% milk in TBST) and α-tubulin (CST, #2144S, 1/1,000 in 5% milk in TBST) were added and incubated overnight at 4 °C. Membrane was washed three times for five minutes with TBST and incubated with Alexa Fluor 647-AffiniPure Donkey Anti-Rabbit IgG (H+L) (Jackson ImmunoResearch Inc., 711-605-152, 1/500 in 5 % milk in TBST) for one hour at r.t. Membrane was washed four times for five minutes with TBST and visualized with a Sapphire Biomolecular Imager (Azure Biosystems).
In situ β5 proteasome activity assay
RPMI 8226 cells were seeded in a 6-well plate (1,000,000 cells/well) in cell culture media supplemented with indicated concentrations of 1, 2, 21 or 22 (1,000x stock in DMSO) or DMSO and cultured for 6 hours. Cells were then collected and lysed as described above. Lysates (1 mg/mL, 25 μL) were dispensed into 100 μL assay buffer (PBS (pH 7.4), MgCl2 (1.5 mM), EDTA (1 mM), EGTA (1 mM), sucrose (250 mM) and freshly added DTT (5 mM), ATP (2 mM) and proteasome substrate Suc-LLVY-AMC (Enzo Life Sciences, 100 μM, 1 μL of 100x stock in DMSO)) in the wells of a black / clear bottom 96-well plate. The plate was incubated at 37 °C for two hours and fluorescence was read at A360ex/A460em using a Synergy H1 hybrid multi-mode microplate reader (Biotek). The protocol was modified from Maher (2014). IC50 values were calculated with GraphPad Prism (V7.02).
Western blot for mono-/poly-Ub and PARP-1
RPMI 8226 cells were seeded in a 6-well plate (1,000,000 cells/well) in cell culture media supplemented with indicated concentrations of 1 (1,000x stock in DMSO) or DMSO and cultured for 6 hours. Cells were then quickly washed twice in PBS and lysed using RIPA buffer (20 mM Tris, 150 mM NaCl, 2 mM EDTA, 0.1 % SDS, 1 % NP-40, 0.25 % deoxycholate, 1 mM PMSF) plus protease inhibitor cocktail (100x, RPI Corp., Cat.No. P50700-1) for five minutes on ice. Cells were quickly sonicated using a needle sonicator, the lysate was spun down in a tabletop centrifuge at 17000 x g for 10 minutes at 4 °C and the cleared lysate was transferred into a new microtube. Protein concentration was measured using the BCA protein assay (Thermo Fisher, Cat No. 23225). SDS-PAGE was performed as previously described and wet transfer to a PVDF membrane was done at 350 mA for one hour. Membrane was blocked with 5 % milk in TBST for one hour. Primary antibodies against respective proteins: PARP-1 (CST, #9532, 1:1,000 in 5 % BSA in TBST), mono- / polyubiquitinated conjugate antibody (Enzo Life Sciences, Prod. No. BML-PW8805, 1:500 in 5 % milk in TBST), and α-tubulin antibody (CST, #2144S, 1:1,000 in 5 % milk in TBST)), were added and incubated overnight at 4 °C. Membrane was washed three times for five minutes with TBST and incubated for one hour at r.t. with Alexa Fluor 647-AffiniPure Donkey Anti-Rabbit IgG (Jackson ImmunoResearch Inc., 711-605-152) or Alexa Fluor 488-AffiniPure Donkey Anti-Mouse IgG (Jackson ImmunoResearch Inc., 715-545-150). Membrane was washed four times for five minutes with TBST and visualized with a Sapphire Biomolecular Imager (Azure Biosystems).
Global MS proteomics profiling of 1 and BTZ
RPMI 8226 cells were seeded in a 6-well plate (1,000,000 cells/well) in cell culture media supplemented with 1 (100 nM, 1,000x stock in DMSO), bortezomib (BTZ, 2.5 nM, 1,000x stock in DMSO) or DMSO and cultured for 6 hours. Cells were then collected and lysed in PBS as described earlier and protein concentration was measured using the Bradford assay. Proteins (30 μg) were then denatured in 6 M urea in 50 mM NH4HCO3, reduced with 10 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) for 30 minutes and alkylated with 25 mM iodoacetamide for 30 minutes in the dark. Samples were diluted to 2 M urea with 50 mM NH4HCO3, and digested with trypsin (Thermo Scientific, 1.5 μL of 0.5 μg/μL) in the presence of 1 mM CaCl2 (100x stock in water). The digestion was performed for 12 hours at 37 °C. Samples were acidified to a final concentration of 5% acetic acid, desalted over a self-packed C18 spin column, and dried. Samples were analyzed by LC-MS/MS (see above), and the MS data were processed with MaxQuant (see above).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed with GraphPad Prism unless noted otherwise. Statistical values including the number of times the data was replicated (“n”) and statistical significance are reported in the Figure legends. Data are represented as mean ± SD or ± SEM and p-values calculated using an unpaired Student’s t-test. LC-MS/MS proteomics experiments were analyzed with MaxQuant using high confidence parameters indicated in the corresponding section. Statistical analysis of the global proteomics dataset was performed with Perseus using indicated parameters in the Figure legend S3.
Supplementary Material
Table S6. Full list of proteins identified in the LC-MS/MS-based global proteomics experiment with the corresponding 1/DMSO and BTZ/DMSO ratios and the p-values. Related to Figure 5 and Figure S3.
Table S5. Full list of 20-enriched proteins from the LC-MS/MS-based competitive pulldown experiment with the corresponding LFQ ratios and SDs. Related to Figure 4.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit-polyclonal antibody against DYKDDDDK (FLAG) | Cell Signaling Technology | Cat#2368S |
| Rabbit-polyclonal antibody against α-tubulin | Cell Signaling Technology | Cat#2144S; UniProt ID P68363 |
| Alexa Fluor 647-AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Inc. | Cat#711-605-152, RRID AB_2492288 |
| Rabbit-monoclonal anti-PARP-1 | Cell Signaling Technology | Cat#9532; UniProt ID P09874 |
| Mouse-monoclonal polyubiquitinylated conjugates antibody (FK1) | Enzo Life Sciences | Cat#BML-PW8805; UniProt ID: P0CG47 (UBB), P0CG48 (UBC), P62979 (RPS27A), P62987 (UBA52) |
| Alexa Fluor 488-AffiniPure Donkey Anti-Mouse IgG | Jackson ImmunoResearch Inc. | Cat#715-545-150; RRID AB_2340846 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RPMI-1640 | Fisher Scientific | Cat#11-875-093 |
| DMEM | Mediatech Inc | Cat#10-013-CV |
| Fetal bovine serum | Life Technologies | Cat#10437028 |
| Penicillin-Streptomycin | Sigma-Aldrich | Cat#P4333 |
| MEM Non-essential Amino Acid | Sigma-Aldrich | Cat#M7145 |
| Chloramphenicol | Fisher Scientific | AC227920250, CAS 56-75-7 |
| Kanamycin sulfate | VWR | A4789.0010; CAS 25389-94-0 |
| IPTG | Cayman Chemical | Cat#15300; CAS 367-93-1 |
| L-(+)-arabinose | Combi Blocks | Cat#QA-6576; CAS 5328-37-0 |
| Compound 1 | This paper | N/A |
| Bortezomib | Cayman Chemical | Cat#10008822; CAS 179324-69-7 |
| Compound 20 | This paper | N/A |
| TAMRA azide | Click Chemistry Tools | Cat# AZ109 |
| tris(2-carboxyethyl)phosphine hydrochloride | Alfa Aesar | Cat#J60316; CAS 51805-45-9 |
| tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine | Sigma Aldrich | Cat#678937; 510758-28-8 |
| Biotin azide | Sigma-Aldrich | Cat#762024; CAS 875770-34-6 |
| Iodoacetamide | Sigma-Aldrich | Cat#I1149; CAS 144-48-9 |
| Streptavidin agarose | ProteoChem | Cat#G4108 |
| Pierce™ Trypsin Protease, MS Grade | Thermo Scientific | Cat#90058 |
| PEI MAX 40000 | Polysciences Inc. | Cat#24765; CAS 49553-93-7 |
| Opti-MEM | Thermo Fisher | Cat#11058021 |
| Compound 2 | This paper | N/A |
| Compound 21 | This paper | N/A |
| Compound 22 | This paper | N/A |
| Suc-LLVY-AMC | Enzo Life Sciences | Cat#BML-P802-0005; CAS 94367-21-2 |
| Protease Inhibitor Cocktail III, Mammalian | Research Products International Corp. | Cat#P50700-1 |
| Critical Commercial Assays | ||
| Bradford Assay | Bio-Rad | Cat#5000006 |
| WST-1 assay | Roche | Cat#CELLPRO-RO |
| BCA protein assay | Thermo Fisher Scientific | Cat#23225 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat#ACS-4500 |
| RPMI 8226 | ATCC | Cat#CCL-155 |
| MM.1R | ATCC | Cat#CRL-2975 |
| Experimental Models: Organisms/Strains | ||
| E. coli BL21(DE3) | Lucigen | Cat#60300-2 |
| Recombinant DNA | ||
| PSMB5 (NM_002797) Human Tagged ORF Clone | Origene | Cat#RC209326 |
| PSMB2 (NM_002794) Human Tagged ORF Clone | Origene | Cat#RC210452 |
| pET-28a(+)-GlbB | (Amatuni and Renata, 2019) | N/A |
| pGro7 | Takara Bio | Cat#3340 |
| Software and Algorithms | ||
| GraphPad Prism (V7.02) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| MaxQuant (V1.6.1.0) | MaxQuant | https://www.maxquant.org/ |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| Perseus (V1.6.0.2) | Perseus | https://maxquant.net/perseus/ |
Acknowledgments
This work is supported, in part, by the National Institutes of Health Grant GM128895 (H.R.). We acknowledge the Bannister lab and the Shen lab for generous access to their instrumentation. We acknowledge Dr. D. Abegg for valuable discussions and proofreading of the manuscript. We thank P. Dickson and the Kodadek lab for providing the cell lines used in this study and for their valuable suggestions.
Footnotes
Declaration of Interests
The authors have applied for a provisional patent for this work.
References
- Abegg D, Frei R, Cerato K, Prasad Hari D, Wang C, Waser J, Adibekian A (2015). Proteome-wide profiling of targets of cysteine reactive small molecules by using ethynyl benziodoxolone reagents. Angew. Chem. Int. Ed 54, 10852–10857. [DOI] [PubMed] [Google Scholar]
- Adams J (2004). The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer 4, 349–360. [DOI] [PubMed] [Google Scholar]
- Altun M, Galardy PJ, Shringarpure R, Hideshima T, LeBlanc R, Anderson KC, Ploegh HL, Kessler BM (2005). Effects of PS-341 on the activity and composition of proteasomes in multiple myeloma cells. Cancer Res. 65, 7896–7901. [DOI] [PubMed] [Google Scholar]
- Amatuni A, Renata H (2019). Identification of a lysine-4-hydroxylases from the glidobactin biosynthesis and evaluation of its biocatalytic potential. Org. Biomol. Chem 17, 1736–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer CR, Groll M, Stein ML, Schellenberg B, Clerc J, Kaiser M, Kondratyuk TP, Pezzuto JM, Dudler R, Bachmann AS (2012). Activity enhancement of the synthetic syrbactin proteasome inhibitor hybrid and biological evaluation in tumor cells. Biochemistry 51, 6880–6888. [DOI] [PubMed] [Google Scholar]
- Berkers CR, Verdoes M, Licthman E, Fiebiger E, Kessler BM, Anderson KC, Ploegh HL, Ovaa H, Galardy PJ (2005). Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat. Methods 2, 357–362. [DOI] [PubMed] [Google Scholar]
- Besse A, Besse L, Kraus M, Mendez-Lopez M, Bader J Xin B-T, de Bruin G, Maurits E, Overkleeft HS, Driessen C (2019). Proteasome inhibition in multiple myeloma: head-to-head comparison of currently available proteasome inhibitors. Cell Chem. Biol 26, 340–351. [DOI] [PubMed] [Google Scholar]
- Blanchard F, Rusiniak ME, Sharma K, Sun X, Todorov I, Castellano MM, Gutierrez C, Baumann H, Burhans WC (2002). Targeted destruction of DNA replication protein Cdc6 by cell death pathways in mammals and yeast. Mol. Biol. Cell 13, 1536–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton M, Lucas MM, Downey SL, Screen M, Pletnev AA, Verdoes M, Tokhunts RA, Amir O, Goddard AL, Pelphrey PM (2009). Selective inhibitor of proteasome’s caspase-like sites sensitizes cells to specific inhibition of chymotrypsin-like sites. Chem. Biol 16, 1278–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Mohan P, Jiang J, Nemirovsky O, He D, Fleisch MC, Niederacher D, Pilarski LM, Lim CJ, Maxwell CA (2014). Spatial regulation of Aurora A activity during mitotic spindle assembly requires RHAMM to correctly localize TPX2. Cell Cycle 13, 2248–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T, Hosono H, Nakagawa K, Asaka M, Takeda H, Matsuda A, Ichikawa S (2014). Total synthesis of syringolin A and improvement of its biological activity. Angew. Chem. Int. Ed 53, 4836–4839. [DOI] [PubMed] [Google Scholar]
- Chondrogianni N, Tzavelas C, Pemberton AJ, Nezis IP, Rivett AJ, Gonos ES (2005). Overexpression of proteasome β5 assembled subunit increases the amount of proteasome and confers ameliorated response to oxidative stress and higher survival rates. J. Biol. Chem 280, 11840–11850. [DOI] [PubMed] [Google Scholar]
- Clerc J, Groll M, Illich DJ, Bachmann AS, Huber R, Schellenberg B, Dudler R, Kaiser M (2009). Synthetic and structural studies on syringolin A and B reveal critical determinants of selectivity and potency of proteasome inhibition. Proc. Natl. Acad. Sci. USA 106, 6507–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc J, Li N, Krahn D, Groll M, Bachmann AS, Florea BI, Overkleeft HS, Kaiser M (2010). The natural product hybrid of syringolin A and glidobactin A synergizes proteasome inhibition potency with subsite selectivity. Chem. Commun 47, 385–387. [DOI] [PubMed] [Google Scholar]
- Clerc J, Schellenberg B, Groll M, Bachmann AS, Huber R, Dudler R, Kaiser M (2010). Convergent synthesis and biological evaluation of syringolin A and derivatives as eukaryotic 20S proteasome inhibitor. Eur. J. Org. Chem 3991–4003. [Google Scholar]
- Cox J; Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Dai C, Stephenson CRJ (2010). Total synthesis of syringolin A. Org. Lett 12, 3453–3455. [DOI] [PubMed] [Google Scholar]
- Fabre B, Livneh I, Ziv T, Ciechanover A (2019). Identification of proteins regulated by the proteasome following induction of endoplasmic reticulum stress. Biochem. Biophys. Res. Commun 517, 188–192. [DOI] [PubMed] [Google Scholar]
- Fehrentz JA, Heitz A, Castro B, (1985). Synthesis of aldehydic peptides inhibiting renin. Int. J. Pept. Protein Res 26, 236–241. [DOI] [PubMed] [Google Scholar]
- Foley MA, Jamison TE (2010). Amide bond formation via reversible, carboxylic acid-promoted lactone aminolysis. Org. Proc. Res. Dev 14, 1177–1181. [Google Scholar]
- Gaich T, Baran PS (2010). Aiming for the ideal synthesis. J. Org. Chem 75, 4657–4673. [DOI] [PubMed] [Google Scholar]
- Groll M, Berkers CR, Ploegh HL, Ovaa H (2006). Crystal structure of the boronic acid-based proteasome inhibitor in complex with the yeast 20S proteasome. Structure 14, 451–456. [DOI] [PubMed] [Google Scholar]
- Groll M, Schellenberg B, Bachmann AS, Archer CR, Huber R, Powell TK, Lindow S, Kaiser M, Dudler RA (2008). A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature 452, 755–758. [DOI] [PubMed] [Google Scholar]
- Guerrero-Garcia TA, Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Mitsiades C, Anderson KC, Richardson PG (2018). The power of proteasome inhibition in multiple myeloma. Expert Rev. Proteomics 15, 1033–1052. [DOI] [PubMed] [Google Scholar]
- Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC (2001). The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 61, 3071–3076. [PubMed] [Google Scholar]
- Hong KU, Park YS, Seong Y-S, Kang D, Bae C-D, Park J (2007). Functional importance of the anaphase-promoting complex-Cdh1-mediated degradation of TMAP/CKAP2 in regulation of spindle function and cytokinesis. Mol. Cell. Biol 27, 3667–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SH, Kim J, Kim J-M, Lee S-Y, Shin D-S, Son K-H, Han DC, Sung YK, Kwon B-M (2007). Apoptosis induction of 2’-hydroxycinnamaldehyde as a proteasome inhibitor is associated with ER stress and mitochondrial perturbation in cancer cells. Biochem. Pharmacol 74, 557–565. [DOI] [PubMed] [Google Scholar]
- Howell LA, Peterson AK, Tomko RJ Jr. (2019). Proteasome subunit α1 overexpression preferentially drives canonical proteasome biogenesis and enhances stress tolerance in yeast. Sci. Rep 9, 12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Rivera TR, Opoku-Ansah J, Ambadi S, Bachmann AS, Pirrung MC Syntheses and cytotoxicity of syringolin B-based proteasome inhibitors. (2011). Tetrahedron 67, 9950–9956. [Google Scholar]
- Illuminati G, Mandolini L (1981). Ring closure reactions of bifunctional chain molecules. Acc. Chem. Res 14, 95–102. [Google Scholar]
- Imker HJ, Krahn D, Clerc J, Kaiser M, Walsh CT, (2010). N-Acylation during glidobactin biosynthesis by the tridomain nonribosomal peptide synthetase module GlbF. Chem. Biol 17, 1077–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya N, Fujita Y, Irreverre F, Witkop B (1965). The synthesis of erythro-g-hydroxy-L-lysine and its nonoccurrence in collagen. Biochemistry 4, 2501–2507. [Google Scholar]
- Kamiński ZJ, Kolesińska B, Sabatino G, Chelli M, Rovero P, Blaszczyk M, Główka ML, Papini AM (2005). N-Triazinylammonium tetrafluoroborates. A new generation of efficient coupling reagents useful for peptide synthesis. J. Am. Chem. Soc 127, 16912–16920. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG (1993). Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 53, 3976–3985. [PubMed] [Google Scholar]
- Kitahata S, Chiba T, Yoshida T, Ri M, Iida S, Matsuda A, Ichikawa S (2016). Design, synthesis, and biological activity of isosyringolin A. Org. Lett 18, 2312–2315. [DOI] [PubMed] [Google Scholar]
- Kitahata S, Yakushiji F, Ichikawa S (2017). Impact of the structures of macrocyclic Michael acceptors on covalent proteasome inhibition. Chem. Sci 8, 6959–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahn D, Ottman C, Kaiser M (2011). The chemistry and biology of syringolins, glidobactins and cepafungins (syrbactins). Nat. Prod. Rep 28, 1854–1867. [DOI] [PubMed] [Google Scholar]
- Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, Orlowski RZ (2007). Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 110, 3281–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan R, Looi K, Omary MB (2015). Absence of keratins 8 and 18 in rodent epithelial cell lines associates with keratin gene mutation and DNA methylation: cell line selective effects on cell invasion. Exp. Cell Res 335, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang X, Renata H (2019). Asymmetric chemoenzymatic synthesis of podophyllotoxin and related aryltetralin lignans. Angew. Chem. Int. Ed 58, 11657–11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Xu B, Li J, Lu H (2009). BAG3 gene silencing sensitizes leukemic cells to bortezomib-induced apoptosis. FEBS Lett. 583, 401–406. [DOI] [PubMed] [Google Scholar]
- Maher P (2014). Proteasome assay in cell lysates. Bio Protoc. 4, e1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manasanch EE, Orlowski RZ (2017). Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol 14, 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin J, Didierjean C, Aubry A, Casimir J-R, Briand J-P, Guichard G (2004). Synthesis of enantiopure 4-hydroxypipecolate and 4-hydroxylysine derivatives from a common 4,6-dioxopiperidinecarboxylate precursor. J. Org. Chem 69, 130–141. [DOI] [PubMed] [Google Scholar]
- Matondo M, Marcellin M, Chaoui K, Bousquet-Dubouch M-P, Gonzalez-de-Peredo A, Monsarrat B, Burlet-Schiltz O (2017). Determination of differentially regulated proteins upon proteasome inhibition in AML cell lines by the combination of large-scale and targeted quantitative proteomics. PROTEOMICS 17, 1600089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melaugh G, Giri N, Davidson CE, James SL, Del Pópolo MG, (2014). Designing and understanding permanent microporosity in liquids. Phys. Chem. Chem. Phys 16, 9422–9431. [DOI] [PubMed] [Google Scholar]
- Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess, Gillette MA, Jaffe JD, Carr SA (2013). Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10, 634–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oerlemans R, Franke NE, Assaraf YG, Cloos J, Van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Votjekova K, Lemos C, van der Heijden JW, Ylstra B, Peters GJ, Kaspers GL, Dijkmans BA, Scheper RJ, Jansen G (2008). Molecular basis of bortezomib resistance: proteasome subunit β5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 112, 2489–2499. [DOI] [PubMed] [Google Scholar]
- Oka M, Nishiyama Y, Ohta S, Kamei H, Konish T, Miyaki T, Oki T, Kawaguchi H (1988). Glidobactins A, B and C, new antitumor antibiotics. I. Production, isolation, chemical properties and biological activity. J. Antibiot 41, 1331–1337. [DOI] [PubMed] [Google Scholar]
- Oka M, Yaginuma K, Numata K, Konishi M, Oki T, Kawaguchi H (1988). Glidobactins A, B and C, new antitumor antibiotics. II. Structure elucidation. J. Antibiot 41, 1338–1350. [DOI] [PubMed] [Google Scholar]
- Oka M, Iwata K-I, Nishiyama Y, Kamei H, Konishi M, Oki T, Kawaguchi H (1988). Chemical modification of the antitumor antibiotic glidobactin. J. Antibiot 41, 1812–1822. [DOI] [PubMed] [Google Scholar]
- Pawar A, Basler M, Goebel H, Alvarez Salinas GO, Groettrup M, Böttcher T (2020). Competitive metabolite profiling of natural products reveal subunit specific inhibitors of the 20S proteasome. ACS Cent. Sci 6, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JT, Andorder MC, Lewis JC (2013). Regioselective arene halogenation using the FAD-dependent halogenase RebH. Angew. Chem. Int. Ed 125, 5379–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrung MC, Biswas G, Ibarra-Rivera TR (2010). Total synthesis of syringolin A and B. Org. Lett 12, 2402–2405. [DOI] [PubMed] [Google Scholar]
- Sabot C, Kumar KA, Meunier S, Mioskowski C (2007). A convenient aminolysis of esters catalyzed by 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) under solvent-free conditions. Tetrahedron Lett. 48, 3863–3866. [Google Scholar]
- Schmidt U, Kleefeldt A, Mangold R (1992). The synthesis of glidobactin A. J. Chem. Soc. Chem. Commun 1687–1689. [Google Scholar]
- Shah SP, Nooka AK, Jaye DL, Bahlis NJ, Lonial S, Boise LH (2016). Bortezomib-induced heat shock response protects multiple myeloma cells and is activated by heat shock factor 1 serine 326 phosphorylation. Oncotarget 7, 59727–59741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji J, Hinoo H, Kato T, Hattori T, Hirooka K, Tawara K, Shiratori O, Terui Y (1990). Isolation of cepafungins I, II and III from Pseudomonas species. J. Antibiot 43, 783–787. [DOI] [PubMed] [Google Scholar]
- Speers AE, Cravatt BF (2004). Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol 11, 535–546. [DOI] [PubMed] [Google Scholar]
- Starheim KK, Holien T, Misund K, Johansson I, Baranowska KA, Sponaas A-M, Hella H, Buene G, Waage A, Sundan A, Bjørkøy G (2016). Intracellular glutathione determines bortezomib cytotoxicity in multiple myeloma cells. Blood Cancer J. 6, e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein ML, Beck P, Kaiser M, Dudler R, Becker CFW, Groll M (2012). One-shot NMR analysis of microbial secretions identifies highly potent proteasome inhibitor. Proc. Natl. Acad. Sci. USA 109, 18367–18371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sund C, Belda O, Wiktelius D, Sahlberg C, Vrang L, Sedig S, Hamelink E, Henderson I, Agback T, Jansson K, Borkakoti N, Derbyshire D, Eneroth A, Samuelsson B Design and synthesis of potent macrocyclic renin inhibitors. (2011). Bioorg. Med. Chem. Lett 21, 358–362. [DOI] [PubMed] [Google Scholar]
- Tamura M, Kochi J (1971) Coupling of Grignard reagents with organic halides. Synthesis 303–305. [Google Scholar]
- Tatsumi Y, Ohta S, Kimura H, Tsurimoto T, Obuse C (2003). The ORC1 cycle in human cells. J. Biol. Chem 278, 41528–41534. [DOI] [PubMed] [Google Scholar]
- Weyburne ES, Wilkins OM, Sha Z, Williams DA, Pletnev AA, de Bruin G, Overkleeft HS, Goldberg AL, Cole MD, Kisselev AF (2017). Inhibition of the proteasome β2 site sensitizes triple-negative breast cancer cells to β5 inhibitors and suppresses Nrf1 activation. Cell. Chem. Biol 24, 218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye B, Jiang P, Zhang T, Ding Y, Sun Y, Hao X, Li L, Wang L, Chen Y, (2018). Total synthesis of the highly N-methylated peptide jahanyne. J. Org. Chem 83, 6741–6747. [DOI] [PubMed] [Google Scholar]
- Yew EHJ, Cheung NS, Choy MS, Qi RZ, Lee AY-W, Peng ZF, Melendez AJ, Manikandan J, Koay ES-C, Chiu L-L, Ng WL, Whiteman M, Kandiah J, Halliwell B (2005). Proteasome inhibition by lactacystin in primary neuronal cells induces both potentially neuroprotective and pro-apoptotic transcriptional responses: a microarray analysis. J. Neurochem 94, 943–956. [DOI] [PubMed] [Google Scholar]
- Zhang X, King-Smith E, Renata H (2018). Total synthesis of tambromycin by combining chemocatalytic and biocatalytic C–H functionalization. Angew. Chem. Int. Ed 57, 5037–5041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S6. Full list of proteins identified in the LC-MS/MS-based global proteomics experiment with the corresponding 1/DMSO and BTZ/DMSO ratios and the p-values. Related to Figure 5 and Figure S3.
Table S5. Full list of 20-enriched proteins from the LC-MS/MS-based competitive pulldown experiment with the corresponding LFQ ratios and SDs. Related to Figure 4.
Data Availability Statement
The published article includes all datasets generated and analyzed during this study. This study did not generate any code.