

Abstract
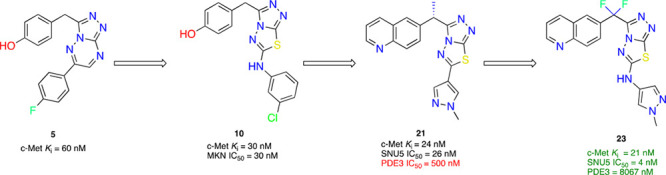
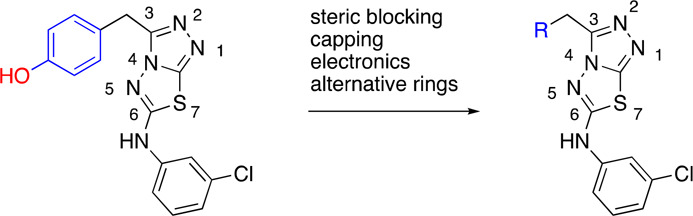
Herein, we report a novel series of highly potent and selective triazolothiadiazole c-Met inhibitors. Starting with molecule 5, we have applied structure-based drug design principles to identify the triazolothiadiazole ring system. We successfully replaced the metabolically unstable phenolic moiety with a quinoline group. Further optimization around the 5,6 bicyclic moiety led to the identification of 21. Compound 21 suffered from PDE3 selectivity issues and subsequent, structurally informed design led to the discovery of compound 23. Compound 23 has exquisite kinase selectivity, excellent potency, favorable ADME profile, and showed dose-dependent antitumor efficacy in a SNU-5 gastric cancer xenograft model.
Keywords: c-Met inhibitor, Structure-based drug design, PDE3
c-Met, also known as hepatocyte growth factor receptor (HGFR), is a member of the receptor tyrosine kinase (RTK) family.1 The natural ligand of c-Met is the hepatocyte growth factor (HGF), also known as scatter factor (SF). c-Met plays an essential role in cellular signaling pathways involving proliferation, invasion, migration, survival, angiogenesis, metastasis, and drug resistance.2 Abnormal activation of the c-Met pathway leads to tumor metastasis, invasion, and recurrence.3 Overexpression of c-Met and/or HGF is often associated with a large number of human cancers such as breast, lung, liver, kidney, stomach, and brain. Therefore, c-Met has been recognized as an attractive target for the development of cancer therapies and was of considerable interest to us during our past effort in oncology.4
In the past two decades, progress has been made toward the development of small-molecule c-Met inhibitors.5 There are two types of ATP-competitive c-Met inhibitors based on binding modes (Figure 1).6 Type 1 inhibitors interact with the hinge residue Met1160 within the U-shaped ATP-binding pocket and also interact with Tyr1230 via a π–π stacking interaction.7 In general, type 1 c-Met inhibitors have good kinase selectivity.8 Crizotinib (1) is a type 1 inhibitor approved by the U.S. Food and Drug Administration (FDA), that inhibits ALK, Ros1, and c-Met.8a Additionally, savolitinib (2) is another type 1 c-Met inhibitor displaying enhanced kinase selectivity which has shown fewer side effects in clinical trials.8b Capmatinib (3) is a newly approved ATP-competitive and selective c-Met inhibitor for the treatment of advanced non-small cell lung cancer.9 Type II inhibitors bind at the deep hydrophobic back pocket between the hinge residue and the solvent-accessible area.10 Those most known type II inhibitors potently inhibit VEGFR as well as other homological kinases.11 Cabozantinib (4) is a type II c-Met inhibitor approved by the FDA.12 Type II inhibitors often have a broader inhibition spectrum which may help in overcoming acquired resistance to type I inhibitors, but they also have tolerability issues in clinical settings.13
Figure 1.

Herein, we describe the discovery and optimization of a new series of triazolothiadiazole compounds as potent and selective c-Met inhibitors.14 Structure-based drug design was used to guide us during lead optimization. Improvements on potency, metabolic stability, and selectivity versus PDE3 are discussed. Finally, we provide in vivo efficacy data on a relevant tumor model.
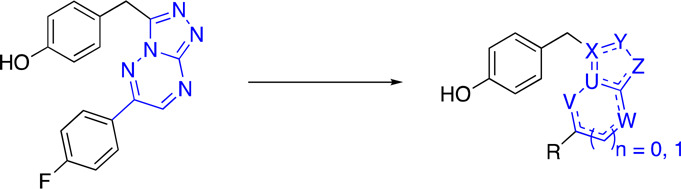
In an effort to identify a potent, selective c-Met inhibitor, we focus on a series of triazolotriazines that showed promising c-Met inhibition activity. One of the promising compounds, 5 (from a Sugen patent),15 displayed c-Met enzymatic Ki and cellular IC50’s of 60 and 32 nM, respectively (Table 1). We began our exploration by systematic replacements of the triazolotriazine core with different [5,5]- and [5,6]-bicyclic cores (Table 1).
Table 1. Exploration of [5,5]- and [5,6]-Bicyclic Triazolotriazine Replacements.
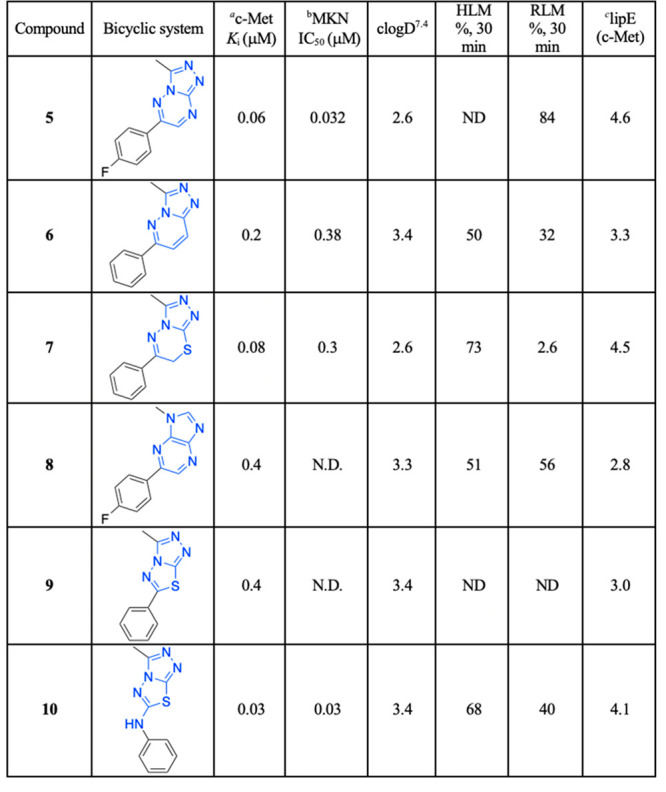
Inhibition of cMet kinase activity (Ki) was determinated by standard radiometric assay.
The cell IC50 was determinated using MKN gastric carcinoma.
LipE value calculated from c-Met pKi.
Five structurally relevant [5,6]- and [5,5]-systems were evaluated for their c-Met inhibition activity (Table 1). Although promising c-Met activity was observed with compound 6 (Ki = 200 nM) and compound 7 (Ki = 80 nM), these two scaffolds were deprioritized quickly due to poor in vitro metabolic stability and poor in vivo pharmacokinetics (PK) in rats (data not shown). The compound 8 revealed lower c-Met kinase activity. Triazolothiadiazole 9 (Ki = 400 nM) was significantly less potent than compound 5 (Ki = 60 nM); however, compound 10 (Ki = 30 nM) demonstrated good potency. The scarcity of the triazolothiadizole bicyclic system in the literature16 and the promising potency prompted us to explore this subseries further. The cocrystal complex of triazolothiadiazole 10 bound to the c-Met kinase domain is shown in Figure 2A. A hydrogen bond is formed between the phenolic oxygen atom and the backbone NH of Met1160. The binding mode also suggests a π-stacking interaction between the triazolothiadiazole core and Tyr1230 and a hydrogen bond interaction between the N1 of the core and the backbone NH of Asp1222. In this co-complex structure, we also observed an interaction between the sulfur of the core ring with the carbonyl of Arg1208. Additionally, the N-linker of compound 10 is unexpectedly engaged in a hydrogen bond to the backbone carbonyl Arg1208.
Figure 2.
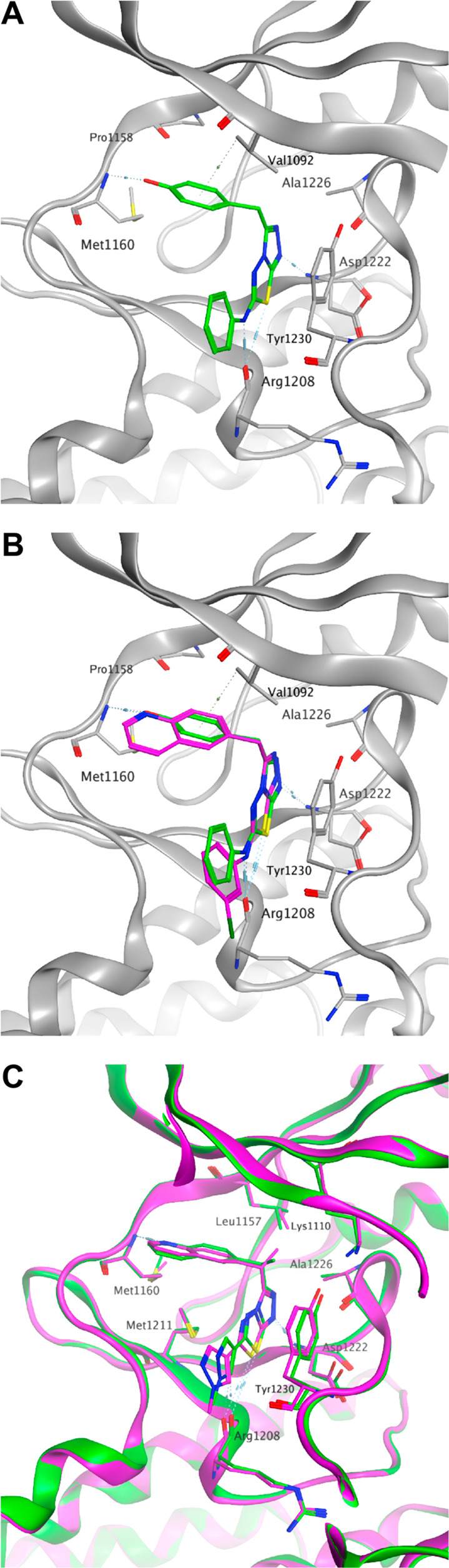
Compound binding modes in the c-Met ATP site pocket and key interactions. (A) Crystal structure of 10 (green). (B) Overlay of quinoline compound 17 (purple) and phenol compound 11 (green). (C) Crystal structures of compound 20 (purple) and 21 (green) superimposed.
Given that the binding mode, as reflected by the cocrystal structure, shows a key interaction between the phenolic moiety and c-Met and that we also observed glucuronidation metabolites with these phenolic compounds (data not shown),17 we next investigated if either modification on the phenol would slow down metabolism or if isosteric phenolic replacements would alleviate undesirable metabolism. As shown in Table 2, moving the hydroxyl group from the 4-position of 11 to the 3-position of 12 disrupted the hydrogen bond interaction, and consequently, the c-Met inhibitory potency dropped by more than 20-fold. Capping the hydroxyl groups with a 1,3-dioxolane group (13) also reduced the potency significantly. Introduction of an ortho-fluoro atom (14) or an ortho-chloro atom (15) also caused a potency erosion. As the nitrogen atoms of quinolines and azaindoles are often used as the hinge binding elements in kinase programs,18 alternative aromatic groups14 as potential hydrogen bond donors were explored. Although the azaindole as the hinge binder 16 caused a sharp potency drop, compound 17 was made and demonstrated good potency and a lipophilic efficiency (c-Met lipE = 3.9) comparable to those of our initial phenolic compound 11. An overlay of quinoline 17 with phenol 11 is shown in Figure 2B. The overlay indicates that the nitrogen atom of the quinoline ring of 17 occupies the same position of the phenolic oxygen of 11. Both formed similar hydrogen bonds with Met1160.
Table 2. Modifications of the Phenolic Moietya.

Inhibition constant (Ki) and cellular IC50 were determined as described in the Supporting Information.
Although compounds 13–17 showed moderate rat hepatocyte stability, high clearances and short half-lives were observed in rat PK experiments (Supporting Information). These data suggested that lower clearance may be obtained by decreasing the lipophilicity of these compounds. Some of our early structure–activity relationship (SAR) studies (not shown)14 indicated that aromatic substitutions at C6 of triazolothiadiazoles were well tolerated, and we believed that modifications of the C6 aromatic groups could modulate the π-stacking interaction with Tyr1230 allowing for increased potency and improved metabolic stability. In this regard, we were pleased to find that heteroaromatics such as pyrazole analogue 18 could maintain potency while increasing our lipophilic efficiency above 5. More importantly, 18 demonstrated improved rat clearance to 24.5 mL/min/kg and a half-life as 1.1 h (Supporting Information).
Since the methylene linker between the quinoline ring and the triazolothiadiazole bicyclic system is easily oxidized by CYP-mediated metabolism,19 the introduction of small groups that can potentially block metabolism was investigated. Given that the cocrystal of compound 17 with c-Met kinase indicated that the methylene linker is adjacent to a pocket composed of Leu1157 and Lys1110, we envisioned that small hydrophobic substitutions might occupy the pocket and enhance kinase inhibition potency. Monomethyl substitution 19 led to a 3-fold improvement in cellular c-Met inhibition. More significantly, 20 and 21 have a 15-fold and 4.5-fold difference in the activity of enzymatic Ki and cellular IC50, respectively, thus indicating the importance of the stereochemistry of the linker substituent.
The enantiomerically pure compounds 20 and 21 were both crystallized in c-Met with the primary structural difference being the chirality at the methylene linker. The (S)-isomer places the methyl group further into a hydrophobic region of the protein flanked by Leu1157, Ala1226, and Lys1110. Additionally, this chiral change shifts the core by ∼0.7 Å toward the entrance of the pocket, leading to ideal distance between Met 1160 and the quinoline nitrogen and moving the methylpyrazole moiety further toward the solvent front while leaving the observed hydrogen bond to Asp1222 unchanged. The overlay is based on the superposition of our internal crystal structures with emphasis on the pocket residues (Figure 2C). The (S)-isomer projects more deeply into the hydrophobic region of this pocket as shown, while not being so bulky as to clash with nearby pocket residues.
Generally speaking, all the compounds in Table 3 have low unbound human and mouse intrinsic clearances, which indicated good physical properties and developability.
Table 3. Substitutions on the Methylene Linker.
| compd | R3, R4 | c-Met Ki (μM) | SNU-5 IC50 (μM) | HepsClintu human, mouse (μL/min/106 cells) | clogD7.4 | lipE (c-Met) |
|---|---|---|---|---|---|---|
| 18 | (H,H) | 0.04 | 0.08 | 4.8, 6.2 | 2.1 | 5.3 |
| 19 | rac-(Me, H) | 0.05 | 0.03 | 3.2, 7.4 | 2.6 | 4.7 |
| 20 | R-(H, Me) | 0.3 | 0.09 | 5.8, 7.0 | 2.6 | 3.9 |
| 21 | S-(Me, H) | 0.02 | 0.02 | 5.1, 7.2 | 2.6 | 5.1 |
The inhibition of c-Met kinase activity was determined in a radiometric assay by a series of titration at 35 μM of ATP. The degree of c-Met inhibition varied linearly with the ATP concentration, indicating competitive inhibition. Compound 21 selectively inhibits c-Met kinase with Ki = 0.025 μM. Generally speaking, all the compounds from the triazolothiadiazole series have shown good kinase selectivity against our in-house panel of 15 kinases including FLT3, GSK3, JAK2, KDR, SRC, SKY, and PI3K with Ki > 4 μM. In addition, compound 21 demonstrates <50% inhibition (at 2 μM) against all 50 kinases included in the upstate/Millipore panel. In a broader panel of 67 receptors, compound 21 was found to inhibit PDE3 with an IC50 of 0.5 μM (Supporting Information). Since PDE3 expression is high in cardiac and vascular myocytes20 and PDE3 inhibition may cause off-target cardiovascular effects, we decided to investigate the selectivity data against PDE3 for some existing compounds within this series (20 and 21) as well as the design of novel analogues aimed at gaining an understanding of the structural requirements to achieve selectivity against PDE3. We overlaid compound 21 with Merck PDE 3 inhibitor “MERCK1”,21 as shown in Figure 3. Compound 21 superimposes well with the published PDE3 inhibitor MERCK1 in a low energy conformation. Hydrogen bonding to Gln988 and hydrophobic contacts to Ile995 and Phe959 are maintained in the PDE3 active site.
Figure 3.
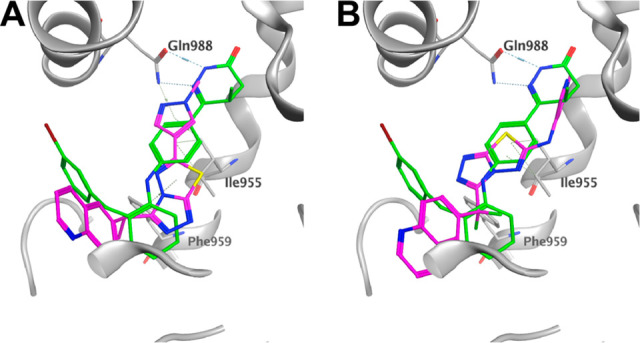
Overlay of 21 (A) and 23 (B) with the published MerckI PDE3 inhibitor (green).
As shown in the Table 4, the stereochemistry at the methylene linker substitution has an influence on the PDE3 selectivity with the (R)-isomers (20 and 24) showing selectivity ratios of 95 and 67 as opposed to ratios of 25 and 33 for their (S)-counterparts, compounds 21 and 25. Introduction of the gem-difluoro linker (data not shown) is well tolerated for cMet activity while also improving the cMet/PDE3 selectivity ratio. With this information in mind, we decided to investigate the possibility of including a NH spacer between the triazolothiadiazole and the C6 bottom pyrazole ring. Much to our delight, compound 22 offered 5 nM cellular potency and a PDE3 selectivity ratio of 600. By combining the gem-difluoro linker and the C6 space, compound 23, which was found to be very potent with 4 nM potency and to have over 2000-fold selectivity against PDE3, was less able to replicate the binding mode of MERCK1. The addition of the nitrogen linker between the core and the methyl pyrazole makes the interaction with Gln988 less conserved in low energy docking poses, potentially removing its ability to make a stable hydrogen bond at that location. Additionally, the proton on the nitrogen linker has no nearby hydrogen bonding partners and a desolvation cost would apply to place it against the nearby hydrophobic surface. It is interesting to note that a similar increase in cellular potency was observed with the introduction of a methyl group on the C3 pyrazole. Despite showing high potency in SNU-5 (IC50 of 3 nM each), compounds 24 and 25 retained potency against PDE3 and therefore have very low selectivity windows and were not pursued further. Docking 23 in the c-Met protein confirmed all key interactions: hydrogen bonding with Met1160, π–π stacking with Tyr1230, and NH linker with Arg1208 (Supporting Information).
Table 4. Selectivity against PDE3 for Compounds 20–25.

Given its very high c-Met potency and high PDE3 selectivity, compound 23 was evaluated in pharmacokinetic studies in multiple species (Table 5). Overall, 23 has favorable pharmacokinetics displaying moderate total clearance and volumes of distribution across all species tested. Following oral dosing of 10 mpk of compound 23 to rat and dog, respectively, the compound was absorbed rapidly in the rat with Cmax occurring at 0.5 h, but in the dog the Cmax occurred at 1.7 h. Exposure in the dog was 2–3-fold higher than that in the rat at this nominal dose. The bioavailability was moderate in the rat and high in the dog.
Table 5. Multiple-Species PK Data for Compound 23.
| ratc | moused | doge | monkeyf | |
|---|---|---|---|---|
| Cl (mL/min/kg)a | 25 | 21 | 14 | 23 |
| Vss (L/g)a | 0.70 | 1.0 | 1.6 | 1.2 |
| T1/2 (h)a | 1.4 | 1.8 | 3.6 | 2.0 |
| Cmax (μg/mL)b | 0.15 | 4.8 | 1.3 | |
| AUC (μg/h/mL)b | 0.60 | 16 | 9.9 | |
| %Fb | 29 | 100 | 66 |
Parameters obtained after IV dosing.
Parameters obtained after PO dosing.
IV dosing: 0.9 mpk; PO dosing: 2.6 mpk with 0.5% MC/0.1% Tween 80 formulation.
IV dosing: 0.8 mpk; PO dosing: 13 mpk with 0.5% MC/0.1% Tween 80 formulation.
IV dosing: 0.8 mpk; PO dosing: 12 mpk with 30% PG/10%Solutol/1% HPMC-AS formulation,
IV dosing: 0.9 mpk.
We next investigated compound 23 in the ectopic human SNU-5 gastric cancer xenograft model, which has amplified c-Met acitivity. The SNU-5 model is a tumor model with amplified c-Met.
As shown in Figure 4, 23 induced tumor regression in a dose-dependent manner with complete regression at 30 mpk (T/Ti of −23.9%) after 38 days post implantation. At the lower dose groups (3 and 10 mpk), significant tumor growth inhibition was observed compared to controls (T/C of 16.6 and 54.2). Additionally, the compound was well-tolerated, with mice showing no significant body weight loss with 30 mpk during the dosing regimen (Figure 4B). 23 alleviated the body weight loss commonly observed in untreated SNU-5 tumor-bearing SCID mice. These results, combined with favorable pharmacokinetic profiles across species, allowed advancement of 23 as a tool compound for preclinical studies.
Figure 4.
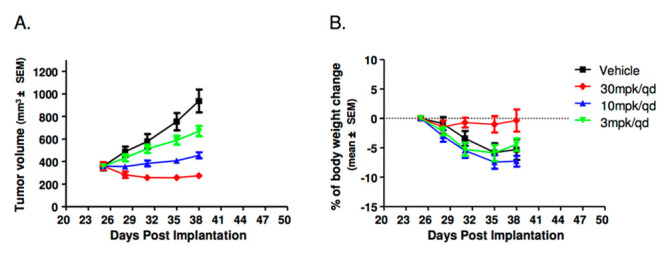
In vivo efficacy of 23 in the ectopic human SNU-5 gastric cancer xenograft model in SCID mice. (A) Tumor growth curve; (B) % change in body weight.
Herein, we have reported a novel series of highly potent and selective triazolothiadiazole c-Met inhibitors. The judicious use of structure-based drug design allowed us to (1) identify the quinoline group as a metabolically stable hinge binding element and (2) dial out the PDE3 liability of our lead triazolothiadiazole scaffold. Compound 23 has exquisite kinase selectivity, excellent potency, and favorable ADME and PK profiles and showed dose-dependent antitumor efficacy in vivo.
Acknowledgments
The authors gratefully acknowledge Dr. Arjun Narayanan for modeling work and Dr. Caroline Chandra Tjin for reviewing and providing comments on the paper.
Glossary
Abbreviations
- cMet
mesenchymal-to-epithelial transition factor
- HGF
hepatocyte growth factor
- HGFR
hepatocyte growth factor receptor
- RTK
receptor tyrosine kinase
- SF
scatter factor
- PDE
pan-phosphodiesterase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00094.
Experimental procedures for the key compounds, biological assay information, kinase selectivity profiles, statistical analysis of compound 23 in SNU-5 xenograft model, selective compound rat IV PK, in vitro pharmacokinetic data of compounds 21 and 23 and computational modeling information (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Giordano S.; Ponzetto C.; Di Renzo M. F.; Cooper C. S.; Comoglio P. M. Tyrosine kinase receptor indistinguishable from the cMet Protein. Nature 1989, 339, 155–156. 10.1038/339155a0. [DOI] [PubMed] [Google Scholar]; b Bottaro D. P.; Rubin J. S.; Faletto D. L.; Chan A. M.; Kmiecik T. E.; Vande Woude G. F.; Aaronson S. A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- Birchmeier C.; Birchmeier W.; Gherardi E.; Vande Woude G. F. Met metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- Dharmawardana P. G.; Giubellino A.; Bottaro D. P. Hereditary papillary renal carcinoma type I. Curr. Mol. Med. 2004, 4, 855–868. 10.2174/1566524043359674. [DOI] [PubMed] [Google Scholar]
- Gschwind A.; Fischer O. M.; Ullrich A. The Discovery of receptor tyrosine kinase: targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- a Cui J. Jean Targeting receptor tyrosine met in cancer: small molecule inhibitors and clinical progress. J. Med. Chem. 2014, 57, 4427–4453. 10.1021/jm401427c. [DOI] [PubMed] [Google Scholar]; b Lv P. C.; Yang Y. S.; Wang Z. C. Recent progress in the development of small molecule cMet inhibitors. Curr. Top. Med. Chem. 2019, 19, 1276–1288. 10.2174/1568026619666190712205353. [DOI] [PubMed] [Google Scholar]; c Parikh P. K.; Ghate M. D. Recent advanced in the discovery of small molecular cMet kinase inhibitors. Eur. J. Med. Chem. 2018, 143, 1103–1138. 10.1016/j.ejmech.2017.08.044. [DOI] [PubMed] [Google Scholar]
- a Dussault I.; Bellon S. F. From concept to reality: the long road to c-Met and RON receptor tyrosine kinase inhibitors for the treatment of cancer. Anti-Cancer Agents. Anti-Cancer Agents Med. Chem. 2009, 9, 221–229. 10.2174/187152009787313792. [DOI] [PubMed] [Google Scholar]; b Liu Y.; Gray N. S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2 (7), 358–364. 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- a Albrecht B. K.; Harmange J.-C.; Bauer D.; Berry L.; Bode C.; Boezio A. A.; Chen A.; Choquette D.; Dussault I.; Fridrich C.; Hirai S.; Hoffman D.; Larrow J. F.; Kaplan-Lefko P.; Lin J.; Lohman J.; Long A. M.; Moriguchi J.; O’Connor A.; Potashman M. H.; Reese M.; Rex K.; Siegmund A.; Shah K.; Shimanovich R.; Springer S. K.; Teffera Y.; Yang Y.; Zhang Y.; Bellon S. Discovery and optimization of triazolopyridazines as potent and selective inhibitors of the c-Met kinase. J. Med. Chem. 2008, 51, 2879–2882. 10.1021/jm800043g. [DOI] [PubMed] [Google Scholar]; b Bellon S. F.; Kaplan-Lefko P.; Yang Y.; Zhang Y.; Moriguchi J.; Rex K.; Johnson C. W.; Rose P. E.; Long A. M.; O’Connor A. B.; Gu G.; Coxon A.; Kim T.-S.; Tasker A.; Burgess T. L.; Dussault I. cMet inhibitors with novel binding mode show activity against several hereditary papillary renal cell carcinoma-related mutations. J. Biol. Chem. 2008, 283, 2675–2683. 10.1074/jbc.M705774200. [DOI] [PubMed] [Google Scholar]
- a Cui J. J.; Tran-Dube M.; Shen H.; Nambu M.; Kung P. P.; Parirish M.; Jia L.; Meng J.; Funk L.; Alton G.; Timofeevski S.; Yamazaki S.; Kania R. S.; Edwards M. P.; et al. Structure based drug design of Crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (Met) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]; b Jia H.; Dai G.; Weng J.; Zhang Z.; Wang Q.; Zhou F.; Jiao L.; Cui Y.; Ren Y.; Fan S.; Zhou J.; Qing W.; Gu Y.; Wang J.; Sai Y.; Su W. Discovery of (S)-1-(1-(imidazo[1,2-a]pyridin-6-yl)ethyl)-6-(1-methyl-1H-pyrazol-4yl)-1H-[1,2,3]triaolo[4,5-b]pyraine (Volitinb) as a highly potent and selective mesenchymal-epithelial transition factor (cMet) inhibitor in clinical development for treatment of cancer. J. Med. Chem. 2014, 57, 7577–7589. 10.1021/jm500510f. [DOI] [PubMed] [Google Scholar]
- a Saad K. M.; Shaker M. E.; Shaaban A. A.; Abdelrahman R. S.; Said E. The c-Met inhibitor Capmatinib alleviates acetaminophen induced. Int. Immunopharmacol. 2020, 81, 106292. 10.1016/j.intimp.2020.106292. [DOI] [PubMed] [Google Scholar]; b Kim S.; Kim T. M.; Kim D. M.; Kim S.; Kim M.; Ahn Y. O.; Keam B.; Heo D. S. Acquired resistance of cMet inhibitor Capmatinib. Cancer Res. Treat. 2019, 51 (3), 951–962. 10.4143/crt.2018.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder G. M.; An Y.; Cai Z.-W.; Chen X.-T.; Clark C.; Cornelius L. A. M.; Dai J.; Gullo-Brown J.; Gupta A.; Henley B.; Hunt J. T.; Jeyaseelan R.; Kamath A.; Kim K.; Lippy J.; Lombardo L. J.; Manne V.; Oppenheimer S.; Sack J. S.; Schmidt R. J.; Shen G.; Stefanski K.; Tokarski J. S.; Trainor G. L.; Wautlet B. S.; Wei D.; Williams D. K.; Zhang Y.; Zhang Y.; Fargnoli J.; Borzilleri R. M. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J. Med. Chem. 2009, 52, 1251–1254. 10.1021/jm801586s. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Wu H.; Wang L.; Liu E.; Knapp S.; Liu Q.-S.; Gray N. S. Exploration of Type II Binding Mode: A Privileged Approach for kinase inhibitor focused drug discovery?. ACS Chem. Biol. 2014, 9, 1230–1241. 10.1021/cb500129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakes F. M.; Chen J.; Tan J.; Yamaguchi K.; Shi Y.; Yu P.; Qian F.; Chu F.; Bentzien F.; Cancilla B.; Orf J.; You A.; Laird A. D.; Engst S.; Lee L.; Lesch J.; Chou Y. C.; Joly A. H. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- Reungwetwattana T.; Liang Y.; Zhu V.; Ou S. I. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017, 103, 27–37. 10.1016/j.lungcan.2016.11.011. [DOI] [PubMed] [Google Scholar]
- a Laufer D.; Aronov A.; Li P.; Deininger D.; Mcginty K.; Stamos D.; Come J.; Steward M.. Inhibitors cMet and Uses Thereof. WO 200706497 A2, 2007.; b Li P.; Waal N.; Ronkin S.; Tang Q.; Lauffer D.. Substituted pyrazole inhibitors of cMet protein Kinases. WO 2010138663 A1, 2010.
- Zhang F.-J.; Vojkovsky T.; Huang P.; Liang C.; Do S. H.; Koenig M.; Cui J.. Preparation of triazolotriazines as cMet modulators for treating cancer. WO 2005010005A1, 2005.
- Amir M.; Kumar H.; Javed S. A. Synthesis and Pharmacological Evaluation of Condensed heterocyclic 6-substituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole derivatives of Naproxen. Bioorg. Med. Chem. Lett. 2007, 17 (16), 4504–4508. 10.1016/j.bmcl.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Wu B.; Basu S.; Meng S.; Wang X.; Hu M. Regioselective sulfation and glucuronidation of phenolics: insight into the structural basis. Curr. Drug Metab. 2011, 12 (9), 900–916. 10.2174/138920011797470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing L.; Klug-Mcleod J.; Rai B.; Lunney E. Kinase hinge binder scaffolds and hydrogen bond patterns. Bioorg. Med. Chem. 2015, 23, 6520–6527. 10.1016/j.bmc.2015.08.006. [DOI] [PubMed] [Google Scholar]
- In our early SAR studies, we found that the introduction of a methyl group at the methylene linker position helps reduce the rat clearance.
- Omori K.; Kotera J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- Scapin G.; Patel S. B.; Chung C.; Varnerin J. P.; Edmondson S. D.; Mastracchio A.; Parmee E. R.; Singh S. B.; Becker J. W.; Van der Ploeg L. H. T.; Tota M. R. Crystal structure of human phosphodiester 3B atomic basis for substrate and inhibitor specificity. Biochemistry 2004, 43, 6091–6100. 10.1021/bi049868i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.