

Abstract
The voltage-gated sodium channel Nav1.7 continues to be a high-profile target for the treatment of various pain afflictions due to its strong human genetic validation. While isoform selective molecules have been discovered and advanced into the clinic, to date, this target has yet to bear fruit in the form of marketed therapeutics for the treatment of pain. Lead optimization efforts over the past decade have focused on selectivity over Nav1.5 due to its link to cardiac side effects as well as the translation of preclinical efficacy to man. Inhibition of Nav1.6 was recently reported to yield potential respiratory side effects preclinically, and this finding necessitated a modified target selectivity profile. Herein, we report the continued optimization of a novel series of arylsulfonamide Nav1.7 inhibitors to afford improved selectivity over Nav1.6 while maintaining rodent oral bioavailability through the use of a novel multiparameter optimization (MPO) paradigm. We also report in vitro–in vivo correlations from Nav1.7 electrophysiology protocols to preclinical models of efficacy to assist in projecting clinical doses. These efforts produced inhibitors such as compound 19 with potency against Nav1.7, selectivity over Nav1.5 and Nav1.6, and efficacy in behavioral models of pain in rodents as well as inhibition of rhesus olfactory response indicative of target modulation.
Keywords: Nav1.7, Nav1.6, ion channel, pain, arylsulfonamide, selectivity, olfaction
The discovery and development of novel nonopioid pain medications has been highlighted as a critical area of unmet medical need in recent years with a number of high-profile reviews and popular articles covering the topic.1 The strong level of genetic validation for the voltage-gated sodium channel Nav1.7 as a novel, nonopioid target for pain relief has afforded an enormous amount of effort in the scientific community focused on the discovery of potent and selective inhibitors of this channel.2 Multimodality efforts across industry and academic groups have produced a number of clinical candidates advancing toward or into human studies.3 However, no selective Nav1.7 inhibitors have demonstrated clinical efficacy in chronic or acute human pain conditions to date.4 This clinical experience has left the scientific field to ponder whether the ligands studied owned the necessary profiles to adequately test the mechanism regarding target engagement or if the translation of the human genetic condition into therapeutics was possible via the current approaches.5 Increasing target inhibition relative to what has been currently achieved in clinical study would add value toward validating or invalidating this important target.
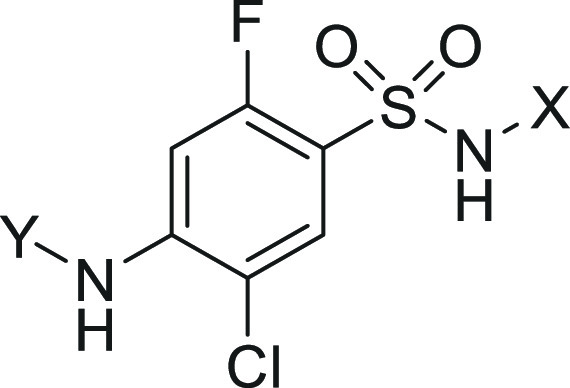
The difficulty with increasing degree of channel blockade relates to the requisite increase in selectivity profile over homologous Nav1.x channels necessary to safely interrogate this hypothesis. High levels of selectivity over Nav1.4 and Nav1.5 blockade, inhibition of which affords known skeletal muscle and cardiac toxicity, respectively, have been achieved within the arylsulfonamide class of Nav1.7 inhibitors represented by compounds 1, 2, and 3 as reported previously (Table 1).6 Lack of central inhibition of Nav1.1 and Nav1.2 has been achieved via limiting CNS penetration with this class of zwitterionic Nav1.7 blocker, affording some safety margin to established CNS-related toxicities.7 However, recent studies from our laboratory revealed significant respiratory inhibition findings potentially related to Nav1.6 blockade in the phrenic nerve, thus necessitating optimization of selectivity over this related channel.8 Notably, published compounds 1, 2, and 3 have limited to no selectivity over Nav1.6 (Table 1). To address the potential increase in channel blockade necessary for efficacy, our efforts focused on establishing in vitro–in vivo correlations (IVIVC) between Nav1.7 potency in different assay protocols to effects in a behavioral model of pain efficacy and a rhesus model of target modulation. The result of these efforts would give us a method to effectively assess increases in on-target potency in vitro related to pharmacodynamic end points. Our efforts also targeted establishing SAR in the arylsulfonamide series that significantly increased selectivity over Nav1.6 to widen our therapeutic index.
Table 1. Mouse Formalin Paw Efficacy and Mouse Nav1.7 Inhibition Relationships under Depolarized and Hyperpolarized Assay Protocols.

Estimated depolarized or hyperpolarized state potency as measured by PatchXpress in HEK293 cells stably expressing mouse Nav1.7, IC50 values are estimated from ≥3 cellular measurements at varying compound concentrations and standard deviation <50% of average value shown.
Mouse formalin paw test performed as described in ref (22).
Estimated hyperpolarized state potency as measured by Qube in HEK293 cells stably expressing human Nav1.7 or Nav1.6; IC50 values are estimated from ≥3 cellular measurements at varying compound concentrations; * = estimated value.
Compounds 1, 2, and 3 in Table 1 were previously reported examples of arylsulfonamide Nav1.7 blockers that display state-dependent channel inhibition. Previous potency values have been reported using a depolarized assay protocol, which placed the channel in a population of states that favor the inactivated state.9 All three compounds have very similar mouse potency in this assay protocol with IC50 values of 13, 18, and 15 nM, respectively. These compounds were studied in a number of assay protocols including a hyperpolarized assay protocol, which placed the channel in a population of states favoring the resting state. Under this protocol, the mouse potency shifted significantly from 17- and 26-fold for 1 and 3 to more than 270-fold in the case of compound 2. All three compounds were evaluated for efficacy in the mouse formalin paw test (mFPT) model, and the unbound concentration in plasma necessary for 90% reversal of the phase 2 effect was related to in vitro potency measures. While shifts were high (17–200-fold) relating phase 2 mFPT efficacy concentrations to depolarized potency, the ratios of mFPT efficacy to hyperpolarized potency were almost unity in all three cases. Notably, this ratio was able to distinguish potency differences between enantiomers 1 and 2 with similar depolarized potency values but disparate hyperpolarized potency values.10Figure 1 demonstrates the relationship between hyperpolarized Nav1.7 mouse potency and unbound concentration for 90% mFPT efficacy across a wide range of potency values within the arylsulfonamide series with an R2 of 0.68. The hyperpolarized assay protocol was also evaluated for Nav1.6, and this value was found to approximate the unbound concentrations necessary for measurable respiratory findings in rodents.8 With this data in hand, the Nav1.7 and Nav1.6 hyperpolarized assay protocols were utilized for further optimization of potency and selectivity.
Figure 1.

In vitro–in vivo correlation between mouse formalin paw test and mouse Nav1.7 potency under hyperpolarized assay protocol.
The hyperpolarized potency values for human Nav1.6 and Nav1.7 for compounds 1, 2, and 3 are displayed at the bottom of Table 1. All compounds have limited to inverted selectivity over Nav1.6, necessitating additional SAR to provide analogues with improved profiles for program advancement. While rodent bioavailability was not an issue for analogues 1–3, limited rat oral bioavailability in the arylsulfonamide class was an issue identified across a variety of analogues in zwitterionic space, potentially due to low permeability.11 An optimization approach was taken to establish SAR to improve selectivity over Nav1.6 while concomitantly exploring space enriched for significant rodent oral bioavailability. A range of physicochemical properties were evaluated to determine potential values that could increase enrichment for rat oral bioavailability within this zwitterionic space for utility in prospective design. The results of four properties with the best enrichment data are shown in Figure 2.
Figure 2.

Distribution of properties (MW, HBD, cLogD, and PSA; Y axis = compound count) of analogues evaluated in rat oral PK studies (%F ≥ 20%, green; %F < 20%, red) and property functions comprising custom oral bioavailability MPO paradigm.
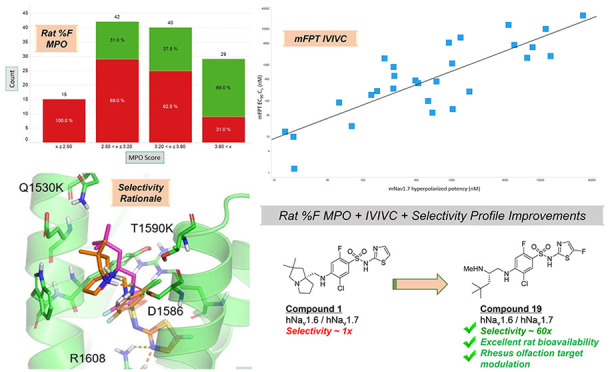
Oral bioavailability (defined as rat %F ≥ 20%: green bars ≥20% and red bars <20%) was generally enriched with reductions in molecular weight (MW), limiting H-bond donor (HBD) count, balancing cLogD in a desirable range, and lowering polar surface area (PSA). These findings were aggregated into a simple four-point scoring system, with each property given a score of 1. The scoring functions are depicted in Figure 2, with a monotonic declining function for MW and PSA, a step function for HBD, and a hump function for cLogD.12 Enrichment in rat bioavailability utilizing this multiparameter optimization (MPO) paradigm is shown in Figure 3. Increased enrichment was observed upon increasing MPO score, with molecules scoring below 2.5, all exhibiting <20% rat oral bioavailability. Increasing bioavailability enrichment was observed between 2.5 and 3.8, and significant enrichment was observed with scores >3.8, with almost 70% of compounds owning significant bioavailability. Compounds 1 and 3 were utilized as leads for optimization with efficacy in mFPT upon oral dosing, low to moderate permeability, and limited selectivity over Nav1.6. Analogue designs were prioritized for synthesis through MPO evaluation and breakthroughs in selectivity over Nav1.6, which could be rationalized via homology modeling (vide infra).
Figure 3.

Categorical enrichment in rat oral bioavailability as a function of MPO score (%F ≥ 20%, green; %F < 20%, red).
Starting with compound 3, opening the [3.3.0] ring system to a pyrrolidine scaffold afforded analogues 4 through 7, which significantly reduced Nav1.7 potency including inactive compounds 6 and 7 (Table 2). Compound 5 was screened for rat pharmacokinetics (PK) and had bioavailability of 14%, consistent with the enrichment for a compound with an MPO of 3.2. Addition of a phenyl ring to compound 5 yielded 8, with a 5-fold improvement in Nav1.7 potency, and importantly, moderate selectivity over Nav1.6. While this analogue had an MPO score of 4.0, bioavailability was only 7%, likely due to high rat clearance (CLint = 5830) potentially due to metabolism.13 The pyrrolidine ring was opened to afford 9 and 10, with reduced Nav1.7 potency but maintained moderate selectivity over Nav1.6 for compound 10. Surprisingly, removing the quaternary center affording 11 increased potency compared to 10 and increased selectivity over Nav1.6 to more than 100-fold. The MPO score of 4.0 for 11 translated to 25% oral bioavailability despite high clearance. Removal of the methyl group from the terminal amine to afford 12 lost all Nav1.7 potency, and addition of a methyl group (13) also reduced on-target potency by 4-fold. Despite the promising selectivity PK profile for compound 11, the 2-aminothiazole sulfonamide was still a risk for potential toxicity via known metabolic activation pathways, which were observed in rat and human microsomal incubations.14 A survey of thiazole replacements that would reduce the potential for metabolic activation revealed very limited options without compromising on target potency.15 Fluorothiazole 14 maintained the overall profile of 11 regarding potency, selectivity, and oral bioavailability while also having the potential to diminish oxidative metabolism on the thiazole ring.16 Thiadiazole 15 also had the potential to reduce oxidative metabolism as on the sulfonamide aryl ring, however, potency and MPO score were reduced through this design element. While 14 was considered a promising new lead, clearance was significantly higher compared with initial lead 3, potentially due to incorporation of the phenyl ring, which increased lipophilicity and offered additional opportunity for metabolism. To further optimize this lead, isosteric replacements for the phenyl ring in 14 were explored.
Table 2. Western SAR and Selectivity over Nav1.6.

Estimated hyperpolarized state potency as measured by Qube in HEK293 cells stably expressing human Nav1.7 or Nav1.6; IC50 values are estimated from ≥3 cellular measurements at varying compound concentrations and standard deviation <50% of average value shown.
Monolayer assay in MDCK cells.
Rat CLint = (84 × rat CL)/[rat fu × (84-rat CL)]; units mL/min/kg; IV, 0.05 mpk cassette dosing or 2 mpk single dosing in DMSO/PEG400/water (20/60/20); PO, 10 mpk in PEG400/Tween90/water (40/10/50).
Replacing the phenyl substituent with cycloalkyl motifs afforded analogues 16 and 17, which both reduced Nav1.7 potency 4- and 14-fold, respectively, as well as reduced MPO score (via reduced lipophilicity; cLogD < 1.5 for each) and permeability as shown in Table 3. Notably, cyclopropane 17 significantly lowered clearance compared to 14 or 16 and maintained excellent oral bioavailability, suggesting that increasing sp3 character could improve PK profiles in the series. Incorporation of a quaternary methyl group in 18 restored potency to within 2-fold of lead 14 and increased permeability. Opening the cyclopropane ring to afford the tert-butyl motif in 19 resulted in slightly improved potency compared to 14, high MPO score, moderate permeability and selectivity, and excellent rat bioavailability. Utilizing 19 as design inspiration, trimethylsilyl analogue 20 provided the most potent Nav1.7 blocker to date in the series with moderate selectivity over Nav1.6, reduced clearance, and good bioavailability.17 As a known phenyl isostere, the bicyclo[1.1.1]pentane was targeted, providing 21 with desired potency and selectivity, but increased clearance was observed; for this reason, 21 was not characterized further. Compound 19 provided improved selectivity over Nav1.6, permeability, and bioavailability compared to initial lead compound 3 and was selected as an exemplar in the series for further characterization.
Table 3. Phenyl Replacements and Selectivity over Nav1.6.


Estimated hyperpolarized state potency as measured by Qube in HEK293 cells stably expressing human Nav1.7 or Nav1.6; IC50 values are estimated from ≥3 cellular measurements at varying compound concentrations and standard deviation <50% of average value shown.
Monolayer assay in MDCK cells.
Rat CLint = (84 × rat CL)/[rat fu × (84-rat CL)]; units mL/min/kg; IV, 0.05 mpk cassette dosing or 2 mpk single dosing in DMSO/PEG400/water (20/60/20); PO, 10 mpk in PEG400/Tween90/water (40/10/50).
An interesting observation was made that compound 19 is essentially an open form of lead compound 1 that increased selectivity over Nav1.6. To explore this selectivity phenomenon, compounds 1 and 19 were docked to the binding site with Glide v7.7 of Schrödinger18 software using a known X-ray crystal structure (PDB 5EK0) containing the human Nav1.7 binding site for the arylsulfonamide class of inhibitors (top of S1–S4 in domain 4 of the channel).19 Also, a homology model of Nav1.6 VSD4 was built using Prime in Maestro v11.4.20 Then, a 100 ns molecular dynamics (MD) simulation was performed for both compounds 1 and 19 in Nav1.7 and Nav1.6 in the presence of explicit water and membrane using Desmond v5.2. Last, 400 snapshots of each simulation were used to estimate binding free energy using prime_mmgbsa in Maestro v11.4. The results were shown in Figure 4.
Figure 4.

Docked models and binding free energy calculations for 1 and 19 in hNav1.7 and hNav1.6.
Not surprisingly, several key interactions were maintained between the two compounds: interaction of the acidic sulfonamide with R1608, terminal basic amine interaction with D1586, and key π–π stacking interaction with Y1537 (not labeled, but central in Figure 4) with the aromatic core of the molecule. The MD simulation also provided slight differences in docked versions of 19 and 1, namely the tert-butyl motif in 19 reached higher into the binding pocket toward the top or loop region of S2, where several amino acid residue differences were observed between human Nav1.7 and Nav1.6 (Q1530K and T1590K shown; others removed for clarity). Steric clashes between 19 and the larger amino acid residues in Nav1.6 could underly the difference in selectivity. Relative binding free energy calculations were also performed for both 1 and 19 using the MM_GBSA method. Compound 19 was calculated to bind approximately 2 kcal/mol more strongly to Nav1.7 than Nav1.6, while compound 1 was predicted to have similar binding energy between the two channels, consistent with the selectivity profiles of both compounds. It should be noted that the standard deviations were overlapping these differences, however, this methodology might represent a prospective method to prioritize analogue designs based on potential for increased selectivity over Nav1.6.
Representative synthetic routes to the compounds above are described in Scheme 1. The synthesis of compound 11 began with a borane reduction of N-Boc-N-methyl-l-phenylalanine (22) followed by Mitsunobu with phthalimide to afford 23. Phthalimide removal afforded 24, which underwent an SNAr with core 1 and acidic deprotection to afford 11. The synthesis of 19 began with the bromination/phosphonate sequence from 25 to afford 26. Horner–Wadsworth–Emmons olefination with pivaldehyde followed by conjugate reduction afforded 27 in modest yield. Reduction with Raney nickel and hydrogen afforded 28, which was engaged in an SNAr reaction with core 2, which afforded 29 and 30 after chiral separation. Deprotection of 30 afforded the hydrochloride salt of 19.21 The identity and absolute stereochemistry of 22 was determined by X-ray crystallography.22
Scheme 1. Synthesis of Compounds 11 (A) and 19 (B).
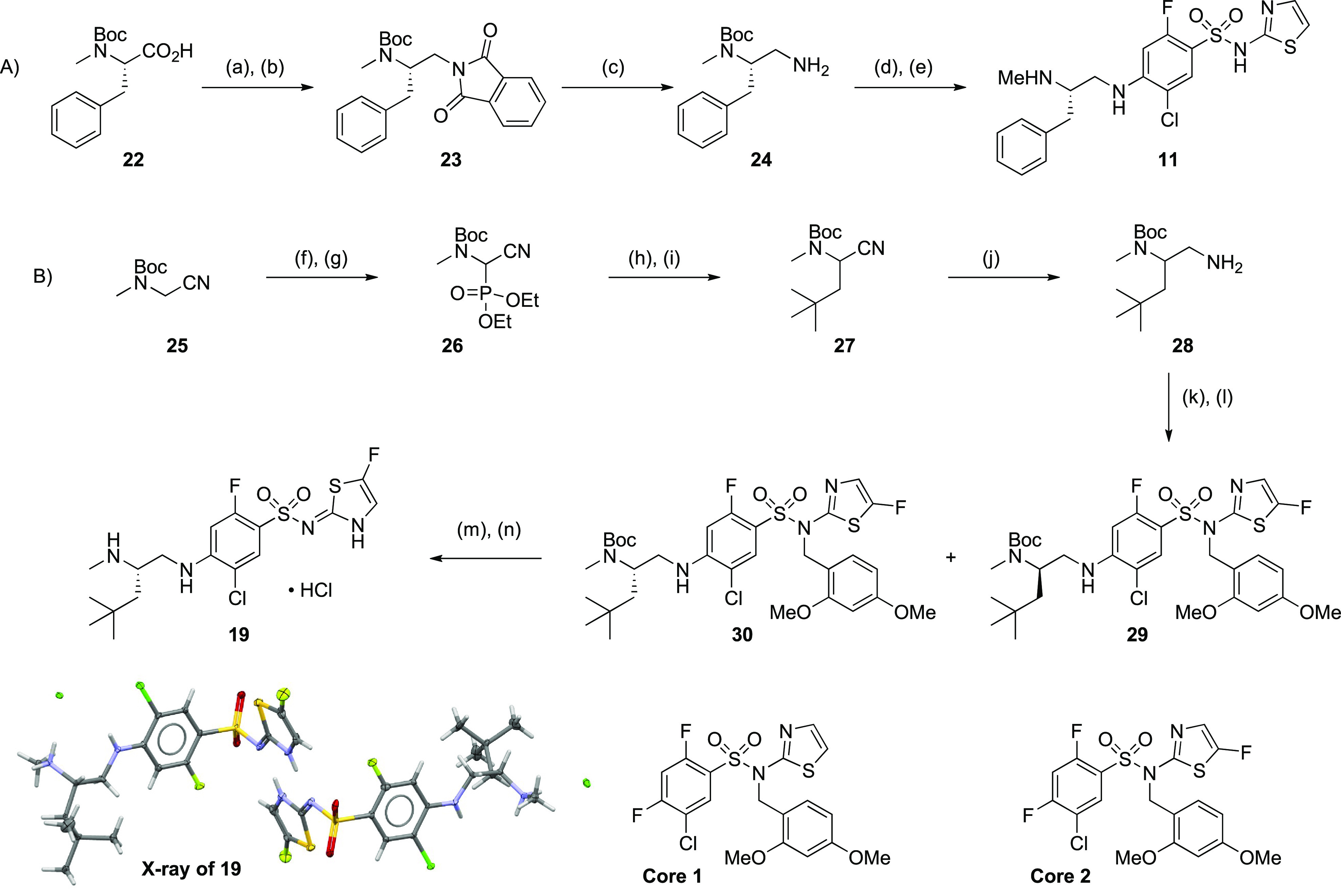
Reagents and reaction conditions: (A) (a) BH3·THF, THF, 0–25 °C, 0.5 h, 97%; (b) phthalimide, DTAD, PS–PPh3, THF, 25 °C, 0.5 h, 78%; (c) hydrazine hydrate, MeOH, reflux, 2 h, 97%; (d) core 1, Hunig’s base, NMP, 100 °C, microwave, 20 min, 77%; (e) TFA, DCM, 25 °C, 3 h, 79%. (B) (f) N-bromosuccinimide, CCl4, 80 °C, 2 h 96%; (g) triethylphosphite, THF, 75 °C, 16 h, 98%; (h) pivaldehyde, tetramethylguanidine, THF, −78 to 25 °C, 9 h, 57%; (i) NaBH4, MeOH, 25 °C, 2 h, 64%; (j) Raney nickel, H2, EtOH, 25 °C, 2 h, 63%; (k) core 2, TEA, DMF, 25 °C, 16 h, 83%; (l) SFC separation, Chiralpak AD-3 150 mm × 4.6 mm I.D.; mobile phase, 2-propanol (0.05% DEA) in CO2 from 5% to 40%; flow rate, 2.4 mL/min; wavelength, 220 nM; enantiomer 1 = 30 (Rt = 5.0 min), eantiomer 2 = 29 (Rt = 5.4 min); (m) TFA, DCM, 25 °C, 1 h; (n) reverse phase HPLC (acetonitrile/0.05% HCl in water), 61% over 2 steps.
Compound 19 underwent additional profiling prior to in vivo evaluation for pain efficacy and target modulation. In preparation for these studies, manual electrophysiology (EP) experiments were performed using the hyperpolarized assay protocol, and potency values were found to be within 3-fold of screening potency values with recapitulation of selectivity profiles over Nav1.6 (60-fold) and limited activity on Nav1.5 (Table 4). The potency of 19 was right-shifted on mouse Nav1.7 (IC = 8.8 μM) and less right-shifted in rhesus experiments (IC50 = 310 nM) compared to human potency consistent with previously evaluated compounds in the series. The physicochemical properties of 19 are within targeted MPO ranges that resulted in high rat bioavailability. This analogue was also a substrate for human and rat P-glycoprotein (Pgp) efflux transporters and, consistent with this finding, limited central penetration was observed upon oral dosing in mice, with a CSF-to-unbound plasma ratio of 0.02. This zwitterionic analogue had good kinetic solubility under low and neutral pH conditions, had limited CYP inhibition potency against three major isoforms, moderate PXR activation potential, and no significant activity in broad off-target profiling. Pharmacokinetics in dog revealed low clearance, a 6 h half-life, and moderate oral bioavailability. Rhesus bioavailability was negligible; however, pharmacokinetics were sufficient to evaluate this compound in our target modulation assay via IV administration. Finally, metabolic profiling in human, rat, dog, and rhesus hepatocytes revealed N-demethylation (metabolite potency; hNav1.7 IC50 = 3.5 μM) as the major route of metabolism with no metabolism of the thiazole motif. This finding suggested that fluorination of the thiazole reduced the potential metabolic liability of the aminothiazole sulfonamide.
Table 4. Additional Profiling of Compound 19d.
| property | compd 19 |
|---|---|
| human Nav1.7/1.6/1.5 IC50 [nM] (fold-selectivity)a | 87/5230 (60×)/ >34000 (>390×) |
| mouse/rhesus Nav1.7 IC50 (nM)a | 8800/310 |
| mouse/rhesus/human PPB | 94%/98%/98% |
| MW/HBD/log Dc/PSA | 453 g/mol/3/1.7/89 Å2 |
| Pgp (BA/AB ratio; h/r) | 4.9/>14 |
| mouse brain/plasma/CSF (μM)e | 0.1/4.9/0.005 |
| pKa (sulfonamide/amine) | 5.4/9.0 |
| solubility (pH 2, 7)b | 170, 148 μM |
| CYP IC50 (3A4/2C9/2D6, μM) | >50, 28, 20 |
| PXR EC50 (μM)/% max | 10/80% |
| Panlabs (97 assays) | no hits <10 μM |
| rat PK: CL, CLint, Vdss, T1/2, Ff | 29, 1670, 2.1, 2.5, 100% |
| dog PK: CL, CLint, Vdss, T1/2, Ff | 4.5, 200, 0.8, 6.0, 25% |
| rhesus PK: CL, CLint, Vdss, T1/2, Ff | 11, 610, 0.3, 1.3, 2% |
Estimated hyperpolarized protocol inhibition potency as measured by manual electrophysiology in HEK293 cells stably expressing human, rhesus, or mouse Nav1.7; IC50 values are estimated from ≥3 cellular measurements at varying compound concentrations and standard deviation <50% of average value shown.
MSD HPLC kinetic solubility assay.
MSD HPLC log D assay.
Monolayer assay in MDCK cells.
100 mpk PO, 1.25 h sampling.
CL(int) units = mL/min/kg, Vdss = L/kg, T1/2 = hours.
To estimate pain efficacy in a rodent behavioral pain assay, compound 19 was evaluated in the mFPT assay and administered orally to mice at three dose levels 15 min prior to formalin injection (Figure 5).23 Plasma sampling was performed 1.5 h after compound administration to coincide with Tmax values provided by satellite PK experiments. Limited reversal of the phase 1 (acute phase) effects of formalin administration were observed at all three dose levels. Significant and full reversal of these effects were observed in phase 2 (tonic phase) of the experiment, with unbound concentrations at EC90 of 360 nM. This represents a ratio of 0.04 (360/8800 nM) when normalizing for mouse potency. Although outliers of this magnitude were observed in Figure 1, this result was surprising given the correlation previously acquired, with most compounds demonstrating approximately a ratio unity comparing mouse hyperpolarized Nav1.7 potency and unbound concentration for full mFPT efficacy. To investigate further, the team decided to assess target modulation orthogonally in a model of rhesus olfaction to further assist with the determination of target concentration for human efficacy.
Figure 5.

Oral efficacy of compound 19 in mouse formalin paw test. Measurement of formalin-induced nociceptive behaviors in mice (C57BL/6 mice) following administration of vehicle (10% Tween 80; dose volume = 5 mL/kg) or rising oral doses of compound 19. Compound 19 was administered 15 min prior to formalin injection and plasma concentrations were evaluated 1.5 h postadministration. Data were analyzed using within-subject ANOVA to determine main effects and 1 sample t test to compare to vehicle (N = 8/group); **P > 0.001.
Loss-of-function mutations in Nav1.7 not only cause insensitivity to painful stimuli but also cause anosmia (lack of sense of smell) through inactivation of Nav1.7 in the olfactory bulb.24 A target modulation assay was developed using functional magnetic resonance imaging (fMRI) to measure odorant-induced olfaction in the olfactory bulb (OB) of anesthetized rhesus monkeys (nonhuman primates, or NHPs), previously reported from our laboratories as a potential translative clinical biomarker.25,26 Briefly, the study was performed with with NHPs each for vehicle control and for compound 19. The fMRI measurement paradigm for odorant-induced olfaction was 1 min for baseline, 1 min for odor stimulation, and 2 min for recovery (4 min total for each fMRI measurement). Thirty fMRI measurements were made for each NHP during a 2 h period. Compound 19 or vehicle delivery was started 1 h after acquisition of data and was continuously infused for the following hour. The strength of fMRI response was quantified by averaging the amplitudes of fMRI signals during the stimulation period. The inhibition on the olfaction was expressed as percentage inhibition of the strength of fMRI response in the control period. Time courses of fMRI signals in olfactory bulb upon a 40 s odor administration (isoamyl acetate: red bars) before/after the vehicle, and compound 19 delivery are shown in parts A and B of Figure 6, respectively. With the vehicle administration, the odorant-induced fMRI activations in the OB are similar before and after the administration, indicating that the vehicle has no significant effect on the olfaction. The odorant-induced fMRI activations in the OB after the administration of 19 were weaker than the fMRI responses before the administration, suggesting that the compound 19 inhibits the odorant-induced olfaction in the NHPs with statistical significance (Figure 6C).
Figure 6.

Target modulation efficacy of compound 19 in rhesus olfaction fMRI. Measurement of odorant-induced olfaction in the olfactory bulb of NHPs following administration of vehicle (30% Captisol, dose volume = ∼8 mL/animal) or compound 19 (19.2 mg/kg). (A) Time courses of fMRI signals which represent the odorant-induced olfaction in the OB of NHPs before and after vehicle administration (mean ± STD, n = 4). (B) Time courses of fMRI signals before and after compound 19 (C′22) administration (mean ± STD, n = 4). (C) Compare the inhibition by compound 22 and the effect from the vehicle (“*” statistical student t test, P < 0.012). Red bars in (A,B): odorant stimulations.
Compound 19 reduced olfactory response by approximately 35%, with an unbound plasma concentration of 1.5 μM and a ratio compared to rhesus potency of 4.8 (1500/310 nM). These results were more consistent with previous compounds studied that demonstrated that full efficacy in mFPT correlated to approximately a 40% reduction in rhesus olfactory response at unbound concentrations approximately equivalent to Nav1.7 potency.27 These disparate behavioral efficacy, mFPT overall correlations and target modulation data made it difficult to project an appropriate dose estimate for efficacy in clinical evaluation. The team chose to bracket human dose predictions using the three efficacy or target modulation data sets. Table 5 displays the estimated BID (“bis en die” or dosing twice per day) human dose projections via allometric scaling for 19 using the three end points to set trough concentration efficacy targets in man.28 While dose projections from behavioral efficacy and IVIVC studies were considered clinically feasible (40–960 mg BID), accessing the top end of the efficacy curve set by rhesus olfaction (4.6 g BID) would likely not be possible. This data suggested the need for significant additional optimization toward clinical candidate identification to confidently interrogate Nav1.7 as a therapeutic target.
Table 5. Estimated Human Dose Projections Based on Changing Ctrough Targets of Compound 19.
| Ctrough target | regimen | dose (mg) |
|---|---|---|
| 0.04× Nav1.7 IC50 | BID | 40 |
| 1× Nav1.7 IC50 | BID | 960 |
| 4.8× Nav1.7 IC50 | BID | 4600 |
In conclusion, a correlation was established between hyperpolarized Nav1.7 and Nav1.6 assay protocols to efficacy and off-target end points: behavioral pain efficacy and target modulation for Nav1.7 and respiratory side effects for Nav1.6. A novel MPO paradigm was developed to enrich design cycles for analogues with adequate rodent bioavailability concomitant with selectivity optimization. This strategy afforded analogues with a more than 100-fold selectivity over Nav1.6 with high rodent oral bioavailability. Molecular dynamics simulations and free energy binding calculations were performed, which provided rationale for selectivity differences between structurally related compounds. These tools could potentially be utilized for prospective design of analogues with increased potency, selectivity, and oral bioavailability. An exemplar compound (19) has demonstrated behavioral pain efficacy in mFPT and target modulation in a rhesus olfaction fMRI assay, however, dose predictions for leading analogues suggest the need for significant optimization toward candidate selection to adequately test the mechanism. Future efforts will focus on the identification of analogues with more aligned ratios in pain efficacy and target modulation compared to potency and reduction of predicted dose estimates via potency and PK optimization. These efforts will be reported in due course.
Acknowledgments
We thank Justin Newman for small-molecule X-ray support. We also thank Wilfredo Pinto for high-resolution mass spectrometry support.
Glossary
Abbreviations
- CNS
central nervous system
- SAR
structure–activity relationship
- HBD
hydrogen bond donor
- cLogD
calculated water/octanol partition coefficient at pH 7.4
- PK
pharmacokinetics
- PSA
polar surface area
- SNAr
nucleophilic aromatic substitution
- THF
tetrahydrofuran
- MeOH
methanol
- DTAD
di-tert-butylazodicarboxylate
- NMP
N-methylpyrrolidinone
- TFA
trifluoroacetic acid
- DCM
dichloromethane
- EtOH
ethanol
- TEA
triethylamine
- DMF
dimethylformamide
- PS
polymer-supported
- CLint
intrinsic clearance
- mFPT
mouse formalin paw test
- NHP
nonhuman primate
- MPO
multiparameter optimization
- fMRI
functional magnetic resonance imaging
- IVIVC
in vitro–in vivo correlation
- BID
“bis in die” or dosing twice per day
- PPB
plasma protein binding
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00218.
Author Present Address
△ (J.E.P.) GlaxoSmithKline, 1250 South Collegeville Road, Collegeville, Pennsylvania, 19426
Author Contributions
All authors have given approval to the final version of the manuscript. Conceived and designed experiments: A.J.R., M.E.L., J.E.P., D.W., F.Z., R.K., R.L.K., A.K.H., and C.S.B. Designed compounds and/or contributed to synthetic route design: A.J.R., M.E.L., J.E.P., M.J.K., III, T.G., X.P., X.W., and H.S. Analyzed data: R.L.K., Y.L., M.C., C.D., A.J., J.B., and D.W. Wrote the paper: A.J.R., D.W., and F.Z. Edited the paper: C.S.B. and M.E.L.
The authors declare the following competing financial interest(s): The authors are or were employees of Merck and Co., Inc. during the period of this work, and may hold stock in those companies.
Supplementary Material
References
- a Garland E. L. Treating chronic pain: the need for non-opioid options. Expert Rev. Clin. Pharmacol. 2014, 7, 545–550. 10.1586/17512433.2014.928587. [DOI] [PubMed] [Google Scholar]; b Dib-Hajj S. D.; Waxman S. G. Translational pain research: lessons from genetic and genomics. Sci. Transl. Med. 2014, 6, 249sr4. 10.1126/scitranslmed.3007017. [DOI] [PubMed] [Google Scholar]; c Chang D. S.; Raghavan R.; Christiansen S.; Cohen S. P. Curr. Opin. Anaesthesiol. 2015, 28, 379–397. 10.1097/ACO.0000000000000216. [DOI] [PubMed] [Google Scholar]; d Busserolles J.; Lolignier S.; Kerckhove N.; Bertin C.; Authier N.; Eschalier A. Replacement of current opioid drugs focusing on MOR-related strategies. Pharmacol. Ther. 2020, 210, 107519. 10.1016/j.pharmthera.2020.107519. [DOI] [PubMed] [Google Scholar]; e Time Staff Writers . The Opioid Diaries. Time Magazine, March 5, 2018, https://time.com/5170231/the-opioid-diaries/. [Google Scholar]
- a Dib-Hajj S. D.; Yang Y.; Black J. A.; Waxman S. G. The Na(V)1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 2013, 14, 49–62. 10.1038/nrn3404. [DOI] [PubMed] [Google Scholar]; b Dib-Hajj S. D.; Rush A. M.; Cummins T. R.; Waxman S. G. Mutation in the sodium channel 1.7 underlie inherited erythromelalgia. Drug Discovery Today: Dis. Mech. 2006, 3, 343–350. 10.1016/j.ddmec.2006.09.005. [DOI] [Google Scholar]; c Faber C. G.; Hoeijmakers J. G.; Ahn H. S.; Cheng X.; Han C.; Choi J. S.; Estacion M.; Lauria G.; Vanhoutte E. K.; Gerrits M. M.; Dib-Hajj S.; Drenth J. P.; Waxman S. G.; Merkies I. S. Gain of function NaV1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 2012, 71, 26–39. 10.1002/ana.22485. [DOI] [PubMed] [Google Scholar]; d Goldberg Y. P.; MacFarlane J.; MacDonald M. L.; Thompson J.; Dube M.-P.; Mattice M.; Fraser R.; Young C.; Hossain S.; Pape T.; Payne B.; Radomski C.; Donaldson G.; Ives E.; Cox J.; Younghusband H. B.; Green R.; Duff A.; Boltshauser E.; Grinspan G. A.; Dimon J. H.; Sibley B. G.; Andria G.; Toscano E.; Kerdraon J.; Bowsher D.; Pimstone S. N.; Samuels M. E.; Sherrington R.; Hayden M. R. Loss-of-function mutations in the NaV1.7 gene underlie congenital indifference to pain in multiple human populations. Clin. Genet. 2007, 71, 311–319. 10.1111/j.1399-0004.2007.00790.x. [DOI] [PubMed] [Google Scholar]; e Gingras J.; Smith S.; Matson D. J.; Johnson D.; Nye K.; Couture L.; Feric E.; Yin R.; Moyer B. D.; Peterson M. L.; Rottman J. B.; Beiler R. J.; Malmberg A. B.; McDonough S. I. Global NaV1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS One 2014, 9, e105895. 10.1371/journal.pone.0105895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a McKerrall S. J.; Sutherlin D. P. NaV1.7 inhibitors for the treatment of chronic pain. Bioorg. Med. Chem. Lett. 2018, 28, 3141–3149. 10.1016/j.bmcl.2018.08.007. [DOI] [PubMed] [Google Scholar]; b Swain N. A.; Batchelor D.; Beaudoin S.; Bechle B. M.; Bradley P. A.; Brown A. D.; Brown B.; Butcher K. J.; Butt R. P.; Chapman M. L.; Denton S.; Ellis D.; Galan S. R. G.; Gaulier S. M.; Greener B. S.; de Groot M. J.; Glossop M. S.; Gurrell I. K.; Hannam J.; Johnson M. S.; Lin Z.; Markworth C. J.; Marron B. E.; Millan D. S.; Nakagawa S.; Pike A.; Printzenhoff D.; Rawson D. J.; Ransley S. J.; Reister S. M.; Sasaki K.; Storer R. I.; Stupple P. A.; West C. W. Discovery of clinical candidate 4-[2-(5-amino-1Hpyrazol-4-yl)-4- chlorophenoxy]-5-chloro-2-fluoro-N-1,3-thiazol-4-ylbenzenesulfonamide (PF-05089771): design and optimization of diary ether aryl sulfonamides as selective inhibitors of NaV1.7. J. Med. Chem. 2017, 60, 7029–7042. 10.1021/acs.jmedchem.7b00598. [DOI] [PubMed] [Google Scholar]; c Graceffa R. F.; Boezio A. A.; Able J.; Altmann S.; Berry L. M.; Boezio C.; Butler J. R.; Chu-Moyer M.; Cooke M.; DiMauro E. F.; Dineen T. A.; Feric Bojic E.; Foti R. S.; Fremeau R. T. Jr.; Guzman-Perez A.; Gao H.; Gunaydin H.; Huang H.; Huang L.; Ilch C.; Jarosh M.; Kornecook T.; Kreiman C. R.; La D. S.; Ligutti J.; Milgram B. C.; Lin M. J.; Marx I. E.; Nguyen H. N.; Peterson E. A.; Rescourio G.; Roberts J.; Schenkel L.; Shimanovich R.; Sparling B. A.; Stellwagen J.; Taborn K.; Vaida K. R.; Wang J.; Yeoman J.; Yu V.; Zhu D.; Moyer B. D.; Weiss M. M. Sulfonamides as selective NaV1.7 inhibitors: Optimizing potency, pharmacokinetics, and metabolic properties to obtain atropisomeric quinolinone (AM-0466) that affords robust in vivo activity. J. Med. Chem. 2017, 60, 5990–6017. 10.1021/acs.jmedchem.6b01850. [DOI] [PubMed] [Google Scholar]; d Sun S.; Jia Q.; Zenova A. Y.; Wilson M. S.; Chowdhury S.; Focken T.; Li J.; Decker S.; Grimwood M. E.; Andrez J.-C.; Hemeon I.; Sheng T.; Chen C.-A.; White A.; Hackos D. H.; Deng L.; Bankar G.; Khakh K.; Chang E.; Kwan R.; Lin S.; Nelkenbrecher K.; Sellers B. D.; DiPasquale A. G.; Chang J.; Pang J.; Sojo L.; Lindgren A.; Waldbrook M.; Xie Z.; Young C.; Johnson J. P.; Robinette C. L.; Cohen C. J.; Safina B. S.; Sutherlin D. P.; Ortwine D. F.; Dehnhardt C. M. Identification of selective acyl sulfonamide-cycloalkylether inhibitors of the voltage-gated sodium channel (NaV) 1.7 with potent analgesic activity. J. Med. Chem. 2019, 62, 908–927. 10.1021/acs.jmedchem.8b01621. [DOI] [PubMed] [Google Scholar]; e Rothenberg M. E.; Tagen M.; Chang J. H.; Boyce-Rustay J.; Friesenhahn J.; Hackos D. H.; Hains A.; Sutherlin D.; Ward M.; Cho W. Safety, tolerability, and pharmacokinetics of GDC-0276, a novel Nav1.7 inhibitor, in a first-in-human, single- and multiple-dose study in healthy volunteers. Clin. Drug Invest. 2019, 39, 873–997. 10.1007/s40261-019-00807-3. [DOI] [PubMed] [Google Scholar]; f Jones H. M.; Butt R. P.; Webster R. W.; Gurrell I.; Dzygiel P.; Flanagan N.; Fraier D.; Hay T.; Iavarone L. E.; Luckwell J.; Pearce H.; Phipps A.; Segelbacher J.; Speed B.; Beaumont K. Clinical micro-dose studies to explore the human pharmacokinetics of four selective inhibitors of human NaV1.7 voltage-dependent sodium channels. Clin. Pharmacokinet. 2016, 55, 875–887. 10.1007/s40262-015-0365-0. [DOI] [PubMed] [Google Scholar]; g Kornecook T. J.; Yin R.; Altmann S.; Be X.; Berry V.; Ilch C. P.; Jarosh M.; Johnson D.; Lee J. H.; Lehto S. G.; Ligutti J.; Liu D.; Luther J.; Matson D.; Ortuno D.; Roberts J.; Taborn K.; Wang J.; Weiss M. M.; Yu V.; Zhu D. X. D.; Fremeau R. T. Jr.; Moyer B. D. Pharmacologic characterization of AMG8379, a potent and selective small molecule sulfonamide antagonist of the voltage-gated sodium channel NaV1.7. J. Pharmacol. Exp. Ther. 2017, 362, 146–16. 10.1124/jpet.116.239590. [DOI] [PubMed] [Google Scholar]; h Adams G. L.; Wang D.; Sun C. From spider toxins to therapeutics-developing selective Nav1.7 peptide inhibitors for pain. RSC Drug Disc. Series. 2017, 59, 411–437. 10.1039/9781788011532-00411. [DOI] [Google Scholar]; i Safina B. S.; McKerrall S. J.; Sun S.; Chen C.-A.; Chowdhury S.; Jia Q.; Li J.; Zenova A. Y.; Andrez J.-C.; Bankar G.; Bergeron P.; Chang J. H.; Chang E.; Chen J.; Dean R.; Decker S. M.; DiPasquale A.; Focken T.; Hemeon I.; Khakh K.; Kim A.; Kwan R.; Lindgren A.; Lin S.; Maher J.; Mezeyova J.; Misner D.; Nelkenbrecher K.; Pang J.; Reese R.; Shields S. D.; Sojo L.; Sheng T.; Verschoof H.; Waldbrook M.; Wilson M. S.; Xie Z.; Young C.; Zabka T. S.; Hackos D. H.; Ortwine D. F.; White A. D.; Johnson J. P. Jr.; Robinette C. L.; Dehnhardt C. M.; Cohen C. J.; Sutherlin D. P. Discovery of acyl-sulfonamide inhibitors GDC-0276 and GDC-310. J. Med. Chem. 2021, 64, 2953–2966. 10.1021/acs.jmedchem.1c00049. [DOI] [PubMed] [Google Scholar]
- a McDonnell A.; Collins S.; Ali Z.; Iavarone L.; Surujbally R.; Kirby S.; Butt R. P. Efficacy of the NaV1.7 blocker PF-05089771 in a randomized, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. Pain 2018, 159, 1465–1476. 10.1097/j.pain.0000000000001227. [DOI] [PubMed] [Google Scholar]; c Siebenga P.; van Amerongen G.; Hay J. L.; McDonnell A.; Gorman D.; Butt R.; Groeneveld G. J. Lack of detection of the analgesic properties of PF-05089771, a selective Nav1.7 inhibitor, using a battery of pain models in healthy subjects. Clin. Transl. Sci. 2020, 13, 318–324. 10.1111/cts.12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy J. V.; Pajouhesh H.; Beckley J. T.; Delwig A.; Du Bois J.; Hunter J. C. Challenges and Opportunities for Therapeutics Targeting the Voltage-Gated Sodium Channel Isoform NaV1.7. J. Med. Chem. 2019, 62, 8695–8710. 10.1021/acs.jmedchem.8b01906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For characterization of the compounds and a general discussion of Nav1.x pharmacology, see:Roecker A. J.; Egbertson M.; Jones K. L. G.; Gomez R.; Kraus R. L.; Li Y.; Koser A. J.; Urban M. O.; Klein R.; Clements M.; Panigel J.; Daley C.; Wang J.; Finger E. N.; Majercak J.; Santarelli V.; Gregan I.; Cato M.; Filzen T.; Jovanovska A.; Wang Y. H.; Wang D.; Joyce L. A.; Sherer E. C.; Peng X.; Wang X.; Sun H.; Coleman P. J.; Houghton A. K.; Layton M. E. Discovery of selective, orally bioavailable, N-linked arylsulfonamide NaV1.7 inhibitors with pain efficacy in mice. Bioorg. Med. Chem. Lett. 2017, 27, 2087–2093. 10.1016/j.bmcl.2017.03.085. [DOI] [PubMed] [Google Scholar]
- a Eijkelkamp N.; Linley J. E.; Baker M. D.; Minett M. S.; Cregg R.; Werdehausen R.; Rugiero F.; Wood J. N. Neurological perspectives on voltage-gated sodium channels. Brain 2012, 135, 2585–2612. 10.1093/brain/aws225. [DOI] [PMC free article] [PubMed] [Google Scholar]; b de Lera Ruiz M.; Kraus R. L. Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J. Med. Chem. 2015, 58 (18), 7093–7118. 10.1021/jm501981g. [DOI] [PubMed] [Google Scholar]
- Klein R. M.; Regan H.; Regan C. P.; Kraus R. L.; Li Y.; Daley C.; Clements M. K.; Greshock T. J.; Roecker A. J.; Pero J. E.; Layton M. E.; Burgey C. B.; Henze D. A.; Houghton A. K.. Association of respiratory failure with inhibition of Nav1.6 in the phrenic nerve. J. Exp. Ther. Pharm. 2020, submitted [DOI] [PMC free article] [PubMed]
- See Experimental Section for details on electrophysiology assay protocols.
- Compound 2 required an estimated 90% inhibition concentration due to the inability to achieve 90% reversal of nociceptive behaviors in phase 2 of the mFPT study. 90% inhibition was modelled from a maximum inhibition of 66% at the oral dose of 30 mg/kg. While Nav1.7 is assumed to be the isoform responsible for mFPT efficacy, the contribution of Nav1.6 inhibition cannot be ruled out. For Figure 1, the equation for a straight line fit is y = 0.7x + 436, very close to a y = x fit for IVIVC purposes.
- Pero J. E.; Rossi M. A.; Lehman H. D. G. F.; Kelly M. J.; Mulhearn J. J.; Wolkenberg S. E.; Cato M. J.; Clements M. K.; Daley C. J.; Filzen T.; Finger E. N.; Gregan Y.; Henze D. A.; Jovanovska A.; Klein R.; Kraus R. L.; Li Y.; Liang A.; Majercak J. M.; Panigel J.; Urban M. O.; Wang J.; Wang Y. H.; Houghton A. K.; Layton M. E. Benzoxazolinone aryl sulfonamides as potent, selective NaV1.7 inhibitors with in vivo efficacy in a preclinical pain model. Bioorg. Med. Chem. Lett. 2017, 27, 2683–2688. 10.1016/j.bmcl.2017.04.040. [DOI] [PubMed] [Google Scholar]
- a Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Central nervous system multiparameter optimization desirability: Application in drug discovery. ACS Chem. Neurosci. 2016, 7, 767–775. 10.1021/acschemneuro.6b00029. [DOI] [PubMed] [Google Scholar]; b Gunaydin H. Probabilistic approach to generating MPOs and its application as a scoring function for CNS drugs. ACS Med. Chem. Lett. 2016, 7, 89–93. 10.1021/acsmedchemlett.5b00390. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bagal S. K.; Bungay P. J. Minimizing drug exposure in the CNS while maintaining good oral absorption. ACS Med. Chem. Lett. 2012, 3, 948–950. 10.1021/ml300378n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- While the MPO paradigm was utilized as a prospective prioritization method, pharmacokinetic parameters were also used to rationalize poor rat bioavailability from a mechanistic perspective.
- Obach R. S.; Kalgutkar A. S.; Ryder T. F.; Walker G. S. In vitro metabolism and covalent binding of enol-carboxamide derivatives and anti-inflammatory agents sudoxicam and meloxicam: insights into the hepatotoxicity of sudoxicam. Chem. Res. Toxicol. 2008, 21, 1890–1899. 10.1021/tx800185b. [DOI] [PubMed] [Google Scholar]
- The published 5- and 6-membered heterocyclic sulfonamides in the Nav1.7 literature were all surveyed on the compound 11 scaffold, and the only motifs with IC50 values < 2 μM are shown in this publication.
- While fluorine can often deactivate aromatic rings towards metabolism, this is not always the case:Pan Y. The dark side of fluorine. ACS Med. Chem. Lett. 2019, 10, 1016–1019. 10.1021/acsmedchemlett.9b00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- While compound 20 was initially more potent than compound 19, manual electrophysiology experiments determined that both compounds were equipotent inhibitors of human Nav1.7. This supported the decision to advance compound 19 as an exemplar for further characterization.
- All programs in this paragraph provided by Schrodinger, LLC, New York, 2017.
- Ahuja S.; Mukund S.; Deng L.; Khakh K.; Chang E.; Ho H.; Shriver S.; Young C.; Lin S.; Johnson J. P. Jr.; Wu P.; Li J.; Coons M.; Tam C.; Brillantes B.; Sampang H.; Mortara K.; Bowman K. K.; Clark K. R.; Estevez A.; Xie Z.; Verschoof H.; Grimwood M.; Dehnhardt C.; Andrez J.-C.; Focken T.; Sutherlin D. P.; Safina B. S.; Starovasnik M. A.; Ortwine D. F.; Franke Y.; Cohen C. J.; Hackos D. H.; Koth C. M.; Payandeh J. Structural basis of NaV1.7 inhibition by an isoform-selective small molecule antagonist. Science 2015, 350, aac5464. 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- Wang D.; Roecker A. J.; Klein R.; Clements M.; Daley C.; Layton M. E.; Greshock T. J.; Kraus R. L.; Wang J.; Majercak J.; Santarelli V.; Gregan I.; Houghton A. K.; Coleman P. J.; Burgey C. S.. Application of binding site mapping, homology modeling and induced docking to the design of potent and selective Nav1.7 inhibitors. Unpublished manuscript.
- For additional synthetic details, please seeRoecker A. J.; Layton M. E.; Greshock T. J.; Pero J. E.; Kelly M. J. III; Zhang T.. N1-phenylpropane-1,2-diamine compounds with selective activity in voltage-gated sodium channels. World Patent WO2017165204, 75 pp.
- The coordinates of the X-ray structural data have been submitted to the Cambridge Structural Database with the deposition number CCDC 2045967.
- Mogil J. S., Wilson S. G., Wan Y.. Assessing nociception in murine subjects. In Methods in Pain Research; Kruger L, Ed.; CRC Press, 2001, Chapter 2. [Google Scholar]
- Heimann D.; Lotsch J.; Hummel T.; Doehring A.; Oertel B. G. Linkage between increased nociception and olfaction via SCN9A haplotype. PLoS One 2013, 8, e68654. 10.1371/journal.pone.0068654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F.; Holahan M. A.; Houghton A. K.; Hargreaves R.; Evelhoch J. L.; Winkelmann C. T.; Williams D. S. Functional imaging of olfaction by CBV fMRI in monkeys: insight into the role of olfactory bulb in habituation. NeuroImage 2015, 106, 364–372. 10.1016/j.neuroimage.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Zhao F.; Holahan M. A.; Wang X.; Uslaner J. M.; Houghton A. K.; Evelhoch J. L.; Winkelmann C. T.; Hines C. D. G. fMRI study of the role of glutamate NMDA receptor in the olfactory processing in monkeys. PLoS One 2018, 13, e0198395. 10.1371/journal.pone.0198395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard J. E.; Pall P.; Vardigan J.; Zhao F.; Holahan M. A.; Kraus R.; Li Y.; Henze D.; Houghton A.; Burgey C. S.; Gibson C. Application of pharmacokinetic-pharmacodynamic modeling to inform translation of in vitro Nav1.7 inhibition to in vivo pharmacological response in non-human primate. Pharm. Res. 2020, 37, 181. 10.1007/s11095-020-02914-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The dose projections were performed using a combination of dog and rat allometry varying the Ctrough concentrations to match the mFPT, IVIVC average, and rhesus olfaction efficacy end points.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.