

Abstract

A series of novel 1,3-oxazole sulfonamides were constructed and screened for their potential to inhibit cancer cell growth. These compounds were evaluated against the full NCI-60 human tumor cell lines, with the majority exhibiting promising overall growth inhibitory properties. They displayed high specificity within the panel of leukemia cell lines versus all other lines tested. When examined in the dose–response assay, GI50 values fell within the low micromolar to nanomolar ranges. 1,3-Oxazole sulfonamide 16 displayed the best average growth inhibition, whereas the 2-chloro-5-methylphenyl and 1-naphthyl substituents on the sulfonamide nitrogen proved to be the most potent leukemia inhibitors with mean GI50 values of 48.8 and 44.7 nM, respectively. In vitro tubulin polymerization experiments revealed that this class of compounds effectively binds to tubulin and induces the depolymerization of microtubules within cells.
Keywords: Small molecules, anticancer drug leads, sulfonamides, heterocycles, oxazoles
The International Agency for Research on Cancer (IARC) estimated that in 2018 there were 17.0 million new cancer cases and 9.5 million deaths due to cancer globally.1 By 2040, the worldwide incidence is forecasted to increase to 27.5 million new cases, with 16.3 million of those cases expected to be fatal. Historically, secondary metabolites isolated from plant, marine, and microbial organisms have proven to be a rich source of compounds with potent anticancer properties, with paclitaxel and vinblastine being two such compounds that have found their way into the clinic. Compounds such as these, however, tend to be plagued by having high molecular weights and poor water solubility, which ultimately restricts their bioavailability and clinical effectiveness. Indeed, clinical efficacy tends to be a major driving force for drug attrition in the oncology world, where the rate of successful drug development is <5%.2 This leaves the door open for synthetic small molecules to make their way onto the scene and pave their own path through the drug discovery process.
Nitrogen-containing heterocycles have long since solidified their place in the pharmaceutical world. Specifically, the 1,3-oxazole motif can be found in a wide variety of drugs and biologically active secondary metabolites, with several having potent anticancer properties such as disorazole C11 and compounds 2–5 (Figure 1).3−13 Similarly, since the discovery of sulfanilamide in the early 1900s, the sulfonamide functional group has found utility in a vast number of small molecules for the treatment of various clinical conditions.14−16 It has been demonstrated that sulfonamide derivatives can affect several important oncogenic signaling pathways like protein kinases, platelet-derived growth factors, and c-kit proteins. The discovery of the potent anticancer 2-anilinopyridine sulfonamide E7010 in the early 1990s solidified this class of compound as a major player in the anticancer agents arena.17 E7010 works by interacting with tubulin via colchicine binding sites and, in effect, inhibiting tubulin polymerization.
Figure 1.

Representative anticancer compounds containing the 1,3-oxazole moiety.
Finally, the incorporation of the cyclopropyl ring into potential drug candidates has proven to be quite fruitful in producing compounds that possess specific therapeutic properties.18 Some of the unique pharmaceutical properties the cyclopropyl fragment can effect are metabolic stability, lipophilicity, bioavailability, receptor subtype selectivity, and brain permeability to name a few. Stockwell and coworkers were the first to report the anticancer activity of a novel 1,3-oxazole sulfonamide bearing a cyclopropyl ring along with its tubulin inhibitory properties.19 Shortly thereafter, Meng and coworkers explored small libraries of similar compounds that featured 1-sulfonyl indoline, benzoxazepine, benzodiazepine, benzothiazepine, and cyclopropyl moieties.20,21 Upon further survey of the literature, however, we were surprised to find that, in general, studies involving 1,3-oxazole sulfonamides remained fairly underrepresented. Our group’s interest in biologically active nitrogen-containing heterocycles, sulfonamides, and strained ring systems in drug design led us to explore the anticancer properties of a set of novel 1,3-oxazole sulfonamide analogues. Our goals were to leverage inexpensive and commercially available building blocks along with simple reaction sequences that minimized cumbersome protecting group steps and that could easily be carried out in the undergraduate research setting. Herein we describe the synthesis, antiproliferative activity, and tubulin polymerization inhibitor properties of a small library of novel 1,3-oxazole sulfonamides.
The synthetic route to 1,3-oxazole sulfonamides (12–19) is shown in Scheme 1. Bromination of acetophenone 6 occurred smoothly to give α-bromoketone 7 in high yield, which was then converted to primary amine salt 8 via the Delépine reaction. Subsequently, 8 was reacted with cyclopropylcarbonyl chloride to give amide 9 and then cyclized with phosphoryl chloride to cleanly give 1,3-oxazole 10. With the desired 1,3-oxazole in hand, the reaction with chlorosulfonic acid and thionyl chloride gave the pivotal benzenesulfonyl chloride 11 needed for the formation of the final sulfonamide linkages and the construction of the initial compound library.
Scheme 1. Synthesis of Sulfonyl Chloride 11 from Acetophenone.

Reagents and conditions: (a) Br2, MTBE, room temperature 85%; (b) i. Hexamethylenetetramine, CHCl3, room temperature; ii. Conc. HCl, EtOH, room temperature, 94%; (c) Cyclopropanecarbonyl chloride, Et3N, CH2Cl2, 0 °C to room temperature, 94%; (d) POCl3, reflux, 82%; (e) HSO3Cl, SOCl2, 0 to 60 °C; 80%.
It is worth mentioning that the benzenesulfonyl chloride derivative 11 was prepared in only five synthetic steps with cheap and commercially available starting materials and with relatively mild reaction conditions. No protecting group chemistry was needed, and only two of the intermediate products required purification. A simple recrystallization of the cyclopropyl amide 9 and chromatography purification of the 1,3-oxazole 10, respectively, provided these products in high yield, whereas intermediates 7, 8, and 11 were suitable to be used directly after aqueous workup. For the construction of the final sulfonamide linkage, we surveyed various reaction conditions and found that the slow addition of the sulfonyl chloride to a solution of the amine in pyridine and chloroform at 0 °C provided the desired 1,3-oxazole sulfonamides in the best yields. We began our initial investigations by synthesizing eight sulfonamides 12–19 (Table 1) using various amine or aniline derivatives and screening these compounds for their anticancer activity.
Table 1. Structures and Yields of Initial Compound Library Constructed from Sulfonyl Chloride 11a.

Reagents and conditions: amine or aniline derivative (1.0 equiv), pyridine (2.5 equiv), CHCl3 (0.20 M), 0 °C to room temperature, 58–79%.
In 1990, the National Cancer Institute (NCI) implemented a screening assay that utilizes 60 different human cancer cell lines for the identification and characterization of novel compounds exhibiting growth inhibition or killing potential of tumor cell lines.22−24 The represented cell lines include leukemia, melanoma, cancers of the lung, colon, brain, ovary, breast, and prostate, and kidney cancers. All compounds submitted for NCI-60 analysis are initially tested at a single high dose (5–10 μM). Compounds that comply with the predetermined threshold inhibition criteria in a minimum number of cell lines are allowed to progress to the full five-dose assay. The results of the high dose screening are presented in terms of the growth percent (GP), which is the growth of the treated culture relative to the untreated culture; for example, a GP of 70% represents 30% growth inhibition. GP values between 0 and 99 represent compounds with cytostatic properties, and GP values between −100 and 0 represent compounds with cytotoxic activity. Table 2 shows average GP data across all cell lines along with data for selected cell lines for each compound in the initial library, and the complete NCI-60 data for each compound can be found in the Supporting Information. From the initial screening assays, we noticed that halogenated anilines (compounds 12, 14, 16, and 18) showed excellent growth inhibitory properties as broad spectrum antiproliferatives. Compound 16 bearing 4-chloro and 3-trifluoromethyl substituents was especially active, with an impressive average GP of 11.4. Of the cell lines tested, 12, 14, 16, and 18 seemed to have specificity for leukemia cell lines, with 16 being the most active, with an average GP of 6.5 (Supporting Information). Compound 17 bearing a 2-naphthylsulfonamide group also exhibited good overall growth inhibition with an average GP of 28.9.
Table 2. Preliminary NCI-60 One-Dose Cell Growth Percentages.
| growth
of selected cell lines (%) |
|||||||
|---|---|---|---|---|---|---|---|
| compound | mean GP (%) | SR | MOLT-4 | COLO 205 | TK-10 | SK-OV-3 | NCI/ADR-RES |
| 12 | 28.1 | 11.6 | 12.7 | 20.2 | 32.5 | 39.7 | –0.9 |
| 13 | 94.2 | 91.3 | 95.3 | 95.0 | 101.0 | 111.7 | 93.5 |
| 14 | 24.3 | 9.9 | 14.2 | 9.0 | 54.2 | 30.2 | 4.4 |
| 15 | 92.2 | 89.3 | 82.5 | 94.5 | 103.5 | 100.8 | 92.3 |
| 16 | 11.4 | 3.5 | 5.2 | 2.7 | 3.0 | 0.8 | 2.9 |
| 17 | 28.9 | 16.5 | 20.2 | 1.5 | 61.9 | 32.7 | 9.7 |
| 18 | 26.9 | 12.7 | 15.8 | 23.9 | 64.1 | 35.5 | –2.2 |
| 19 | 94.1 | 80.3 | 94.1 | 94.4 | 99.1 | 104.7 | 94.1 |
Cytotoxic data obtained from a preliminary NCI-60 screening assay. Mean GP represents the average cell growth of 60 cancer cell lines treated with compounds 12–19 at 10 μM concentration.
Given their initial activity, compounds 14, 16, 17, and 18 progressed to the dose–response screening assay to further evaluate their growth inhibitory properties against the 60-cell panel at five concentrations (ranging from 0.01 to 100 μM). In the five-dose experiment, three dose–response parameters (GI50, TGI, and LC50) are calculated for each compound. GI50 is the drug concentration resulting in a 50% reduction of the net protein increase, TGI is the drug concentration resulting in total growth inhibition, which signifies cytostatic effects, and finally, LC50 is the drug concentration resulting in a 50% reduction in the measured protein at the end of the drug treatment as compared with that at the beginning and is used to signify cytotoxic effects. We were delighted to find that our compounds exhibited GI50 values in the micromolar to submicromolar range across many of the cell lines tested (Table 3). Of the four compounds tested, 16 possessed the best overall growth inhibition with an average GI50 of 0.655 μM and many individual GI50s in the nanomolar concentration range. 14 and 18 bearing 4-chloro and 4-bromosulfonamide groups, respectively, had GI50 values in either the single digit or submicromolar ranges in virtually all cell lines tested. Although 2-naphthylamine is a known carcinogen, we found it interesting that 17 showed good growth inhibitory properties without being overtly toxic to the cell lines. Over half of the cell lines tested required the concentration of 17 to be >100 μM to exert cytotoxic effects. Once again, we noticed that leukemia cell lines were particularly sensitive to the sulfonamide derivatives tested. LC50 values for 14, 16, 17, and 18 were found to be >100 μM in all leukemia cell assays, suggesting that these substrates might be more cytostatic in nature versus cytotoxic. Armed with the structure–activity (SAR) data generated from the screening of our initial compound library, we concluded that compounds containing an aniline moiety were much more active than their alkyl amine counterparts. On the basis of this observation, we elected to synthesize a second-generation library with the goal of exploring how the substituent identity and placement along the distal aromatic ring would affect activity.
Table 3. Average GI50 and LC50 Data Obtained for Compounds 14, 16, 17, and 18 after Testing in the Full NCI-60 Dose–Response Assaya.
| compound | mean GI50 | mean LC50 |
|---|---|---|
| 14 | 1.43 | 57.43 |
| 16 | 0.655 | 55.40 |
| 17 | 1.84 | 78.58 |
| 18 | 2.75 | 53.96 |
Values shown are in μM.
The library was synthesized from intermediate sulfonyl chloride 11 following the general reaction in Table 1, and the structures of the compounds from the second-generation library are shown in Figure 2. Given that virtually all of the most active compounds from the first-generation library possessed a halogen, we included a number of halogenated aniline derivatives in which halogen identity and placement on the ring were varied. We also elected to include aniline derivatives with electron-donating and electron-withdrawing substituents. Compounds in our second-generation library were submitted for NCI-60 testing in the same manner as before, and the average growth percentage data are summarized in Table 4.
Figure 2.
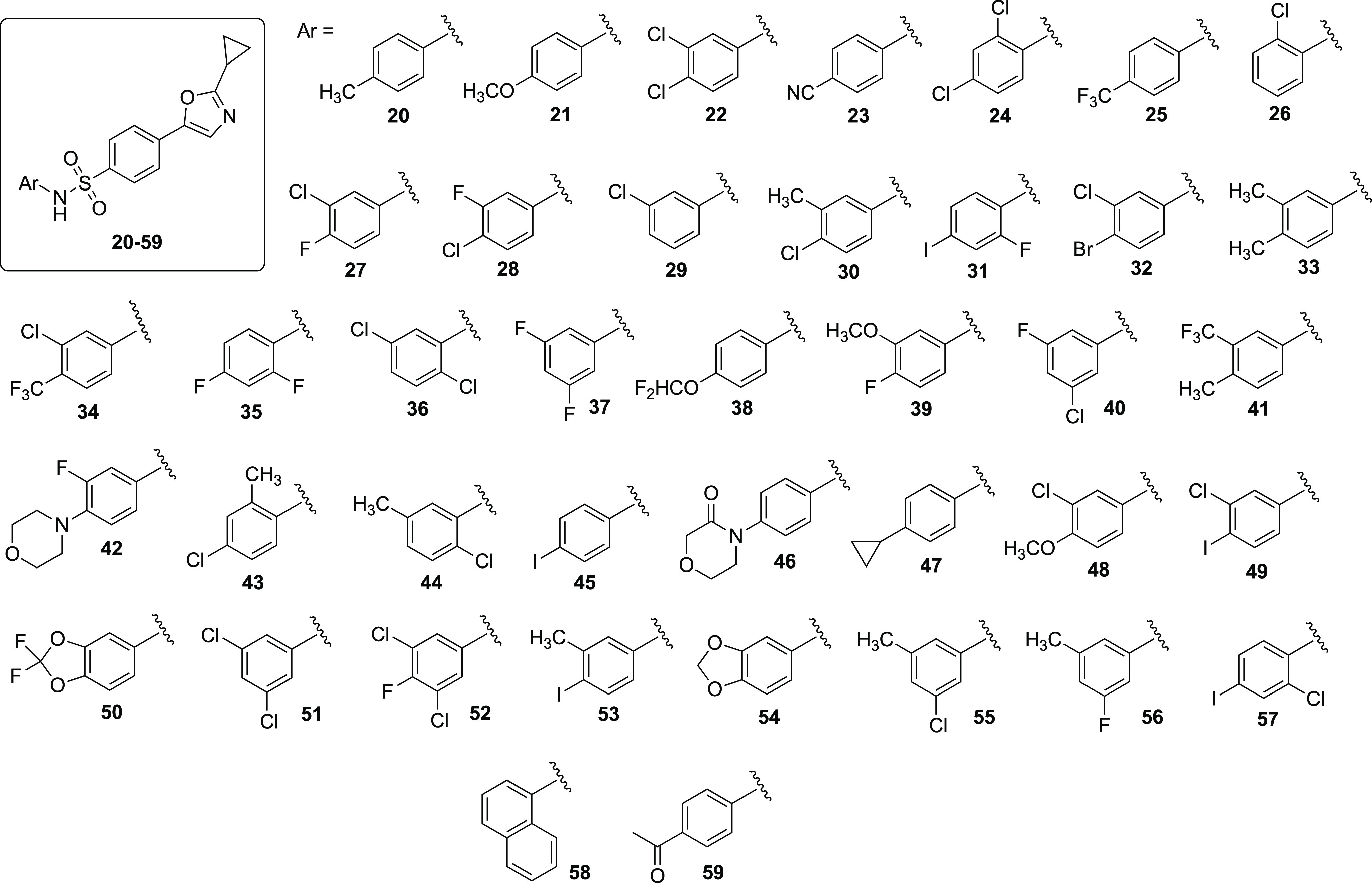
Structures of the second-generation library of aryl sulfonamide 1,3-oxazoles. All compounds were prepared from sulfonyl chloride 11 according to the procedure outlined in Table 1.
Table 4. Preliminary NCI-60 One-Dose Cell Growth Percentages of the Second-Generation 1,3-Oxazole Sulfonamide Library.
| compound | mean GP (%) | compound | mean GP (%) |
|---|---|---|---|
| 20 | 33.1 | 40 | 30.3 |
| 21 | 56.1 | 41 | 60.5 |
| 22 | 25.3 | 42 | 92.2 |
| 23 | 95.7 | 43 | 24.0 |
| 24 | 25.0 | 44 | 25.3 |
| 25 | 94.5 | 45 | 57.3 |
| 26 | 27.4 | 46 | 96.7 |
| 27 | 23.0 | 47 | 63.4 |
| 28 | 23.8 | 48 | 32.58 |
| 29 | 24.2 | 49 | 29.8 |
| 30 | 21.7 | 50 | 41.7 |
| 31 | 49.8 | 51 | 36.9 |
| 32 | 21.5 | 52 | 37.1 |
| 33 | 22.8 | 53 | 35.9 |
| 34 | 77.1 | 54 | 35.8 |
| 35 | 29.1 | 55 | 31.6 |
| 36 | 34.6 | 56 | 28.1 |
| 37 | 35.4 | 57 | 39.5 |
| 38 | 63.1 | 58 | 28.6 |
| 39 | 25.9 | 59 | 93.5 |
Cytotoxic data obtained from a preliminary NCI-60 screening assay. Mean GP represents the average cell growth of 60 cancer cell lines treated with compounds 20–59 at 10 μM concentration.
Many of the compounds in our second-generation library exhibited good overall growth inhibition, albeit none were as effective as compound 16. In general, we observed that halogenated 1,3-oxazole sulfonamides seemed to be the most active substrates among compounds tested in the second library. The exceptions were para-methyl sulfonamide 20, 3,4-dimethyl sulfonamide 33, and 1-naphthylsulfonamide derivative 58. Compound 58 piqued our interest due to the activity of its 2-naphthyl counterpart 17, and we observed almost identical mean growth inhibition along with similar inhibition profiles within individual cell lines. The para-methyl substrate 20 showed good overall growth inhibition; however, when a second methyl group was added to the three-position of the aniline ring of 33, even better inhibitory activity was observed. Compounds 23 and 25 bearing electron-withdrawing cyano and trifluoromethyl groups, respectively, were found to be inactive with an average growth inhibition of <10%. Compounds that contained electron-donating groups were observed to give mixed results. The 4-methoxy 21 and 3,4-methylenedioxy 54 substrates had modest growth inhibitory properties, but 46, which contained a morpholin-3-one group, was inactive altogether.
Given the activity of 33, we were surprised that growth inhibition greatly decreased by connecting the two methyl groups to incorporate a second cyclopropyl ring on 47. With respect to the halogenated derivatives, our goal was to explore how the identity of the halogen, the position, and the number of halogen atoms on the distal aniline ring affected the activity. We observed that placing the chlorine atom at either the two- or three-position resulted in similar growth inhibition as our original 4-chloro analogue 14. Furthermore, adding a second chlorine atom to the two- or three-position of the aniline ring, as in the case of 22 and 24, resulted in virtually the same average growth inhibition as 14. Placing the chlorines in either a 2,5- or 3,5-relationship to one another, as in 36 and 51, resulted in a slight decrease in activity. Difluorinated analogues 35 and 37 showed good growth inhibition, although the activity was greater with the fluorines in a 2,4-relationship. As can be seen in Table 4, mixed halogenated derivatives performed exceptionally well, with most average growths below 25%. Compounds 27, 28, 40, and 52 that contained both chlorine and fluorine atoms were active, however the growth inhibition was optimal when the chlorine and fluorine were in a 3,4-relationship relative to one another, as in 27 and 28. 32, which contained 3-chloro and 4-bromo substituents, displayed exceptional activity, with an average growth of only 21%. With respect to sulfonamides that contained iodine, we found that the activity was in large part dependent on the identity and placement of the cohalogen. If iodine was used as the only halogen, as in 45, then the growth inhibition was low. For 57, which contained a chlorine at the two-position, the activity was respectable, with an average growth of 39%; however, when the chlorine was relocated to the three-position for 49, the growth was further suppressed to only 29%. We had high hopes for 31, which contained the popular 2-fluoro-4-iodoaniline oncogenic pharmacaphore; however, when it was incorporated onto our 1,3-oxazole sulfonamide backbone, we observed only minimal anticancer properties.25,26
With the final subclass of compounds in our second-generation library, we integrated both a halogen and an alkyl group onto the aniline ring. To our delight, most of the derivatives possessed good to excellent growth inhibition. Among the compounds tested, 30, which contained 3-methyl and 4-chloro substituents, had the best overall inhibition profile and suppressed cell growth by nearly 80%. When the methyl group was moved to the two-position (43), the activity slightly decreased; however, the mean growth remained below 25%. When the chloro and methyl groups were altered on the aniline ring to either a 2,5- (44) or 3,5-relationship (55), the growth remained low. The overall activity was retained when we exchanged a chlorine for a fluorine atom and oriented the methyl/fluoro groups in a 3,5-relationship (56). When iodine was used as the halogen (53), the activity decreased in comparison with the chloro-methyl and fluoro-methyl analogues. Given the exceptional activity of 16 in our first-generation library, we opted to switch the chloro and trifluoromethyl groups on the aniline ring to the three- and four-positions, respectively; however, when the substitution pattern was changed to give sulfonamide 34, the activity was lost altogether. When we included a methoxy group in conjunction with a halogen on the distal ring, the identity of the halogen mattered. Substrate 39, which contained a fluorine atom, was found to better inhibit cell growth versus its chloro-containing counterpart 48.
Given the initial screening results of the compounds in our second-generation library, several progressed to the full NCI-60 dose–response assay. As with the compounds in our first round of testing, those in our second library exhibited the same specificity for leukemia cell lines. Table 5 contains mean GI50, TGI, and LC50 data for leukemia cell lines for compounds selected for five-dose testing. (Full NCI-60 data for all cell lines can be found in the Supporting Information.) Of the 20 compounds from our second-generation library selected for the five-dose experiments, all but 4 (36, 37, 40, and 49) displayed submicromolar average GI50 values against leukemia cell lines. Even the aforementioned compounds, however, showed low single-digit micromolar activity. We were encouraged to find that even though GI50 values were low, toxicity concentrations were high, with most LC50s being above the 100 μM assay threshold. The 2-chloro-5-methyl (44) and 1-naphthyl (58) analogues proved to be the most potent inhibitors, with mean GI50 values of 48.8 and 44.7 nM respectively. Compounds 55 and 56 had submicromolar growth inhibitory properties but were the most intriguing due to their single-digit micromolar TGI values and relatively low lethal effects. From a global perspective, the data suggest that 1,3-oxazole sulfonamides derived from halogenated and alkyl-substituted anilines are potent and selective inhibitors of leukemia cell lines. Furthermore, we observed that the overall growth inhibition could be retained with a wide range of halogens and substitution patterns on the aniline ring itself. It is worth mentioning that even though 44 and 58 proved to be exceptionally potent, the CYP450-mediated hydroxylation and subsequent para-iminoquinone metabolite formation could be of concern.
Table 5. NCI-60 Dose–Response Data for Leukemia Cell Lines.
| mean
NCI-60 data for leukemia cell lines |
|||
|---|---|---|---|
| compound | mean GI50 | mean TGI | mean LC50 |
| 22 | 0.416 | 62.11 | >100 |
| 24 | 0.481 | 74.5 | >100 |
| 26 | 0.429 | 49.70 | >100 |
| 27 | 0.260 | 73.70 | >100 |
| 28 | 0.380 | 63.03 | >100 |
| 29 | 0.477 | 54.16 | >100 |
| 30 | 0.216 | 33.36 | >100 |
| 32 | 0.491 | 70.32 | >100 |
| 33 | 0.420 | >100 | >100 |
| 35 | 0.356 | 66.05 | >100 |
| 36 | 1.02 | >100 | >100 |
| 37 | 2.14 | 80.00 | >100 |
| 39 | 0.328 | 75.74 | >100 |
| 40 | 1.44 | 77.47 | >100 |
| 43 | 0.277 | 54.72 | >100 |
| 44 | 0.0488 | 52.75 | >100 |
| 49 | 1.01 | 19.62 | >100 |
| 55 | 0.175 | 7.04 | 84.9 |
| 56 | 0.138 | 7.25 | 78.05 |
| 58 | 0.0447 | 22.47 | >100 |
Average GI50, TGI, and LC50 data obtained after testing in the full NCI-60 dose–response assay. Values shown are in μM.
For our last SAR study, we elected to explore the importance of the N–H hydrogen bond donor group of the sulfonamide. For this study, we synthesized compounds 60–62, which contained a para-halogen and in which we replaced the hydrogen on the sulfonamide with a methyl group. These substrates were prepared according to the same experimental protocol outlined in Table 1 from sulfonyl chloride 11. Table 6 contains mean growth percentage data from the initial screening assay along with mean GI50 values for compounds selected for the dose–response experiments. Average growth percentages hovered around the 30% mark for all three compounds tested, with the 4-bromo and 4-fluoro derivatives satisfying the criteria needed to progress to the full NCI-60 five-dose experiments. (Complete NCI-60 data for 60–62 can be found in the Supporting Information.) In the dose–response assay, 60 exhibited submicromolar to nanomolar GI50 values across the majority of cell lines while simultaneously having low-toxicity effects, with most LC50 values being >100 μM. 62 showed similar trends but overall was not as active as its 4-fluoro counterpart. With respect to the leukemia cell lines, the identity of the halogen did not seem to matter, with both substrates having nanomolar GI50s and LC50s greater than 100 for all lines tested. From a cursory perspective, the data suggest that the N–H donor group of halogenated 1,3-oxazole sulfonamides is not essential to maintain overall antiproliferative activity.
Table 6. Structures and NCI-60 Data for N-Methylated 1,3-Oxazole Sulfonamides 60–62.

| compound | mean GP | mean GI50 |
|---|---|---|
| 60 | 31.76 | 1.18 |
| 61 | 30.57 | N/A |
| 62 | 31.39 | 3.92 |
Values shown are in μM.
The COMPARE analysis is an in silico platform developed by NCI as a tool for comparing cytotoxic parameters in the five-dose assays.27 If patterns of sensitivity in the 60-cell-line panel exist (e.g., GI50, LC50, TGI) between experimental agents and known compounds, then it is possible that the compounds have common targets or growth inhibitory mechanisms of action. Upon analyzing several of our substrates with COMPARE, we found the strongest correlation to microtubule inhibitors (MTIs), suggesting that these novel 1,3-oxazole sulfonamides might also be microtubule-targeting agents. To test this hypothesis, we utilized an in vitro tubulin polymerization assay using purified porcine tubulin. The assay is based on adaptation of an original method described by Shelanski and Lee, which demonstrated that under buffered conditions, tubulin monomer will self-polymerize to microtubules and increase light scattering to 340 nm.28,29 Drugs such as vinblastine and colchicine are known to inhibit tubulin self-polymerization in this assay, whereas drugs like paclitaxel, a microtubule-stabilizing agent, accelerate polymerization. For our study, we selected compounds 16, 22, 30, and 32 due to their overall growth inhibitory properties and their activity in the five-dose assay. The tubulin polymerization effects of our compounds were tested at concentrations of 10, 2, 0.4, and 0.08 μM, respectively. Figure 3 shows the results of the microtubule polymerization assay for 22, and the data for 16, 30, and 32 are recorded in the Supporting Information. When we added paclitaxel to the reaction mixture, it accelerated tubulin polymerization, as expected, and showed faster kinetics than the untreated control (Figure 3). Compounds 16, 22, 30, and 32 were all found to inhibit self-polymerization, which suggests that they bind directly to tubulin and induce the depolymerization of microtubule networks within cells. From the assay data, we were able to estimate IC50 values for our four compounds and found 16 to have an IC50 of 0.22 μM. 22, 30, and 32 were more potent, and all had IC50 values <0.08 μM.
Figure 3.

In vitro microtubule polymerization assay for 22 across four concentration ranges. The polymerization of tubulin was monitored by an increase in the absorbance at 340 nm for 30 min at 37 °C. The data suggest that compounds 16, 22, 30, and 32 are microtubule-destabilizing agents. Paclitaxel was evaluated in parallel at one concentration as the assay positive control.
In summary, a novel series of 1,3-oxazole sulfonamide derivatives having a cyclopropyl ring were synthesized based on a rational drug design strategy and screened for their anticancer activity. Compounds containing a halogenated aniline derivative were found to be the most active, with most examples inhibiting cancer cell growth by at least 75%. The most potent substrates possessed GI50 values in the submicromolar to nanomolar ranges while simultaneously displaying low overall toxicity, as evidenced by high LC50 values. After an in silico analysis of the most active compounds in NCI’s COMPARE algorithm, 16, 22, 30, and 32 were evaluated in a tubulin polymerization assay. The results suggest that our compounds could be binding to tubulin and inducing the depolymerization of microtubules in cells. Of the eight MTIs currently in clinical use, all are natural product derivatives that violate Lipinski’s rule of five, with the exception of colchicine. The serious side effects, poor solubility, and synthetic challenges associated with these drugs still remain problematic, even though they remain in wide chemotherapeutic use. Anticancer agents derived from synthetic small molecules like those reported here may be able to hurdle the shortcomings of the current MTIs; however, we recognize that caution should be exercised with molecules such as those described here because sulfonamides can trigger adverse reactions if the patient has a sulfa allergy. Further SAR studies with respect to the 1,3-oxazole sulfonamides reported here and the nature of the specificity to leukemia cell lines are ongoing. Furthermore, we have also begun studies involving the physiochemical properties of our compound library. A potential driving force for toxicity and cell-line specificity could be the compound’s ability to enter a given cell line rather than substitution patterns on the distal sulfonamide ring.
Acknowledgments
We thank Lieutenant Commander Doug Marks for his valuable support of this project. We thank Prof. Tom Bell and Dr. Trang Brennan for their helpful advice and suggestions. These opinions, recommendations, findings, and conclusions do not necessarily reflect the views or policies of NIST or the United States Government. Certain commercial products are identified in order to adequately specify the procedure; this does not imply endorsement or recommendation by NIST, nor does it imply that such products are necessarily the best available for the purpose.
Glossary
Abbreviations
- SAR
structure–activity relationship
- MTI
microtubule inhibitor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00219.
Author Present Address
§ K.L.B.: 5581 Russell Drive, Milton, Florida 32570, United States.
Author Contributions
K.L.B. was the primary investigator and was responsible for the compound synthesis and characterization. E.S. provided assistance with the HR-MS measurements.
Support for this research was generously provided by the United States Naval Academy Department of Chemistry.
The authors declare no competing financial interest.
Dedication
This work is dedicated to the late Jeanette “Nettie” Barnes. Her life, legacy, and brave battle against cancer continue to be the inspiration that drives oncology research in our group.
Supplementary Material
References
- Bray F.; Ferlay J.; Soerjomataram I.; Siegel R. L.; Torre L. A.; Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Ca-Cancer J. Clin. 2018, 68, 394–424. 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Kola I.; Landis J. Can the Pharmaceutical Industry reduce attrition rates?. Nat. Rev. Drug Discovery 2004, 3 (8), 711–715. 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- Hopkins C. D.; Wipf P. Isolation, Biology and Chemistry of the Disorazoles: New Anti-Cancer Macrodiolides. Nat. Prod. Rep. 2009, 26, 585–601. 10.1039/b813799b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsch D.; Schadt O.; Stieber F.; Meyring M.; Grädler U.; Bladt F.; Friese-Hamim M.; Knühl C.; Pehl U.; Blaukat A. Identification and Optimization of Pyridazinones as Potent and Selective c-Met Kinase Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1597–1602. 10.1016/j.bmcl.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Wang L.; Mei X.; Wang C.; Zhu W. Biomimetic Semi-Synthesis of Fradcarbazole A and its Analogues. Tetrahedron 2015, 71, 7990–7997. 10.1016/j.tet.2015.08.065. [DOI] [Google Scholar]
- Lintnerová L.; García-Caballero M.; Gregán F.; Melicherĉík M.; Quesada A. R.; Dobiaŝ J.; Lác J.; Saliŝová M.; Boháĉ A. A Development of Chimeric VEGFR2 TK Inhibitor Based on Two Ligand Conformers from PDB: 1Y6A Complex-Medicinal Chemistry Consequences of a TK’s Analysis Eur. Eur. J. Med. Chem. 2014, 72, 146–159. 10.1016/j.ejmech.2013.11.023. [DOI] [PubMed] [Google Scholar]
- Rafferty S. W.; Eisner J. R.; Moore W. R.; Schotzinger R. J.; Hoekstra W. J. Highly selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase Inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2444–2447. 10.1016/j.bmcl.2014.04.024. [DOI] [PubMed] [Google Scholar]
- Ichiba T.; Yoshida W. Y.; Scheuer P. J.; Higa T.; Gravalos D. G. Hennoxazoles, Bioactive Bisoxazoles from a Marine Sponge. J. Am. Chem. Soc. 1991, 113, 3173–3174. 10.1021/ja00008a056. [DOI] [Google Scholar]
- Shin-ya K.; Wierzba K.; Matsuo K.; Ohtani T.; Yamada Y.; Furihata K.; Hayakawa Y.; Seto H. Telomestatin, a Novel Telomerase Inhibitor from Streptomyces anulatus. J. Am. Chem. Soc. 2001, 123, 1262–1263. 10.1021/ja005780q. [DOI] [PubMed] [Google Scholar]
- Sasse F.; Steinmetz H.; Höfle G.; Reichenbach H. Rhizopodin, a New Compound from Myxococcus stipitatus (myxobacteria) Causes Formation of Rhizopodia-Like Structures in Animal Cell Cultures: Production, Isolation. J. Antibiot. 1993, 46, 741–748. 10.7164/antibiotics.46.741. [DOI] [PubMed] [Google Scholar]
- Gronewold T. M.; Sasse F.; Lünsdorf H.; Reichenbach H. Effects of Rhizopodin and Latrunculin B on the Morphology and on the Actin Cytoskeleton of Mammalian Cells. Cell Tissue Res. 1999, 295, 121–129. 10.1007/s004410051218. [DOI] [PubMed] [Google Scholar]
- Hagelueken G.; Albrecht S. C.; Steinmetz H.; Jansen R.; Heinz D. W.; Kalesse M.; Schubert W. D. The Absolute Configuration of Rhizopodin and Its Inhibition of Actin Polymerization by Dimerization. Angew. Chem., Int. Ed. 2009, 48, 595–598. 10.1002/anie.200802915. [DOI] [PubMed] [Google Scholar]
- Zhang H. Z.; Zhao Z. L.; Zhou C. H. Recent Advances in Oxazole-Based Medicinal Chemistry. Eur. J. Med. Chem. 2018, 144, 444–492. 10.1016/j.ejmech.2017.12.044. [DOI] [PubMed] [Google Scholar]
- Scozzafava A.; Owa T.; Mastrolorenzo A.; Supuran C. T. Anticancer and Antiviral Sulfonamides. Curr. Med. Chem. 2003, 10, 925–953. 10.2174/0929867033457647. [DOI] [PubMed] [Google Scholar]
- Khan F. A.; Mushtaq S.; Naz S.; Farooq U.; Zaidi A.; Bukhari S. M.; Rauf A.; Mubarak M. S. Sulfonamides as Potential Bioactive Scaffolds. Curr. Org. Chem. 2018, 22, 818–830. 10.2174/1385272822666180122153839. [DOI] [Google Scholar]
- Zhao C.; Rakesh K.P.; Ravidar L.; Fang W.-Y.; Qin H.-L. Pharmaceutical and Medicinal significance of sulfur (SVI)-Containing Motifs for Drug Discovery: A Critical Review. Eur. J. Med. Chem. 2019, 162, 679–734. 10.1016/j.ejmech.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino H.; Ueda N.; Niijima J.; Sugumi H.; Kotake Y.; Koyanagi N.; Yoshimatsu K.; Asada M.; Watanabe T.; Nagasu T.; et al. Novel Sulfonamides as Potential, Systemically Active Antitumor Agents. J. Med. Chem. 1992, 35, 2496–2497. 10.1021/jm00091a018. [DOI] [PubMed] [Google Scholar]
- Talele T. T. The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 2016, 59, 8712–8756. 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- Yang W. S.; Shimada K.; Delva D.; Patel M.; Ode E.; Skouta R.; Stockwell B. R. Identification of Simple Compounds with Microtubule-Binding Activity that Inhibit Cancer Cell Growth with High Potency. ACS Med. Chem. Lett. 2012, 3, 35–38. 10.1021/ml200195s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Zhou S.; Ji L.; Zhang C.; Yu S.; Li Z.; Meng X. Synthesis and Structure-Activity Relationship of 4-Azaheterocycle Benzenesulfonamide Derivatives as New Microtubule-Targeting Agents. Bioorg. Med. Chem. Lett. 2014, 24, 5055–5058. 10.1016/j.bmcl.2014.09.016. [DOI] [PubMed] [Google Scholar]
- Yang J.; Yang S.; Zhou S.; Lu D.; Ji L.; Li Z.; Yu S.; Meng X. Synthesis, Anti-Cancer Evaluation of Benzenesulfonamide Derivatives as Potent Tubulin-Targeting Agents. Eur. J. Med. Chem. 2016, 122, 488–496. 10.1016/j.ejmech.2016.07.002. [DOI] [PubMed] [Google Scholar]
- Alley M. C.; Scudiero D. A.; Monks P. A.; Hursey M. L.; Czerwinski M. J.; Fine D. L.; Abbott B. J.; Mayo J. G.; Shoemaker R. H.; Boyd M. R. Feasability of Drug Screening with Panels of Human Tumor Cell Lines Using a Microculture Tetrazolium Assay. Cancer Res. 1988, 48, 589–601. [PubMed] [Google Scholar]
- Shoemaker R. H.; Monks A.; Alley M. C.; Scudiero D. A.; Fine D. L.; McLemore T. L.; Abbott B. J.; Paull K. D.; Mayo J. G.; Boyd M. R. Development of Human Tumor Cell Line Panels for use in Disease-Oriented Drug Screening. Prog. Clin. Biol. Res. 1988, 276, 265–286. [PubMed] [Google Scholar]
- Shoemaker R. H. The NCI60 Human Tumour Cell Line Anticancer Drug Screen. Nat. Rev. Cancer 2006, 6, 813–823. 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- L M. Allosteric MEK1/2 inhibitors for the treatment of cancer: an overview. J. Drug Res. Dev. 2015, 1, 101–109. 10.16966/2470-1009.101. [DOI] [Google Scholar]
- Abe H.; Kikuchi S.; Hayakawa K.; Iida T.; Nagahashi N.; Maeda K.; Sakamoto J.; Matsumoto N.; Miura T.; Matsumura K.; Seki N.; Inaba T.; Kawasaki H.; Yamaguchi T.; Kakefuda R.; Nanayama T.; Kurachi H.; Hori Y.; Yoshida T.; Kakegawa J.; Watanabe Y.; Gilmartin A. G.; Richter M. C.; Moss K. G.; Laquerre S. G. Discovery of a highly potent and selective MEK inhibitor: GSK1120212 (JTP-74057 DMSO solvate). ACS Med. Chem. Lett. 2011, 2, 320–324. 10.1021/ml200004g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull K. D.; Shoemaker R. H.; Hodes L.; Monks A.; Scudiero D. A.; Rubinstein L.; Plowman J.; Boyd M. R. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: Development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 1989, 81 (14), 1088–1092. 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- Shelanski M. L.; Gaskin F.; Cantor C. R. Microtubule assembly in the absence of added nucleotides. Proc. Natl. Acad. Sci. U. S. A. 1973, 70 (3), 765–768. 10.1073/pnas.70.3.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. C.; Timasheff S. N. In vitro reconstitution of calf brain microtubules: Effects of solution variables. Biochemistry 1977, 16 (8), 1754–1762. 10.1021/bi00627a037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.