

Abstract

Signal transducer and activator of transcription 3 (STAT3) is an attractive cancer therapeutic target. We report herein our extensive in vitro and in vivo evaluations of SD-91, the product of the hydrolysis of our previously reported STAT3 degrader SD-36. SD-91 binds to STAT3 protein with a high affinity and displays >300-fold selectivity over other STAT family protein members. SD-91 potently and effectively induces degradation of STAT3 protein and displays a high selectivity over other STAT members and >7000 non-STAT proteins in cells. A single administration of SD-91 selectively depletes STAT3 protein in tumor tissues with a persistent effect. SD-91 achieves complete and long-lasting tumor regression in the MOLM-16 xenograft model in mice even with weekly administration. Hence, SD-91 is a potent, highly selective, and efficacious STAT3 degrader for extensive evaluations for the treatment of human cancers and other diseases for which STAT3 plays a key role.
Keywords: STAT3, PROTAC, Degrader, Cancer
The IL-6/JAK/STAT3 pathway plays a critical role in human cancers and other human diseases. Targeting different components of the IL-6/JAK/STAT3 pathway has been pursued as therapeutic strategies.1 To date, inhibitors of IL-6, the IL-6 receptor, or JAKs have received regulatory approval for treatment of several human diseases.1
STAT3 is a transcriptional factor and has long been considered as an attractive therapeutic target. However, successful targeting STAT3 using small molecules or biological approaches has proven to be challenging.1 It has been initially proposed that dimerization of STAT3 through its Src homology domain 2 (SH2) is required for its transcriptional activity.2 Therefore, small-molecule inhibitors have been developed to bind to the STAT3 SH2 domain and to block the dimerization of STAT3.1,3 Unfortunately, many STAT3 SH2 domain inhibitors lack the desired selectivity for STAT3 over other STAT family protein members and/or do not have a clearly defined cellular mechanism of action.1,3 Furthermore, it has been shown that monomeric STAT3 protein can still function as a transcriptional factor, and thus blocking the STAT3 dimerization will only partially inhibit the transcriptional activity of STAT3.4 Antisense oligonucleotides (ASOs) have been developed to target STAT3 but ASO molecules are mostly concentrated in the liver in vivo with systematic administration and have very limited distributions in other tissues.1,5,6 Therefore, there is a clear need to develop new and more effective therapeutic strategies in order to successfully target STAT3.
Recently, we have reported the discovery of SD-367,8 as the first potent, selective, and highly efficacious small-molecule degrader of STAT3 designed using the proteolysis targeting chimaera (PROTAC) strategy.9,10 SD-36 potently induces the degradation of STAT3 protein in cells and is highly selective over other STAT proteins and >5000 other proteins. A single intravenous administration of SD-36 induces complete degradation of STAT3 protein in normal tissues and xenograft tumor tissue in mice and is capable of achieving complete tumor regression. Importantly, extensive toxicity testing showed that SD-36 is well tolerated in both immune-competent and immune-compromised mice.8
Further testing showed that SD-36 is not stable under certain conditions, and its difluoro methylene group of the difluoro phosphoric mimetic undergoes hydrolysis and converts into a ketone group. In the present study, we report our extensive evaluations of SD-91, a product of the hydrolysis of SD-36 as a potent, selective, and highly efficacious degrader of STAT3 with excellent chemical stability.
During purification of SD-36 by HPLC, we found that when the separated fraction of the HPLC eluent containing SD-36 was left at room temperature overnight, a trace amount of a new compound was detected, which has a molecular weight of 1135, 22 less than that for SD-36. On the basis of the molecular weight, we proposed that hydrolysis of the gem-difluoride in SD-36 may have occurred and the gem-difluoride group was transformed to a carbonyl group (Figure 1A). To confirm this hypothesis, we synthesized the proposed compound SD-91 (Figure 1A), which is indeed identical to the hydrolyzed product from SD-36.
Figure 1.
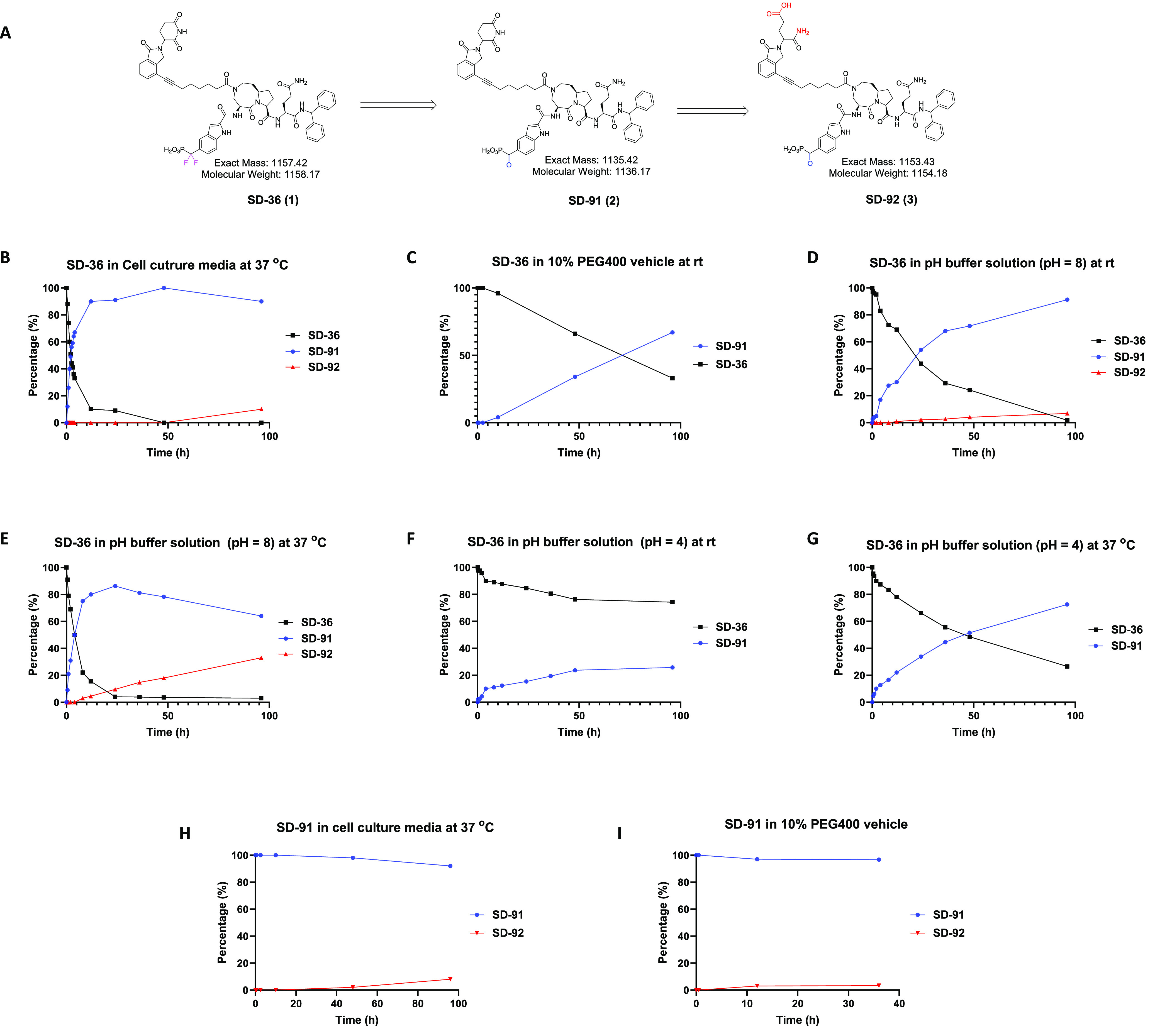
Evaluation of in vitro conversion of SD-36 to SD-91. (A) Chemical structures of SD-36 and SD-91. Percentages of the indicated compounds at the indicated time points after incubation of SD-36 in cell culture at 37 °C (B); in 10% PEG400 vehicle at room temperature (C); in pH buffer solution (pH = 8) at room temperature (D) and at 37 °C (E); in pH buffer solution (pH = 4) at room temperature (F) and at 37 °C (G); percentages of the indicated compounds at the indicated time points after incubation of SD-91 in cell culture at 37 °C (H), in 10% PEG400 vehicle at room temperature (I).
We investigated the stability of SD-36 further in the cell culture media used in our cellular experiments and in the dosing vehicle used in our previous in vivo experiments. Surprisingly, SD-36 was found to convert into SD-91 in the cell culture media rapidly, with nearly 50% of conversion observed at 2 h and 90% of conversion at 24 h (Figure 1B). Similar conversion rates from SD-36 to SD-91 were observed in cell culture media only and cell culture media with cells (Supporting Information (SI), Figure S1). Additionally, SD-92, which is the glutarimide ring opening product of SD-91 (Figure 1A), was detected in the cell culture media and approximately 10% of SD-92 was also observed at 96 h (Figure 1B).
In the dosing vehicle (10% PEG400/90% phosphate buffered saline), the conversion of SD-36 to SD-91 is much slower than that in the cell culture media (Figure 1C). No obvious conversion of SD-36 to SD-91 was observed at 2.5 h, and only 4% of conversion was detected after 10 h. In our animal experiments, SD-36 was prepared fresh and was used within a few hours. Therefore, we concluded that when SD-36 was dosed in the animal experiments, no significant amount of SD-91 was presented in the dosing solution.
We further tested SD-36 for its stability in pH buffer solutions at different pH values at either room temperature or 37 °C (Figure 1D–G). At pH = 8 and room temperature, the conversion of SD-36 to SD-91 is relatively fast (t1/2 = ∼22 h) (Figure 1D) and is greatly accelerated at 37 °C (t1/2 = 4 h) (Figure 1E). At pH = 4 and room temperature, much slower conversion from SD-36 to SD-91 was observed with only 25% at 96 h (Figure 1F).
At pH = 4 and 37 °C, approximately 50% of the conversion from SD-36 to SD-91 was observed after 2 days (Figure 1G). SD-92 was detected at pH = 8 but not at pH = 4 (Figure 1D-G). The conversion of SD-36 to SD-91 is thus both pH and temperature dependent and accelerated with elevated temperatures or at higher pH values.
We evaluated the stability of SD-91 under different conditions. SD-91 was found to be stable in both the cell culture media (Figure 1H) and the in vivo dosing vehicle (Figure 1I). Of note, <10% of SD-92 was detected after 96 h in both the cell culture media (Figure 1H) and the in vivo dosing vehicle (Figure 1I). Taken together, these data showed that SD-91 has excellent stability in the cell culture media and the in vivo dosing vehicle.
As shown in Figure 1, SD-36 can convert to SD-91 in both acidic and basic conditions, however, the conversion of SD-36 to SD-91 is fairly rapid in basic condition but has a much slower rate in acidic condition. Upon the basis of these stability data, we proposed two possible mechanisms of conversion of SD-36 to SD-91 in basic and acidic conditions (Figure 2).
Figure 2.
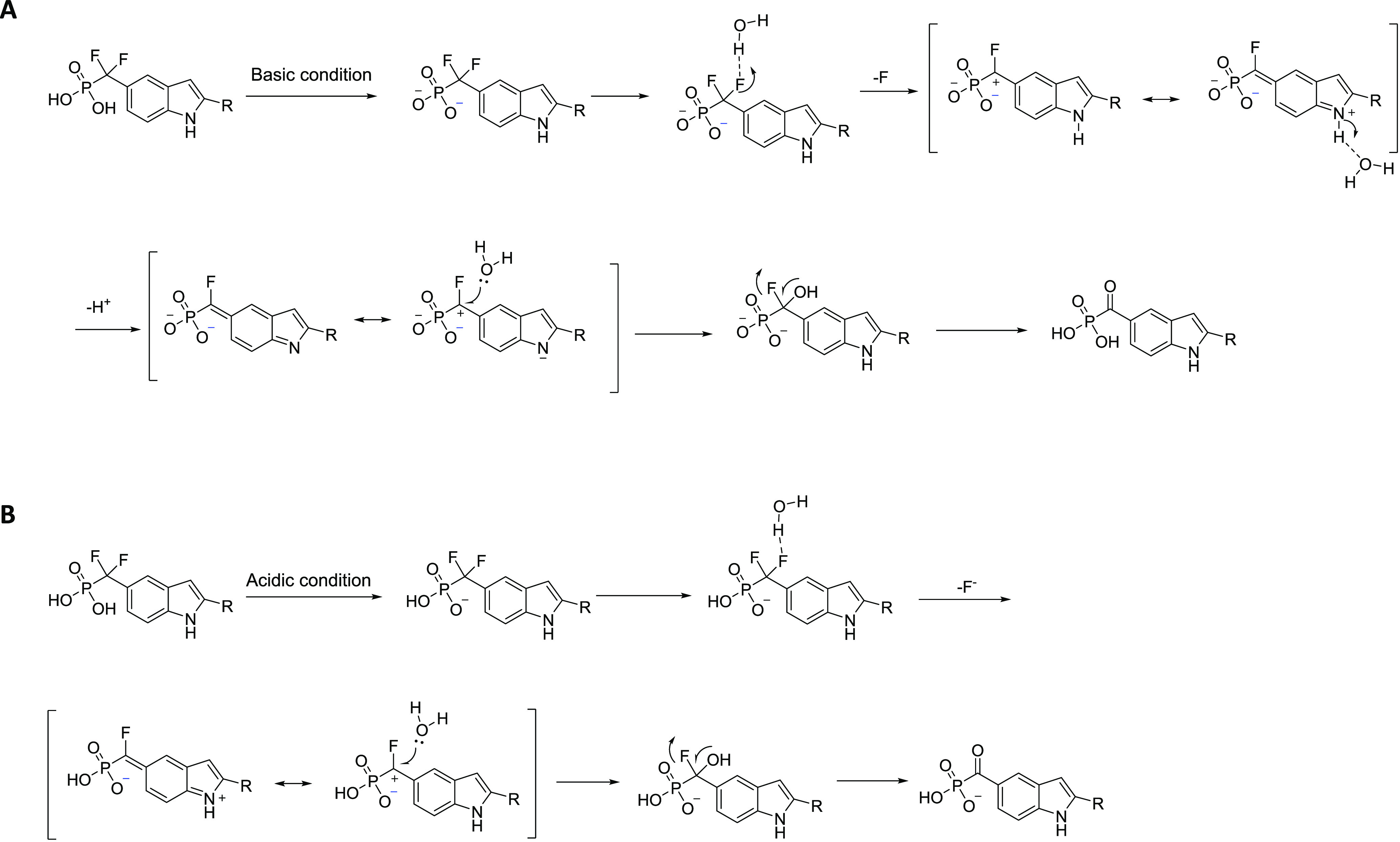
Proposed mechanism of the hydrolysis of the gem-difluoride in SD-36 in (A) basic or neutral conditions and (B) acidic conditions.
In both cases, a water molecule associates with one F in the difluoromethyl phosphonate and induces the cleavage of the C–F bond to form a carbocation intermediate by release of HF. The carbocation intermediate is captured by a water molecule and subsequent cleavage of another C–F bond leads to the formation of carbonylphosphonic acid. Notably, the indole ring facilitates the cleavage of C–F bond by stabilizing the carbocation transition state in both cases.
The double-ionized phosphonate formed under basic or near neutral conditions (pH > 5.7) (Figure 2A), whereas under weak acidic conditions (2 > pH > 5.7), single-ionized phosphonate forms (Figure 2B). The double-ionized phosphonate may contribute to the stabilization of the carbocation transition state. Additionally, the more stable transition state formed by loss the indole NH proton (Figure 2A) may explain the much faster conversion rate of SD-36 to SD-91 in basic conditions than that in acidic conditions.
We evaluated SD-91 for its binding affinity to STAT3. SD-91 binds to STAT3 with a Ki value of 5.5 nM in our competitive fluorescence-polarization assay, whereas SD-36 has a Ki value of 11.7 nM to STAT3 (Figure 3A). Hence, both SD-91 and SD-36 bind to STAT3 with a high affinity to STAT3 with SD-91 being 2× more potent than SD-36.
Figure 3.

SD-91 potently and selectively binds to recombinant human STAT3 protein over other recombinant human STAT family proteins. (A) Representative competitive binding curves of SD-91 and SD-36 to recombinant human STAT3 protein using a competitive fluorescence polarization assay. (B) Kinetic binding sensorgrams of SD-91 with concentrations ranging from 200 nM with 2× dilution and the steady-state fitting of the equilibrium responses vs compound concentrations based on 1:1 binding model by biolayer interferometry (BLI) assay. (C) Kinetic binding sensorgrams of SD-91 to other recombinant human STAT proteins (STAT1, STAT2, STAT4, STAT5A, STAT5B, and STAT6) by biolayer interferometry (BLI) assay with concentrations ranging from 200 nM with 2× dilution.
The STAT family members, namely STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6 share a conserved SH2 domain. We employed a biolayer interferometry (BLI) assay to evaluate the binding affinities of SD-91 to recombinant human STAT proteins containing their respective SH2 domain, with the data summarized in Figure 3B,C.
In the BLI assays, SD-91 binds to STAT3 with a Kd value of 28 nM (Figure 3B), while it displays a very weak affinity (Kd > 10 μM) (Figure 3C) to all other STAT proteins (STAT1, STAT2, STAT4, STAT5A, STAT5B, and STAT6). Thus, SD-91 exhibits a high binding affinity to STAT3 and >300-fold selectivity over other STAT family members. In our previous study, SD-36 binds to STAT3 with a Kd value of 44 nM and has Kd values of 1086 and 804 nM to STAT1 and STAT4, thus displaying approximately 20-fold selectivity for STAT3 over STAT1 and STAT4.8 Hence, SD-91 displays a much higher binding selectivity than SD-36 for STAT3 over other STAT proteins.
We tested SD-91 for its ability to induce degradation of STAT3 and its selectivity over other STAT proteins in cells, with SD-36 as a positive control. Additionally, we have synthesized SI-191 (Figure 4A), which is the corresponding inhibitor of SD-91. We included SI-191 and SI-109 as additional control compounds in our cellular experiments.
Figure 4.

SD-91 selectively degrades STAT3 protein in cells. (A) Chemical structures of two control STAT3 inhibitors SI-191 and SI-109. (B) Western blotting of STAT proteins in MOLM-16 cells treated with different compounds at indicated concentrations for 4 h. GAPDH was used as the loading control. (C) Densitometry of STAT3 and GAPDH proteins in the MOLM-16 cell line treated with SD-91 or SD-36 for 4 h. (D) Western blotting of STAT proteins in the SU-DHL-1 cells. (E) Western blotting of STAT proteins in the SUP-M2 cells. (F) Quantitative proteomics analysis of MOLM-16 cells treated with 1 or 10 μM of SD-91 for 4 h.
Our previous study showed that SD-36 induces rapid degradation of STAT3 in the MOLM-16 cell line.7,8 We thus treated the MOLM-16 cells for 4 h with SD-91 and SD-36 and evaluated different STAT proteins by Western blot (Figure 4B). Our data showed that SD-36 and SD-91 effectively reduce the levels of STAT3 protein in a dose-dependent manner and achieve complete degradation at 0.4 μM. The estimated DC50 values by densitometry for SD-36 and SD-91 are 0.38 and 0.12 μM, respectively, in the MOLM-16 cell line (Figure 4C). Both compounds have no significant effect on other STAT proteins at all concentrations tested (25 nM to 100 μM), demonstrating >300× degradation selectivity for STAT3 over other STAT proteins.
Our previous study showed that SD-36 has a slower kinetics in inducing degradation of STAT3 in the SU-DHL-1 and SUP-M2 lymphoma cell lines than that in the MOLM-16 cell line.7,8 We thus treated the SU-DHL-1 and SUP-M2 lymphoma cell lines with SD-91 and SD-36 for 14 h to evaluate their ability to degrade STAT3 and their selectivity over other STAT proteins, with the data shown in Figure 4C–E. In the SU-DHL-1 cell line, SD-36 and SD-91 effectively induced degradation of STAT3 protein in a dose-dependent manner (Figure 4D) and achieved DC50 values of 61 nM and 17 nM, respectively (Figure 4C). Consistent with our previous data, both SD-91 and SD-36 induce upregulation of STAT4 protein in the SU-DHL-1 lymphoma cell lines, but both compounds have no effect on other STAT proteins in all concentrations tested (25 nM to 100 μM).
In the SUP-M2 cell line, SD-36 and SD-91 also effectively induce degradation of STAT3 protein in a dose-dependent manner (Figure 4E) and achieve DC50 values of 2.5 μM and 1.2 μM, respectively (Figure 4C). In the SUP-M2 cell line, both SD-36 and SD-91 have no effect on other STAT proteins in all concentrations tested (25 nM to 100 μM).
We next performed proteomic analysis to assess the proteome-wide selectivity of SD-91 in the MOLM-16 cell line. As shown in Figure 4F, STAT3 was the only protein whose level was significantly decreased by SD-91 with both 1 μM and 10 μM for 4 h. These data show that SD-91 is a potent STAT3 degrader with high selectivity over >7000 proteins in cell.
SI-191 and SI-109, the two corresponding inhibitors of SD-91 and SD-36, have no effect on each of these STAT proteins at both 25 and 100 μM in these three cell lines (Figure 4).
Hence, in each of these three cell lines, SD-91 is more potent than SD-36 in inducing degradation of STAT3 protein, and both SD-91 and SD-36 demonstrate very high degradation selectivity in these three cell lines tested. Of note, no “hook effect” was observed for SD-91 at concentrations as high as 100 μM in all three tested cell lines.
We tested SD-36 and SD-91 for their cell growth inhibition activity in the MOLM-16 acute leukemia, SU-DHL-1 and SUP-M2 lymphoma cell lines, with two inhibitors SI-109 and SI-191 as controls (Table 1). Our data showed that SD-36 has IC50 values of 0.18, 3.4, and 0.53 μM, respectively, in the MOLM-16 acute leukemia, SU-DHL-1, and SUP-M2 lymphoma cell lines. In comparison, SD-91 has IC50 values of 0.17, 2.6, and 0.46 μM, respectively, in the MOLM-16 acute leukemia, SU-DHL-1, and SUP-M2 lymphoma cell lines. Of note, the similar potencies of SD-36 and SD-91 in cell growth inhibition in each of these cell lines is perhaps not surprising, given the rapid conversion of SD-36 to SD-91 in the cell culture media.
Table 1. Cell Growth Inhibitory Activities of SD-36, SI-109, SD-91, and SI-191 in the Molm-16, SU-DHL-1, and SUP-M2 Lymphoma Cell Lines.
| compd | MOLM-16 (IC50, μM) | SU-DHL-1 (IC50, μM) | SUP-M2 (IC50, μM) |
|---|---|---|---|
| SD-36 | 0.18 ± 0.15 | 3.4 ± 3.1 | 0.53 ± 0.44 |
| SD-91 | 0.17 ± 0.13 | 2.6 ± 2.6 | 0.46 ± 0.39 |
| SI-109 | >10 | >10 | >10 |
| SI-191 | >10 | >10 | >10 |
We next performed the pharmacodynamics study of SD-91 in mice bearing the MOLM-16 xenograft tumors, with SD-36 included as the control.
A single intravenous administration of SD-91 at 50 mg/kg in mice essentially depletes STAT3 protein in the MOLM-16 xenograft tumor tissue at the 1 h time-point, with the effect persisting for at least 24 h (Figure 5A). In comparison, a single intravenous administration of SD-36 at 50 mg/kg has the same effect on STAT3 protein as compared to that for SD-91.
Figure 5.

Pharmacodynamic and pharmacokinetic studies of SD-91 and SD-36 in xenograft tumor tissues in mice. (A) SCID mice bearing Molm-16 were treated intravenously with a single dose of SD-36, SD-91, or dosing vehicle as indicated. Tumor lysates were analyzed by immunoblotting. (B) Concentrations of SD-36 and SD-91 in mouse plasma and tumor after the mice were treated with a single dose of SD-36. (C) Concentrations of SD-91 in mouse plasma and tumor after the mice were treated with a single dose of SD-91. (D) SCID mice bearing Molm-16 or SU-DHL-1 tumors were treated intravenously with a single dose of SD-91 or dosing vehicle as indicated. Tumor lysates were analyzed by immunoblotting.
We examined the effect of SD-36 and SD-91 on STAT1 and STAT6 proteins in the MOLM-16 tumor tissue. Both SD-36 and SD-91 have no or minimal effect on the levels of STAT1 and STAT6 proteins (Figure 5A).
Although SD-36 was quite stable in the dosing vehicle used in our in vivo experiments, we hypothesized that SD-36 may convert into SD-91 in animals. We thus investigated the in vivo conversion of SD-36 to SD-91 in mice bearing the MOLM-16 xenograft tumors, with the data summarized in Figure 5B. Our analysis showed that upon an intravenous administration of SD-36 at 50 mg/kg in mice, a high plasma concentration of SD-36 (∼100 000 ng/mL) was detected at 1 h but decreased to approximately 1000 ng/mL at 6 h and to a very low level (58 ng/mL) at 24 h. A very high concentration (∼50 000 ng/mL) of SD-91 was found in the plasma at 1 h, and a sustained plasma drug concentration of ∼25 000 ng/mL was detected at the 6 h time point.
In the tumor tissue, similarly high concentrations of both SD-36 (∼7700 ng/g) and SD-91 (∼6350 ng/g) were found at the 1 h time point. At the 6 h t ime-point, SD-91 has a higher concentration than SD-36 (1750 vs 581 ng/g). SD-91 and SD-36 have similar drug concentrations in the tumor tissue at the 24 h time point (396 ng/g for SD-91 vs 275 ng/g for SD-36).
Upon the basis of these analytical data and rapid kinetics of STAT3 protein degradation in the MOLM-16 tumors, we concluded that when mice were administered with SD-36, the observed induced degradation of STAT3 protein in the tumor tissue is a combined effect of both the parent compound SD-36 and the hydrolyzed product SD-91.
When animals were dosed with a single intravenous administration of SD-91 at 50 mg/kg, a very high concentration (∼85 000 ng/mL) of SD-91 was detected in plasma at 1 h (Figure 5C). Furthermore, a sustained plasma drug concentration of ∼49 000 ng/mL was observed at 6 h and an appreciable drug concentration of ∼1300 ng/mL was detected at the 24 h time point (Figure 5C). In the tumor tissue, a high concentration (∼16 000 ng/g) of SD-91 was found at the 1 h time point and the concentration of SD-91 in the tumor tissue decreased to ∼4780 ng/g at the 6 h and ∼1800 ng/g at the 24 h time point.
We further evaluated SD-91 for its pharmacodynamics effect on both the phosphorylated (p-STAT3Y705) and total STAT3 proteins in the MOLM-16 xenograft tumors in mice (Figure 5D). A single administration of SD-91 at 50 mg/kg essentially eliminated all of the phosphorylated STAT3 protein as well as the total STAT3 protein in the MOLM-16 tumor tissue for at least 48 h.
We next determined the ability of SD-91 to reduce the total STAT3 and the phosphorylated STAT3 protein in the SU-DHL-1 xenograft tumors in mice (Figure 5D). A single administration of SD-91 at 50 mg/kg is also very effective in reducing the levels of the phosphorylated STAT3 protein as well as the total STAT3 protein in the SU-DHL-1 xenograft tumor tissue for at least 24 h.
Upon the basis of the strong PD effect for SD-91, we evaluated SD-91 for its antitumor activity in the MOLM-16 xenograft model in mice (Figure 6).
Figure 6.
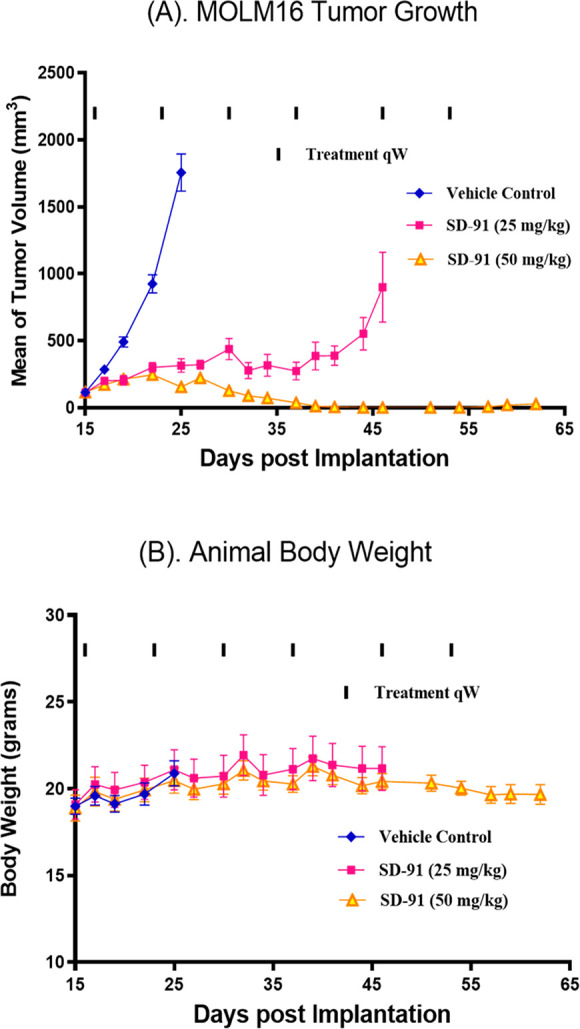
Antitumor activity of SD-91 in the MOLM-16 xenograft model in mice. SCID mice bearing the MOLM-16 tumors were treated with SD-91 at the indicated dosing schedules were treated at the indicated dosing schedules, and tumor volumes (A) and mice body weight (B) were measured every 2 days.
Because a single dose of SD-91 is capable of depleting both pSTAT3Y705 and total STAT3 proteins by >90% for at least 48 h in the MOLM-16 tumor tissue, we evaluated its efficacy with weekly administration (Figure 6A). SD-91 at 25 mg/kg weekly dosing inhibited tumor growth by 87% after only two doses over the vehicle control treatment. SD-91 at 50 mg/kg weekly administration was capable of achieving complete tumor regression after only three doses. It is significant that SD-91 did not induce any weight loss or any other signs of toxicity in mice during or after the treatment (Figure 6B).
Hence, our in vivo efficacy experiment thus demonstrated that SD-91 is highly efficacious in the MOLM-16 xenograft model even with weekly dosing, similar to the efficacy results we have obtained previously for SD-36.
The synthesis of the α-ketophosphonic acid has been reported previously.11,12 However, the precursor of α-ketophosphonic acid, α-ketophosphonic ester, reacts rapidly with nucleophiles which lead to the rupture the hosphorus-carbon (C–P) bond of α-ketophosphonic ester,11,12 which makes the synthesis of α-ketophosphonic acid from α-ketophosphonic ester very challenging. To address this issue, we have developed a practical method for synthesis of STAT3 degraders with a novel phosphotyrosine mimetic containing α-ketophosphonic acid.
As we developed an efficient method for the synthesis of the intermediate 4, we investigated the possibility of employment 4 as our starting point for the synthesis of SD-91, as shown in Scheme 1.
Scheme 1. Synthesis of SD-91.

Reagents and conditions: (a) Pentachlorophenol, DCC, DMAP, DMF; (b) TMSI, BSTFA, DCM; (c) TFA, CH3CN, H2O; (d) 8, HOBt, DIEA, DMF.
The pentachlorophenyl carboxy ester is an active carboxyester, and the pentachlorophenyl carboxy ester has been reported to be stable to the treatment of TMSI, which was used to hydrolyze the diethyl phosphate group.13 Esterification of compound 4 led to a pentachlorophenyl carboxyester 5, which was hydrolyzed by TMSI to afford compound 6. After exploring the reaction conditions, we found that the difluoromethy phosphonic acid 6 was able to be transformed to α-ketophosphonic acid 7 with the treatment of TFA in CH3CN and H2O at 60 °C. The active carboxyester 7 reacts smoothly with amine 8 to yield compound SD-91.
In summary, investigations into the hydrolysis of SD-36 showed that its gem-difluoride can convert into a ketone, which generates SD-91. Our extensive in vitro and in vivo data clearly demonstrate that SD-91 is a potent, selective, and highly efficacious STAT3 degrader with excellent chemical and metabolic stability. Our study further revealed that because SD-36 can convert into SD-91 in vitro and in vivo, the observed in vitro activity in cells and in vivo PD and efficacy for SD-36 is a combined effect of SD-36 itself and SD-91. Taken together, SD-91 represents a promising STAT3 degrader with excellent chemical stability for extensive in vitro and in vivo evaluations as a new therapeutic agent for the treatment of human cancers and other diseases in which STAT3 plays a key role.
Acknowledgments
We thank G.W. A. Milne for his critical reading and editing of the manuscript. This work is supported in part by the National Institutes of Health/National Cancer Institute (5R01CA244509 to SW), Oncopia Therapeutics, Inc. (to SW), and the Rogel Cancer Center Core Grant from the National Cancer Institutes, NIH (P30 CA046592).
Glossary
Abbreviations
- STAT3
signal transducer and activator of transcription 3
- SH2
Src homology domain 2
- PROTAC
proteolysis targeting chimaera
- BLI
bio-layer interferometry
- PD
pharmacodynamics.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00155.
Stability test of SD-36 in cell culture media; chemistry methods, and biochemistry and biology assay methods; 1H NMR and UPLC-MS spectrum for SD-91 (PDF)
Author Contributions
H.Z., L.B., and R.Q. contributed equally.The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): The University of Michigan has filed a number of patent applications on this class of STAT3 degraders, which have been licensed to Oncopia Therapeutics Inc. S. Wang, H. Zhou, R. Xu, L. Bai, and D. McEachern are co-inventors on one or more of these patents, and receive royalties on these patents from the University of Michigan. S. Wang is a co-founder of Oncopia and is a paid consultant of Oncoipa. The University of Michigan and S. Wang own stock in Oncopia. Oncopia has provided a research contract to the University of Michigan for which S. Wang is the principal investigator.
Supplementary Material
References
- Johnson D. E.; O’Keefe R. A.; Grandis J. R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D. E.; Darnell J. E. STATs: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Beebe J. D.; Liu J. Y.; Zhang J. T. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we?. Pharmacol. Ther. 2018, 191, 74–91. 10.1016/j.pharmthera.2018.06.006. [DOI] [PubMed] [Google Scholar]
- Yang J.; Stark G. R. Roles of unphosphorylated STATs in signaling. Cell Res. 2008, 18, 443–451. 10.1038/cr.2008.41. [DOI] [PubMed] [Google Scholar]
- Hong D.; Kurzrock R.; Kim Y.; Woessner R.; Younes A.; Nemunaitis J.; Fowler N.; Zhou T. Y.; Schmidt J.; Jo M. J.; Lee S. J.; Yamashita M.; Hughes S. G.; Fayad L.; Piha-Paul S.; Nadella M. V. P.; Mohseni M.; Lawson D.; Reimer C.; Blakey D. C.; Xiao X. K.; Hsu J.; Revenko A.; Monia B. P.; MacLeod A. R. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. 10.1126/scitranslmed.aac5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi C.; Wood M. J. A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21. 10.1038/nrneurol.2017.148. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Bai L.; Xu R.; Zhao Y.; Chen J.; McEachern D.; Chinnaswamy K.; Wen B.; Dai L.; Kumar P.; Yang C. Y.; Liu Z.; Wang M.; Liu L.; Meagher J. L.; Yi H.; Sun D.; Stuckey J. A.; Wang S. Structure-Based Discovery of SD-36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein. J. Med. Chem. 2019, 62, 11280–11300. 10.1021/acs.jmedchem.9b01530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L.; Zhou H.; Xu R.; Zhao Y.; Chinnaswamy K.; McEachern D.; Chen J.; Yang C. Y.; Liu Z.; Wang M.; Liu L.; Jiang H.; Wen B.; Kumar P.; Meagher J. L.; Sun D.; Stuckey J. A.; Wang S. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511. 10.1016/j.ccell.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; e417
- Deshaies R. J. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 2020, 580, 329–338. 10.1038/s41586-020-2168-1. [DOI] [PubMed] [Google Scholar]
- Sun X.; Gao H.; Yang Y.; He M.; Wu Y.; Song Y.; Tong Y.; Rao Y. PROTACs: great opportunities for academia and industry. Signal Transduct. Target Ther. 2019, 4, 64. 10.1038/s41392-019-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horatscheck A.; Wagner S.; Ortwein J.; Kim B. G.; Lisurek M.; Beligny S.; Schutz A.; Rademann J. Benzoylphosphonate-based photoactive phosphopeptide mimetics for modulation of protein tyrosine phosphatases and highly specific labeling of SH2 domains. Angew. Chem., Int. Ed. 2012, 51, 9441–9447. 10.1002/anie.201201475. [DOI] [PubMed] [Google Scholar]
- Wagner S.; Schutz A.; Rademann J. Light-switched inhibitors of protein tyrosine phosphatase PTP1B based on phosphonocarbonyl phenylalanine as photoactive phosphotyrosine mimetic.. Bioorg. Med. Chem. 2015, 23, 2839–2847. 10.1016/j.bmc.2015.03.074. [DOI] [PubMed] [Google Scholar]
- Mandal P. K.; Liao W. S.; McMurray J. S. Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3. Org. Lett. 2009, 11, 3394–3397. 10.1021/ol9012662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.