

Ion mobility spectrometry-mass spectrometry offers the potential to reveal structures of biological molecules not amenable to established biophysical methods. It is, however, still controversial if ion mobility truly reveals biologically relevant structures. This is because the measurement takes place in the gas phase but it is not known for how long the native structure survives in this environment.1 Ion mobility spectrometry is thus a non-equilibrium method2 and successful only to the extent that native structures are metastable within the measurement time-scale.3,4 Ergo, the critical question is, how much “heat” do ions take up from electric fields in an ion mobility spectrometer?
In reference 5, Morsa et al. investigated ion heating in a trapped ion mobility spectrometer (TIMS). The authors claim that “Using the lowest possible transmission voltages […the authors] obtained vibrational effective temperatures Teff,vib of 512 K” for a thermometer ion which accounts for a “high [axial electric field] E/N above the low-field limit”, from which the authors conclude that “the ion transport regimes in traditional [drift-tube ion mobility spectrometry] and modern […] TIMS instrumentations differ therefore advising caution when comparing data obtained on the different platforms.”
If true, these claims made in reference 5 would have far-reaching, adverse implications on the use of TIMS for structural studies: if the ion separation process in TIMS truly heated molecules to ~500 K, then ion mobilities would be unreliable and biological molecules would structurally denature in TIMS.
Indeed, reference 5 expresses such concerns by “questioning the adequacy of the strategy [of keeping the ions stationary employed in TIMS to improve the mobility resolving power] for native mass spectrometry applications targeting fragile analytes associated with low activation energy barrier Ea such as small molecules or noncovalent complexes.”
These claims, however, stand in stark contrast to experiences made by us and others that ion mobilities obtained from TIMS measurements are accurate6,7 and—when carefully tuned—spectra from TIMS and tandem-TIMS resemble “soft” drift-tube spectra for fragile non-covalent peptide assemblies,8 monomeric proteins,2,9–11 and even indicate solvent to be trapped inside protein assemblies.12
Here, we clarify (a) that the ion heating to ~500 K reported in reference 5 is not intrinsic to TIMS and (b) that TIMS enables native mass spectrometry applications:
When discussing the low-field limit and vibrational ion heating in ion mobility spectrometry, all forces that contribute to the center-of-mass collision energy must be considered. Hence, for TIMS, one must consider contributions from the axial DC electric field, the radial RF electric field, and space-charge effects/ion-ion interactions (Figure 1).
Reference 5 attributes ion heating in TIMS exclusively to the axial electric field but does not consider ion heating due to space-charge effects/RF power absorption. The latter can, however, readily be minimized by instrument tuning10 whereas only heating caused by the axial electric field is intrinsic to TIMS.
Reference 5 operates under TIMS settings for which space-charge effects prevail: such effects were observed10,13 already at accumulation times lower than the 100–300 ms used in reference 5.
The TIMS settings used in reference 5 are too “harsh” to retain “native” protein folds, as evidenced by prior literature.11,14,15 In contrast, using “softer” TIMS settings, we successfully retain native-like protein structures.
Differences in instrumentation or tunability between TIMS instruments used in reference 5 and those used by us are negligible and do not explain the ion heating reported in reference 5 (Figure 2).
In sum: Morsa et al. measure the effective ion temperature under conditions that are known to be “hot” but claim that the observed heating is an intrinsic property of the TIMS method.
Figure 1.
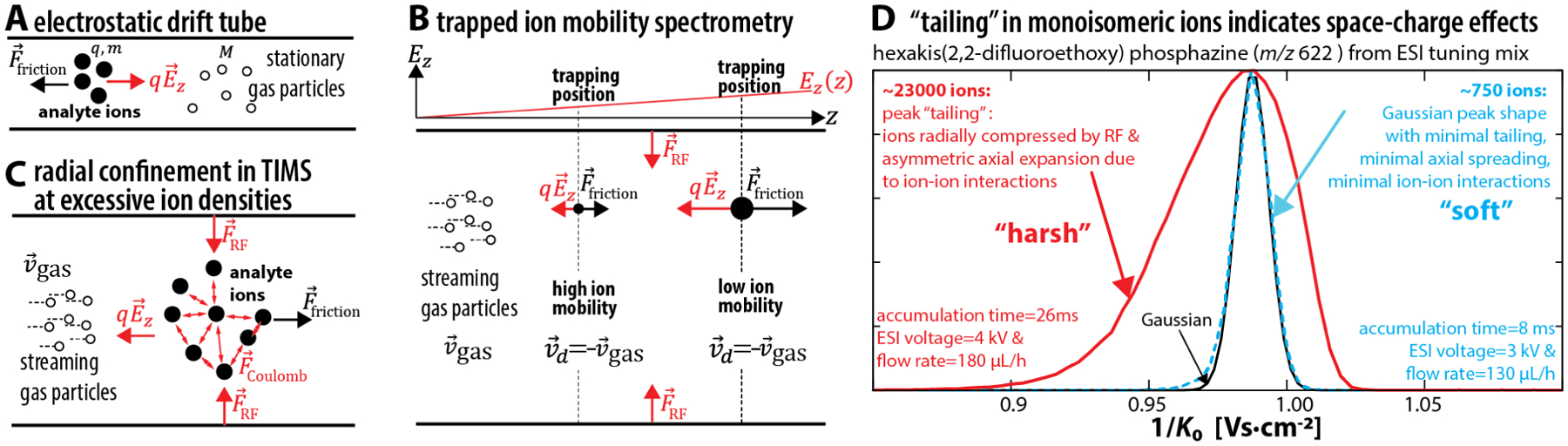
(a) Drift tubes operate at constant electric field strength Ez. (b) TIMS traps ions axially and radially. Axially, ions are trapped along an electric field gradient at different equilibrium positions z where the force on the ion due to Ez is offset by the friction caused by collisions with the gas particles. Relative to the gas flow at velocity , all trapped ions drift with the same velocity . Radially, ions are trapped by an RF electric field. (c) At high ion densities, long-range ion-ion repulsion and power absorption from the RF electric field contribute to the collision energy 〈ε〉. (d) Extremely high ion densities may result in axial spread of the trapped ions in analogy to Figure 2 of reference 5, here shown for hexakis(2,2-difluoroethoxy) phosphazine from Agilent ESI tuning mix (red trace). Low ion densities (blue trace) yield Gaussian peak shapes (black trace). Note that, depending on sample concentrations and electrospray settings, space-charge effects can already be present at accumulation times of 26 ms whereas reference 5 operates at ≥ 100 ms.
Figure 2.

TIMS and tandem-TIMS instruments in our lab (A)-(C) and that used in reference 5 (D). Reference 5 uses a longer TIMS tunnel (D) than used by us in (A) and (B). We recently constructed an orthogonal tandem-TIMS instrument (C) from a commercial timsTOF Pro, which comprises two TIMS devices identical to the one used in reference 5. Note that ion separation occurs in the TIMS analyzer tunnels (key 5) and is comparable for all instruments shown in (A)-(D). (E) Ion mobility spectra for the tetrameric glycoprotein complex avidin recorded under native conditions (3003BCm; 100 mM ammonium acetate) recorded on the coaxial (black trace) and orthogonal (red trace) tandem-TIMS instruments. Notice the strong agreement between the spectra and with cross sections reported from a drift tube (4150 and 4160 Å2, respectively, for charge states 16+ and 17+) and the expected cross section of the x-ray structures (3650–4300 Å2). This demonstrates that also the TIMS design used in reference 5 is sufficiently “soft” for native mass spectrometry. (Differences at the peak bases in (E) do not indicate ion heating because avidin does not compact upon activation; see reference 12.)
The low-field limit in ion mobility spectrometry
Consider the ion swarm in Figure 1a, pulled through a buffer gas with mass M by a static and uniform axial electric field, Ez The motion r(t) of these ions is governed by Boltzmann’s transport equation. It is equivalent, but much more intuitive, to follow Langevin and consider the forces acting on an ion with mass m and charge : q
| (1) |
Here, represents the friction, the stochastic fluctuations, qEz the force due to Ez, and Fext comprises all other forces on the ion that do not contribute to the motion in direction of Ez (e.g. ion-ion interactions, RF-electric field). For a static electric field Ez, ions attain a steady-state drift-velocity vd with
| (2) |
where N (N0) is the (reduced) gas number density and the (reduced) ion mobility K(K0) is a proportionality factor between the drift velocity vd and the applied electric field strength Ez.
The basis for interpreting the ion mobility in terms of the microscopic ion structure is the Einstein relation: it connects, through the diffusion constant, the mobility of the ion to its momentum transfer cross section, Ω.16–18 For this to hold, ions must undergo only binary ion-neutral collisions, but never ternary or ion-ion collisions.16,17 Moreover, the translational energy of the ion must follow a Boltzmann-distribution at the gas temperature Tgas, i.e. the mean center-of-mass collision energy must be approximately thermal (vrel is the relative ion-neutral velocity and R the gas constant).16–18 Hence, K yields structural information only if)
| (3) |
where we defined the center-of-mass translational ion temperature Ttrans.
For purely electrostatic drift tubes (Fext =0), it is trivial to express 〈ε〉 in terms of the drift velocity vd:17,18
| (4) |
By inserting (2) into (4), it is obvious that purely electrostatic drift tubes operate at sufficiently low 〈ε〉 in the limit of
| (5) |
This is the “low-field limit in ion mobility spectrometry”.
We stress: (1) even for purely electrostatic drift tubes (Fext=0), the focus on only the field strength Ez/N is not correct: what is a high field for an ion with high mobility may still be a low field for an ion with low mobility.18 (2) It is the collision energy 〈ε〉, but not Ez/N, that matters: 〈ε〉 must be close to thermal for the Einstein relation to hold. Therefore, when measuring ion mobilities using instruments with Fext ≠ 0, such as TIMS, one must consider all forces that accelerate ions and contribute to 〈ε〉 when discussing the low-field limit.
Only ion heating caused by the axial electric field is intrinsic to the resolving power in TIMS
Figure 1b outlines the forces adding to 〈ε〉 for ions trapped in TIMS. Axially, gas particles streaming through TIMS with velocity push the ions towards the exit. Ions are then trapped at an axial position z, when the axial electric field Ez(z) counterbalances the friction experienced by the ions. Radially, ions are confined by an RF electric field. Hence, generally, the collision energy 〈ε〉 in TIMS arises from forces due to the axial DC and radial RF electric fields and ion-ion interactions.
We stress the significance of the distinction between ion heating caused by the axial electric field versus ion heating due to space-charge effects/RF confinement in TIMS: The resolving power in TIMS is proportional to the gas velocity vgas (Equation 22 in reference 20). Therefore, ion heating caused by the gas velocity electric field Ez(z) is intrinsic to the ion separation process in TIMS: ion heating caused by the axial electric field cannot be reduced without also reducing the TIMS resolving power. On the other hand, ion heating caused by RF / ion-ion interactions can readily be minimized by reducing the RF amplitude and the ion density of the ions trapped in TIMS (for example, by reducing the ion accumulation time, the electrospray voltage, and the desolvation gas temperature as shown in reference 10). Doing so does not diminish the TIMS resolving power.
Through collisional energy transfer,19 the vibrational energy of ions in TIMS arises from contributions of the axial and radial electric fields and ion-ion interactions. This is important when interpreting the vibrational ion temperatures of ~500 K reported in reference 5: Morsa et al probe solely the vibrational ion energy (Teff,vib) of their thermometer ions but do not discuss how the vibrational energy partitions into contributions from ion-ion interactions, or the axial and radial electric fields.
Axial field heating in TIMS
How strongly does the axial electric field contribute to ion heating in TIMS? The axial electric field in TIMS contributes to the collision energy 〈ε〉 analogously to that in a drift tube.16,20,21 When trapped at their equilibrium positions, the ions drift relative to the gas particles at velocity (Figure 1b). Thus, the contribution of the axial field strength to the collision energy can be estimated from the field-heating term in Equation (4), . Because does not depend on the ion, the contribution of the axial field to the collision energy is determined by the gas velocity and comparable for all ions. Further, the operator controls the extend of field heating by controlling the gas velocity through a valve that sets the pressure difference across the tunnel, typically ~0.5–1.0 mbar.22 Hence, subject to technical limitations, the operator controls how strongly the axial electric field contributes to 〈ε〉 by controlling the gas velocity .
Basic considerations reveal that effective ion temperatures reported in reference 5 cannot arise from the axial electric field alone. Vibrational ion temperatures of ~500 K due to the axial electric field would require gas velocities on the order of ~420 m/s, in Equation 4. That is, supersonic gas flow at ~Mach 1.2 through the entire TIMS analyzer. While supersonic molecular beams are known, these are constructed from pressure differentials of >1 atm to ~10−6 mbar through an actual aperture (diameter <1mm) and are only observed in proximity to the aperture.23 By contrast, in TIMS, the pressure drops from ~2–3 mbar to ~1–2 mbar across a ~5–10 cm tunnel with a diameter of ~8 mm.
Indeed, prior work suggests that the contribution of the axial field to 〈ε〉 is generally minor. Park calculated, and experimentally corroborated, that gas velocities in TIMS are on the order of ~120–150 m/s for typical pressure settings.22 These gas velocities suggest that the axial electric field contributes to the translational ion temperature Ttrans by ~15–25 K in nitrogen, in Equation (4). Such minor contributions to the collision energy in prior work are supported by the facts that mobilities calibrated in TIMS2,6,7,10 are within the error of drift tube mobilities and that we succeeded in developing a sample-independent calibration for TIMS.7
RF-confinement and space-charge effects in TIMS
How strongly do space-charge effects and RF power absorption contribute to ion heating in TIMS? Prior work10,13,21 indicates that ion heating due to space-charge effects and the radial confinement strongly increase with the ion (charge) density and the amplitude of the applied RF electric potential.
Previous work13 indicates that space-charge effects prevail at accumulation times of ~100 ms in a TIMS device comparable to the one used in reference 5. In line with this report, we observed that charge state 7+ of the protein ubiquitin unfolds due to space-charge effects/RF power absorption when accumulating ~2000 ions in the TIMS analyzer (see Figure 6 and Table S2 in reference 10). We stress that this unfolding due to RF-heating was observed at accumulation times of ~70 ms. Consistent with these results, proteomics studies using timsTOF instruments use accumulation times of ~50–100 ms.24,25
In the context of these prior reports, the accumulation times of 100–300 ms used in reference 5 strongly indicate the presence of space-charge effects. Another indication for space-charge effects is the strong peak tailing in Figure 2 of reference 5 for the benzylpyridinium ions. It is known that peak tailing in the TIMS spectra for monoisomeric ions can arise from axial spread of the trapped ions in response to a strong radial confinement.21 We illustrate this peak tailing in Figure 1d for a calibrant ion from Agilent tuning mix when trapping about ~23000 identical ions in TIMS. Hence, depending on sample concentration and electrospray settings, such “overfilling” of TIMS can occur already at accumulation times of 26 ms (Figure 1d).
For these reasons, we are concerned by the fact that reference 5 does not consider if space-charge effects/RF power absorption contribute to the reported ion temperatures and neither reports details as to the RF confinement, such as RF amplitude and frequency or ion counts.
Additionally, caution is warranted when interpreting the rate constants kobs as conducted in reference 5. The rate constants kobs in Figure 4 of reference 5 are obtained from measuring the precursor ion survival yield as a function of the trapping time in TIMS. It is, however, well-known that ions “evaporate” from a trap operated at ~1 mbar; the rate of this ion loss strongly depends on, e.g. the ion mobility and the RF amplitude.26,27 This effect has also been discussed for TIMS.21,28 With respect to reference 5, this means that the survival yield may decrease even in the absence of any fragmentation. Reference 5 does not, however, consider this ion loss in their interpretation of the rate constants kobs.
Reference 5 does not retain native-like protein structures in TIMS
On the basis of Figure 5d of reference 5, the authors claim to retain “compact ‘native’ folds” of cytochrome c for charge state 8+. The spectrum shows a main peak at ~2150 Å2. However, the cross section calculated for the x-ray structure of cytochrome c in nitrogen gas15 is ~1565 Å2. This cross section is consistent with nitrogen data recorded in drift-tubes for charge states 6+ (1490 Å2, 1477 Å2) and 7+ (1590 Å2, 1536 Å2), respectively, produced from native electrospray.14,15 Thus, the predominant peak observed for cytochrome c in reference 5 is ~40% larger than what is expected for the native state and hence reflects denatured structures. Furthermore, prior work using TIMS exhibits a main peak at ~1700 Å2 for cytochrome c 8+ (Figure 3 in reference 11), which indicates that the TIMS settings used in reference 5 are not appropriate to study biological compounds.
Differences in TIMS instrumentation or tunability do not rationalize ion heating reported in reference 5
It is unlikely that particularities in design or tunability would cause the TIMS device used in reference 5 to produce intrinsically “hotter” ions than the single-TIMS and tandem-TIMS instruments used in prior work.
Figures 2A–D show the various TIMS instruments used in our laboratory and that of reference 5. The ion deflector, entrance and exit ion funnels are identical in all instruments. Differences exist only in the TIMS tunnels: the TIMS designs in Figures 2A and 2B used previously by us2,7–10,12,16 and others11,22,28 have a shorter TIMS tunnel (46 mm) and perform ion accumulation and mobility analysis sequentially in time. Our recently constructed orthogonal tandem-TIMS (Figure 2C) has two TIMS devices with a longer tunnel (96 mm). These two TIMS devices are identical to the one used in reference 5 (Figure 2D). The increased length of the tunnel increases the duty cycle by enabling ion accumulation in the first half of the tunnel concurrently with mobility analysis in the second half.
The TIMS analyzer tunnels (Figure 2, key (5)) are essentially the same in all instruments: mobility analysis occurs in a region of segmented quadrupolar electrodes; the axial electric field gradients are equivalent; the gas velocity can be adjusted by a valve; mobility-calibration can be conducted at different gas velocities. Hence, we do not see a fundamental, substantiated argument why the TIMS design used in reference 5 should be intrinsically “hotter” than any of the other TIMS instruments.
We note substantial differences in “soft” settings used in our lab2,8,10 and reference 5 for small monomeric proteins (Table 1): we operate TIMS at (1) lower desolvation gas temperature to prevent thermal denaturation of the proteins; (2) shorter accumulation times to reduce the ion density in TIMS and avoid space-charge effects; (3) lower DC bias across the entrance funnel to minimize energetic collisions (Figure 4 in reference 10). Overall, low ion densities and low RF-electric fields are crucial for “soft” TIMS experiments, because this minimizes the contribution of ion-ion interactions and the RF electric field to the collision energy 〈ε〉, thereby minimizing the vibrational energy of the ions in TIMS (Figure 1).
Table 1.
Key differences between “soft” TIMS settings employed in our laboratory and by reference 5
Using our “soft” settings from Table 1, our TIMS and tandem-TIMS instruments shown in Figures 2A and 2B produce spectra consistent with “soft” drift tubes and x-ray/NMR structures.2,7,9,10,12 Tandem-TIMS reproduces “soft” drift tube cross sections and relative abundances for the protein ubiquitin in different solvents.2,7,9,10 In addition, computational analysis indicates that the protein ubiquitin detected by tandem-TIMS strongly resembles the native NMR structure.2 For the nonapep-tide bradykinin, tandem-TIMS reveals assemblies in accord with a “soft” drift tube.8 Indeed, tandem-TIMS is sufficiently “soft” so that solvent particles are retained in the interior of the avidin tetramer.12
Does the TIMS design used in reference 5 (Figure 2D) and in our orthogonal tandem-TIMS (Figure 2C) produce substantially “hotter” ions, rendering these instruments inadequate for native mass spectrometry applications? Figure 2E demonstrates that this is highly unlikely.
We recorded spectra for the homotetrameric glycoprotein complex avidin on our co-axial12 (Figure 2B) and orthogonal (Figure 2D) tandem-TIMS instruments and compare them in Figure 2E. The spectra reveal no significant consequence of performing two consecutive ion mobility separations in TIMS devices that are identical to the one used in reference 5. Further, the cross sections obtained by both tandem-TIMS instruments (see Figure 2E) are consistent with the expected cross sections for the x-ray structures12 (3650–4300 Å2) and those measured for avidin in a drift tube14 (4150 and 4160 Å2, respectively, for charge state 16+ and 17+). These results demonstrate that the TIMS design used in reference 5 is sufficiently “soft” to enable native mass spectrometry applications.
CONCLUSIONS
Here, we clarified (a) that the ion heating to ~500 K reported in reference 5 is not intrinsic to TIMS and (b) that TIMS enables native mass spectrometry applications.
ACKNOWLEDGEMENTS
This work was supported by the National Science Foundation and the National Institutes of Health under CHE-1654608 and R01GM135682 (C.B.) C.B. acknowledges a Joint Development Agreement with Bruker Daltonics (Billerica, MA).
REFERENCES
- (1).Breuker K; McLafferty FW Proc. Natl. Acad. Sci 2008, 105 (47), 18145–18152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bleiholder C; Liu FC J. Phys. Chem. B 2019, 123 (13), 2756–2769. [DOI] [PubMed] [Google Scholar]
- (3).Wyttenbach T; Bowers MT J. Phys. Chem. B 2011, 115 (42), 12266–12275. [DOI] [PubMed] [Google Scholar]
- (4).Koeniger SL; Merenbloom SI; Sevugarajan S; Clemmer DE J. Am. Chem. Soc 2006, 128 (35), 11713–11719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Morsa D; Hanozin E; Eppe G; Quinton L; Gabelica V; Pauw ED Anal. Chem 2020, 92 [DOI] [PubMed] [Google Scholar]
- (6)., Naylor CN; Reinecke T; Ridgeway ME; Park MA; Clowers BH J. Am. Soc. Mass Spectrom 2019, 30 (10), 2152–2162. [DOI] [PubMed] [Google Scholar]
- (7).Chai M; Young MN; Liu FC; Bleiholder C Anal. Chem 2018, 90 (15), 9040–9047. [DOI] [PubMed] [Google Scholar]
- (8).Kirk SR; Liu FC; Cropley TC; Carlock HR; Bleiholder C J. Am. Soc. Mass Spectrom 2019, 30 (7), 1204–1212. [DOI] [PubMed] [Google Scholar]
- (9).Liu FC; Ridgeway ME; Park MA; Bleiholder C Analyst 2018, 143 (10), 2249–2258. [DOI] [PubMed] [Google Scholar]
- (10).Liu FC; Kirk SR; Bleiholder C Analyst 2016, 141 (12), 3722–3730. [DOI] [PubMed] [Google Scholar]
- (11).Molano-Arevalo JC; Jeanne Dit Fouque K; Pham K; Miksovska J; Ridgeway ME; Park MA; Fernandez-Lima F Anal. Chem 2017, 89 (17), 8757–8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Liu FC; Cropley TC; Ridgeway ME; Park MA; Bleiholder C Anal. Chem 2020, 92 (6), 4459–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ridgeway ME; Lubeck M; Jordens J; Mann M; Park MA Int. J. Mass Spectrom 2018, 425, 22–35. [Google Scholar]
- (14).Bush MF; Hall Z; Giles K; Hoyes J; Robinson CV; Ruotolo BT Anal. Chem 2010, 82 (22), 9557–9565. [DOI] [PubMed] [Google Scholar]
- (15).May JC; Jurneczko E; Stow SM; Kratochvil I; Kalkhof S; McLean JA Int. J. Mass Spectrom 2018, 427, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bleiholder C Int. J. Mass Spectrom 2016, 399–400, 1–9. [Google Scholar]
- (17).Revercomb HE; Mason EA Anal. Chem 1975, 47 (7), 970–983. [Google Scholar]
- (18).Transport Properties of Ions in Gases. In Transport Properties of Ions in Gases; Mason EA, McDaniel EW., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, FRG, 2005; pp 487–497. [Google Scholar]
- (19).Rapp D; Kassal T Chem. Rev 1969, 69 (1), 61–102. [Google Scholar]
- (20).Michelmann K; Silveira JA; Ridgeway ME; Park MA J. Am. Soc. Mass Spectrom 2015, 26 (1), 14–24. [DOI] [PubMed] [Google Scholar]
- (21).Hernandez DR; DeBord JD; Ridgeway ME; Kaplan DA; Park MA; Fernandez-Lima F Analyst 2014, 139 (8), 1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Silveira JA; Michelmann K; Ridgeway ME; Park MA J. Am. Soc. Mass Spectrom 2016, 27 (4), 585–595. [DOI] [PubMed] [Google Scholar]
- (23).Scoles G Atomic and Molecular Beam Methods; Oxford University Press, 1988; Vol. 1. [Google Scholar]
- (24).Meier F; Beck S; Grassl N; Lubeck M; Park MA; Raether O; Mann M J. Proteome Res. 2015, 14 (12), 5378–5387. [DOI] [PubMed] [Google Scholar]
- (25).Vasilopoulou CG; Sulek K; Brunner A-D; Meitei NS; Schweiger-Hufnagel U; Meyer SW; Barsch A; Mann M; Meier F Nat. Commun 2020, 11 (1), 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tolmachev AV; Chernushevich IV; Dodonov AF; Standing KG Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At 1997, 124 (1), 112–119. [Google Scholar]
- (27).Zhang X; Garimella SVB; Prost SA; Webb IK; Chen T-C; Tang K; Tolmachev AV; Norheim RV; Baker ES; Anderson GA; Ibrahim YM; Smith RD Anal. Chem 2015, 87 (12), 6010–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ridgeway ME; Silveira JA; Meier JE; Park MA Analyst 2015, 140 (20), 6964–6972. [DOI] [PubMed] [Google Scholar]