

Abstract
Purpose:
Epithelial-to-mesenchymal transition (EMT) is important in cancer progression and metastasis. We and others have previously reported that lysyl oxidase (LOX) is overexpressed in aggressive cancers, is associated with increased mortality, and regulates EMT. However, the mechanism by which LOX mediates EMT is unknown. In this study, we investigated the effect of LOX on mediators of EMT.
Experimental Design:
We used chromatin immunoprecipitation and promoter luciferase assays to determine the target gene of LOX. To determine the effects of SNAI2 in vivo, we used our metastatic anaplastic thyroid cancer (ATC) mouse model. To investigate the effects of LOX and SNAI2 on MMPs and TIMPs, protein arrays were used. Primary tumors from patients with metastatic, breast and colon cancer, and tissue array for thyroid cancer were assessed for SNAI2 and TIMP4 expression by immunohistochemistry.
Results:
We found that LOX knockdown decreases SNAI2 expression in cancer cell lines. Furthermore, knockdown of LOX reduced SNAI2 expression in a metastatic mouse model of thyroid cancer. We also demonstrated that LOX binds and transactivates the SNAI2 promoter. We found a direct correlation in thyroid and breast cancer samples between LOX and SNAI2 expression. To understand how LOX/SNAI2 axis mediates these effects, we performed a comprehensive analysis of MMPs/TIMPs. LOX and SNAI2 depletion reduced TIMP4 secretion. Analysis of SNAI2 and TIMP4 expression showed overexpression of both proteins in aggressive thyroid, colon, and breast tumors.
Conclusions:
Our findings provide new evidence that LOX regulates SNAI2 expression and that SNAI2-mediated TIMP4 secretion plays a role in cancer progression.
Introduction
Locally invasive and metastatic cancer causes significant morbidity and mortality and is often resistant to multimodal therapy in many epithelial human solid malignancies. An omnipresent feature of such advanced cancers is dedifferentiation and epithelial-to-mesenchymal transition (EMT). One prototypical cancer is the rare, but uniformly lethal, anaplastic (undifferentiated) thyroid cancer (ATC; refs. 1, 2). Activation of EMT is a key feature of anaplastic transformation. The initial stage of metastasis progression is dependent on EMT. EMT is characterized by cellular, morphologic changes, loss of polarity, in addition to loss of cell– adhesion, and gain of migratory and invasive properties which is associated with increased cell motility (3, 4).
Transcription factors such as SNAIL1, SNAI2, ZEB1, and TWIST1 have been found to act as oncogenic transcription factors and have been implicated in EMT. SNAI2 has five zinc finger domains that play a pivotal role during embryo development and mesenchymal tumorigenesis (5). Upregulation of SNAI2 has been associated with inhibition of E-cadherin expression, lymph node metastasis, and shorter survival time in some cancers (6, 7). However, our understanding of the regulation of EMT transcription factors in aggressive cancers and cancer progression is incomplete, and a broader understanding could provide important insights into new targets for cancer therapy.
Lysyl oxidase (LOX) is an extracellular copper-dependent amine oxidase that catalyzes the exchange of an amine to aldehyde group on a peptidyl lysine, producing hydrogen peroxide and ammonia as by-products of catalytic activity. One of the well-characterized functions of LOX is the covalent cross-linkage of collagens or elastin to increase extracellular matrix (ECM) tensile strength. A growing body of evidence indicates that LOX promotes tumor progression and metastasis by regulating collagen cross-linking and stiffness in breast cancer, colorectal cancer, and lung cancer (8-12). We have previously shown that LOX is upregulated in ATC, promotes tumor growth and metastasis in ATC, and is associated with higher mortality rate (13). Remodeling of the ECM and changes in cell interactions with the ECM are essential in the initiation and progression of EMT and are associated with dysregulation of matrix metalloproteinases (MMP) and tissue inhibitors of matrix metalloproteinases (TIMP), which can lead to the degradation of the ECM (14). Overexpression of MMPs has been shown in several human malignancies and has been associated with cancer initiation and progression by directly affecting cellular adhesion and by enhancing cancer cell invasion and migration (15). The classic function of TIMPs is to regulate the proteolytic activity of MMPs. TIMPs are involved in several processes, including cell invasion and migration, cell proliferation and apoptosis, and angiogenesis (16).
In this study, we determined the role of LOX in the regulation of EMT-inducing transcription factors. Our in vitro and in vivo data identified SNAI2 as a target of LOX. These results led us to investigate the role of the LOX/SNAI2 axis in the regulation of MMPs and TIMPs in cancer cells. We found that LOX and SNAI2 regulate TIMP4 secretion, which in turn modulates VEGF expression.
Materials and Methods
Cell culture, transfection, and drug treatment
The 8505c ATC cell line (BRAF V600E, TP53, EGFR, PIK3R1, PIK3R2 mutations) was obtained from the European Collection of Cell Cultures (17). The THJ-16T ATC cell line (TP53, RB, PIKCA) was kindly provided by Dr. John A. Copland (Mayo Clinic; ref. 18). The HeLa cell line was purchased from ATCC. The MDA-MB231 [estrogen repceptor (ER)−, progesterone receptor (PR)−, HER2−] and MCF-7 (ER+, PR+/−, HER2−) breast cancer cell line was provided by the cell repository of the National Cancer Institute (NCI, Frederick, MD; refs. 19, 20). The BCPAP papillary thyroid cancer cell line (BRAF V600E, TP53 mutations) was obtained from the Leibniz-Institute DSMZ (21). The cell lines were maintained in DMEM with d-glucose (4,500 mg/L), l-glutamine (2 mmol/L), and sodium pyruvate (110 mg/L), supplemented with 10% fetal calf serum (FCS), penicillin (10,000 U/mL), streptomycin (10,000 U/mL), and fungizone (250 mg/mL), all in a standard humidified incubator at 37°C, in a 5% CO2 atmosphere. 8505C, THJ-16T, MDA-MB231 were used at early passages and we performed the cell line authentication using Short Tandem Repeat assay (STR). MCF-7 were used at early passages (less than 6 months after resuscitation of the original cells), the cell line authentication was done by NCI Frederick Cell repository using Applied Biosystems AmpFISTR Identifier testing with PCR amplification (Applied Biosystems, Thermofisher Scientific). BCPAP cells were used at very early passages (less than 6 months after resuscitation of the original cells), the cell line authentication was done by DSMZ using multiplex PCR of minisatellite markers. HeLa cells obtained from ATCC were used at early passages (less than 6 months after resuscitation of the original cells), the authentication was performed by the company using the STR method.
Tissue samples and patient information
Thyroid tissue was obtained at the time of surgical resection, snap-frozen, and stored at −80°C. Serial tissue sample sections were used for RNA extraction, stained with hematoxylin and eosin, and reviewed by a pathologist to confirm the diagnosis and ensure a tumor nuclei content of more than 80%. The study was approved by the Office for Human Research Protections, at the Department of Health and Human Services. Patient information was collected prospectively under an Institutional Review Board–approved protocol at the NIH (Bethesda, MD) after obtaining written informed consent.
Chromatin immunoprecipitation assay
A chromatin immunoprecipitation (ChIP) assay was performed using the Chromatin Immunoprecipitation Assay Kit (Magna ChIP; Millipore) according to the manufacturer's instructions. Briefly, the cells were cross-linked by 1% formaldehyde for 10 minutes. The formaldehyde was quenched using 2 ml of 10X glycine for 5 minutes at room temperature before harvesting. Cells were collected by centrifugation in PBS containing protease inhibitors and were lysed in SDS lysis buffer. Soluble chromatin was prepared after sonication to a DNA length of 200 to 500 base pairs. Fragmented chromatin was immunoprecipitated using antibodies against LOX (Abcam) or IgG control overnight at 4°C on a rotating platform. The magnetic beads were washed, chromatin extracted, and protein–DNA complexes reverse cross-linked. DNA was purified and analyzed by PCR using the specific primers for SNAI2 promoter. The total input was the supernatant from the no-antibody control.
Cell transfection
siRNA LOX (siLOX), siRNA SNAI2 (siSNAI2), and siRNA TIMP4 (siTIMP4) and their corresponding negative control, siControl (siC; Life technologies, Thermo Fisher Scientific), were transiently transfected into 2 × 105 cells in 6-well plates, using the transfection reagent RNAiMax (Invitrogen, Thermo Fisher Scientific) according to the manufacturer's protocol.
Cancer cells were seeded into a 96-well plate (15,000 cells per well). After 24 hours, the cells were co-transfected with a GoClone reporter vector containing the promoter region of SNAI2 (Switch-Gear Genomics, Active Motif) or mutant vector (generated by GENEWIZ using the SNAI2 promoter from SwitchGear Genomics) and the siLOX (Applied Biosystems, Thermo Fisher Scientific), using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. Luciferase activity was measured 24 hours after transfection, using the LightSwitch Luciferase Assay Reagent (SwitchGear Genomics) according to the manufacturer's protocol.
RNA extraction
Total RNA was extracted from snap-frozen tissues and cell lines using TRIzol reagent (Invitrogen, Thermo Fisher Scientific) according to the manufacturer's protocol. RNA yield was determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific).
Real-time PCR
Gene expression levels were measured using specific primers and probes. Briefly, 500 to 1,000 ng of total RNA was reverse transcribed using a High Capacity Reverse Transcription cDNA kit (Cat #4374967, Applied Biosystems, Thermo Fisher Scientific), and the resulting cDNA was diluted and amplified according to the manufacturer's instructions. GAPDH was used as an endogenous control. Gene expression levels were calculated using SDS 2.3 software (Applied Biosystems, Thermo Fisher Scientific).
Cell invasion and migration assays
Cell invasion and migration were assessed using a Transwell chamber assay (Corning), according to the manufacturer's protocol, with and without Matrigel, respectively. The medium in the lower chamber of the plate was supplemented with 10% FBS as a chemoattractant. After 22 hours of incubation at 37°C, the cells attached to the upper side of the Transwell were cleaned with cotton swabs, and the cells invading through the bottom surface of the inserts were fixed, stained with Diff-Quik (Dade Behring), and photographed and counted using ImageJ software (NIH).
VEGF ELISA test
Four cancer cell lines were transfected with either SiC, siLOX, or siSNAI2 in 6-well plates and grown for 48 hours; next, they were washed in PBS and incubated in a low-serum medium (DMEM, 0.5% FBS) for 24 hours. The conditioned medium was then centrifuged at 1,500 rpm for 5 minutes, and the supernatants were collected, whereas the cells were harvested in cell lysis buffer. The concentration of VEGF in the supernatants was determined by ELISA using a Quantikine Human VEGF Immunoassay kit (R&D Systems), according to the manufacturer's protocol. The color intensity was determined by measuring the absorbance in a SpectraMax M5 spectrophotometer (Molecular Devices) at a wavelength of 450 nm with correction at 540 nm. The VEGF concentration was normalized to the total protein. VEGF release was then expressed as μg of VEGF/mg of total protein.
In vitro tubule formation
Cancer cells (30,000 cells) were incubated in 24-well plates coated with Matrigel (Matrigel Basement Membrane Matrix, Corning). The tubes made by the epithelial cancer cells on the Matrigel were photographed using a Zeiss microscope at a magnification of 10×.
Immunohistochemical analysis
Sections were deparaffinized and rehydrated, and antigen retrieval was performed with citrate buffer in a water bath at 120°C. The sections were incubated with the anti-LOX antibody (1:100; Abcam), anti-SNAI2 or anti-TIMP4 (1:100, Abcam) overnight at 4°C, followed by incubation with a biotinylated secondary antibody for 1 hour at room temperature. The slides were developed with diaminobenzidine [DAB; EnVision+ Kit system HRP (DAB), Dako] and counterstained with hematoxylin. The slides were scanned at a 20× magnification using a ScanScope XT digital slide scanner (Aperio Technologies, Leica) to create whole-slide image data files at a resolution of 0.5 μm/pixel, and they were viewed using ImageScope software (Aperio Technologies).
Tissue microarrays were purchased from US Biomax (#TH641). These arrays included duplicates of six follicular adenomas, six follicular thyroid carcinomas, six papillary thyroid carcinomas, six anaplastic thyroid carcinomas, and 16 normal tissues from lung, thyroid, and testis.
Protein extraction and Western blot assay
Cells were lysed in a buffer containing 10 mmol/L Tris and 1% SDS, protease inhibitor cocktail (Sigma Aldrich), and phosphatase inhibitors (PhosphoSTOP, Roche). The lysates were quantified for protein concentrations using a BCA Protein Assay kit (Pierce, Life technologies). Cell lysates were analyzed in SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Invitrogen, Thermo Fisher Scientific). The membranes were blocked with 5% BSA in TBS-Tween buffer and then incubated with primary antibodies overnight at 4°C. The membranes were then incubated with the HRP-conjugated secondary antibodies. Proteins bands were analyzed using enhanced chemi-luminescence (ECL) reagent (Pierce, Thermo Fisher Scientific).
Antibody arrays
To assess the expression and secretion of MMPs/TIMPs and angiogenesis-related protein in supernatant and cell lysates, we used angiogenesis-related protein MMPs/TIMPs array (Abcam) and angiogenesis array (R&D systems).
Metastatic mouse models
The animal studies were approved by the Institutional Animal Care and Use Committee of the National Cancer Institute. The mice were maintained according to guidelines set forth by Animal Research Advisory Committee of the NIH. For the metastatic mouse model, 8505C-Luc cells (n = 30,000) transfected with SiSNAI2 and its corresponding control were injected into the tail veins of Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (22). The mice were injected intraperitoneally with 30 mg/mL of luciferin. Following anesthesia, the mice were imaged, and the images were analyzed using IVIS Living Image software (Caliper Life).
The surface area and the number of metastatic sites per animal were counted on hematoxylin and eosin (H&E) slides using Aperio ImageScope v12.1.0.5029 (Leica).
Results
LOX regulates SNAI2 expression
We previously observed that LOX regulates cancer progression and metastasis but does not affect the expression of N-cadherin, E-cadherin, and vimentin in ATC cells. Thus, we asked whether LOX mediates its effect by regulating EMT transcription factors. One of the most common features associated with EMT is the overexpression of the transcription factors SNAIL, SNAI2, ZEB-1, and TWIST-1. These proteins are involved in EMT during embryonic development as well as in cancer progression and metastasis. To explore the role of LOX in the regulation of these EMT transcription factors, we compared their expression level in cells transfected with either siRNA LOX or siRNA control. LOX knockdown did not significantly alter the mRNA levels of SNAI1, ZEB-1, or TWIST-1 (Fig. 1A). However, LOX knockdown showed a significant decrease in SNAI2 mRNA and protein expression levels (Fig. 1A and B). The pretreatment of cancer cells in culture with 100 μmol/L of β-aminopropionitrile (BAPN), an inhibitor of LOX catalytic activity, for 48 hours did not alter the expression of SNAI2 protein (Fig. 1C). These data suggest that LOX regulates SNAI2 expression and that this effect is not mediated by the catalytic activity of LOX.
Figure 1.
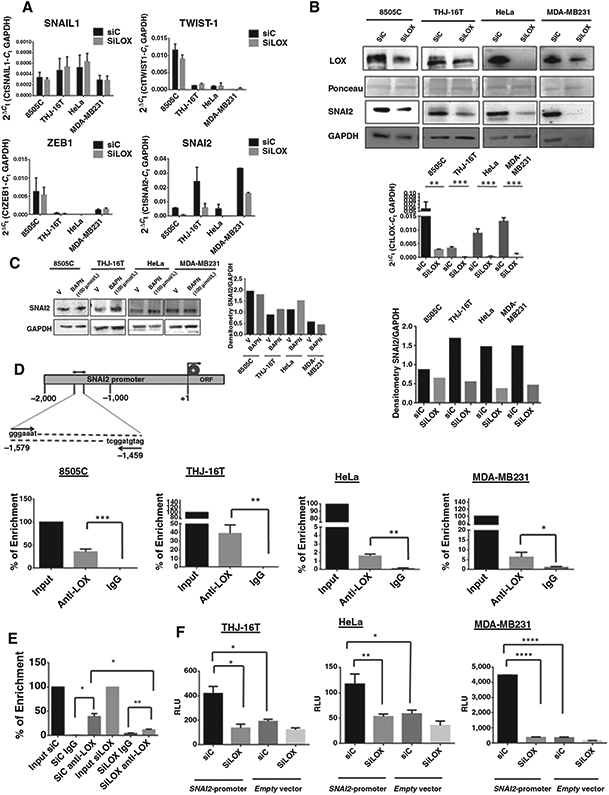
LOX directly binds to SNAI2 promoter and regulates SNAI2 expression. A, knockdown of LOX in four cancer cell lines decreased SNAI2 transcript 72 hours after transfection. There was no significant difference in SNAIL1, TWIST-1, and ZEB1 mRNA expression with LOX knockdown. B, LOX knockdown inhibits LOX protein and mRNA and SNAI2 protein expression in cancer cell lines 72 hours after transfection. Bar graph of SNAI2 expression quantified by densitometry and normalized to GAPDH. C, inhibition of LOX catalytic activity by 100 μmol/L BAPN for 48 hours does not significantly alter SNAI2 protein expression, as shown by Western blotting and by band densitometry of SNAI2 normalized to GAPDH. D, top, schematic representation of the promoter region of human SNAI2. Binding sites of the primers used for analysis by hIP are indicated relative to the transcription start site. Bottom, semiquantitative RT-PCR of ChIP samples. Fragmented chromatin was immunoprecipitated using monoclonal antibody anti-LOX or IgG control. Data analysis shows DNA binding of LOX to SNAI2 promoter in 8505C, THJ-16T, MDA-MB231, and HeLa cells. E, SNAI2 binding was decreased following LOX knockdown in THJ-16T. F, LOX knockdown represses SNAI2 promoter activity in cancer cell lines. The three cancer cell lines have been transfected with SiLOX or siControl in combination with a human SNAI2 promoter reporter construct, and luciferase activity was measured. All experiments have been performed at least three times. Errors bars represent mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
We next asked whether LOX directly regulates the transcription of SNAI2. We performed ChIP assays of the promoter region of SNAI2 using monoclonal anti-LOX antibody. Quantitative PCR showed an enrichment of SNAI2 promoter with anti-LOX antibody as compared with IgG control in the cancer cell lines (Fig. 1D). Confirming the specificity of the assay and the antibody binding, no enrichment was observed when LOX was depleted using siRNA LOX (Fig. 1E). To determine whether SNAI2 promoter activity was regulated by LOX, we performed a luciferase reporter assay. The cancer cell lines were transiently co-transfected with siLOX and a reporter construct in which the human SNAI2 promoter region was cloned upstream of the luciferase reporter. Inhibition of LOX expression decreased SNAI2 promoter activity, as compared with that of the SiControl (Fig. 1F). To further validate LOX binding the SNAI2 promoter and regulating transcription of SNAI2, an SNAI2 promoter mutant vector was generated and transfected in MCF-7 cells. Disruption of SNAI2 promoter sequence significantly reduced luciferase activity (Supplementary Fig. S1). These results suggest that LOX binds to the SNAI2 promoter and regulates the transcriptional activity of the SNAI2 promoter.
Analysis of LOX–SNAI2 interaction in pre-EMT cell lines MCF7 and BCPAP using a ChIP assay showed an enrichment of SNAI2 promoter with anti-LOX antibody (Supplementary Fig. S2A). Furthermore, using the SNAI2 reporter assay, we observed a significant decrease of luciferase activity in siLOX-transfected cells compared with siControl (Supplementary Fig. S2B). The inhibitory effects of LOX depletion on SNAI2 promoter activity was more pronounced in MCF-7 than in BCPAP cells. This is consistent with our finding that LOX depletion did not impair SNAI2 mRNA expression in BCPAP cell line (Supplementary Fig. S2C and S2D). Taken together, these data suggest that the low level of SNAI2 in the pre-EMT cell lines underlies a modest regulation of SNAI2 by LOX.
SNAI2 is overexpressed in aggressive thyroid cancer and directly correlates with LOX expression in thyroid and breast cancers
To determine the expression profile of SNAI2 in cancer, we analyzed SNAI2 mRNA expression in a cohort of patients with differentiated thyroid cancer (papillary thyroid cancer, PTC), poorly differentiated thyroid cancer (PDTC), and undifferentiated thyroid cancer (ATC) by RT-PCR. PTC and ATC exhibited a significantly higher expression of SNAI2 mRNA, as compared with normal thyroid tissue samples (Fig. 2A). Furthermore, SNAI2 expression was significantly higher in ATC than in PTC (Fig. 2A). We also found a significant positive correlation between SNAI2 and LOX expression in our cohort of patients with thyroid cancer (Fig. 2B). In addition, we performed the same analysis in a dataset from the publicly available Genome Expression Omnibus (GEO) database and found a significant positive correlation between SNAI2 and LOX expression in cohorts of patients with thyroid cancer and breast cancer (Fig. 2C and D). We also determined, by immunohistochemistry, the expression level of SNAI2 and LOX protein in patients with aggressive thyroid cancer (tall-cell variant of papillary thyroid carcinoma). Strong LOX and SNAI2 stainings were observed in cancer tissue, whereas both proteins were weakly expressed in adjacent normal tissue (Fig. 2E). Taken together, these data further support our in vitro findings that LOX regulates SNAI2 expression.
Figure 2.

SNAI2 expression correlates with LOX expression and is higher in aggressive thyroid cancers. A, scatter plot of SNAI2 mRNA expression in different histologic types of thyroid cancer. Expression level of SNAI2 was normalized to GAPDH. B–D, LOX and SNAI2 mRNA expression directly correlate in human cancer samples. B, scatter plots showing positive correlation between LOX and SNAI2 mRNA expression in a cohort of patients with thyroid cancer, by RT-PCR. C, LOX and SNAI2 mRNA expression in thyroid cancer samples from publicly available GEO dataset (GSE33630). D, LOX and SNAI2 mRNA expression in breast cancer samples from publicly available GEO dataset (GSE65194). E, representative immunohistochemical staining in a tall-cell variant of PTC (top) and normal thyroid tissue (bottom) for LOX and SNAI2 (magnification, 20×). F, correlative analysis of LOX and SNAI2 expression and the percentage of stromal cells in thyroid cancer samples from the publicly available TCGA dataset. G, SNAI2 is highly expressed in thyroid tumors with extrathyroidal invasion from the publicly available TCGA dataset. *, P < 0.05; **, P < 0.01. ns, not significant.
The tumor microenvironment is an important factor in cancer progression and EMT (23). Stromal cells are involved in the metastatic process and play a major role in promoting primary tumor growth. To evaluate whether LOX and SNAI2 are also associated with stromal cell infiltration, we used The Cancer Genome Atlas (TCGA) database for thyroid cancer and found a significant positive correlation between LOX and SNAI2 mRNA expression levels and the percentage of stromal cells in tumor samples (Fig. 2F). To determine whether SNAI2 is also associated with other pathologic phenotypic features of EMT, we used the same TCGA dataset and found that SNAI2 mRNA expression was significantly higher in patients with extrathyroidal invasion, an important prognostic factor in patients with thyroid cancer (Fig. 2G). This finding supports our results that suggest that both LOX and SNAI2 are involved in cancer progression and metastasis as they are associated with features in the tumor microenvironment that promote EMT.
SNAI2 recapitulates the effects of LOX on cancer metastasis in vitro and in vivo
To determine whether SNAI2 can recapitulate the effects of LOX in cancer metastasis, we performed functional knockdown studies of SNAI2. SNAI2 depletion reduced cellular invasion and migration in vitro (Fig. 3A and B). To further investigate the role of SNAI2 in a mouse model of metastatic thyroid cancer, we injected transiently transfected 8505C ATC cell lines (8505C-Luc-SiControl, 8505C-Luc-SiSNAI2, and 8505C-Luc-SiLOX) into the tail vein of immunodeficient mice. Mice injected with 8505C-Luc-SiControl had more and larger lung and liver metastasis than those injected with 8505C-Luc-siSNAI2 cells (Fig. 3C-E). Previously, we reported that LOX depletion resulted in decreased lung metastases levels in mice. Histologic analysis of lung tissues from each mouse revealed a decline in SNAI2 expression in the 8505C-Luc-siLOX mice group (Fig. 3F). Collectively, these results demonstrate that SNAI2 depletion mimics the effects of LOX on cancer metastasis (13).
Figure 3.

SNAI2 knockdown reduces cancer cell invasion and metastasis. A and B, left, invading and migrating cells through the inserts were photographed (images) and quantitated (graphs). RT-PCR and Western blot analysis of SNAI2 expression in siSNAI2-transfected cells compared with siControl (A, right). Experiments were performed at least 3 times and the values represent the mean ± SD from triplicate wells. C, representative bioluminescent imaging of SiC and SiSNAI2 mice 3 weeks after tail vein injection of 8505C cell line and quantitation of the luciferase signal in siC and siSNAI2 mice per groups. n = 4 mice per group. The values represent the mean ± SEM. D, surface area of lung tumors and number of lung tumor sites per mouse were measured using Aperio ImageScope software from H&E slides. E, ex vivo images of lung and liver metastasis from siC and siSNAI2 mouse. F, representative immunohistochemistry for SNAI2 protein in lung metastasis tumors of siC and siLOX mice. Knockdown of LOX in vivo decreased the expression of SNAI2 in lung tissues by immunohistochemistry. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
LOX and SNAI2 regulate TIMP4 secretion
To gain insight into how LOX and SNAI2 regulate cancer invasion and metastasis, we analyzed their effects on MMPs and TIMPs, as these factors are widely known as key mediators in cancer invasion and metastasis. Protein analysis in cell culture supernatant showed that TIMP4 was reduced in siLOX and siSNAI2 samples, as compared with control, whereas other proteins were either undetectable or not significantly changed (Fig. 4A). We confirmed by ELISA that TIMP4 secretion was decreased in cancer cells transfected with siLOX or siSNAI2 (Fig. 4B). There was no significant difference in TIMP4 protein levels in the total lysate from cells transfected with either siLOX or siSNAI2 (Supplementary Fig. S3A). Also, no significant difference in TIMP4 intracellular levels was seen by Western blot analysis (Supplementary Fig. S3B). In pre-EMT cell lines, a decrease of TIMP4 secretion with knockdown of LOX and SNAI2 was only observed in MCF-7 cells (Supplementary Fig. S3C). Taken together, these data suggest that SNAI2 and LOX modulate the secretion of TIMP4 from cancer cells.
Figure 4.
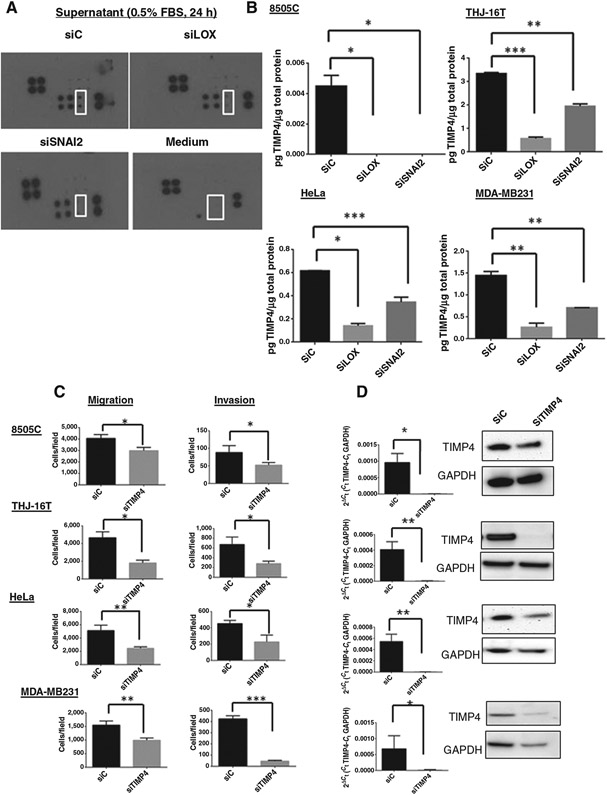
Knockdown of LOX and SNAI2 decreases TIMP4 secretion. A, array blots of MMPs and TIMPs in cell culture supernatant in THJ-16T cells transfected with either siC, siLOX, or siSNAI2. Control medium with 0.5% FBS and with no cells was used as a negative control. Duplicates of TIMP4 are indicated (white square). Positive control proteins are in the top left and lower right corners of each panel. B, analysis by ELISA of TIMP4 in culture media of cells transfected with siLOX and siSNAI2 72 hours posttransfection. Mean ± SD from triplicate samples. *, P < 0.05; **, P < 0.01; ***, P < 0.001. TIMP4 regulates cellular migration and invasion. C, knockdown of TIMP4 inhibits cellular migration and invasion 72 hours posttransfection. Experiments were performed at least 3 times and the values represent the means ± SD from triplicate wells. D, TIMP4 mRNA and TIMP4 protein expression level were evaluated 72 hours posttransfection. All the experiments were performed at least 3 times. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
TIMP4 regulates cellular invasion and migration and VEGF expression and secretion in cancer cells
Given the previous studies on the role of TIMPs in cancer metastasis, we aimed to investigate whether TIMP4 is also important in cancer invasion. Because TIMP4 levels were high in the cancer cell lines, we used a knockdown strategy to study the effect of TIMP4 on cellular invasion and migration. TIMP4 knockdown resulted in lower cellular invasion and migration, as compared with control (Fig. 4C and D, Supplementary Fig. S4A-S4D).
To gain insight into whether TIMP4 regulates angiogenesis, we performed an angiogenesis array. Analysis of the effects of TIMP4 inhibition showed altered secretion and expression of several proteins including VEGF (Supplementary Fig. S5A and S5B). We further analyzed VEGF expression in cancer cells transfected with siTIMP4 or siControl because both the secretion and expression in cell lysates were reduced. ELISA and RT-PCR revealed that TIMP4 knockdown resulted in lower VEGF-A secretion and expression in cancer cells indicating that TIMP4 regulates the proangiogenic factor VEGF (Fig. 5A). Analysis of the regulation of VEGF in nonaggressive cell lines showed that in MCF-7 cells inhibition of SNAI2 and TIMP4 decreased the secretion of VEGF (Supplementary Fig. S5C and S5D). These data confirm our findings in the more aggressive cancer cell lines. However, in BCPAP cells only depletion of TIMP4 decreased the VEGF secretion which is consistent with the modest effect of siSNAI2 on TIMP4 secretion (Supplementary Fig. S3C). Autocrine production of VEGF-A has been shown to be a major player in the functional plasticity of aggressive cancer cells forming vascular networks (24,25). Using a tube formation assay, we observed that siTIMP4 cells were not able to form elongated structures (Fig. 5B). These data suggest that, by inhibiting VEGF, TIMP4 impairs cancer cell plasticity. To validate our in vitro data, we asked whether the knockdown of SNAI2 inhibits TIMP4 expression in our metastatic mouse model. Immunohistochemical staining showed lower TIMP4 and VEGF staining in lung tissue tumor cells from siSNAI2 mice, as compared with the control group (Fig. 5C). Taken together, these data demonstrate that SNAI2 inhibits VEGF by regulating TIMP4 secretion.
Figure 5.

TIMP4 regulates VEGF expression and secretion. A, VEGF mRNA and VEGF secretion were determined at 72 hours after transfection using RT-PCR and ELISA test. B, tubule-like structure formation in cancer cells is inhibited with the knockdown of TIMP4. C, representative immunohistochemical staining for TIMP4 and VEGF with knockdown of SNAI2. All experiments were performed at least 3 times. Serial sections of lung metastasis from siC and SiSNAI2 mice were subjected to H&E or immunohistochemical staining with anti-TIMP4 and anti-VEGF. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
SNAI2 and TIMP4 are highly expressed in thyroid, breast, and colon cancers
Given that we discovered that LOX regulates SNAI2 expression and that SNAI2 reduces TIMP4 secretion and VEGF expression and secretion, we next asked whether SNAI2 and TIMP4 are co-expressed in cancer samples. We examined the expression and localization of TIMP4 and SNAI2 in different histologic types of thyroid cancer and in primary tumors from patients with metastatic breast and colon cancer. We found higher SNAI2 staining in thyroid cancer than in normal tissue or benign thyroid lesions (Fig. 6A). SNAI2 expression was also significantly higher in breast and colon tumors, as compared with adjacent normal tissue (Fig. 6B and C). SNAI2 staining was positive in the nucleus and cytoplasm of cancer cells. TIMP4 expression showed stronger staining in thyroid, and colon cancer tissue, and in a pattern that was similar to that of SNAI2, however no difference in the expression level of TIMP4 was seen in breast tumors (Fig. 6A-C).
Figure 6.

SNAI2 and TIMP4 are overexpressed in thyroid, breast, and colon cancers. A, SNAI2 and TIMP4 are co-expressed in thyroid cancer tissue, as compared with normal tissue and benign lesion (magnification, 4.6×). FA, follicular adenoma; N, normal; PTC, papillary thyroid cancer. The inset images show magnification at 20×. B and C, nuclear and cytoplasmic staining of SNAI2 and TIMP4 in primary tumors from metastatic breast and colon cancer (magnification, 40×).
Discussion
EMT is a complex process through which epithelial cancer cells acquire a reversible change in phenotype. Several studies have established a strong link between high levels of EMT transcription factors and increased cancer cell metastasis (26, 27). SNAI2 is overexpressed in numerous cancers and also promotes invasion in lung adenocarcinoma, glioma, ovarian, cervical, and pancreatic cancers and is a prognostic marker in some cancers (7, 28, 29). A recent study found overexpression of SNAI2 in aggressive thyroid cancer (30). The role of LOX family members, such as LOX and LOX-like protein 2 (LOXL2), in cancer metastasis has been shown in several human malignancies; however, the relationship between LOX and EMT transcription factors has not been previously reported (8, 31, 32). Here we show a novel role of LOX in that it transcriptionally regulates SNAI2 expression. We observed a significant positive correlation between SNAI2 and LOX in thyroid and breast cancer samples, leading us to investigate the interaction between LOX and SNAI2. We found that LOX interacts with the SNAI2 promoter and regulates SNAI2 promoter activity. Knockdown of LOX in different cancer cell lines showed a decrease in SNAI2 transcript and protein expression. Furthermore, knockdown of LOX inhibited SNAI2 expression in metastatic tumor sites in vivo. Moon and colleagues observed that LOXL2 interacts with SNAIL at the protein level to mediate EMT and that it increases the invasive ability of breast cancer cell lines (33). Because of the homology between LOX and LOXL2 proteins, these data support our finding that the role of LOX in cancer metastasis is due to its interactions with EMT transcription factors, such as SNAIL and SNAI2.
Since we have previously shown that the knockdown of LOX does not affect canonical EMT markers such as N-cadherin, E-cadherin, and vimentin protein expression in ATC (13), we sought to determine the mechanism by which LOX-SNAI2 mediates its effect in cellular migration/invasion and cancer progression and metastasis. ECM remodeling is an essential process during cancer invasion, angiogenesis, and metastasis. MMPs are a family of zinc-dependent endopeptidases that degrade all ECM components and play a key role in these processes (34,35). The expression of MMPs is highly regulated at many levels. TIMPs regulate MMPs after their secretion into the extracellular environment. Thus, we postulated that LOX–SNAI2 axis mediates its effect by modulating the MMP/TIMP levels. Using a MMPs/TIMPs array, we found reduced TIMP4 secretion with LOX or SNAI2 knockdown in multiple cancer cell lines, suggesting that the LOX–SNAI2 axis regulates TIMP4 secretion. Cancer cells interact with their microenvironment through MMPs to modify the ECM. TIMPs can inhibit the activities of multifunctional MMPs (36). MMPs play an important role in tumor progression, and there has been growing evidence that they may be good therapeutic targets for cancer therapy (37, 38). Several studies indicate that TIMPs display anticancer activities by reducing tumor cell invasion and metastasis (39). TIMP4 expression has been associated with longer survival in patients with rectal cancer. In addition, elevated levels of TIMP4 have been found in patients with breast, cervical, and prostate cancers, whereas low levels have been found in patients with pancreatic cancer (40-42). These divergent expression profiles in cancer samples could be due to tissue-specific differences and tumor stages. The mechanisms of action of TIMP4 in carcinogenesis are still unknown. TIMP4 is an MMP2 and MMP9 inhibitor, and our array data in both culture medium and cellular protein lysates did not show differences in MMP-2 and −9 secretion and expression. These data support the hypothesis that TIMP4 plays a role in cancer that is likely independent of MMPs. In the present study, we showed a strong staining of SNAI2 and TIMP4 in thyroid, breast, and colon cancers, as compared with adjacent normal tissue. Since we have shown an overexpression of LOX in aggressive thyroid cancer and previous studies have reported a significant increase of LOX expression in colorectal and metastatic breast cancer, these data support an association between LOX and SNAI2 expression in solid malignancies (8, 11). TIMP4 immunoreactivity was observed in the cytoplasm and nuclei of cancer cells, and, in some cases, with stronger nuclear staining, which supports our data and suggests that TIMP4 functions independently of MMP and plays a protumoral and proinvasive role.
The proangiogenic effects of other TIMP family members have been reported. TIMP1 overexpression was associated with increased VEGF expression and neovascularization in an in vivo model of rat breast carcinoma (43). In addition, an antiangiogenic effect of TIMP1, that has been described to alter migration of endothelial cells, was reported to be both MMP-dependent and -independent (43, 44). Since the signaling pathways leading to the antiapoptotic functions of both TIMP1 and TIMP4 are similar, we propose that, like TIMP1, TIMP4 can be a positive regulator of angiogenesis (45). Indeed, the inhibition of SNAI2 or TIMP4 in vitro reduced VEGF expression and secretion. Moreover, metastatic tumors resulting from cancer cells with depleted SNAI2 exhibited reduced VEGF expression. Thus, by regulating TIMP4 secretion, SNAI2 regulates VEGF expression. The signaling pathways mediating VEGF expression have been extensively studied; however, the mechanisms of regulation of VEGF by TIMPs are still unknown. Further studies are needed to investigate the MMP-independent effect of TIMP4 on angiogenesis and VEGF secretion.
Communications between cancer cells and their stromal cells play a central role in tumorigenesis and cancer metastasis by creating a microenvironment that supports angiogenesis and cancer cell invasion and migration (46, 47). The significant positive correlation between LOX–SNAI2 and the percentage of stromal cells in tumor further support the role of this axis in cancer invasion and metastasis. Accumulation of cancer-associated fibroblasts induced by TIMP1 has been shown in colon and prostate cancer; therefore, it is possible that TIMP4 secreted by tumors overexpressing LOX and SNAI2 may promote stromal cells infiltration in aggressive tumors (48).
We believe that the role of the LOX–SNAI2 axis in cells undergoing EMT is not dependent on the cancer type. However, the genetic and or mutation profile of thyroid, breast, and colon cancers are different, so it is possible that mutations that have been associated with aggressive tumor phenotype such as BRAF V600E mutation, TP53 mutation, or estrogen and progesterone receptor status may influence the regulation of the LOX–SNAI2 axis. This is consistent with the high levels of LOX we found in the cell lines used in our studies, which have driver mutations that are common in the cancer types we studied.
LOX and SNAI2 as a prognostic marker could help predict poor outcome in patients once validated in large cohort studies. Furthermore, we believe our findings support that LOX is a target for therapy in patients with aggressive cancers as inhibition of LOX may impair or reverse the EMT process to improve patient outcome.
In summary, we show for the first time that LOX transcriptionally regulates the expression of the EMT transcription factor SNAI2. Moreover, SNAI2 knockdown studies recapitulate the phenotypic effects (cellular migration and invasion, and metastasis) of LOX in vitro and in vivo, suggesting a role for LOX–SNAI2 axis in cancer progression, which are strongly correlated in human cancer samples. The effect of the LOX–SNAI2 axis in cancer progression is mediated, at least in part, by regulating TIMP4 secretion.
Supplementary Material
Translational Relevance.
Epithelial-to-mesenchymal transition (EMT) is a complex process through which epithelial cancer cells acquire a reversible change in phenotype. We investigated the role of LOX in the regulation of EMT transcription factors. We found that LOX transcriptionally regulates SNAI2 expression. Moreover, SNAI2 knockdown studies recapitulate the phenotypic effects of LOX in vitro and in vivo, suggesting a LOX-SNAI2 axis in cancer progression. In addition, our study shows that the effect of the LOX-SNAI2 axis in cancer progression is mediated by TIMP4 secretion. We also found overexpression of SNAI2 and TIMP4 proteins in aggressive thyroid, breast, and colon cancer. Our findings provide new evidence that LOX regulates SNAI2 expression and that SNAI2-mediated TIMP4 secretion plays a role in cancer progression. The findings have important consequences for developing strategies to block this axis and control cancer progression.
Acknowledgments
The authors thank the Molecular Pathology Unit at the National Cancer Institute for their help scanning the immunohistochemical slides.
Grant Support
This research was supported by the intramural research program of the Center for Cancer Research, National Cancer Institute, NIH (grant number 1ZIABC011275-06).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Nikiforov YE. Editorial: anaplastic carcinoma of the thyroid–will aurora B light a path for treatment? J Clin Endocrinol Metab 2005;90:1243–5. [DOI] [PubMed] [Google Scholar]
- 2.Smallridge RC. Approach to the patient with anaplastic thyroid carcinoma. J Clin Endocrinol Metab 2012;97:2566–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thiery JP, Adoque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139:871–90. [DOI] [PubMed] [Google Scholar]
- 4.Sleeman JP, Thiery JP. Snapshot: the epithelial-mesenchymal transition. Cell 2011;145:162.e1. [DOI] [PubMed] [Google Scholar]
- 5.Savagner P, Karavanova I, Perantoni A, Thiery JP, Yamada KM. Slug mRNA is expressed by specific mesodermal derivatives during rodent organogenesis. Dev Dyn 1998;213:182–7. [DOI] [PubMed] [Google Scholar]
- 6.Shioiri M, Shida T, Koda K, Oda K, Seike K, Nishimura M, et al. Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. Br J Cancer 2006;94:1816–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shih JY, Tsai MF, Chang TH, Chang YL, Yuan A, Yu CJ, et al. Transcription repressor slug promotes carcinoma invasion and predicts outcome of patients with lung adenocarcinoma. Clin Cancer Res 2005; 11: 8070–8. [DOI] [PubMed] [Google Scholar]
- 8.Erler JT, Bennewith KL, Nicolau M, Domhofer N, Kong C, Le QT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006;440: 1222–6. [DOI] [PubMed] [Google Scholar]
- 9.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009;139:891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker AM, Bird D, Lang G, Cox TR, Erler JT. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene 2013;32:1863–8. [DOI] [PubMed] [Google Scholar]
- 11.Baker AM, Cox TR, Bird D, Lang G, Murray GI, Sun XF, et al. The role of lysyl oxidase in SRC-dependent proliferation and metastasis of colorectal cancer. J Natl Cancer Inst 2011;103:407–24. [DOI] [PubMed] [Google Scholar]
- 12.Gao Y, Xiao Q, Ma H, Li L, Liu J, Feng Y, et al. LKB1 inhibits lung cancer progression through lysyl oxidase and extracellular matrix remodeling. Proc Natl Acad Sci U S A 2010;107:18892–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boufraqech M, Nilubol N, Zhang L, Gara SK, Sadowski SM, Mehta A, et al. miR-30a inhibits LOX expression and progression of anaplastic thyroid cancer. Cancer Res 2014;75:367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nistico P, Bissell MJ, Radisky DC. Epithelial-mesenchymal transition: general principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb Perspect Biol 2012;4:pii: a011908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ray JM, Stetler-Stevenson WG. Gelatinase A activity directly modulates melanoma cell adhesion and spreading. EMBO J 1995;14:908–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta 2010;1803:55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L, Zhang Y, Mehta A, Boufraqech M, Davis S, Wang J, et al. Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget 2015;6:9073–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marlow LA, D'Innocenzi J, Zhang Y, Rohl SD, Cooper SJ, Sebo T, et al. Detailed molecular fingerprinting of four new anaplastic thyroid carcinoma cell lines and their use for verification of RhoB as a molecular therapeutic target. J Clin Endocrinol Metab 2010;95:5338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 2010;12:R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006;10:515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fabien N, Fusco A, Santoro M, Barbier Y, Dubois PM, Paulin C. Description of a human papillary thyroid carcinoma cell line. Morphologic study and expression of tumoral markers. Cancer 1994;73:2206–12. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Gaskins K, Yu Z, Xiong Y, Merino MJ, Kebebew E. An in vivo mouse model of metastatic human thyroid cancer. Thyroid 2014;24:695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung YY, Lee YK, Koo JS. Expression of cancer-associated fibroblast-related proteins in adipose stroma of breast cancer. Tumour Biol 2015;36:8685–95. [DOI] [PubMed] [Google Scholar]
- 24.Vartanian A, Stepanova E, Grigorieva I, Solomko E, Baryshnikov A, Lichinitser M. VEGFR1 and PKCalpha signaling control melanoma vasculogenic mimicry in a VEGFR2 kinase-independent manner. Melanoma Res 2011; 21:91–8. [DOI] [PubMed] [Google Scholar]
- 25.Wang JY, Sun T, Zhao XL, Zhang SW, Zhang DF, Gu Q, et al. Functional significance of VEGF-a in human ovarian carcinoma: role in vasculogenic mimicry. Cancer Biol Ther 2008;7:758–66. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004;117:927–39. [DOI] [PubMed] [Google Scholar]
- 27.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 2007;7:415–28. [DOI] [PubMed] [Google Scholar]
- 28.Yang HW, Menon LG, Black PM, Carroll RS, Johnson MD. SNAI2/Slug promotes growth and invasion in human gliomas. BMC Cancer 2010;10:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang B, Kim J, Jeong D, Jeong Y, Jeon S, Jung SI, et al. Klotho inhibits the capacity of cell migration and invasion in cervical cancer. Oncol Rep 2012;28:1022–8. [DOI] [PubMed] [Google Scholar]
- 30.Jung CW, Han KH, Seol H, Park S, Koh JS, Lee SS, et al. Expression of cancer stem cell markers and epithelial-mesenchymal transition-related factors in anaplastic thyroid carcinoma. Int J Clin Exp Pathol 2015;8:560–8. [PMC free article] [PubMed] [Google Scholar]
- 31.Kirschmann DA, Seftor EA, Fong SF, Nieva DR, Sullivan CM, Edwards EM, et al. A molecular role for lysyl oxidase in breast cancer invasion. Cancer Res 2002;62:4478–83. [PubMed] [Google Scholar]
- 32.Yang X, Li S, Li W, Chen J, Xiao X, Wang Y, et al. Inactivation of lysyl oxidase by beta-aminopropionitrile inhibits hypoxia-induced invasion and migration of cervical cancer cells. Oncol Rep 2013;29:541–8. [DOI] [PubMed] [Google Scholar]
- 33.Moon HJ, Finney J, Xu L, Moore D, Welch DR, Mure M. MCF-7 cells expressing nuclear associated lysyl oxidase-like 2 (LOXL2) exhibit an epithelial-to-mesenchymal transition (EMT) phenotype and are highly invasive in vitro. J Biol Chem 2013;288:30000–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stamenkovic I Matrix metalloproteinases in tumor invasion and metastasis. Semin Cancer Biol 2000;10:415–33. [DOI] [PubMed] [Google Scholar]
- 35.Song C, Zhu S, Wu C, Kang J. Histone deacetylase (HDAC) 10 suppresses cervical cancer metastasis through inhibition of matrix metalloproteinase (MMP) 2 and 9 expression. J Biol Chem 2013;288:28021–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun J Matrix metalloproteinases and tissue inhibitor of metalloproteinases are essential for the inflammatory response in cancer cells. J Signal Transduct 2010;2010:985132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bourboulia D, Stetler-Stevenson WG. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): positive and negative regulators in tumor cell adhesion. Semin Cancer Biol 2010;20:161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Birkedal-Hansen H, Yamada S, Windsor J, Pollard AH, Lyons G, Stetler-Stevenson W, et al. Matrix metalloproteinases. Curr Protoc Cell Biol 2008; Chapter 10:Unit 10.8. [DOI] [PubMed] [Google Scholar]
- 39.Brand K Cancer gene therapy with tissue inhibitors of metalloproteinases (TIMPs). Curr Gene Ther 2002;2:255–71. [DOI] [PubMed] [Google Scholar]
- 40.Hilska M, Roberts PJ, Collan YU, Laine VJ, Kossi J, Hirsimaki P, et al. Prognostic significance of matrix metalloproteinases-1, -2, -7 and -13 and tissue inhibitors of metalloproteinases-1, -2, -3 and -4 in colorectal cancer. Int J Cancer 2007;121:714–23. [DOI] [PubMed] [Google Scholar]
- 41.Bister V, Skoog T, Virolainen S, Kiviluoto T, Puolakkainen P, Saarialho-Kere U. Increased expression of matrix metalloproteinases-21 and -26 and TIMP-4 in pancreatic adenocarcinoma. Mod Pathol 2007;20:1128–40. [DOI] [PubMed] [Google Scholar]
- 42.Lizarraga F, Espinosa M, Maldonado V, Melendez-Zajgla J. Tissue inhibitor of metalloproteinases-4 is expressed in cervical cancer patients. Anticancer Res 2005;25:623–7. [PubMed] [Google Scholar]
- 43.Yoshiji H, Harris SR, Raso E, Gomez DE, Lindsay CK, Shibuya M, et al. Mammary carcinoma cells over-expressing tissue inhibitor ofmetalloproteinases-1 show enhanced vascular endothelial growth factor expression. Int J Cancer 1998;75:81–7. [DOI] [PubMed] [Google Scholar]
- 44.Fernandez HA, Kallenbach K, Seghezzi G, Grossi E, Colvin S, Schneider R, et al. Inhibition of endothelial cell migration by gene transfer of tissue inhibitor of metalloproteinases-1. J Surg Res 1999;82:156–62. [DOI] [PubMed] [Google Scholar]
- 45.Stetler-Stevenson WG. Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase-independent biological activities. Sci Signal 2008;1:re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005;8:211–26. [DOI] [PubMed] [Google Scholar]
- 47.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007;449:557–63. [DOI] [PubMed] [Google Scholar]
- 48.Gong Y, Scott E, Lu R, Xu Y, Oh WK, Yu Q. TIMP-1 promotes accumulation of cancer associated fibroblasts and cancer progression. PLoS One 2013;8: e77366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.