

Abstract
The failure of anti-CD20 antibody (Rituximab) as therapy for lupus may be attributed to the transient and incomplete B cell depletion achieved in clinical trials. Here, using an alternative approach, we report that complete and sustained CD19+ B cell depletion is a highly effective therapy in lupus models. CD8+ T cells expressing CD19-targeted chimeric antigen receptors (CARs) persistently depleted CD19+ B cells, eliminated autoantibody production, reversed disease manifestations in target organs, and extended life spans well beyond normal in the (NZB × NZW) F1 and MRLfas/fas mouse models of lupus. CAR T cells were active for 1 year in vivo and were enriched in the CD44+CD62L+ T cell subset. Adoptively transferred splenic T cells from CAR T cell–treated mice depleted CD19+ B cells and reduced disease in naive autoimmune mice, indicating that disease control was cell-mediated. Sustained B cell depletion with CD19-targeted CAR T cell immunotherapy is a stable and effective strategy to treat murine lupus, and its effectiveness should be explored in clinical trials for lupus.
INTRODUCTION
Systemic lupus erythematosus (SLE) is an autoimmune disease that is difficult to diagnose and treat because of the episodic nature of flare-ups, the involvement of multiple organ systems and variable presentation, and severity (1). Autoantibodies to DNA and nuclear protein autoantigens are consistently present in lupus and contribute to pathogenesis by forming immune complex deposits in various tissues (2, 3). Additional B cell contributions to lupus include antigen presentation to self-reactive T cells and the secretion of inflammatory cytokines. Approaches that inactivate or deplete B cells offer attractive therapies for lupus, and efforts to identify promising targets on B cells merit continued attention (4–6).
B cell depletion with a monoclonal antibody against the B cell surface marker CD20, Rituximab, has shown therapeutic promise in rheumatoid arthritis (7) and multiple sclerosis (8) but was unsuccessful in two separate clinical trials for SLE (9, 10). Patients whose B cell depletion in peripheral blood occurred faster, was more complete, or lasted longer experienced the most notable improvements in disease presentation in the Lupus Nephritis Assessment with Rituximab (LUNAR) trial (10). However, disease scores and lupus flares did not distinguish between treatment and placebo groups, such that neither the Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER) (9) nor LUNAR (10) clinical trials met predetermined end points for clinical efficacy. Studies in lupus-prone mice showed that high concentrations of endogenous immune complexes blocked macrophage function and required a higher dose of anti-CD20 antibody to achieve B cell depletion (11–13). Incomplete B cell depletion of tissue-resident B cells (10), or the transient nature of the treatment, may have contributed to the failure of the initial Rituximab trials to attain satisfactory outcomes.
Administration of anti-CD20 antibodies has additional limitations as treatment for lupus. Upon injection, monoclonal antibodies follow drug-like pharmacokinetics with exponential decay that requires repeated administrations to achieve and maintain a therapeutic dose (14). Over time, resistance to the treatment arises in the host and further limits use (15). Cancer immunotherapy to target malignant transformed B cells has led to an alternative approach that can deplete B cells. Cytotoxic T cells, which express transduced genes for surface receptors against CD19, kill the targeted cells upon binding (16, 17). Autologous T cells with such CD19-targeted chimeric antigen receptors (CARs) have shown remarkable efficacy in a growing number of B cell malignancies because they retain potency over time and can lead to lasting remission (18). Cell-based therapies provide further advantages over antibodies because CAR T cells require only a single administration, migrate to multiple lymphoid tissues and organs in the recipient, and develop into both effector and memory cell populations.
Here, we test whether permanent and profound B cell depletion by CD19-targeted CAR T cells can lead to lasting remission of experimental lupus. In two mouse strains that are reliable models of SLE and that differ in the underlying genetic mechanisms leading to autoimmunity (19), sustained CD19+ B cell depletion prevented auto-antibody production, alleviated manifestations of lupus pathogenesis, and lengthened life spans. In addition, B cell depletion affected the distribution of T cell populations, preventing the disease-associated bias toward CD4+ lymphocytes (20).
RESULTS
Experimental design and implementation
The experimental design for testing of CD19-targeted CAR T cells in mouse models of lupus was closely related to a standard CAR approach, except that we used purified CD8+ T cells because of concern with the potential disease-enhancing effect of autoreactive CD4+ T cells (Fig. 1, A to D). We used the 1D3 CD19-targeted CAR (21), which consists of the 1D3 extracellular single-chain Fv (heterodimer of the variable region of the light and heavy chains) domain linked to the CD28 transmembrane and cytoplasmic signaling domains and a variant CD3ζ C terminus (Fig. 1A). This CD3ζ variant contains alanine residues in place of the conserved tyrosine residues in two of the three cytoplasmic immunoreceptor tyrosine-based activation motif (ITAM) domains. These mutations reduce activation and mitigate the subsequent exhaustion of the CAR T cells, thus contributing to increased persistence of the engineered T cells in vivo (21, 22). Recipient mice were conditioned by irradiation to facilitate engraftment of the transferred T cells by transiently depleting the rapidly dividing endogenous lymphoid and myeloid cell populations.
Fig. 1. CAR design and transduction.
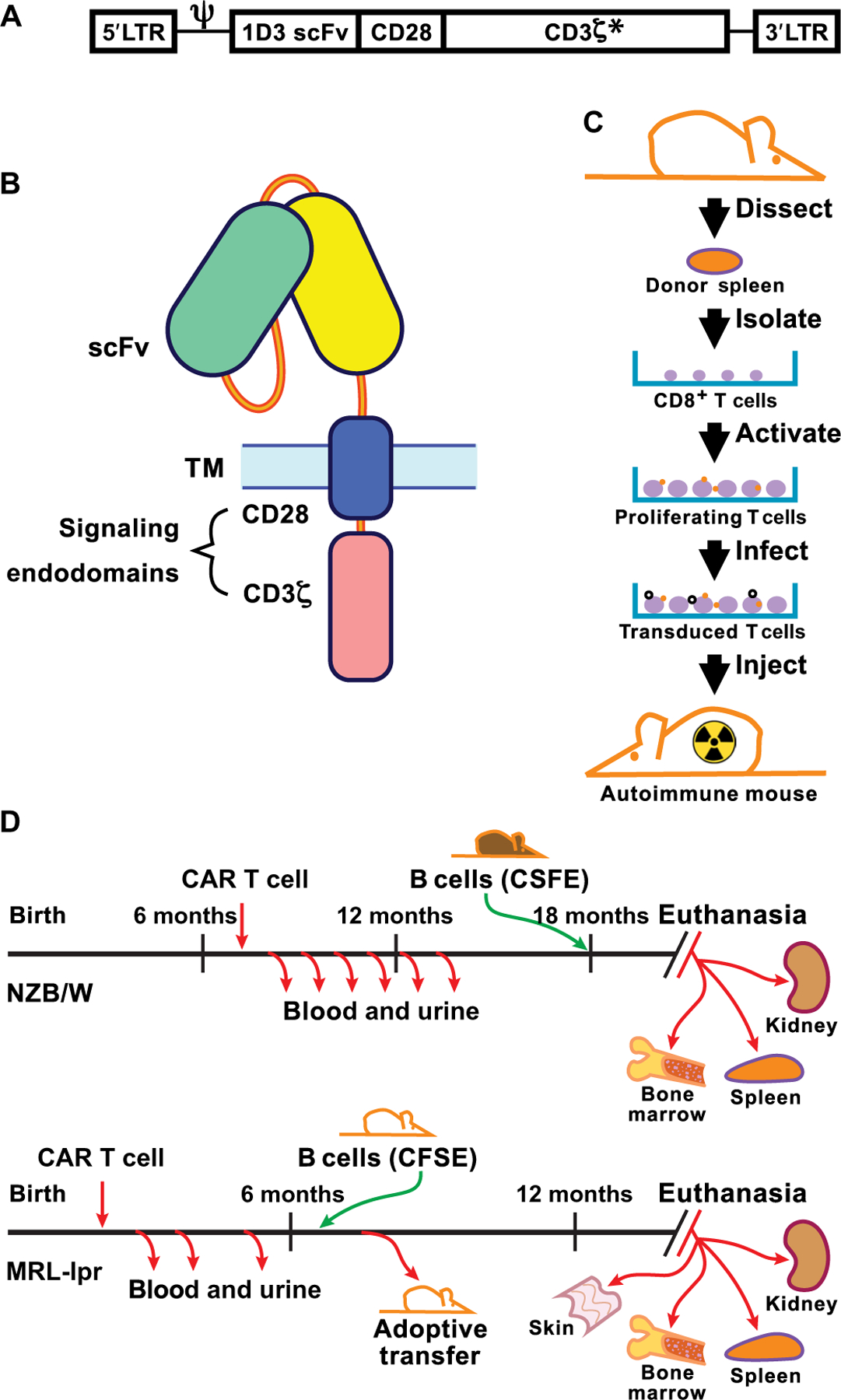
(A) The structural gene for 1D3–28Z.1–3 is shown downstream of the MSGV 5′ long terminal repeat (LTR) and splice acceptor (Ψ). The asterisk indicates that the first and third ITAMs in CD3ζ have tyrosine to alanine replacement mutations. (B) Diagram of CAR protein in the cell membrane showing the extracellular single-chain Fv (scFv) domain and the transmembrane (TM) and signaling domains that combine CD28 and CD3ζ C termini. (C) Steps involved in the isolation, activation, and transduction of CD8+ splenocytes with virus before infusion into a recipient mouse that was irradiated ahead of T cell transfer to achieve transient myeloablation. (D) Timeline of CAR T cell treatment and sample collection in NZB/W and MRL-lpr mice. CFSE, carboxyfluorescein diacetate succinimidyl ester.
To evaluate gene delivery methods, we transduced the CD19-targeted CAR into splenic CD8+ T cells via different virus transduction methods. In the (NZB × NZW) F1 (NZB/W) lupus model, 7-month-old female mice were used as donors of CD8+ T cells for transduction and as recipients of the CD19-targeted CAR T cells. We chose female mice of that age because they manifest overt signs of autoimmune disease (19). The three virus transductions achieved between 5 and 15% expression in primary T cells and more than 90% expression in cells from the human Jurkat T cell line (fig. S1). After 5 days of cell culture, we injected 1.2 × 106 transduced CD8+ T cells per mouse from one of the three tested viral vector groups (table S1) and an equal number of mock-transduced T cells into control mice.
The different viral vectors achieved different efficiencies of B cell depletion (Fig. 2A and table S1). CD19+ B cells were depleted in 8 of 10 NZB/W mice receiving CD8+ T cells transduced with the amphotropic murine leukemia virus (A-MLV) retrovirus, in 4 of 9 mice receiving CD8+ T cells transduced with vesicular stomatitis virus G glycoprotein (VSV-G)–pseudotyped MLV, and in only 2 of 10 mice that received CD8+ T cells transduced with VSV-G–pseudotyped lentivirus (G-LV). These results defined two comparison groups for follow-up studies, those that were CD19+ B cell depleted (CD19-d) and those in which CD19+ B cells recovered within 3 weeks after the transient myelodepletion. The CD19+ B cell depletion phenotypes were stable thereafter because only one mouse that showed CD19+ B cell depletion at 4 weeks after CAR T cell infusion regained CD19+ B cells by 8 weeks after treatment (fig. S2). All other CAR T cell–treated mice, in which the CAR T cell infusion depleted CD19+ B cells, exhibited CD19+ B cell aplasia throughout the course of the experiments, more than 1 year from the administration of transduced T cells.
Fig. 2. Flow cytometry of blood lymphocytes from CAR T cell–treated mice and controls.
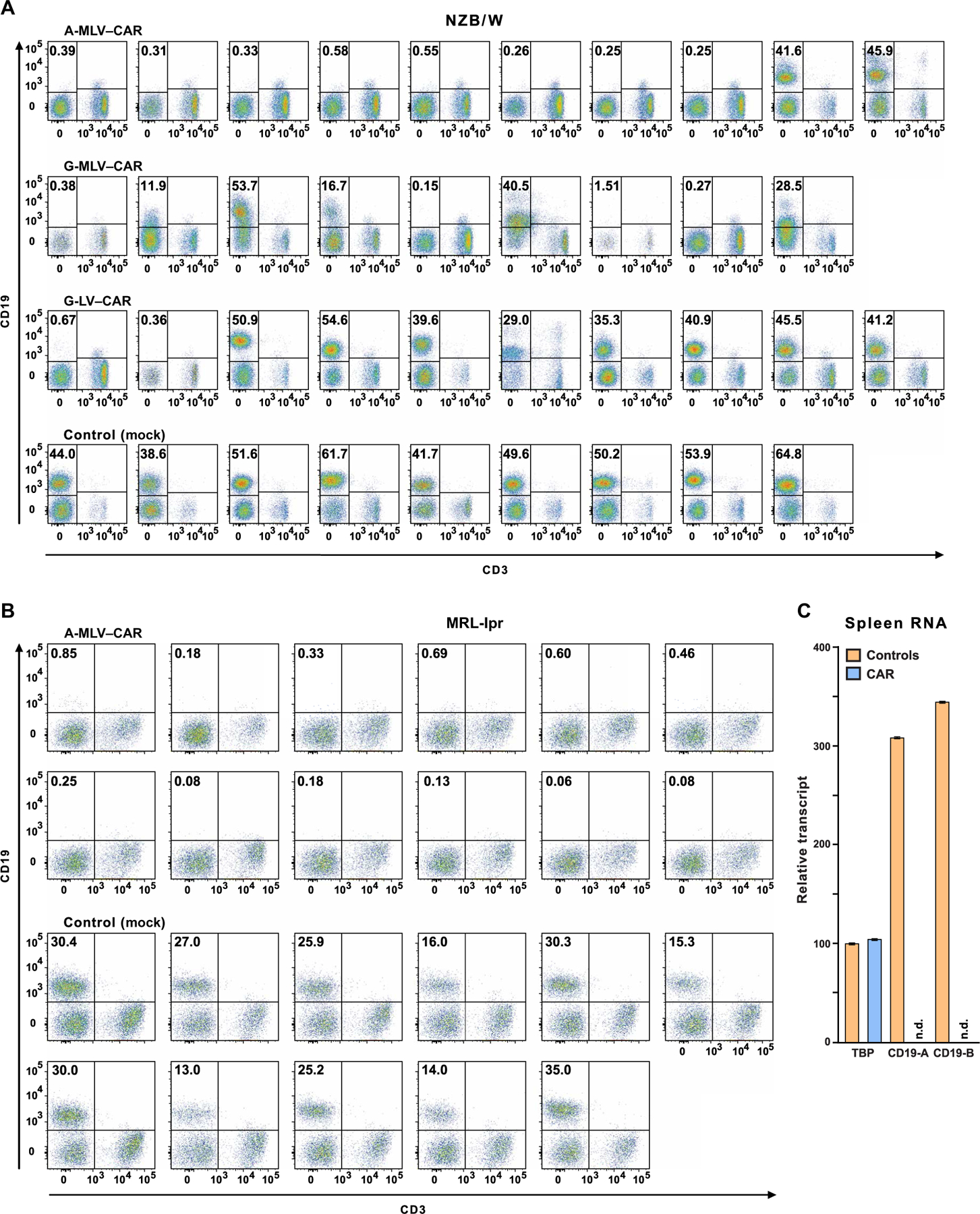
Mice were administered CD19-targeted CAR CD8+ T cells transduced by one of three different viruses, as indicated above the plots. CD19+ B cell depletion was assessed 2 months after treatment. Pseudocolor plots indicate CD19 and CD3 expression on lymphocytes, as defined by forward and side scatter in blood from individual mice. Frequencies of CD19+ B cells are shown in the top left quadrant. (A) Fourteen NZB/W mice in the three transduction groups that had less than 1% CD19+ lymphocytes were designated as CD19-d, whereas control mice and 15 of the CAR T cell–treated mice that had 12 to 60% CD19+ B cells were combined into the CD19-sufficient (CD19-s) experimental group. (B) MRL-lpr mice were injected with A-MLV–CAR–transduced CD8+ T cells or with nontransduced CD8+ T cells (control), and frequencies of CD19+ lymphocytes were determined. (C) Total RNA from the spleens of four control and four CAR T cell–treated mice was purified to measure transcripts coding for CD19 and TATA binding protein (TBP). CD19 message, as measured by reverse transcription polymerase chain reaction (RT-PCR) at two exon-intron boundaries (CD19-A or CD19-B; see table S2 for details), was determined in CAR T cell–treated mice and controls. n.d., nondetectable.
Encouraged by the initial success of the CD19-targeted CAR approach in NZB/W female mice, we expanded the study to include MRLfas/fas (MRL-lpr) mice. These mice develop a lupus-like disease that is T cell–driven and manifests more quickly than the disease of NZB/W mice (19). Two-month-old MRL-lpr female mice received CD19-targeted CD8+ T cells from same-age donor mice after transduction with the A-MLV retrovirus. This virus, along with an improvement of injection procedures (see Materials and Methods), led to the depletion of CD19+ B cells from 12 of 12 MRL-lpr mice (Fig. 2B). As controls, we used MRL-lpr female mice infused with mock-transduced syngeneic CD8+ T cells. To test whether CD19-targeted CAR T cells select for structural variants of CD19 that would not be detected by flow cytometry, we quantified CD19 message in splenic RNA from CAR T cell–treated and control mice (Fig. 2C). Probes for exons 5 and 6 and exons 14 and 15 of CD19 confirmed absence of CD19 gene expression in spleens of treated mice 1 year after CAR T cell transfer.
Serology of CAR T cell–treated mice
We determined total plasma IgM (immunoglobulin M), IgG, and anti-DNA autoantibody titers using enzyme-linked immunosorbent assay (ELISA) in CAR T cell–treated and control mice (Fig. 3). The results revealed statistically significant differences between CAR T cell–treated mice that were CD19-d versus treated but CD19 sufficient (CD19-s) mice and control mice. Total serum IgM and IgG were decreased in CD19-d mice from 8 to 11 weeks to 18 to 19 weeks after CAR T cell injections (for NZB/W and MRL-lpr, respectively; Fig. 3, A and B). Anti-DNA IgG and IgM that were detectable before CAR T cell injections in both NZB/W and MRL-lpr mice were decreased to below the detection limit and remained low to undetectable in most of the CD19-d NZB/W and CAR T cell–treated MRL-lpr mice (Fig. 3, A and B). In contrast, anti-DNA IgG and IgM autoantibodies remained elevated or increased in CD19-s and control mice, consistent with the progression of autoimmunity in both strains.
Fig. 3. Serologic analysis of IgM and IgG concentrations and anti-DNA reactivities at different times after CAR T cell infusion.
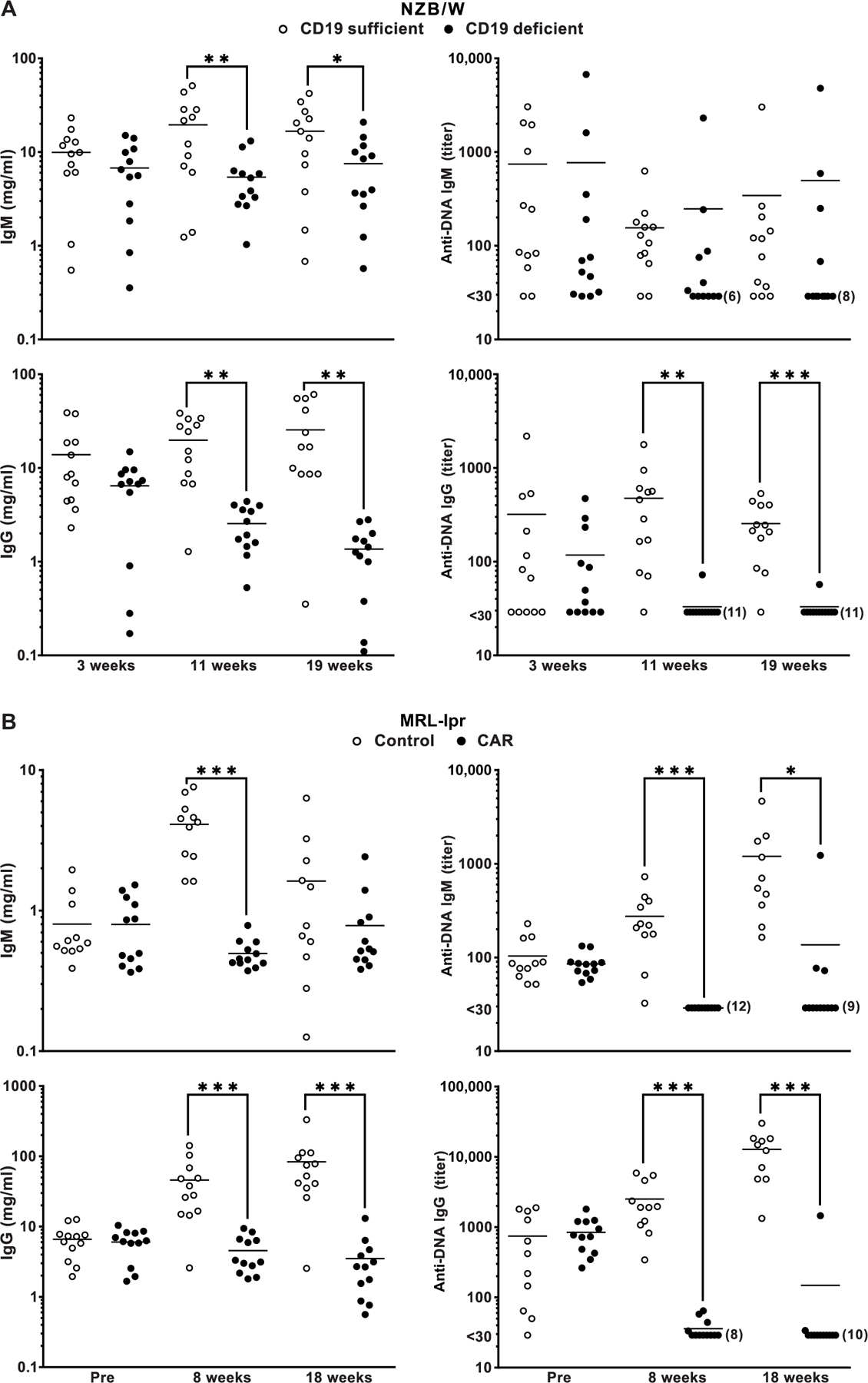
(A) NZB/W and (B) MRL-lpr plasma were tested using ELISA for total IgM and IgG concentration and IgM and IgG anti-DNA titers. Values for CD19-d and CD19-s and control mice are indicated. IgM and IgG standards were used to generate standard curves for estimating IgM and IgG concentrations. Times of plasma collection for the relevant groups are indicated. Horizontal lines are means. Differences between measurements taken at the indicated time points were compared by one-tailed t test, and significance is indicated by *P < 0.05, **P < 0.01, and ***P < 0.001.
Effects of CAR T cell treatment on lupus pathogenesis and survival
The most notable effect of CD19-targeted CAR treatment on disease pathogenesis was the increase in life spans of both NZB/W (Fig. 4A) and MRL-lpr mice (Fig. 4B). Most CD19-d NZB/W mice lived about 1 year after CAR T cell injection, attaining 18 months of age. This was a highly significant extension of life spans (P = 0.0019) when compared to CD19-s NZB/W mice, all of which had been euthanized because of advanced disease by that time. Similarly, 8 of 12 CAR T cell–treated MRL-lpr mice lived for 1 year after treatment, outliving control MRL-lpr mice and attaining 14 months of age at the termination of the experiment. The CD19-targeted CAR T cell treatment considerably extended life spans, as treated mice reached an age that is quite unusual for these lupus-prone strains of mice (19). This result provided unambiguous confirmation that CAR T cell therapy profoundly affects autoimmune disease progression in both NZB/W and MRL-lpr mice.
Fig. 4. Impact of CD19-targeted CAR T cell treatment on lupus pathogenesis and survival.
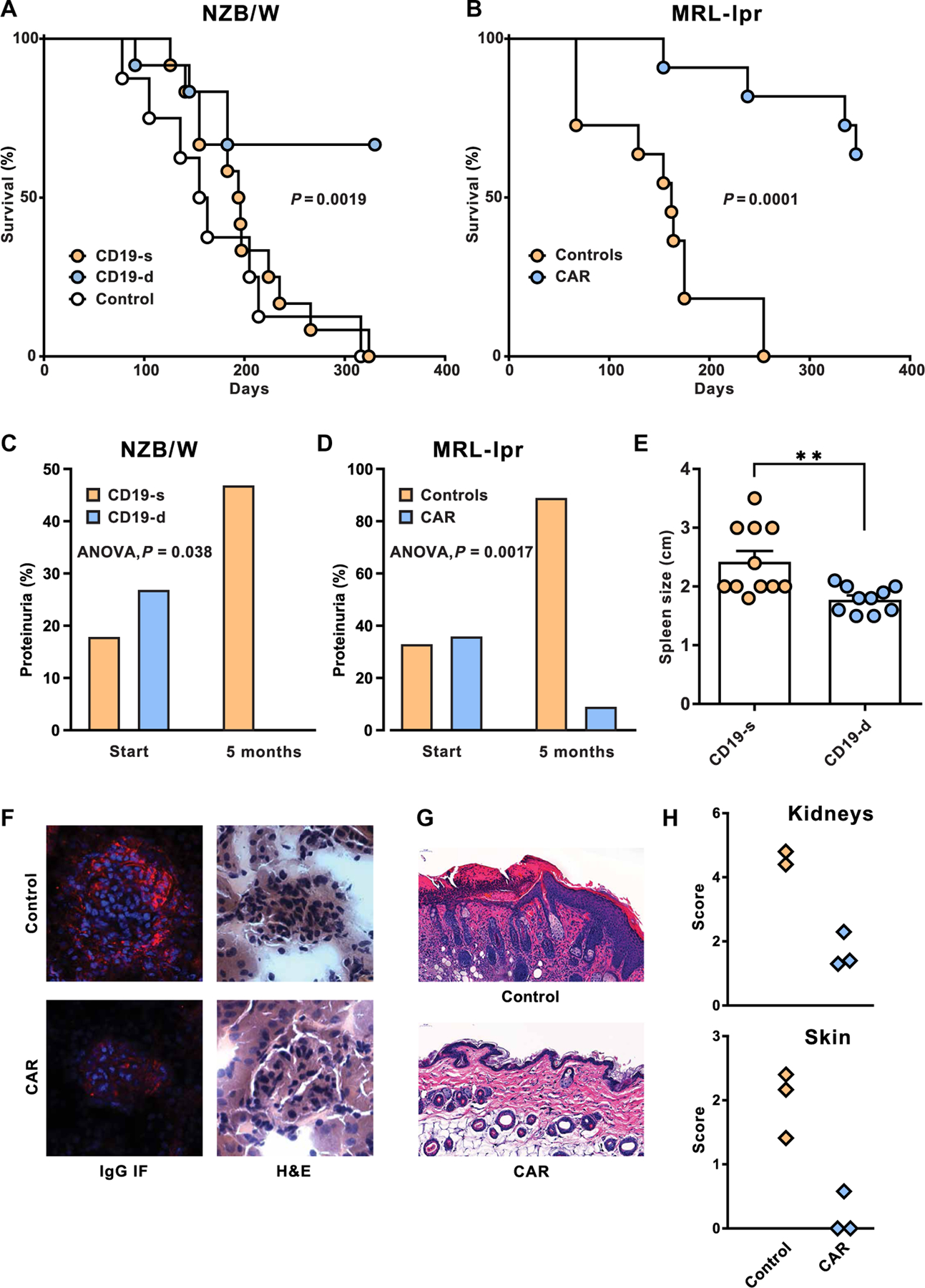
(A) Survival curves of NZB/W mice after CAR T cell infusion into 7-month-old recipients. Survival of CD19-d mice (n = 12) was compared relative to CD19-s mice (n = 12), and significance was determined by log rank (Mantel-Cox; P = 0.0019). For comparison, the survival of control mice (n = 8) is plotted. (B) Survival curves of CAR T cell-treated and control MRL-lpr mice. CAR T cell–treated mice (n = 11) were compared to control MRL-lpr mice (n = 11), and survival of mice in the two groups was evaluated by long rank (Mantel-Cox; P = 0.0001). (C and D) High-grade proteinuria (1 mg/ml and higher) was determined before treatment (start) and at 5 months after CAR T cell treatment. (C) Proteinuria in CD19-s (n = 12) and CD19-d (n = 11) NZB/W mice was analyzed by single-factor analysis of variance (ANOVA) (P = 0.038). (D) High-grade proteinuria in CAR T cell–treated MRL-lpr mice (n = 11) and controls (n = 9) was similarly compared (P = 0.0017). (E) Lengths of spleens in CD19-d and CD19-s NZB/W mice were measured at euthanasia (P < 0.005). Bars are means, and horizontal lines are SEMs. (F) Kidneys from NZB/W mice were sectioned and analyzed for cellularity and morphology by H&E staining and for mouse IgG deposits by immunofluorescence (IF). (G) MRL-lpr skin sections were stained by H&E to compare the epidermis of control and CAR T cell–treated mice. (H) Pathology scores of MRL-lpr kidney and skin sections were determined as described in Materials and Methods.
To elucidate the extended survival, we evaluated the effects of B cell depletion on disease manifestations. Because anti-DNA autoantibodies are linked to glomerulonephritis in lupus mice, we measured proteinuria as an indication of kidney function. At 7 months of age, before administration of CAR T cells, 6 of 28 NZB/W mice had high-grade proteinuria. Five months after CAR T cell administration, 0 of 11 CD19-d mice had high-grade proteinuria, compared to 8 of 17 CD19-s mice (Fig. 4C). Although 2-month-old MRL-lpr mice are generally considered prediseased, 7 of 20 MRL-lpr mice in our experiment had high-grade proteinuria before CAR T cell treatment. Five months after treatment, high-grade proteinuria was detected in only 1 of 11 CAR T cell–treated mice but was manifested in 8 of 9 control mice (Fig. 4D).
The CD19+ B cell depletion significantly reduced splenomegaly in CAR T cell–treated NZB/W mice relative to untreated control mice (P < 0.005; Fig. 4E). Histopathology of NZB/W kidney sections confirmed that CAR T cell treatment reduced the size and cellular infiltrate in glomeruli and largely prevented IgG deposits in both autoimmune strains of mice (Fig. 4F and fig. S3). In MRL-lpr mice, autoimmune disease also manifests as a skin disorder that begins with alopecia and progresses to skin lesions and scarring (23). To examine the skin pathology in CAR T cell–treated and control mice, we compared hematoxylin and eosin (H&E)–stained skin sections by microscopy. At 8 months, when the control mice had cell infiltrates and inflammation progressing to acanthosis and hyperkeratosis, the CD19-d MRL-lpr mice had nearly normal skin architecture (Fig. 4G) and minimal alopecia (fig. S4). The notable differences in pathology scores between CAR T cell–treated and control MRL-lpr mice (Fig. 4H) confirmed that CD19-targeted CAR T cells alleviate diverse lupus manifestations.
Effects of CAR T cell treatment on B and T lymphocyte populations
To investigate a possible source of the persistent plasma IgM in mice that had a near-complete depletion of circulating CD19+ B cells, we evaluated blood of CD19-d mice for the presence of surface IgM+ B cells. Control mice of both strains had IgMhi and IgMlo subpopulations of surface IgM+ B cells (Fig. 5A). The IgMlo, but not the IgMhi, subpopulation of B cells was also present in CAR T cell–treated NZB/W and MRL-lpr mice, although these B cells had undetectable CD19. This persisting IgMlo, CD19− population may contribute to the continued IgM production.
Fig. 5. Effect of CD19-targeted CAR T cell treatment on lymphocyte phenotype and plasma proteome.
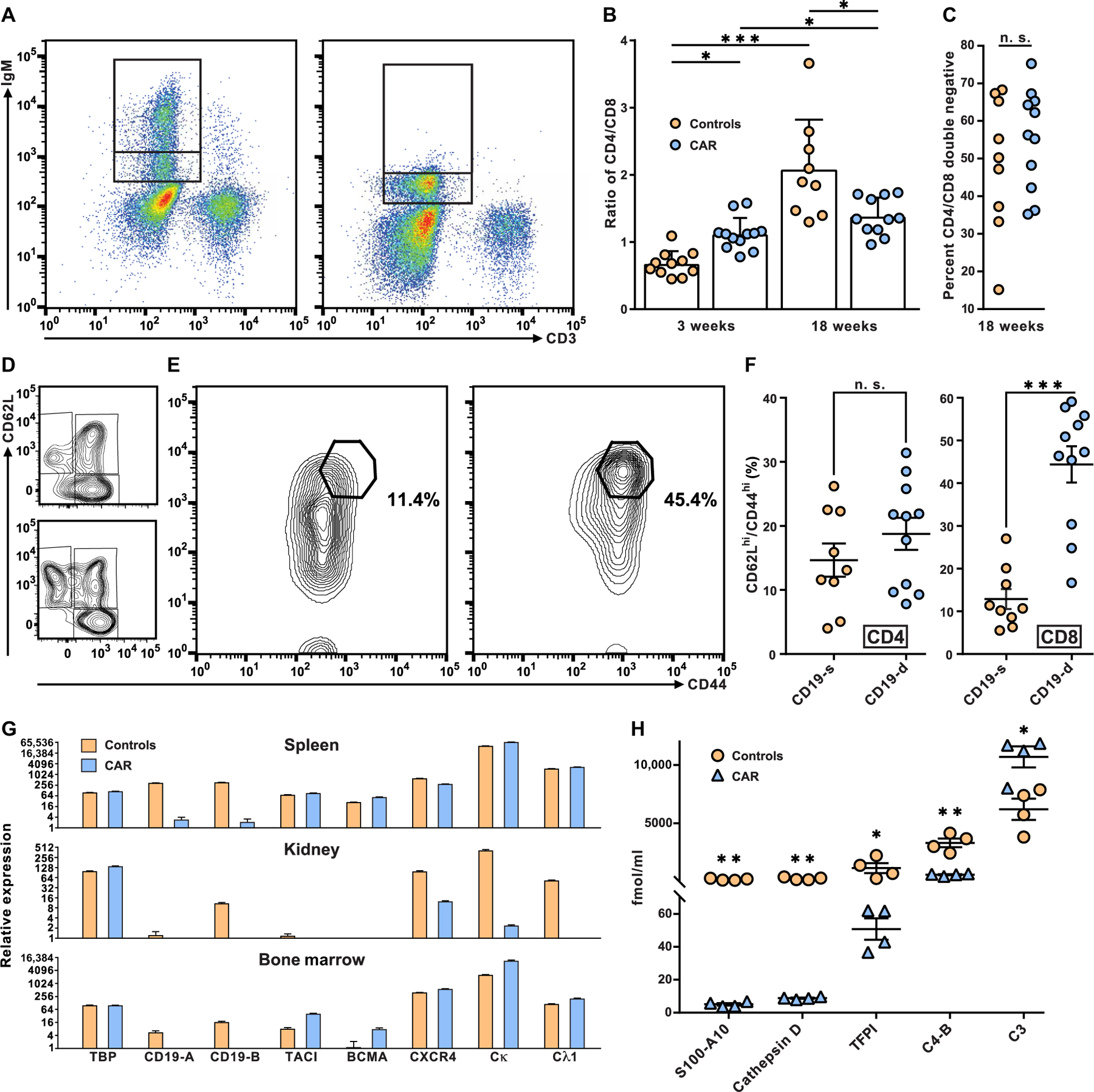
(A) The expression of surface IgM on peripheral blood B lymphocytes from control MRL-lpr mice (left) was used to define an intermediate and a high IgM+ population (indicated by stacked gates). These gates were used to assess expression of IgM on B cells from CAR T cell–treated MRL-lpr mice (right). (B) The ratio of CD4 to CD8 T cells was determined in 18-week-old MRL-lpr mice treated with CAR T cells and controls. Each symbol represents data from a single mouse, bars are means, and horizontal lines are SDs. Differences between CAR T cell–treated and control mice were examined by two-tailed t test and noted by *P < 0.05 and ***P < 0.001. (C) Proportions of CD4−CD8− DN T cells as the percentage of CD3+ cells from CAR T cell–treated and control MRL-lpr mice were plotted (n. s., no significant differences). (D) Effector memory (CD62LloCD44hi), central memory (CD62LhiCD44hi), and naïve (CD62LhiCD44lo) CD4+ T cells from representative control (top) and CAR T cell–treated MRL-lpr mice (bottom) were determined (for complete CD4+ T cell phenotype assessment, see table S3). (E) The CD62LhiCD44hi population of CD8+ T cells from a representative control mouse (left) and CAR T cell–treated MRL-lpr mouse (right) are shown, and the percentage of CD8+ T cells in the CD62LhiCD44hi gates was displayed. (F) Overall proportions of CD62LhiCD44hi cells among CD8+ or CD4+ T cells in CAR T cell–treated and control mice were determined and plotted. Each symbol represents data from a single mouse, horizontal lines are means, and offset lines are SDs. Differences between CAR T cell–treated and control mice were examined by two-tailed t test and marked by ***P < 0.001. (G) Relative RNA expression in spleens, kidneys, and bone marrow from four CAR T cell–treated and four control NZB/W mice were measured by RT-PCR for transcripts originating from TBP, a house keeping gene, two regions of CD19, TACI, BCMA, CXCR4, and the light-chain constant regions of Igκ and λ1 (see Materials and Methods and table S2). Values were plotted using ln2. Means and SE are displayed. (H) Plasma was prepared from CAR T cell–treated and control MRL-lpr mice, and plasma proteins were analyzed by mass spectrometry. Horizontal lines are means, and offset lines are SDs. Concentrations were compared by two-tailed t test, and significance was indicated by *P < 0.05, **P < 0.01.
We next considered whether the severe depletion of CD19 B cells may delay the progression of disease, in part, by modulating the T cell contribution to pathogenesis. In MRL-lpr mice, CD4+ T cells accumulate with age. The CAR T cell treatment prevented the twofold increase in CD4-to-CD8 T cell ratio that was evident in control mice by 7 months of age (Fig. 5B). As consequence of the lpr mutation, the MRL-lpr strain also accumulates CD4−CD8− double-negative (DN) T cells that do not directly contribute to disease (24); as expected from a T cell–intrinsic genetic defect, we observed no differences between DN T cells from CAR T cell–treated versus control MRL-lpr mice (Fig. 5C). Treatment with CD19-targeted CAR T cells did not appreciably affect the relative proportions of CD62LloCD44hi effector memory CD4+ T cells or CD62LhiCD44hi central memory CD4+ T cells (Fig. 5D), although CD62LhiCD44lo naïve CD4+ were more abundant in CAR T cell–treated MRL-lpr mice (table S4). CD62LhiCD44hi central memory CD8+ T cells constituted nearly half of the CD8+ T cells in most CAR T cell–treated MRL-lpr mice (Fig. 5E), thus indicating a highly significant (P < 0.001) increase in central memory CD8+ T cells relative to age-matched controls and suggesting that this cell population may harbor the CAR.
To test for the presence and functional status of B cells that may remain in the spleen, kidneys, or bone marrow of CAR T cell–treated mice, we used quantitative PCR (qPCR) to determine relative RNA expression for genes that are implicated in the migration and growth factor response of B cells and plasma cells (Fig. 5G). Although CD19 expression was present in spleens, kidneys, and bone marrow samples from control mice, CD19 expression was not detected in any of the three tissues from treated mice, presumably reflecting the efficacy of CD19-targeted CAR T cells. Despite the strong reduction in CD19 expression, RNA coding for Ig light chains and two growth factor receptors found on B cells, transmembrane activator and CamI interactor (TACI) and B cell maturation antigen (BCMA), differed by only twofold or less in spleens from CAR T cell-treated mice versus controls (Fig. 5G). In the bone marrow of treated mice, transcripts for light chains, TACI, and BCMA were expressed at comparable or higher amounts relative to controls, indicating the continued presence of antibody-producing cells in that lymphoid tissue. In contrast, transcripts for Ig light chains were greatly reduced in kidneys from CAR T cell–treated mice relative to control mouse kidneys, suggesting the effective depletion of antibody-producing cells from kidneys of the treated NZB/W mice.
To assess systemic markers of inflammation and autoimmunity in CAR T cell–treated and control MRL-lpr mice, we analyzed the plasma proteome by mass spectrometry. CAR T cell–treated mice had a distinct proteome, in which S100-A10, cathepsin D, tissue factor pathway inhibitor, and complement C4-B were reduced in comparison to control MRL-lpr mice (Fig. 5H). Complement protein C3 was increased in the plasma of CAR T cell–treated mice. The S100-A10 protein is a marker for chronic inflammation (25), and the remaining proteins correlate with lupus pathogenesis in both mice and humans (26–28). The fact that CAR T cell treatment altered the abundance of these five plasma proteins provides further evidence for the amelioration of disease in CAR T cell–treated mice.
Functional tests of CD19-targeted CAR T cells
Because we were surprised at the apparent year-long persistence of CAR T cells in mice, we sought to find evidence of their continued cytotoxic efficacy. To test whether CD19-targeted CAR T cells continue to deplete B cells, we adoptively transferred syngeneic, CFSE-labeled B cells into MRL-lpr mice that had received CAR T cells 4 months earlier. Whereas labeled B cells were present in the blood of control mice 6 days after transfer, they were depleted from circulation in the CAR T cell–treated MRL-lpr mice (Fig. 6A). Active depletion of B cells continued at 11 months after infusion of CAR T cells into NZB/W mice. Syngeneic, CFSE-labeled B cells were undetectable 5 days after transfer to CD19-d NZB/W mice, yet they were present in the blood of CD19-s mice (fig. S5). A third test of CD19-targeted CAR T cell function was carried out by transferring purified CD8+ T cells from CAR T cell–treated MRL-lpr mice to a new cohort of untreated MRL-lpr recipients. An MRL-lpr mouse that had received CAR T cells 7 months earlier served as a donor of splenic CD8+ T cells that were purified and injected into four recipients. Four months later, the MRL-lpr recipients lacked CD19+ B cells (Fig. 6B), and they had acquired a prominent CD62L/CD44 double-positive CD8+ T cell population (Fig. 6C). The CD62L+CD44+ double-positive CD8+ T cells from the spleen of one secondary recipient mouse were enriched by fluorescence-activated cell sorting and were determined to express CD19-targeted CAR transcripts to about four-fold higher concentrations than the CD62LloCD44lo CD8+ T cells (fig. S6). These data firmly established that the CD19-targeted CAR T cells remained viable and functional for several months after injection into NZB/W or MRL-lpr recipients.
Fig. 6. Tests of CAR T cell function in recipient mice.
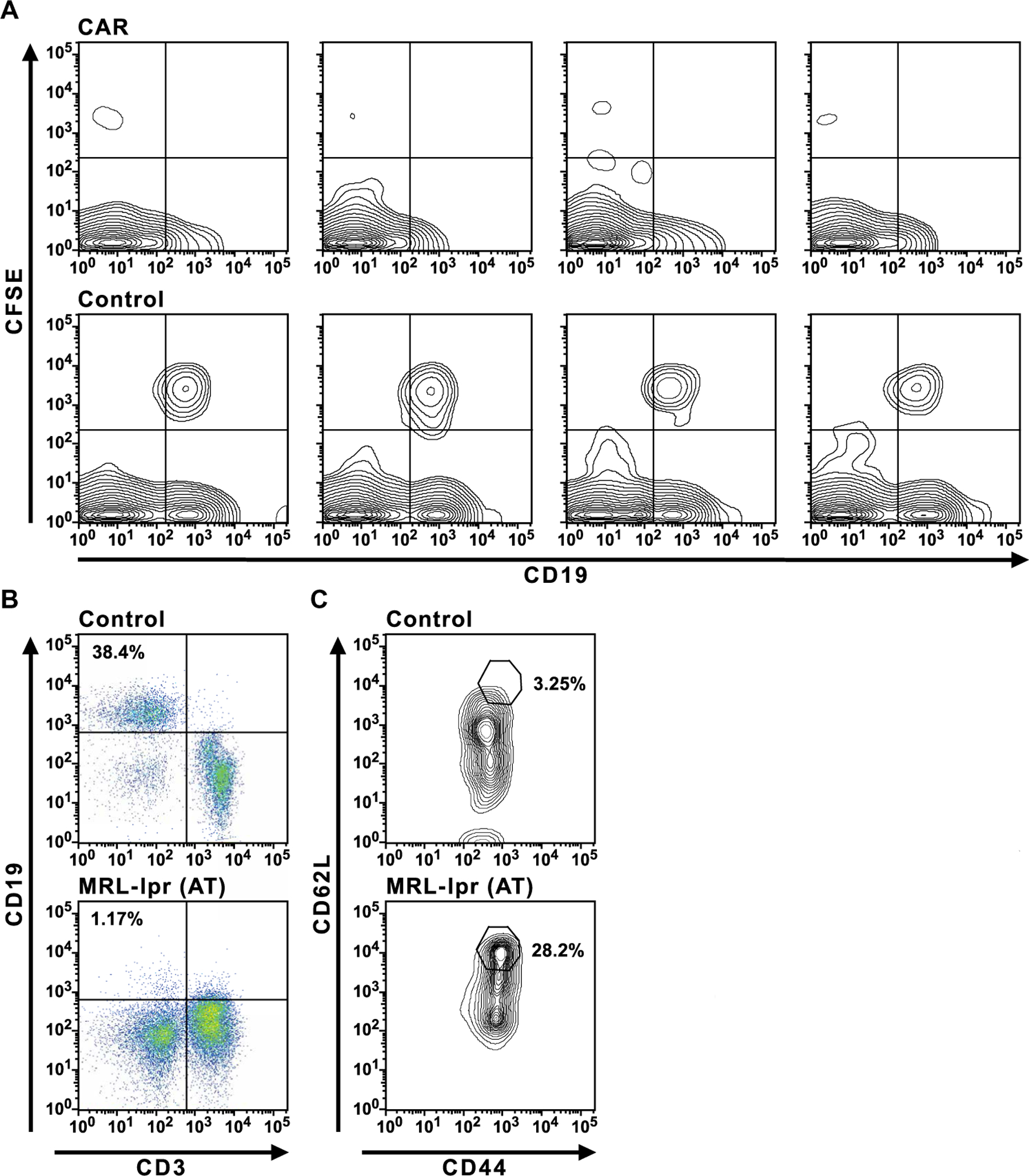
(A) Injections of CFSE-labeled B cells into control or CAR T cell–treated MRL-lpr mice were performed 4 months after CAR T cell administration. Six days after injection into control or CAR T cell–treated mice, peripheral blood was assayed for labeled B cells by flow cytometry. (B and C) Adoptive transfer (AT) of purified splenic CD8+ T cells from an MRL-lpr mouse that was treated 7 months earlier with CD19-targeted CAR T cells into a second group of previously untreated, 2-month-old MRL-lpr mice. The secondary MRL-lpr recipients (MRL-lpr AT) and control MRL-lpr mice were assayed for CD19+ B cells (B) in peripheral blood 4 months after transfer and CD44hiCD62Lhi CD8+ T cells (C). The data in (B) and (C) are representative of four MRL-lpr AT mice observed in two experiments.
DISCUSSION
The depletion of CD19+ B cells by CD19-targeted CAR CD8+ T cells effectively eliminated autoantibody production and deferred or reversed disease manifestations of experimental lupus in two mouse models. These results contrast with previous results in the same mouse models, which showed resistance to anti-CD20 antibody-mediated B cell depletion (11–13). We propose that CD19-targeted CAR T cells have superior efficacy because cytotoxic T cells induce target cell death by a direct mechanism, whereas antibody-mediated cytotoxicity requires the buildup of bound antibody for complement-dependent target cell lysis, antibody-dependent cellular cytotoxicity, or clearance by phagocytes. Previous studies indicated that in models of lupus, the increased abundance of endogenous antibodies and immune complexes impairs B cell depletion by macrophages (12). Thus, anti-CD20 antibody was only effective if given repeatedly and at high doses to autoimmune mice. CD19-targeted CAR T cells, in contrast, kill B cells without the need for an accessory cell type and, thus, deplete B cells more effectively.
In addition, CD19-targeted CAR T cells may have achieved a more sustained B cell depletion due to the specificity and type of treatment used here. CD19 is a B cell surface marker that is intimately involved in B cell signaling and contributes to marginal zone B cell development in the spleen (29). CD19 is critical for B-1 (30) and B-2 (31) B cell signaling, proliferation, and differentiation in germinal centers (32). Moreover, CD19 is expressed on activated B cells, including dividing plasmablasts and early plasma cells (33). Hence, CD19-targeted CAR T cells may target B cells that are stimulated by autoantigens and that are directly responsible for autoantibody production. The depletion of CD19+ B cells did not eliminate all IgM+ B cells, and CAR T cell–treated MRL-lpr mice retained pretreatment expression of plasma IgM and IgG. CAR T cell–treated NZB/W likewise retained plasma IgM and substantial, albeit reduced, IgG.
Potential parallels exist with patients with cancer treated with CD19-targeted CTL019 CAR T cells, who often demonstrate persistence of IgM and IgG in serum, despite the continued presence of CTL019 (34, 35). Patients maintained production of IgM and IgG, including protective antibodies to vaccine antigens (35). The absence of IgM and IgG anti-DNA in CD19-targeted CAR T cell–treated mice was notable and somewhat unexpected. Both B-1 (36) and B-2 (37, 38) cells produce anti-DNA antibodies.
In lupus, autoantibody-secreting cells may arise as short-lived plasmablasts in the spleen and develop into long-lived plasma cells that survive in the bone marrow (39–41). In addition, plasma cells that are linked to the severity of lupus symptoms may accumulate in the kidneys (42). Our RNA analysis indicates the continued expression of Ig light chains in spleens and bone marrow of CAR T cell–treated NZB/W mice, suggesting that CD19-targeted CAR T cells do not deplete all splenic B cell populations or long-lived plasma cells in the bone marrow. The marked reduction of Ig light-chain message in kidneys and the depletion of circulating anti-DNA antibodies in treated mice support the potential of CD19-targeted CAR T cell treatment to mitigate one particular type of disease-related plasma cell yet spare other long-lived plasma cells in the bone marrow.
Limitations of our study include the incomplete characterization of the plasma cell populations and the limited analysis of the specificity and functions of the residual IgMlo B cells in the CAR T cell–treated mice. Similarly, the long-term potential of CD19-targeted CAR T cells may not yet have been fully revealed, in part, because of the short life span of the host species used here. However, the results of the current study provide a blueprint to address most of these limitations.
CD19-targeted CAR T cells may alleviate lupus manifestations over an extended period of time because CAR T cells proliferate in the host and thus offer the potential for a permanent suppression of CD19 B cell production (43). In addition, cell-based therapies ensure that CD19 B cells are depleted from different tissues to which CAR T cells have access, such as the spleen and bone marrow of treated mice. Thus, we presume that B cell depletion starts as soon as CD19-positive, pro–B cells arise in the bone marrow and extend to more mature CD19+ B cells present in the spleen or the blood. This complete and sustained eradication of all CD19+ cells may lead to a more effective B cell depletion than that obtained with monoclonal antibody therapy. We confirmed that the CAR T cells remained functional for several months because CAR T cell–treated mice rapidly depleted transferred autologous CD19+ B cells, whereas such cells persisted in control animals. Moreover, we were able to transfer splenic CD8+ T cells from a CAR T cell–treated MRL-lpr mouse to a second cohort of recipient mice and determine that the transferred T cells depleted CD19+ B cells in the recipients. Together, these results validated the anti-CD19 CAR T cell approach, which ensured a stringent and complete CD19+ B cell depletion and decreased anti-DNA antibodies by one to two orders of magnitude relative to control mice.
The sustained suppression of autoantibody production may have contributed to the observed recovery in kidney function because proteinuria that was initially present in recipient mice returned to baseline. Although experiments with TLR-9−/− MRL-lpr mice implicate autoantibody specificities other than for DNA or chromatin in glomerulonephritis (44), anti-DNA antibodies that bind directly to glomerular basement membrane initiate glomerulonephritis in the absence of other autoimmune specificities (45). Therefore, it is likely—albeit unproven—that elimination of anti-DNA antibody-producing cells contributed to the reduction of proteinuria and mitigated lupus pathogenesis in both MRL-lpr and NZB/W mice.
Before this study, it was unclear whether a CAR transgene could function appropriately in T cells from an autoreactive mouse strain. The long-term efficacy of B cell depletion in our study may have benefited from the use of a second-generation CAR in which the CD3ζ signaling domain was attenuated. Previous studies had demonstrated that dampened CAR signaling extended in vivo persistence of CAR T cells (21, 22). In addition, we also determined that A-MLV retrovirus is a preferable means of CAR gene delivery to mouse CD8+ T cells as compared to VSV-G–pseudotyped MLV or LV. The two features, in combination, may have resulted in stable CD19+ B cell depletion that continued for more than 1 year in both NZB/W and MRL-lpr mice. Using these approaches, we established that CD8+ T cells from NZB/W and MRL-lpr mice correctly carry out the cytotoxic function of the CD19-directed CAR.
Over time, the CAR T cell–treated mice accumulated a CD62LhiCD44hi double-positive CD8+ T cell population, which we observed in the initial and in the secondary recipients of the transduced T cells. In humans, this combination of markers serves to identify a stem cell–like T cell memory population, which has the capacity for self-renewal and differentiation into effector T cells (46, 47). Because central memory CD8+ T cells maintain cytotoxic activity over an extended time, we predict that the CD62L+CD44+ double-positive cells identified here are particularly relevant for the efficacy of our treatment. In support, we showed that CD62LhiCD44hi double-positive CD8+ T cells express the CD19-targeted CAR, which argues for their contribution to sustained B cell depletion.
In summary, advances in CAR T cell technology promise new and more effective treatment options for autoimmune diseases. An antigen-directed CAR that depleted desmoglein-specific B cells in a mouse model of pemphigus was recently described (48). The efficacy of the CD19-targeted CAR T cell approach used here illustrates the potential of CAR therapies in polyantigenic diseases such as lupus. Therapeutic B cell depletion by CD19-targeted CAR T cells for lupus does carry risks: Patients will potentially be susceptible to humoral immunosuppression. Yet, current therapies also suppress the immune system and yield an increased risk of infections. On the basis of 8 years of clinical trial experience with CAR T cells in humans, however, many of the risks associated with CAR T therapy may be manageable. Cancer survivors treated with CD19-targeted CAR T cells retain circulating, protective antibodies and B cells (35). In addition, strategies are available to mitigate toxicity and serious side effects of CD19-targeted CAR T cells. Applications of CD19-targeted CAR T cells to lupus may take advantage of methods for T cell transfer that do not require aggressive patient conditioning (49) that treat excessive cytokine release (50) and that use self-inactivating versions of CAR T cells (51). Confounding risks that are particular to lupus include the potential for autoreactive T cells to be carriers of the activating CAR gene. For the lifetime of recipient mice in our experiments, CAR T cells remained effective without overt side effects. We suggest that CD19-targeted CAR T cells represent a viable strategy for treatment of lupus that may overcome current limitations inherent in the use of monoclonal antibodies. Although caution in extending observations from animal models to humans is essential, our data encourage initial application of CD19-targeted CAR T cells in the most difficult to treat patients with lupus.
MATERIALS AND METHODS
Study design
The goal of this study was to test CD19-targeted CAR T cell treatment in two autoimmune mouse models. Figure 1 shows the CAR protocols. The efficiencies of the treatments are compared in table S1. Mice were followed over time to compare the efficacy of treatments by flow cytometry for CD19+ B cells in blood and by isotype and anti-DNA serology. The effects of the treatment on lupus manifestations were evaluated by overall survival, circulating lymphocyte populations, proteinuria at the beginning and after 5 months of treatment, splenomegaly and kidney immunofluorescence in NZB/W F1 mice, and skin pathology in MRL-lpr mice. At the termination of the experiment, kidney pathology in both strains and gene transcripts in target organs of NZB/W mice were analyzed, and plasma proteomics of MRL-lpr mice was performed. The long-term potential for CAR T cells to deplete CD19+ B cells was tested by transfer of fluorophore-labeled CD19+ B cells into previously CAR T cell–treated recipients and by adoptive transfer of CD8+ T cells from CAR T cell–treated MRL-lpr mice into naïve MRL-lpr mice. Primary data are reported in data file S1.
Animals
Animal welfare was monitored, and euthanasia was induced in strict accordance with University of Tennessee Health Science Center (UTHSC) Institutional Animal Care and Use Committee–approved protocol (15–007). Female NZB/W (at 7 months of age; the Jackson Laboratory) or MRL-lpr mice (7 to 10 weeks old; the Jackson Laboratory) were used in all studies. Two days before the injection of CAR virus injected or control T cells, mice were irradiated with 5 grays to achieve transient myeloablation.
Antibodies
To phenotype mouse lymphocytes in blood or tissues, we used phycoerythrin (PE)–conjugated anti-mouse CD3ɛ (no. 50–0031, Tonbo Biosciences), anaphase-promoting complex (APC) anti-mouse CD8a (no. 20–0081, Tonbo Biosciences), PE-Cy7 anti-mouse CD4 (no. 60–0041, Tonbo Biosciences), redFluor 710 anti-mouse CD19 (no. 80–0193, Tonbo Biosciences), PE-CF594 anti-mouse CD197 (no. 563596, BD Horizon), BV605 anti-mouse CD62L (no. 563252, BD Horizon), anti-mouse IgM (no. 1021–01, SouthernBiotech), and APC-Cy7 anti-mouse CD44 (no. 560568, BD Horizon). To detect CD19-targeted CAR, we used Alexa Fluor 488–conjugated AffiniPure mouse anti-rat Fab-fragment specific reagent (no. 121–545-106, Jackson ImmunoResearch).
Construction and use of retrovirus and lentivirus transfer vectors
All recombinant DNA protocols were approved by the UTHSC Institutional Biosafety Committee. All plasmid constructs were grown in recombination-deficient Escherichia coli (K12 derivative strains) using standard molecular biology techniques. The MSGV-1D3–28Z.1–3 retroviral transfer plasmid (GenBank account no. HM754222) was constructed, as previously described (21). Briefly, this transfer genome contains a CAR consisting of the anti-mouse CD19 scFv derived from the 1D3 IgG2a/κ rat hybridoma linked in frame to a transmembrane domain of mouse CD28 and to two intracellular signaling domains, one derived from CD28, the other, an optimized version of the CD3ζ signaling domain. The FEW 1D3–28Z.1–3 lentiviral transfer plasmid was constructed for this study as follows. The FUWG plasmid was obtained from Addgene and its PacI—AgeI restriction fragment containing a ubiquitin C promoter was replaced by a PacI—AgeI fragment containing a human elongation factor 1α (EF-1α) promoter to create the FEWG plasmid. The sequences downstream of the EF-1α promoter were then replaced with the entire anti-mouse CD19 CAR to create the lentiviral transfer plasmid FEW 1D3–28Z.1–3.
MLV-based retroviral vectors were produced by cotransfection of 293FT cells (Invitrogen) with the MSGV-1D3–28Z.1–3 retroviral transfer plasmid and two additional plasmids that complement in trans the assembly of the virus. One plasmid, the pCAGGS-Gag-Pol, encoded a codon-optimized gag-pol gene from the retrovirus Moloney MLV, the second encoded either the surface glycoprotein of amphotropic 4070A MLV (gift of M. Eiden, National Institutes of Health) or the glycoprotein G of vesicular stomatitis virus. Lentiviral vectors were produced by cotransfection of the FEW 1D3–28Z.1–3 transfer plasmid and the ViraPower packaging mix (Invitrogen). Transfections of 293FT cells were carried out using Lipofectamine 2000 (Invitrogen). Virus was collected at 24, 48, and 72 hours after transfection, and cell supernatants were passed through a 0.45-μm sterile filter units and then further concentrated to yield high-titer stocks. Retroviral stocks were prepared using sterilized Centricon Plus-70 concentration devices as recommended by the manufacturer. Aliquots of concentrated virus were stored at −80°C, and all retroviral stocks tested negative for replication-competent virus. Lentivirus were centrifuged at 28,000 rpm at 4°C for 90 min and resuspended in sterile phosphate-buffered saline (PBS) at 1/200 of the original volume, and aliquots were stored at −80°C until used for transductions.
Purification of CD8+ T cells from spleen
Spleens were harvested from age- and strain-matched donor mice, minced, and dispersed into single cells in RPMI-10 (RPMI-1640 with 10% fetal bovine serum) by pressing between sterile frosted glass slides. Cells were filtered through a 40-μm pore-size cell strainer and washed in RPMI-10, and red blood cells (RBCs) were lysed using the mouse erythrocyte lysis kit (catalog no. WL2000, R&D Systems). CD8+ T cells were enriched by negative selection on a MagCellect mouse CD8+ T cell isolation kit (catalog no. MAGM203, R&D Systems) as instructed by the manufacturer. The yield of CD8+ T cells was about 107 per spleen, and 95% of the isolated cells expressed CD8a. The cells were cultured at 1 million/ml of RPMI-10 medium with recombinant mouse interleukin-2 (IL-2) (no. 402-ML, R&D Systems) at 30 IU/ml and CD28/CD3 beads (Mouse T-activator anti-CD3/CD28 Dynabeads, Gibco) that were added at a 1:1 ratio with the CD8+ T cells.
Transduction of CAR transgenes
The T cells were exposed to two consecutive virus infections. The first infection was carried out at 6 hours after plating of cells with activator beads, and the second infection was at 24 hours. Six-well tissue culture plates were coated with 2.5 ml per well of RetroNectin (no. T100B, Clontech) in sterile PBS, without Ca and Mg, at a final concentration of 9 μg/ml. After coating, plates were washed with sterile PBS and blocked with 2% bovine serum albumin (BSA) in PBS for 30 min. After another wash, plates were incubated with 1.5 ml of RPMI-10 per well to which 40 μl of 200× concentrated virus were added just before dispensing into wells. The plate was centrifuged at 2000 rpm at room temperature for 2 hours.
After centrifugation, about 0.5 ml of the media was left in the well, and 1 ml of the activated T cells along with the activator beads was added. The second infection was performed exactly as the first, with freshly prepared tissue culture plates. Cells were fed fresh media containing 30 IU of IL-2 every other day and reached a fivefold expansion from the initial cell numbers on the 5th day of culture. At that time, remaining activator beads were magnetically removed and discarded, and CD8+ T cells were washed, counted, and suspended at 1.2 × 107/ml in RPMI-1640 containing 10% syngeneic mouse serum just before injections into recipients. An aliquot of the cells was assayed for CAR expression by anti-rat Fab antibody, and 5 to 15% of cells were stained positive (fig. S1). About 100 μl of cell suspension was injected into the retro-orbital sinus of each mouse with appropriate sedation and administration of analgesics. Immediately after the injection, the mice were administered 45,000 IU of IL-2 into the scruff of their neck.
Flow cytometry
After the injection of CD8+ T cells, mice were bled at periodic intervals from the retro-orbital plexus, starting at about 3 weeks after T cell injection, and blood was collected into heparinized tubes. Seventy microliters of blood was used to phenotype circulating lymphocytes using antibodies for CD3e, CD4, CD8a, and CD19, following standard procedures. Cells were incubated with 2 μl of mouse Fc block (no. 14–0161, Affymetrix), before addition of antibody cocktail. After labeling, RBCs were lysed with 750 μl of high-yield lyse solution (no. HYL250, Life Technologies). Cytometry was performed on a Bio-Rad ZE5. CAR expression on primary T cells was determined by staining with Alexa Fluor 488–conjugated mouse anti-rat Fab fragment–specific antibodies (catalog no. 121–545-106, Jackson ImmunoResearch). Ghost Dye Red 710 (no. 13–0871, Tonbo Biosciences) was used to differentiate between live and dead cells.
Serology and ELISA
Plasma was prepared by centrifugation of blood for 10 min at 2000g and used to determine total IgG, IgM, and anti-DNA antibodies. Total amounts of immunoglobulins in the mouse plasma were determined using ELISA in 96-well plates coated with AffiniPure goat anti-mouse IgG (H + L) (no. 115–005-003, Jackson ImmunoResearch) or, alternatively, with goat anti-mouse IgM (H + L) (SouthernBiotech) in coating buffer [0.1 M NaHCO3 (pH 9.6)] at 4°C overnight. Coated plates were washed and incubated for 1 hour at room temperature in blocking buffer (1% BSA in PBS with 0.02% NaN3).
For mouse IgG and IgM detection, 10-fold serial dilutions of the plasma were prepared in blocking buffer (range 10−2 to 10−7), added to the plates, and incubated at room temperature for 3 hours. The plate was washed four times with PBS containing 0.05% Tween 20 (PBS-T). The detection antibody (alkaline phosphatase–labeled goat anti-mouse IgG, or anti-mouse IgM, at 1:1000 dilution in blocking buffer) was added and incubated for 1 hour at room temperature. The plates were washed four times with PBS-T. Phosphatase detection substrate (no. S0942, Sigma-Aldrich) at 1 mg/ml was added for color development. Absorption in plates was read at 405 nm. With each ELISA, a positive standard mouse IgG (no. 015–000-0030, the Jackson Laboratory), or mouse IgM was used to control for inter-experimental variation. Anti-DNA IgG and IgM antibody titers were determined using ELISA plates coated with calf thymus DNA (no. D3664, Sigma-Aldrich) at 10 μg/ml of PBS, supplemented with 1 mM EDTA. Titers were calculated as the inverse of dilutions at which optical density equaled twice background readings. The detection of bound antibodies was performed as for isotype ELISAs except that threefold serial dilutions were prepared starting at a 1:30 initial dilution (range, 1:30 to 1:7290).
Plasma proteome analysis
Plasma was collected from CAR T cell–treated and control MRL-lpr mice and depleted of the most abundant mouse plasma proteins by chromatography on mouse ProSep 10 columns (Sigma-Aldrich), as instructed by the manufacturer. After depletion, 50 μl of plasma samples were mixed with 10 μl of 50 mM NH4HCO3 and 25 μl of 0.2% RapiGest SF (no. 186001861, Waters). The samples were heated at 80°C for 15 min, 2.5 μl of 100 mM dithiothreitol solution was added, and the samples were incubated at 60°C for 30 min. After cooling to room temperature, 2.5 μl of 300 mM iodoacetamide solution was added and incubated for 30 min in the dark. After protein denaturation and alkylation, the samples were mixed with 10 μl of sequencing-grade trypsin solution (0.05 mg/ml; no. V511A, Promega). Trypsin digestion was carried out overnight at 37°C. The digested samples were acidified by addition of 1 μl of concentrated trifluoroacetic acid (TFA) and dried in a speed vacuum centrifuge. The dried peptide digests were reconstituted with a solution of H2O/ACN (acetonitrile)/TFA (98:2:0.1) and desalted with a C18 micro tip column (ZipTip, Millipore). The peptides were diluted 2:1 (v/v) with an internal standard solution containing 37.5 fmol/ml of yeast alcohol dehydrogenase (ADH) tryptic digest (Waters). All peptide separations were performed on a nanoUPLC liquid chromatography system (Waters ACQUITY M-Class UPLC System). Mobile phase A was H2O containing 0.1% formic acid; mobile phase B was ACN containing 0.1% formic acid. Peptide samples were injected, 2 μl of partial loop injection onto a Symmetry C18 (5 mm, 180 μm × 20 μm trap column; Waters). After preconcentration, the peptides were eluted from the trap column to the analytical column, HSS T3 C18 (1.8 mm, 75 μm × 250 mm; Waters), and separated with a gradient of 2 to 40% mobile phase B over 90 min at a flow rate of 0.3 ml/min. The column temperature was maintained at 45°C. The eluted peptides were analyzed online on a Synapt G2-Si (Waters) high-resolution ion mobility mass spectrometer (IMS). The lock mass peptide, [Glu1]-fibrinopeptide B, was delivered by infusion through the instrument fluidics system. The quadrupole radio frequency was set for efficient transmission of ions with mass/charge ratio of 300 to 2000. Data were acquired in a data-independent acquisition mode with ion mobility separation and optimized transfer collision energy, ultra-definition MS [UDMS(E)] (52). The time-of-flight analyzer was operated in resolution mode (resolution of ≥20,000 full width at half maximum, with average mass error of 5 parts per million or less), a scan time of 0.6 ms, MS(E) range of 50 to 2000 Da, and IMS separation. Protein identification and label-free quantification was performed with Progenesis QI (Nonlinear Dynamics/Waters) (53). The MS(E) data were searched against a mouse protein database (no. UP000000589, UniProt) with static modification on cysteine (carbamidomethylation), variable modification on methionine (oxidation), and allowing up to one missed cleavage per peptide. Label-free quantification was performed using the top three Hi-N method (54) with ADH as internal standard.
Tissue preparation and pathology
Kidneys and spleens were flash-frozen in optimal cutting temperature (OCT) compound (no. 23–730-571, Fisher Biotec) and sectioned at 5-μm thickness on a cryotome. Frozen sections were thawed, blocked in blocking buffer, and incubated with goat anti-mouse IgG isotype antibodies for fluorescence microscopy. Skin sections were formalin-fixed, paraffin-embedded, sectioned, and stained with H&E stain to evaluate skin pathology. Renal pathology was assessed as described by Singh et al. (55) based on the histology of kidneys fixed in 4% paraformaldehyde and embedded in paraffin. Sections were stained with H&E and scored on a 0 to 3 scale, indicating no to severe manifestations. Separate scores were assigned to glomerular hypercellularity, necrosis, karyorrhexis, cellular crescents, and hyaline deposits to indicate acute lesions in glomeruli. Similarly, interstitial inflammation, tubular cell necrosis, and tubular cell casts were scored to determine acute tubule-interstitial disease. Chronic kidney lesions were scored for glomerulosclerosis, glomerular scars, fibrous crescents, tubular atrophy, and interstitial fibrosis. The scores were averaged to obtain a mean score for glomerular, tubule-interstitial, and chronic components, and the three subscores were added to generate an overall kidney biopsy score. Skin sections were evaluated for acanthosis, hyperkeratosis, cellular infiltration, vascular dilation, ulceration, and scar formation, scored on a scale from 0 to 3 for severity; scores were averaged over all categories, expressed for head versus body; and average scores were computed. Proteinuria was measured by placing a 5-μl drop of freshly collected urine onto an Albustix indicator strip (no. 2191, Siemens) and comparing the observed color to a standard color scale provided by the manufacturer. The classification proposed by Adelman et al. (56) was used to define high-grade proteinuria.
RT-PCR and qPCR
To detect CAR T cell expression in various tissues, we isolated total RNA from spleen, bone marrow, kidney, and blood using TRIzol reagent (no. 15596018, Invitrogen). Kidney and spleen RNA were also prepared from kidneys and spleens of four CD19-d and CD19-s NZB/W mice that were stored frozen in OCT. Those samples were obtained by sectioning tissues into 50-μm-thick sections with a cryotome, removing the OCT by a quick rinse in ice-cold PBS and transferring the slices to tubes containing TRIzol reagent. Contaminating DNA was removed by deoxyribonuclease treatment, and total RNA was quantified using spectrophotometry. The RNA was converted to complementary DNA (cDNA) using transcriptor first-strand cDNA kit (no. 04379012001, Roche), and qPCR was performed on a Lightcycler 480 instrument (Roche) using gene-specific primers and probes designed using Roche’s Universal ProbeLibrary Assay Design Center. For details of primers and probes used in this study, see table S2.
Statistical analysis
Data for the CAR T cell–treated and control experimental groups were analyzed by one or two-tailed t test, as appropriate for pairwise comparison of groups with divergent sample distributions. Proteinuria was measured between treated and control mice by one-way ANOVA, and survival curves were compared by Mantel-Cox log-rank test. The statistics features of Microsoft Excel and GraphPad Prism data analysis software suites were used.
Supplementary Material
Fig. S1. Analysis of CAR expression at the cell surface.
Fig. S2. Transient depletion of CD19+ B cells from one NZB/W mouse.
Fig. S3. Immunofluorescence images of representative glomeruli from CAR T cell–treated and control MRL-lpr mice.
Fig. S4. Comparison of alopecia and skin lesions between CAR T cell–treated and negative control MRL-lpr mice.
Fig. S5. Analysis of CAR T cell cytotoxicity in vivo.
Fig. S6. qPCR of transcripts expressed in sorted CD8+ T cells.
Fig. S7. Reverse transcription qPCR of CAR RNA in bone marrow and spleen of CD19+ depleted and CD19+ intact NZB/W mice.
Fig. S8. Typical gating strategy.
Table S1. Comparison of viral vectors used to transduce T cells.
Table S2. List of oligonucleotide primers used for RT-PCR.
Table S3. CD4+ T cell subsets.
Data file S1. Primary data.
Acknowledgments:
We thank H. Haecker (St. Jude’s Children’s Research Hospital), M. Shlomchik (University of Pittsburgh), and D. Pattanaik (UTHSC) for constructive comments on the manuscript. The technical assistance of S. Barnett and T. Costello with animal care, D. Daria with flow cytometry, T. J. Hollingsworth with tissue imaging, and T. Higgins with graphics are appreciated.
Funding: This work was supported by the Lupus Research Alliance of New York, NY to M.R. Funding was also provided by the CORNET mechanism (UTHSC) and by the ORR fund to M.R. Portions of the work were supported by PHS NIH award R01CA081171 to L.M.A.
Footnotes
Competing interests: UTHSC filed a patent application, no. 65022–274929, titled “Use of Anti-CD19 CAR T Cells in Lupus.” (M.R., inventor). J.N.K. has patent applications for CARs targeting human CD19.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/11/482/eaav1648/DC1
REFERENCES AND NOTES
- 1.Rahman A, Isenberg DA, Systemic lupus erythematosus. N. Engl. J. Med 358, 929–939 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Shlomchik MJ, Activating systemic autoimmunity: B’s, T’s, and tolls. Curr. Opin. Immunol 21, 626–633 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sang A, Zheng Y-Y, Morel L, Contributions of B cells to lupus pathogenesis. Mol. Immunol 62, 329–338 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y, Chen F, Putt M, Koo YK, Madaio M, Cambier JC, Cohen PL, Eisenberg RA, B cell depletion with anti-CD79 mAbs ameliorates autoimmune disease in MRL/lpr mice. J. Immunol 181, 2961–2972 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallagher S, Yusuf I, McCaughtry TM, Turman S, Sun H, Kolbeck R, Herbst R, Wang Y, MEDI-551 treatment effectively depletes B cells and reduces serum titers of autoantibodies in mice transgenic for Sle1 and human CD19. Arthritis Rheumatol 68, 965–976 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Touma Z, Gladman DD, Current and future therapies for SLE: Obstacles and recommendations for the development of novel treatments. Lupus Sci. Med 4, e000239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW, Hessey EW, Shaw T, Totoritis MC; REFLEX Trial Group, Rituximab for rheumatoid arthritis refractory to anti–tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum 54, 2793–2806 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Calabresi PA, B-cell depletion — A frontier in monoclonal antibodies for multiple sclerosis. N. Engl. J. Med 376, 280–282 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, Utset TO, Gordon C, Isenberg DA, Hsieh H-J, Zhang D, Brunetta PG, Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum 62, 222–233 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez Mendez LM, Cascino MD, Garg J, Katsumoto TR, Brakeman P, Dall’Era M, Looney RJ, Rovin B, Dragone L, Brunetta P, Peripheral blood B cell depletion after rituximab and complete response in lupus nephritis. Clin. J. Am. Soc. Nephrol 13, 1502–1509 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bekar KW, Owen T, Dunn R, Ichikawa T, Wang W, Wang R, Barnard J, Brady S, Nevarez S, Goldman BI, Kehry M, Anolik JH, Prolonged effects of short-term anti-CD20 B cell depletion therapy in murine systemic lupus erythematosus. Arthritis Rheum 62, 2443–2457 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahuja A, Teichmann LL, Wang H, Dunn R, Kehry MR, Shlomchik MJ, An acquired defect in IgG-dependent phagocytosis explains the impairment in antibody-mediated cellular depletion in lupus. J. Immunol 187, 3888–3894 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Shlomchik MJ, Depletion of B cells in murine lupus: Efficacy and resistance. J. Immunol 179, 3351–3361 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Reddy V, Cambridge G, Isenberg DA, Glennie MJ, Cragg MS, Leandro M, Internalization of rituximab and the efficiency of B cell depletion in rheumatoid arthritis and systemic lupus erythematosus. Arthritis Rheumatol 67, 2046–2055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Looney RJ, Anolik JH, Campbell D, Felgar RE, Young F, Arend LJ, Sloand JA, Rosenblatt J, Sanz I, B cell depletion as a novel treatment for systemic lupus erythematosus: A phase I/II dose-escalation trial of rituximab. Arthritis Rheum 50, 2580–2589 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Cooper LJN, Topp MS, Serrano LM, Gonzalez S, Chang W-C, Naranjo A, Wright C, Popplewell L, Raubitschek A, Forman SJ, Jensen MC, T-cell clones can be rendered specific for CD19: Toward the selective augmentation of the graft-versus-B–lineage leukemia effect. Blood 101, 1637–1644 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Brentjens RJ, Latouche J-B, Santos E, Marti F, Gong MC, Lyddane C, King PD, Larson S, Weiss M, Rivière I, Sadelain M, Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med 9, 279–286 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Lim WA, June CH, The principles of engineering immune cells to treat cancer. Cell 168, 724–740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Theofilopoulos AN, Dixon FJ, Murine models of systemic lupus erythematosus. Adv. Immunol 37, 269–390 (1985). [DOI] [PubMed] [Google Scholar]
- 20.Koh D-R, Ho A, Rahemtulla A, Fung-Leung W-P, Griesser H, Mak T-W, Murine lupus in MRL/lpr mice lacking CD4 or CD8 T cells. Eur. J. Immunol 25, 2558–2562 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA, Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood 116, 3875–3886 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, Sadelain M, Eshhar Z, Rosenberg SA, Morgan RA, A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J. Immunol 183, 5563–5574 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bribes E, Galiegue S, Bourrie B, Casellas P, Involvement of the peripheral benzodiazepine receptor in the development of cutaneous pathology in Mrl/Lpr mice. Immunol. Lett 85, 13–18 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Jevnikar AM, Grusby MJ, Glimcher LH, Prevention of nephritis in major histocompatibility complex class II-deficient MRL-lpr mice. J. Exp. Med 179, 1137–1143 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salle V, Cordonnier C, Schmidt J, Mazière C, Smail A, Attencourt C, Mabille MP, Mazière JC, Makdassi R, Choukroun G, Diouf M, Duhaut P, Ducroix JP, Vascular expression of annexin A2 in lupus nephritis. J. Clin. Pathol 69, 533–536 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Phi NC, Chien DK, Binh VV, Gergely P, Cathepsin D-like activity in serum of patients with systemic lupus erythematosus. J. Clin. Lab. Immunol 29, 185–188 (1989). [PubMed] [Google Scholar]
- 27.Roldan V, Marco P, Fernandez C, Pascual E, Levels of tissue factor pathway inhibitor in lupus patients correlate with lupus activity and endothelial damage markers. Haematologica 87, 1231–1232 (2002). [PubMed] [Google Scholar]
- 28.Bao L, Haas M, Quigg RJ, Complement factor H deficiency accelerates development of lupus nephritis. J. Am. Soc. Nephrol 22, 285–295 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.You Y, Zhao H, Wang Y, Carter RH, Cutting edge: Primary and secondary effects of CD19 deficiency on cells of the marginal zone. J. Immunol 182, 7343–7347 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato S, Miller AS, Howard MC, Tedder TF, Regulation of B lymphocyte development and activation by the CD19/CD21/CD81/Leu 13 complex requires the cytoplasmic domain of CD19. J. Immunol 159, 3278–3287 (1997). [PubMed] [Google Scholar]
- 31.Wang Y, Brooks SR, Li X, Anzelon AN, Rickert RC, Carter RH, The physiologic role of CD19 cytoplasmic tyrosines. Immunity 17, 501–514 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Carter RH, CD19 regulates B cell maturation, proliferation, and positive selection in the FDC zone of murine splenic germinal centers. Immunity 22, 749–761 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Pracht K, Meinzinger J, Daum P, Schulz SR, Reimer D, Hauke M, Roth E, Mielenz D, Berek C, Côrte-Real J, Jäck HM, Schuh W, A new staining protocol for detection of murine antibody-secreting plasma cell subsets by flow cytometry. Eur. J. Immunol 47, 1389–1392 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, Brogdon JL, Pruteanu-Malinici, Bhoj V, Landsburg D, Wasik M, Levine BL, Lacey SF, Melenhorst JJ, Porter DL, June CH, Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med 377, 2545–2554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhoj VG, Arhontoulis D, Wertheim G, Capobianchi J, Callahan CA, Ellebrecht CT, Obstfeld AE, Lacey SF, Melenhorst JJ, Nazimuddin F, Hwang W-T, Maude SL, Wasik MA, Bagg A, Schuster S, Feldman MD, Porter DL, Grupp SA, June CH, Milone MC, Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood 128, 360–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayakawa K, Hardy RR, Honda M, Herzenberg LA, Steinberg AD, Herzenberg LA, Ly-1 B cells: Functionally distinct lymphocytes that secrete IgM autoantibodies. Proc. Natl. Acad. Sci. U.S.A 81, 2494–2498 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marion TN, Bothwell AL, Briles DE, Janeway CA Jr., IgG anti-DNA autoantibodies within an individual autoimmune mouse are the products of clonal selection. J. Immunol 142, 4269–4274 (1989). [PubMed] [Google Scholar]
- 38.Shlomchik M, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, Weigert M, Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J. Exp. Med 171, 265–292 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hiepe F, Alexander T, Voll RE, Plasma cells. Z. Rheumatol 74, 20–25 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Hale M, Rawlings DJ, Jackson SW, The long and the short of it: Insights into the cellular source of autoantibodies as revealed by B cell depletion therapy. Curr. Opin. Immunol 55, 81–88 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahévas M, Michel M, Weill J-C, Reynaud C-A, Long-lived plasma cells in autoimmunity: Lessons from B-cell depleting therapy. Front. Immunol 4, 494 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Espeli M, Bokers S, Giannico G, Dickinson HA, Bardsley V, Fogo AB, Smith KGC, Local renal autoantibody production in lupus nephritis. J. Am. Soc. Nephrol 22, 296–305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davila ML, Kloss CC, Gunset G, Sadelain M, CD19 CAR-targeted T cells induce long-term remission and B cell aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLOS ONE 8, e61338 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ, Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25, 417–428 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Krishnan MR, Wang C, Marion TN, Anti-DNA autoantibodies initiate experimental lupus nephritis by binding directly to the glomerular basement membrane in mice. Kidney Int 82, 184–192 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu F, Zhang W, Shao H, Bo H, Shen H, Li J, Liu Y, Wang T, Ma W, Huang S, Human effector T cells derived from central memory cells rather than CD8+ T cells modified by tumor-specific TCR gene transfer possess superior traits for adoptive immunotherapy. Cancer Lett 339, 195–207 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Gattinoni L, Speiser DE, Lichterfeld M, Bonini C, T memory stem cells in health and disease. Nat. Med 23, 18–27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, Milone MC, Payne AS, Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353, 179–184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ, Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 119, 4133–4141 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, Przepiorka D, Farrell AT, Pazdur R, FDA approval summary: Tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. Oncologist 23, 943–947 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paszkiewicz PJ, Fräßle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, Sadelain M, Liu L, Jensen MC, Riddell SR, Busch DH, Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Invest 126, 4262–4272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Distler U, Kuharev J, Navarro P, Levin Y, Schild H, Tenzer S, Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods 11, 167–170 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Sedic M, Gethings LA, Vissers JPC, Shockcor JP, McDonald S, Vasieva O, Lemac M, Langridge JI, Batinić D, Pavelić SK, Label-free mass spectrometric profiling of urinary proteins and metabolites from paediatric idiopathic nephrotic syndrome. Biochem. Biophys. Res. Commun 452, 21–26 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Silva JC, Gorenstein MV, Li G-Z, Vissers JPC, Geromanos SJ, Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol. Cell. Proteomics 5, 144–156 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Singh RR, Saxena V, Zang S, Li L, Finkelman FD, Witte DP, Jacob CO, Differential contribution of IL-4 and STAT6 vs STAT4 to the development of lupus nephritis. J. Immunol 170, 4818–4825 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adelman NE, Watling DL, McDevitt HO, Treatment of (NZB x NZW)F1 disease with anti-I-A monoclonal antibodies. J. Exp. Med 158, 1350–1355 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Analysis of CAR expression at the cell surface.
Fig. S2. Transient depletion of CD19+ B cells from one NZB/W mouse.
Fig. S3. Immunofluorescence images of representative glomeruli from CAR T cell–treated and control MRL-lpr mice.
Fig. S4. Comparison of alopecia and skin lesions between CAR T cell–treated and negative control MRL-lpr mice.
Fig. S5. Analysis of CAR T cell cytotoxicity in vivo.
Fig. S6. qPCR of transcripts expressed in sorted CD8+ T cells.
Fig. S7. Reverse transcription qPCR of CAR RNA in bone marrow and spleen of CD19+ depleted and CD19+ intact NZB/W mice.
Fig. S8. Typical gating strategy.
Table S1. Comparison of viral vectors used to transduce T cells.
Table S2. List of oligonucleotide primers used for RT-PCR.
Table S3. CD4+ T cell subsets.
Data file S1. Primary data.