

Abstract
The association between inflammation, infection, and venous thrombosis has long been recognized; yet, only in the last decades have we begun to understand the mechanisms through which the immune and coagulation systems interact and reciprocally regulate one another. These interconnected networks mount an effective response to injury and pathogen invasion, but if unregulated can result in pathologic thrombosis and organ damage. Neutrophils, monocytes, and platelets interact with each other and the endothelium in host defense and also play critical roles in the formation of venous thromboembolism. This knowledge has advanced our understanding of both human physiology and pathophysiology, as well as identified mechanisms of anticoagulant resistance and novel therapeutic targets for the prevention and treatment of thrombosis. In this review, we discuss the contributions of inflammation and infection to venous thromboembolism.
Keywords: Inflammation, venous thrombosis, venous thromboembolism, thromboinflammation, coagulation, neutrophil extracellular traps, platelets, endothelium
Subject Terms: Endothelium/Vascular Type/Nitric Oxide, Peripheral Vascular Disease, Inflammation
Venous thromboembolism (VTE), encompassing both pulmonary embolism (PE) and deep vein thrombosis (DVT), is a leading cause of morbidity and mortality worldwide. VTE is responsible for 300,000 deaths annually in the United States alone and is the third most common cause of cardiovascular mortality1. Following an initial VTE, patients remain at risk of recurrent thrombosis, which is only partially mitigated by treatment with anticoagulants. The fatality rate of PE continues to increase, most notably in adults younger than 65 years of age2. Similarly, patients with acute or chronic inflammatory diseases, including sepsis, inflammatory bowel disease, and autoimmune disease, are at higher risk of initial and recurrent VTE3, 4. These concerning trends are despite the introduction of direct oral anticoagulants as well and improved use of VTE prophylaxis in high-risk situations. Taken together, these observations highlight the need for a deeper understanding of the molecular and cellular mechanisms that culminate in VTE in order to identify novel therapeutic approaches.
Virchow’s triad of factors predisposing to thrombosis- altered blood flow or stasis, changes in composition of blood (hypercoagulability), and vessel wall damage, have been the foundation for our understanding of the pathophysiology of venous thrombosis. However, inflammatory molecules and immune cells are now recognized as central contributors to thrombosis. In many clinical contexts, the risk factors for VTE, including inflammatory bowel disease, systematic lupus erythematosus, obesity, surgery, cancer, and acute and chronic infection are characterized by dysregulated immune networks that intersect with coagulation3. Immune dysregulation and disruption of the natural antithrombotic blood:vessel interface adds contextual elements to Virchow’s triad which may more accurately reflect the modern understanding of VTE5, 6. Current therapeutics for VTE target coagulation factors and are associated with a risk of bleeding, but don’t address the key inflammatory processes that lead to thrombosis and leave a significant gap in treatments available to prevent and treat VTE effectively.
The relationship between inflammation and plasma coagulation is an intricate network that balances an organism’s response to injury and pathogen invasion with maintaining homeostasis. Perturbation of this balance can tip physiologic responses toward pathological thrombosis and organ damage. The molecular interactions between inflammation and coagulation have been elegantly reviewed by Foley et al7. In this Review, we focus on the contributions of inflammation to the pathogenesis of VTE by exploring the nuanced cellular interactions between immune responses and coagulation that result in VTE. We begin by defining central concepts in the relationship between thrombosis and inflammation, followed by a review of the vascular immune cell-based model of venous thrombogenesis.
Hemostasis, immunothrombosis, and deranged thromboinflammation
Hemostasis is a protective process to reduce blood loss after injury through a series of highly regulated steps, initially by sealing the damaged blood vessel and ultimately by supporting vessel repair. Central to this process is the controlled generation of thrombin and fibrin, which is contemporaneously described using a cell-based model of coagulation (Figure 1). Platelets, leukocytes, and endothelial cells are the primary cellular components and they localize coagulation reactions, ensuring activation of coagulation factors occurs at areas of injury. Pathologic thrombosis in veins or arteries occurs when coagulation is triggered either in the absence of vascular injury, or if unchecked coagulation occurs at sites of injury. In the deep veins, the majority of thrombi form on grossly intact endothelium and frequently within valve pockets of deep veins, where lower blood oxygen levels and blood stasis occur8. Thrombi in the pulmonary circulation can occur in situ, or as pulmonary emboli when venous thrombi become unstable, detach, embolize to the lungs, and occlude pulmonary circulation.
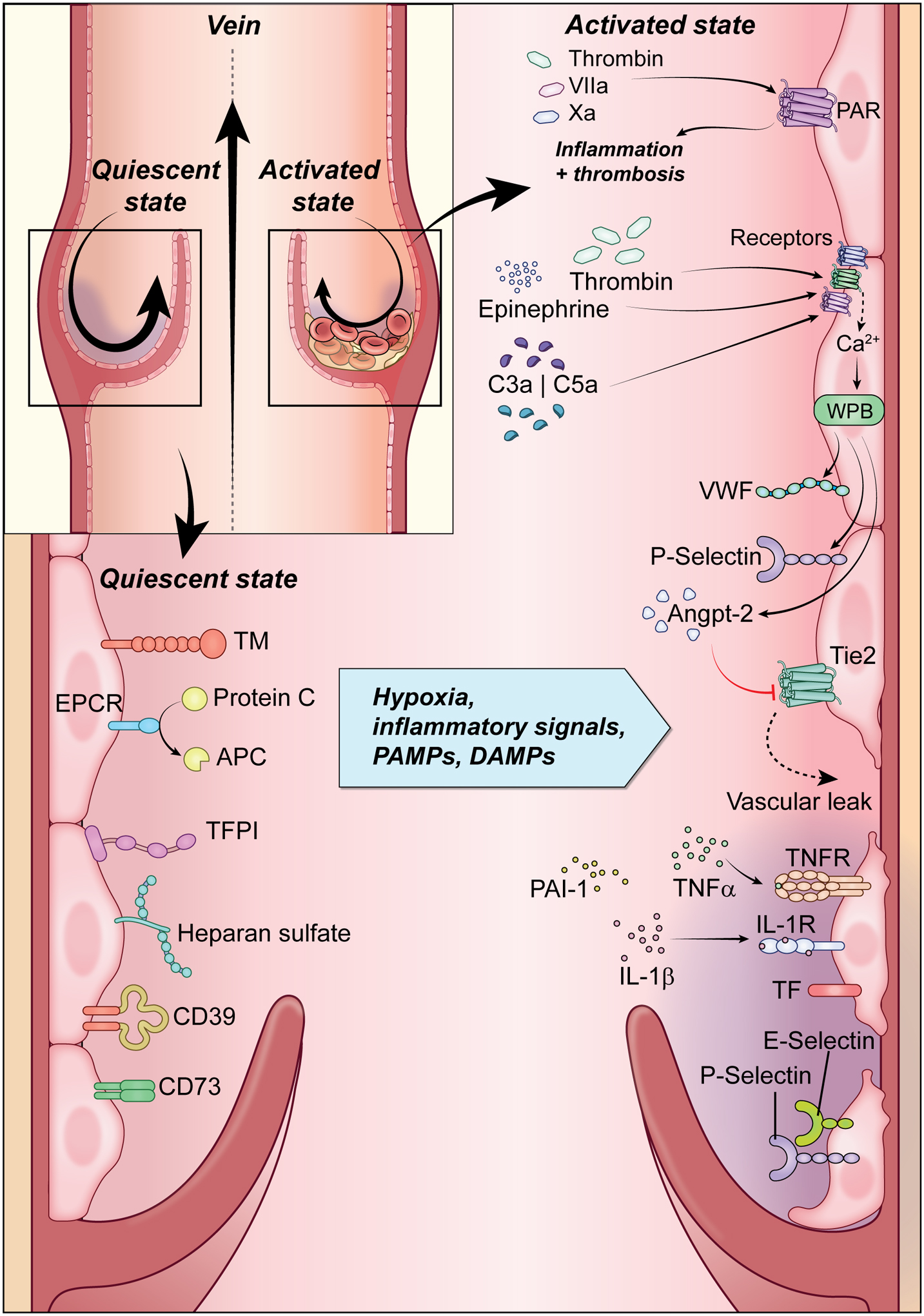
Figure 1: Endothelial Activation.
The quiescent endothelium shown on the left wall of the vein is an antithrombotic surface. Endothelial cells prevent or limit coagulation through expression of heparan sulfate, tissue factor pathway inhibitor (TFPI), CD39, and CD73, as well as release of tissue plasminogen activator (t-PA), prostacyclin and nitric oxide. Additionally, expression of endothelial protein C receptor (EPCR) and thrombomodulin (TM) exert anti-inflammatory effects and negatively regulate thrombin generation. Shown on the right wall of the vein, endothelial cells, in response to injury or functional perturbations, endothelial cells undergo shift to prothrombotic surface. There are increased expression leukocyte adhesion molecules (P- and E-selectin) and tissue factor (TF), loss of vascular integrity and expression of antithrombotic molecules, and production of cytokines. (Illustration credit: Erina He).
Similarly, inflammation, an early defense mechanism of the immune system in response to infection, irritation, or injury is necessary for host defense. Characterized by increases in local blood flow, recruitment of leukocytes, and release of chemokines and cytokines, inflammation contains and eliminates pathogens, and facilitates wound healing. Immunothrombosis, the result of reciprocal actions of the immune and coagulation systems, is an emergent response to the entry of infectious pathogens into the vasculature. Following pathogen recognition, local thrombosis and microvascular occlusion aid in control of infection dissemination and pathogen elimination9. This thrombotic mechanism of controlling infections is evolutionarily conserved, and ancient organisms that lack an adaptive immune system, such as Horseshoe crabs, rely on clotting factors and a single type of circulating blood cell to ward off pathogen invasion through coagulation of hemolymph10. Infectious diseases have exerted external pressures that influenced the evolution of the human hemostatic system. For example, the acquisition by Group A Streptococcus of the ability to produce a streptokinase that specifically activates human plasminogen provides a window into the pathogen-host “arms race” in physiologic coagulation factor interactions11, 12. Distinct from physiologic immunothrombosis, exaggerated and uncontrolled activation of the immune and coagulation systems can lead to microvascular and macrovascular thrombosis called “thromboinflammation” and result in tissue damage13. The historical separation of the inflammatory and coagulation systems has curtailed a more complete understanding of the mechanisms of hypercoagulability described in Virchow’s triad.
Endothelial activation and local blood:vessel barrier dysfunction
The layer of endothelial cells at the blood:vessel interface is a physical barrier between blood-borne clotting factors and prothrombotic extracellular matrices. The intact endothelium tonically maintains blood fluidity and vascular homeostasis through anticoagulant and anti-inflammatory properties, including the production of prostacyclins, nitric oxide (NO), and the ADP-dissipating ectonucleotidase CD3914, 15 (Figure 1). The single cell layer endothelium also exert a fibrinolytic counterbalance to physiologic thrombosis, by producing tissue-type plasminogen activator (tPA)16 and urokinase plasminogen activator (uPA)17. The endothelial lining actively resists inflammation by controlling the expression of adhesion and signaling molecules at its luminal surface that facilitate leukocyte recruitment.
Natural anticoagulants on the endothelium that also reduce inflammation include the glycosaminoglycans (principally heparan sulfate (HS) and chondroitin sulfate (CS)), tissue factor pathway inhibitor (TFPI), endothelial cell protein C receptor (EPCR), and thrombomodulin (TM)18, 19.
Following injury or local inflammation, the endothelium loses its anticoagulant and anti-inflammatory properties to eliminate the insult and initiate repair. This phenotypic shift can be triggered by hemodynamic flow pattern changes, cytokine signals, hypoxia, tissue injury, endotoxemia, or pathogen recognition5, 18, 20, 21. Left unchecked, these physiologic responses exert a pathologic effect. Rapid loss of anticoagulant molecules and vascular integrity are accompanied by increases in the expression of leukocyte adhesion molecules and cytokine production. In response to metabolic and cytotoxic insults such as hyperglycemia, endotoxemia and sepsis, matrix metalloproteases dissolve the antithrombotic endothelial glycocalyx coated with HS and CS, resulting in the loss of binding sites for the serine protease inhibitor, antithrombin III (ATIII)22,23. The associated shedding of EPCR and TM further breaks down the endothelial anticoagulant and anti-inflammatory shield by reducing its capacity to activate protein C. The importance of the APC pathway in both inflammation and thrombosis has been reviewed previously7, 24.
The endothelium is also involved in immune cell trafficking and modulating inflammatory responses. Simultaneous with the denudation of the vascular anticoagulant surface, adhesive glycoproteins (P-selectin (P-sel), E-selectin (E-sel), intracellular adhesion molecule-1 (ICAM-1), venous cellular adhesion molecule-1 (VCAM-1)) are rapidly mobilized to the surface of activated endothelial cells and tether leukocytes through ligand-receptor interactions to initiate leukocyte-endothelial crosstalk. P-sel is stored in subplasmalemmal Weibel-Palade bodies (WPB) in endothelial cells, and in the α granules of platelets25, 26. Rapid exocytosis of these intracellular storage depots can be triggered by extracellular signals such as thrombin, histamine from mast cells, bradykinin, tissue necrosis factor-α (TNF-α), bacterial toxins, epinephrine, anaphylatoxins, or disturbed blood flow, and results in translocation of P-selectin to the cell surface27–29. These adhesion molecules are important coordinators of thrombo-inflammation. Intravital microscopy studies have identified that early leukocyte-endothelium interactions in veins are facilitated by selectins, ICAM-1, and VCAM-1. P-sel has been proposed as a sensitive and specific clinical biomarker for diagnosis of VTE, and improves prospective VTE risk stratification in patients with cancer who are undergoing chemotherapy30, 31. In animal models, P-sel inhibition is effective in preventing and treating VTE32, 33, and crizanlizumab, a monoclonal antibody that targets P-sel, is used to reduce vaso-occlusive crises in patients with sickle cell disease34, 35. P-sel and E-sel inhibition are now under investigation as treatments for patients with COVID-19 associated vasculopathy36, 37.
Early in endothelial activation, exocytosis of WPB also delivers von Willebrand Factor (VWF) to the endothelial plasma membrane, where it binds to platelets via the glycoprotein Ibα (GPIbα) receptor, an interaction that is essential for thrombus formation38. Under low-shear conditions, VWF also mediates leukocyte recruitment via direct binding to β2 integrins and P-selectin glycoprotein ligand (PSGL)-1, and increases endothelial permeability that may promote leukocyte extravasation39. The important role of VWF in inflammation and thrombosis is highlighted by recent observations of elevated levels of VWF and FVIII in diet-induced obese mice and protection against obesity-induced thrombosis by inhibition of VWF40. In large, cohort studies, gene variants associated with plasma VWF levels segregate with incident VTE, and a novel Vwf locus was included in a recently validated polygenic risk score for VTE41, 42. Given its rapid mobilization following WPB release, the level of VWF in circulation is often used as a marker of endothelial activation and vascular inflammation. Indeed, in patients hospitalized with COVID-19, circulating levels of VWF and TM were associated with severity of illness and with mortality43.
Endothelial activation and subsequent alterations in hemostatic balance occur in response to diverse infectious and sterile-inflammatory conditions, including bacterial and viral infections such as SARS-CoV-2, as well as in chronic inflammatory states such as sickle cell disease, diabetes, obesity, lupus, and others44, 45. Circulating pathogen- and damage-associated molecular patterns (PAMPs, DAMPs), cytokines (e.g. TNF-α and IL-1β), and autoantibodies secreted from leukocytes and platelets bind to receptors on the endothelial surface, triggering intracellular signaling cascades that activate the transcription factors NFκB and activator protein-1 and produce cell adhesion glycoproteins46, 47. In a positive feedback loop, activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) also increases the expression of numerous procoagulants, chemokines, and cytokines including C-X-C Motif Chemokine Ligand 2 (CXCL2) and CXCL4, TNF-α, IL-1β, IL-18, and IL-6 amongst other signaling molecules48, 49. Additionally, in in vitro studies of endothelial cells, ROS increases the expression of tissue factor (TF) – the receptor and cofactor for factor VII/VIIa necessary to activate the extrinsic pathway of coagulation, overwhelming anticoagulant molecules50.
In the context of chronic and acute inflammatory illnesses including COVID-19, a similar pattern of endothelial activation occurs with the shedding of cell surface anticoagulant proteins, release of WBP contents, and expression of cell adhesion molecules, resulting in a multi-organ, systemic thrombo-inflammatory phenotype. These changes facilitate platelet and leukocyte recruitment, dismantle the normal anticoagulant and anti-inflammatory phenotype of the vessel wall and support thrombosis at the blood:vessel interface43, 47, 51. Chronic and acute inflammatory conditions may also contribute to VTE by compounding regional differences in endothelial physiology.
It is worth highlighting that endothelial cell phenotypes can be heterogeneous based on vessel size (macrovascular versus microvascular), and vascular bed (venous, arterial, and lymphatic). The distinct endothelium can be characterized by differential expression of cell surface adhesion molecules and signaling receptors, intracellular signaling molecules and response to stimuli. Endothelial cells from post-capillary venules are particularly response to inflammatory cytokines, which induces expression of adhesion molecules and uniquely supports leukocyte interactions52–54. In the venous system, the endothelium in the vein valve sinus is exposed to hypoxia and disturbed blood flow, and maintains a compensatory environment that is carefully tuned to prevent thrombosis55. Stasis of blood flow or the introduction of inflammatory stimuli in the valve sinus can overwhelm this delicate antithrombotic balance, denuding natural anticoagulants and promoting an inflammatory milieu that results in thrombosis21, 55, 56.
Leukocyte Contributions to Venous Thrombosis
Glycoprotein adhesion molecules on activated endothelial cells support rapid leukocyte recruitment to sites of injury or inflammation. While not classic components of the coagulation system, leukocytes are critical for VTE formation and facilitate initiation of coagulation on the endothelium. In a murine flow restricted model of DVT, turbulent blood flow activates the uninterrupted endothelium, enabling leukocyte binding and local inflammation, with subsequent thrombogenesis and leukocyte migration (Figure 2). Similar to human DVT, circulating leukocytes are major cellular components in this model38, 57. Neutrophils and monocytes adhere to intact endothelium within hours of flow reduction57, a process dependent on endothelial-derived P-sel binding to PSGL-1 on the leukocyte surface58, 57, 59, and on leukocyte C-X-C Motif Chemokine Receptor 2 (CXCR2) which engages with CXCL1 on endothelial cells60. Platelets and myeloid cells release extracellular vesicles that carry coagulants and inflammatory mediators to remote sites61–63. Ultimately, thrombus initiation and propagation under flow restriction are dependent on both classic immune and hemostatic factors, including P-sel, accumulation of neutrophils and monocytes, platelet activation, VWF, and blood-cell derived TF57, 60, 64, 65.
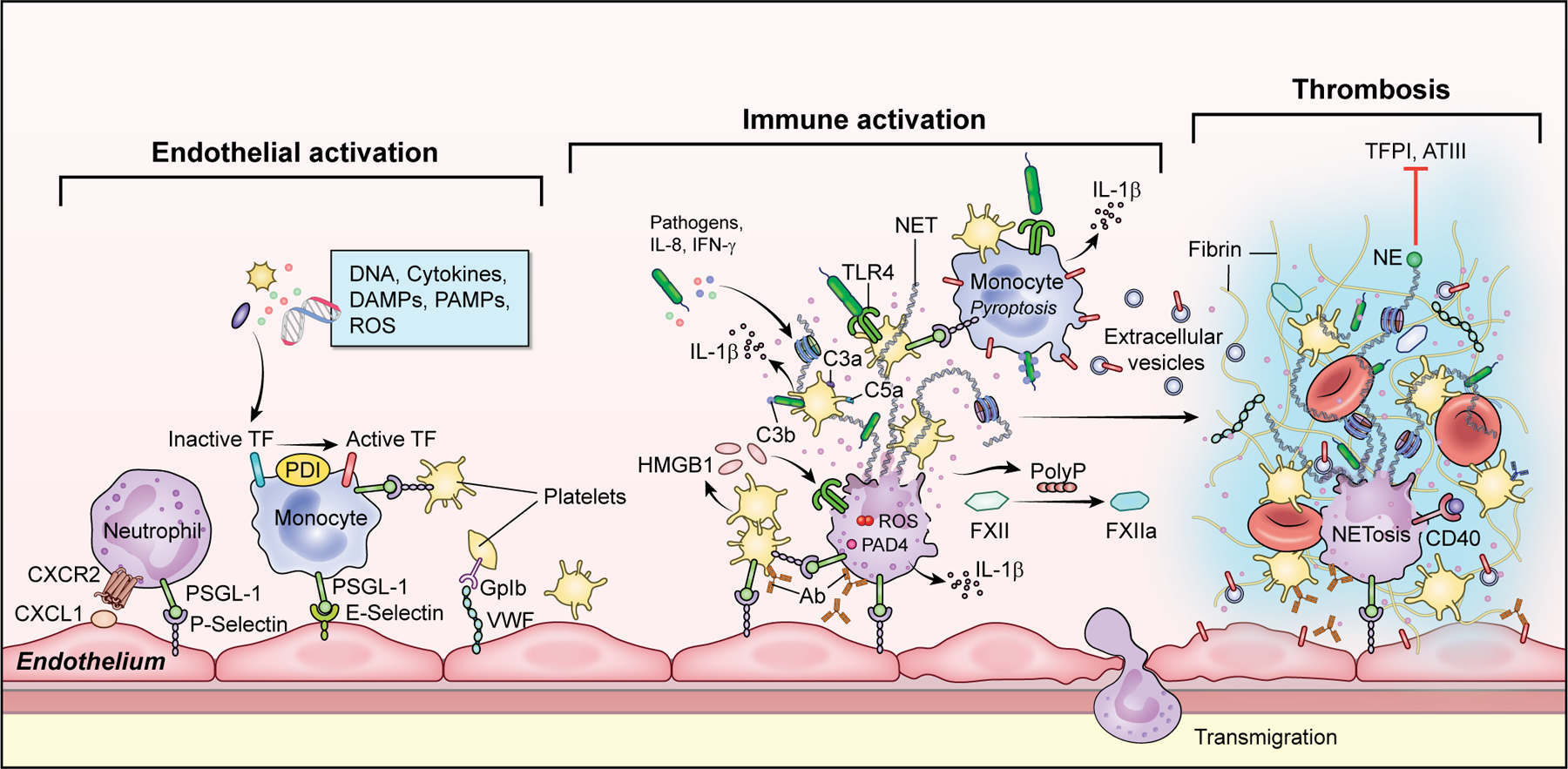
Figure 2: Leukocyte and Platelet Recruitment and Activation.
Increased adhesion molecules on endothelium including C-X-C Motif Chemokine Ligand 1(CXCL1), P-selectin and von Willebrand Facotr (VWF) support leukocyte and platelet rolling and binding. Inflammatory cytokines, pathogen- and damage associated molecular patterns (PAMPs and DAMPs), reactive oxygen species (ROS), and growth factors activate induce tissue factor (TF) expression on monocytes with resulting initiation of coagulation. Diverse stimuli including autoantibodies, cytokines, activated platelets with release of platelet factor 4 (PF4) and high mobility group protein B1 (HMGB1), and pathogens trigger neutrophils to undergo NETosis. On the scaffold of decondensed chromatin decorated with cytosolic and granule protein, there is VWF and platelet binding, erythrocyte entrapment, and activation of coagulation through presentation of TF, thrombin and factor XI (FXI), and auto-activation of FXII with resulting fibrin polymerization and thrombosis. There is simultaneous negatively regulate fibrinolysis through degradation of tissue factor pathway inhibitor (TFPI) and antithrombin (ATIII). (Illustration credit: Erina He).
Neutrophils and neutrophil extracellular traps link prototypical host defense mechanisms and venous thrombosis
Neutrophils have historically been defined by their role in innate immunity, in which pathogen recognition through toll-like receptors (TLRs), nucleotide oligomerization domain (NOD)-like receptors and C-type lectin receptors triggers a swift immune response to contain and eliminate invading pathogens through phagocytosis, degranulation, and formation of neutrophil extracellular traps (NETs)66–69. Neutrophils orchestrate inflammatory and immune responses in venous thrombosis at least in part by releasing NETs, sticky web-like structures comprised of a scaffold of decondensed chromatin decorated with cytosolic and granule proteins. These neutrophil-derived proteins, including histones, cytokines, alarmins, defensins, proteases, and myeloperoxidase, have antimicrobial and other functions70. In the absence of NETosis, activated neutrophils release cytokines and other molecules relevant in thrombosis which will be discussed later in this review.
NETs are able to directly activate coagulation, bridging the immune and coagulation systems through immunothrombosis. Although they serve as a vital host defense response to contain infections70, when dysregulated, NETs contribute to sterile inflammation, autoimmunity, arterial and venous thrombosis57, 71–74. Extracellular trap production has also been described in monocytes, eosinophils, and basophils, but neutrophils are believed to be the dominant trap producers in venous thrombosis75.
Neutrophils and NETs have been implicated in a variety of systemic immune and infectious disorders, many of which are accompanied by high risk of thrombosis such as antiphospholipid syndrome (APS), thrombotic thrombocytopenic purpura (TTP), heparin-induced thrombocytopenia (HIT), preeclampsia, autoimmune diseases, COVID-19, and the recently described SARS-CoV-2 vaccine-induced immune thrombotic thrombocytopenia (VITT)71, 76–84. Evidence of NETs can be detected in human venous, arterial, and microvascular thrombi57, 85. Murine models have confirmed the importance of neutrophils and NETosis in venous thrombogenesis. Depletion of neutrophils, degradation of NETs by administration of DNase, or inability to produce NETs due to deficiency in protein arginine deiminase (PAD4) are all protective against thrombosis in flow-restricted conditions57, 71, 76, 86, 87.
Diverse stimuli including autoantibodies, cytokines, activated platelets, and a cache of bacteria, fungi, and viruses can initiate signaling cascades that result in NET production in an environment- and stimulus-dependent fashion71, 88–91. Although the intermediate signals that direct neutrophils to undergo NETosis rather than other functional responses remain to be fully elucidated, the steps in neutrophil activation are beginning to be defined92, 93. NETosis is a tightly regulated process that involves activation of surface integrins, receptors, and mechanosensors that trigger intracellular calcium mobilization, ROS generation, shape change, and PAD4-catalyzed citrullination of proteins including histones which maintain chromatin structure94. Cytoskeletal changes then disassemble the nuclear envelope, leading to release of decondensed chromatin into the cytosol. Acute cytotoxic stress responses result in shedding of microvesicles and vesiculation of the endoplasmic reticulum. These processes culminate in rupture of the plasma membrane and extrusion of unraveled DNA into the extracellular compartment as NETs93, 95.
Critical activation steps in NETosis can occur in multiple intracellular locations. Neutrophil interactions with antiphospholipid antibodies or S. aureus stimulate the production of mitochondrial ROS and subsequent NETosis96, 97. Mitochondrial ROS production in neutrophils from patients with lupus is also triggered by activation of the endoplasmic reticulum stress sensor, inositol-requiring enzyme 1α (IREα), which is known to perpetuate inflammation in chronic diseases and may represent an intermediate step between autoantibody signaling and ROS production73. Although citrullination of histones by PAD4 in the nucleus was initially considered indispensable for NETosis, recent studies demonstrating PAD4-independent NETosis suggest this paradigm is species- and stimulus-dependent98–100.
Recent studies have triggered interest in the transmembrane protein, Solute Carrier Family 44 Member 2 (Slc44a2) receptor for its potential role in VTE. Slc44a2 is highly expressed on neutrophils and believed to contribute to VTE, particularly in inflammatory settings. GWAS identified an association between expression of the human neutrophil antigen 3b (HNA-3b) epitope (rs2288904, 461G>A; Arg154Gln) on the Slc44a locus and a 30% lower risk of VTE101. Slc44a2 has subsequently been identified as a receptor for VWF102 and αIIbβ3 on VWF-primed platelets103. Experimentally, Slc44a2 deficient mice develop smaller thrombi and recruit significantly fewer neutrophils to inflamed mesenteric venules104, 105. Mechanistically, the HNA-3a epitope on neutrophils is required for slow rolling and interacting with VWF at venous shear rates, which is further augmented by LPS105. Taken together, these findings suggest that under venous flow rates and inflammatory conditions, Slc44a2/HNA-3a may be important for neutrophil adhesion and NETosis and warrants further investigation.
Once released, NETs can activate coagulation and limit fibrinolysis by tPA106. The NET chromatin scaffold serves as a surface for fibrin polymerization, VWF binding, platelet adhesion, and erythrocyte entrapment106. Fibrin formation may also be amplified by direct fibrin-FXIIa interactions107. The components of NETs additionally contribute to thrombosis by presenting coagulation factors (eg. TF, FXII, FXI, thrombin), highly charged molecules (eg. histones H3 and H4, nucleosomes), and myeloperoxidase and serine proteases that degrade natural anticoagulants108–111. Neutrophils, NETs, and NET degradation products can also serve as counterpoints to balance thrombin generation. Neutrophil elastase and cathepsin G, which cleave PSGL-1 from the neutrophil surface, limit neutrophil interactions with the endothelium and platelets112. Extracellular DNA and sterile breakdown products bind to TLR9 on both monocytes and neutrophils, resulting in negative regulation of their prothrombotic functions113. These observations highlight the nuanced diversity of neutrophil functions in thromboinflammation, which may differ by stimulus, inflammatory milieu, and temporal kinetics.
As NETs are themselves degraded by endogenous DNases, histones and other liberated components may become available to activate endothelial cells and platelets, impair protein C activation, promote thrombin generation, and interfere with thrombin inhibition by antithrombin114, 115. Intriguingly, studies of blood samples from patients with lupus, antiphospholipid syndrome, and COVID-19 have shed light on a new paradigm wherein anti-DNase and “anti-NET” antibodies bind and stabilize NETs by “shielding” them from catalytic degradation by DNases109, 116, 117. Such anti-NET antibodies represent an additional layer of control in thromboinflammation and may also trigger an Fc receptor-mediated immune response, perpetuating the cycle of inflammation118.
Certain neutrophil subpopulations may have a proclivity to form NETs. Aged neutrophils in circulation are more likely to release NETs, rapidly homing to sites of inflammation or infection to induce an immediate effector response119. Additionally, some chronic autoimmune diseases are characterized by a higher proportion of circulating low-density granulocytes (LDGs) that exhibit a proinflammatory secretory phenotype and heightened capacity to form NETs120. Finally, in acute inflammatory conditions, emergent granulopoiesis leads to rapid mobilization of immature neutrophils from the bone marrow. The effect of immature neutrophils on inflammation, NETosis, and thrombosis remains to be determined121.
Neutrophils also contribute to thrombosis independent of NETs by releasing cytokines, DAMPs, and extracellular vesicles (EVs) that result in neutrophil interactions with other cells7, 122. Restricting neutrophil-platelet and neutrophil-endothelial crosstalk in mice by deletion of the P-sel ligand, PSGL-1, reduces venous thrombosis59. The myeloid alarmin heterodimer calprotectin (S100A8/9) is an example of a neutrophil effector with diverse functions. Calprotectin modulates calcium signaling, functions as a chemoattractant, and triggers cellular responses by binding to the scavenger and danger signal receptors CD36, TLR4, and receptor for advanced glycation end products (RAGE)123. In preclinical studies, mice lacking the myeloid alarmin protein S100A9 are protected from venous thrombosis124. In patients with severe COVID-19, in whom microvascular and macrovascular thrombosis can result in organ failure, circulating levels of calprotectin were elevated and were predictive of respiratory failure81, 125, 126.
Neutrophils have become an interesting target for therapeutic modulation given their multi-faceted role in thrombo-inflammation. Although systemic depletion of neutrophils compromises host defense and is therefore not a viable therapeutic approach, multiple strategies to inhibit the effects of NETs including by sequestering cell-free DNA, nucleosomes, or histones; inhibiting serine proteases; degrading NETs using recombinant DNases; and targeting NET-associated coagulation factors are being investigated as potential treatments for thrombotic and inflammatory diseases9, 127. Precise targeting of PAD4 and arginine deamination has proven to be challenging for clinical translation and a different approach using antibodies to inhibit histone citrulline residues appears more promising as a means to reduce neutrophil activation128. The urgent need for therapeutics to inhibit NETosis has led to a resurgence of interest in repurposing existing drugs including colchicine, dipyridamole, and fostamatinib129, 130.
Monocytes, tissue factor, and inflammasome effectors in thrombogenesis
Monocytes, while classically defined by their phagocytic role in innate immunity, contribute to venous thrombogenesis in multiple ways131. Animal models of DVT differ in their dependence on monocytes for acute venous thrombogenesis. Depletion of all monocytes with clodronate reduces DVT in a FeCl3 model, while inducible depletion of CD11b+ monocytes affected clot resolution but not acute thrombogenesis132, 133. In flow-dependent models of DVT, monocytes interact with the endothelium and recruit platelets to the site within hours. Although the complete contributions of monocytes continue to be elucidated, the most well-defined roles for monocytes in coagulation are to initiate coagulation through presentation of TF and to potential inflammation through inflammasome activation57, 65.
TF roles in coagulation and immune responses
The transmembrane glycoprotein TF is the primary initiator of coagulation under most physiologic and pathologic conditions, as well as an important mediator in the physiologic response to injury or infection. Concordant with its role as a central regulator of inflammation and thrombosis, preformed TF can rapidly shift functions from cell-signaling to coagulation by conformational activation in a complement-dependent, thiol-disulfide exchange catalyzed by protein disulfide isomerase (PDI) (Figure 3)134. TF initiates proinflammatory pathways, primarily through activation of the G-protein coupled protease-activated receptors (PARs), which are variably expressed on platelets, leukocytes, and endothelial cells. Activation of PARs initiates diverse inflammatory signals through release of cytokines (eg. TNF-α, IL-1, IL-6) and growth factors135 and induction of adhesion molecules that result in recruitment and activation of leukocytes136. The release of cytokines and DAMPs further increases TF expression on monocytes, initiating a self-reinforcing loop with sustained or escalating TF expression and activation of complement.
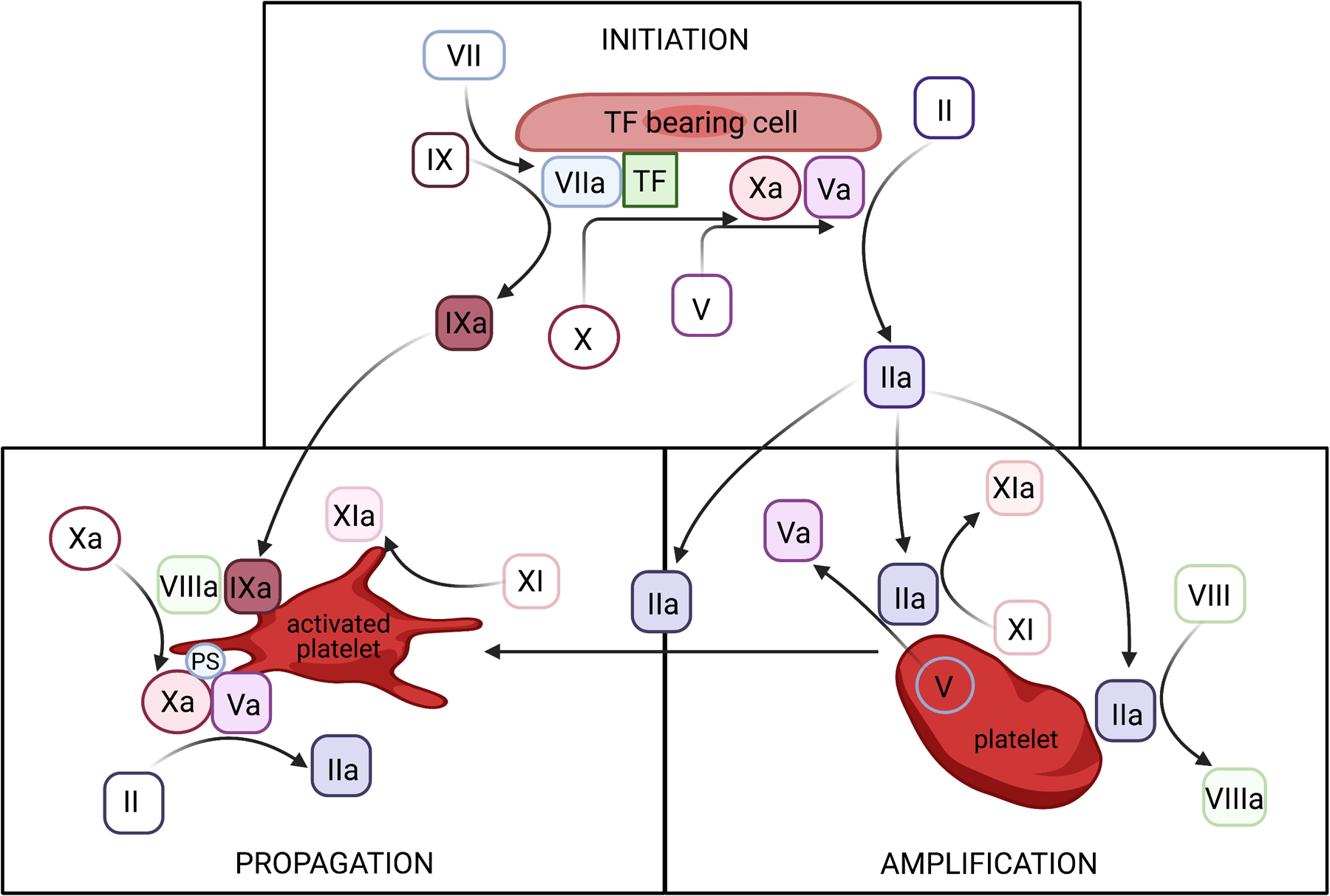
Figure 3: Cell-based model of coagulation.
Initiation phase: On the surface of a tissue factor (TF)-bearing cell, small amounts of thrombin (II) and factor IXa (IXa) are generated that then migrate to the surface of platelets. Amplification phase: Factors V, VIII, XI and platelets are activated. Propagation: On surface of activated platelet, activated factors assemble and generate thrombin burst. PS – phosphatidylserine. (Illustration made using Biorender)
Activated TF exposed to blood complexes with FVIIa, increasing FVII activity two-million-fold137 and resulting in thrombin generation. While three circulating pools of TF exist in the vasculature: cell-associated, EV-associated, and an inactive soluble form that lacks a cytoplasmic tail, TF on monocytes/macrophages and the EVs they generate are indispensable for thrombus accretion in flow-dependent models of DVT7, 57, 131. Under physiologic conditions, coagulation initiation is under tight negative regulation by endogenous anticoagulants including ATIII, the protein C pathway, and TFPI. During venous thrombogenesis, monocyte TF is activated in response to stimuli from cytokines, chemokines, cell adhesion molecules, pathogens, DAMPs, hypoxia, and tissue damage. Circulating platelet-monocyte aggregates also trigger monocyte TF expression by facilitating transcellular communication through P-sel and αIIbβ3.138 While constitutive TF deletion is embryonically lethal in mice, mice with inducible TF deletion or constitutive expression of low levels of human TF may have a bleeding diathesis but are protected from thrombosis57, 65. Conversely, exaggerated TF activity is responsible for catastrophic thrombosis and lethality following pathogen stimulation65. Elevated levels of circulating TF-positive EVs have been detected in patients following surgery or with pancreatic cancer and are associated with thrombosis risk or mortality131. Viral infections with influenza A, HIV and Ebola virus are also associated with elevated circulating TF131. Most recently, studies during the pandemic have revealed that increased circulating monocyte TF expression, and TF-positive EVs are associated with mechanical ventilation and death138, 139. Intriguingly, TF mRNA and protein have been detected in neutrophils from patients with COVID-19, and were reduced by complement factor 3 (C3) blockade140. Inhibition of TF is now being evaluated in a clinical trial (NCT04655586) as a treatment to block thrombo-inflammation in patients with COVID-19. Altogether, these clinical and experimental findings demonstrate the importance of TF for physiologic hemostasis and immune function, but the potential for overwhelming thrombotic and inflammatory damage if there is loss of the natural brakes in thromboinflammation.
Inflammasome activation and venous thrombosis
Proximate to TF activity, monocytes bridge inflammation, infection and venous thrombosis by coordinating inflammasome activation and coagulation. Assembly of the canonical NOD-like receptor family pryrin domain containing 3 (NLRP3) inflammasome in response to DAMPs and PAMPs, and the resulting prototypical inflammatory cytokine IL-1β are key steps in the initiation and propagation of venous thrombosis141. Through transcellular communication, activated neutrophils and NETs can license macrophages to produce IL-1β143 and a bidirectional interdependence between macrophage inflammasome activation and NET formation is plausible142. TF expression in monocytes can be triggered by pyroptosis, an inflammasome-directed form of lytic cell death in response to pathogens and PAMPs65, 143. The specific canonical or non-canonical inflammasome pathway activated diverges by stimulus, with gram negative rod proteins leading to caspase-1 activation, while LPS triggers a caspase-11 pathway of pyroptosis and TF expression144. However, both pathways appear to converge on gasdermin D as the final common element required for inflammasome-dependent TF production. Additional, consequences of inflammasome activation and subsequent pyroptosis include release of extracellular histones, caspases, ASC speck proteins, gasdermin D, and other effectors capable of eliciting cis and trans cellular responses integral in host defense and venous thrombosis144.
Although monocytes have been attributed as the major source of IL-1β, recent studies point to neutrophils, endothelium, and platelets as additional cellular compartments for inflammasome activation and production of IL-1β and IL-18145–147. As neutrophils outnumber monocytes in the leukocyte milieu during acute venous thrombosis, it is conceivable that monocytes and neutrophils cooperate in host defense and thrombosis by regulating NETosis through an inflammasome-operated “molecular switch”, which may be internally or externally derived. Endogenous thromboinflammatory checkpoints align the multiplicity of mechanisms that lead to thrombosis. Reduced expression of the ectonucleotidases CD39 or CD73, or activation of hypoxia-inducible factor alpha (HIF-1α) can release the brakes on inflammasome activation, NETosis, and vascular quiescence, resulting in thrombosis64, 141, 148, 149. Indeed, immune checkpoint inhibitors widely used to treat patients with cancer have recently been associated with an increased risk of VTE150. These are important areas that warrant further investigation as the therapeutic use of checkpoint inhibitors and inflammasome modulators are advanced in patient care.
Platelet roles in inflammation and infection
Platelets are abundant, circulating anuclear cells derived from megakaryocytes that have conventionally been recognized for their prominent roles in hemostasis and arterial thrombosis. They aggregate at sites of endothelial injury to support hemostasis by recruiting other platelets, forming an adhesive platelet plug as a physical barrier, facilitating the generation of thrombin, and serving as a surface for coagulation to prevent blood loss151. Defects in platelet function such as storage pool disorders or thrombocytopenia confer patients with a higher risk of bleeding complications, whereas hyperactive platelets are associated with arterial thrombotic conditions such as myocardial infarction and stroke152, 153. However, a more complex role for platelets in maintaining homeostasis has emerged in the past two decades, with platelets being identified as important mediators in a variety of biologic functions including inflammation, immunity, and venous thrombosis153–155.
Platelets contribute to the pathogenesis and sequelae of inflammatory diseases that are associated with a higher risk of VTE, including psoriasis, cancer metastases, transplant rejection, and infectious diseases such as sepsis, malaria, dengue and SARS-CoV-2 infection153, 156–158. During thromboinflammation, platelets not only provide a surface for and regulate coagulation, but they also interact with the immune and complement systems and release cytokines relevant in thrombogenesis. The essential role for platelets in venous thrombosis is demonstrated in experimental models of DVT, in which depletion of platelets, or isolated deficiency of either GpIb or VWF eliminates thrombosis in blood flowing through venous circuits38, 57. The clinical significance of platelets in VTE is further emphasized by the reduction in recurrent VTE in patients treated with an antiplatelet agent153, 159, 160.
Following endothelial disruption during normal hemostasis, platelets adhere to specialized receptors on the underlying matrix and initiate signals to achieve hemostasis and alert the immune system (reviewed by van de Meijden and Koupenova)152, 153. The sequences in platelet activation that follow include calcium influx and cytoskeletal rearrangements that lead to a shape change, and enrichment of surface receptors for adhesion and clotting proteins. During venous thrombosis, platelets directly adhere to collagen via α2β1 integrin (GPIa/IIa) and glycoprotein VI, and to the endothelium through VWF-GpIb and P-selectin-PSGL-1 interactions. Indirect platelet-endothelial associations occur when platelet-leukocyte aggregates form through receptor-ligand interactions eg. platelet surface C3 binding to leukocyte CD11b/CD18161, 162. Platelet receptor-specific stimuli leads to release or expression of a cache of cytokines, coagulation, and signaling molecules including a range of bioactive eiconasoids152. The primary platelet storage depots are α-granules, dense granules, and lysosomes. The biorepository content of α-granules includes coagulation factors, chemokines, adhesion proteins and regulators of angiogenesis, while the cargo of dense granules includes ADP, ATP, calcium, serotonin, and other mediators. Following recruitment to the endothelium, platelets promote inflammation and subsequent venous thrombosis through several signaling pathways including mammalian target of rapamycin complex 1 (mTORC1), ROS, and activation of the inflammasome complex resulting in release of the cytokines IL-1β and IL-18146, 147, 163, 164. Platelet aggregation and release of granular contents also localize platelets to leukocytes adherent to the endothelium and licenses leukocyte signaling that can amplify venous thrombosis.
As the interactions between the hemostatic and immune functions of platelets are unraveled, they provide growing insight into the mechanisms by which platelets participate in venous thrombogenesis during acute and chronic inflammatory conditions. In autoimmune diseases such as rheumatoid arthritis and psoriasis, in which dysregulated thrombosis can occur, platelets are hyperactivated and induce inflammation at the vessel wall and in circulating leukocytes151, 156. In patients with glioblastoma, platelet cell-surface lectin-like receptor type-C2 (CLEC-2) interactions with podoplanin can trigger platelet aggregation and venous thrombosis165. In the context of inflammation, platelets are recruited and activated by circulating PAMPs and DAMPs through TLRs and purinergic receptors at the plasma membrane21, 166. In turn, platelets release their own stores of DAMPs including high mobility group box 1 (HMGB1) and S100A9 that amplify the normal hemostatic and immune response124, 167. HMGB1 recruits monocytes and additional platelets to sites of venous thrombus accretion and stimulates NETosis which can potentiate thrombus propagation168. Platelets also stimulate NETosis through the CD40L-CD40 receptor axis89, 169, and through the release of inorganic polyphosphates (polyP) from platelet dense granules111. Potent activation of NETosis and the inflammasome complex by platelets can aid in control of pathogens, but can also create a hypercoagulable nidus90, 170.
PolyP are linear polymers of inorganic phosphates, derived from endogenous pools such as platelets and exogenous pools such as bacteria. Although polyP were only recently discovered in mammalian systems, a developing body of research has shown that polyP has diverse biological functions, including procoagulant and proinflammatory signaling171. Platelet-derived polyP are released from dense granules as short-chain polymers, 60–100 phosphates in length similar to those released by mast cells and basophils. Platelet-sized polyP can activate the contact pathway (via factor XII and factor XI with thrombotic and inflammatory sequelae), activate factor V, enhance the activity of thrombin-activatable fibrinolysis inhibitor (TAFI) which delays clot lysis, modulate migration and function of immune cells including neutrophils, monocytes, and macrophages, and regulate endothelial cell function172–175. Deficiency of polyP such as occurs in the dense granule storage disorder, Hermansky-Pudlack syndrome are associated with defective FXII-dependent clotting and thrombosis173. PolyPs also activate canonical inflammatory pathways through NF-κB and β-catenin signaling, increasing endothelial permeability, expression of cell adhesion molecules and apoptosis176. Notably, both mammalian- and pathogen-derived polyP have size dependent-functions ranging from acting as a potent procoagulant to an effective immune cell modulator171. As a counterpoint to its roles in coagulation and inflammation, platelet-sized polyP can also inhibit complement activation177. The molecular mechanisms that regulate polyP synthesis, degradation, and its divergent effects on biological systems remain to be fully investigated.
During bacterial and viral infections, platelet-leukocyte interactions may precede platelet aggregation. This suggests that a parallel pathway to that found in autoimmune disease exists in pathogen responses to facilitate rapid cellular communication and amplify the immune response and trigger thrombosis. When encountering pathogens, platelets rapidly mobilize host defense responses and aggregate on the surface of bacteria. These interactions trigger the release of alpha granules which contain antimicrobial proteins such as β-defensin 1, platelet basic protein, and connective tissue-activating peptide 3, as well as the inflammatory mediators that are released during sterile inflammation178. Severe, viral infections, such as with the influenza H1N1 or the SARS-CoV-2 virus, are accompanied by markedly increased risk for microvascular and macrovascular thrombosis in both, venous and arterial circuits81. The platelet transcriptome in COVID-19 is directed toward a hyperimmune response marked by antigen presentation and mitochondrial dysfunction158. During these infections, the increase in circulating platelet-monocyte and platelet-neutrophil aggregates correlates with increases in platelet activation, tissue factor activity, NET formation, and severity of illness158. Similarly, virus particle engulfment by platelets can trigger secretion of complement component C3 and GM-CSF, which trigger NETosis in healthy neutrophils162. Pathogen-antibody immune complexes, or dysregulated autoantibodies can trigger thrombin generation through platelet FcγRIIA signaling179. Platelets also release platelet factor 4 (PF4), which binds to glycosaminoglycans on the pathogen surface180. Following adhesion to glycoproteins, PF4 undergoes a conformational change and exposes a new epitope for antibody binding. Pathogen-bound or PF4-bound antibodies are presented to platelets in a self-amplifying loop, to phagocytes for clearance, and to B-cells for maturation and clonal expansion. Platelet-activating antibodies to PF4 have been detected in patients with bacterial periodontal disease, sepsis, and acute COVID-19157, 181, 182. Engagement of this host defense response paradigm is also believed to occur in the recently described VITT following exposure to certain SARS-CoV-2 vaccines183, 184. Exposure of PF4 to highly negative-charged constituents of the vaccine may trigger conformational changes in PF4 and antibody binding similar to the cycle of platelet activation and coagulation observed following bacterial infection or in HITT83. Although rare, understanding the immune mechanisms that lead to VITT will be important to inform autoantibody-driven thrombosis in other diseases.
Bidirectional interactions between platelets and complement additionally contribute to thromboinflammation. Both C3a and C5a bind to activated platelets, triggering the release of alpha granules185. Additionally, C3 is required for platelet activation and mice deficient in C3 have prolonged bleeding after vascular injury, have decreased fibrin and platelet deposition, and are protected against DVT186. Notably, C5-deficient mice do not have abnormalities in platelet activation, platelet deposition nor hemostasis, however, they had reduced fibrin formation and DVT. Platelets reciprocally regulate complement activation. C3, C1 inhibitor, and factor H are stored in α-granules186. C1 inhibitor, an acute phase protein, inactivates C1r and C1s in the classical pathway, as well as associates with P-selectin and interferes with leukocyte rolling187, 188. Additionally, polyP enhances the interaction of C1 inhibitor with C1s, slowing C2 and C4 activation189. Factor H binds to the active and inactive forms of GpIIb-IIIa on platelets and regulates activation of the alternative complement pathway190. Finally, platelets also express complement regulatory proteins which limit excessive complement activation191. These findings highlight possible differences in therapeutic effects and risks associated with different complement factor inhibitors in the treatment of thromboinflammatory diseases192, 193. Roles for complement signaling have also been established in arterial disease. In atherosclerotic plaques, de-differentiated smooth muscle cells (SMC) overexpress C3, activate the anaphylatoxin arm of the complement cascade, and induce vascular inflammation194. Notably, despite the presence of opsonin C3b on their surface, the SMCs avoid immune detection by expressing high levels of the “don’t-eat-me” signal, CD47 during M1 macrophage skewing. Blockade of CD47 in patients with cancer using a monoclonal antibody appears to reduce carotid artery inflammation195. Clinically, elevated C3 levels are associated with both an increased risk of venous and arterial thrombosis, and future studies will determine whether complement pathways integrate venous and arterial disease194, 196
By connecting multiple, conserved host defense response mechanisms, platelets responding to “danger” or pathogen signals aid in host defense, but can also fuel a disastrous cycle that culminates in thrombus initiation and propagation.
Extracellular vesicles
EVs, sub-micron sized phospholipid vesicles shed from a variety of cell types, have become a topic of significant interest for their ability to deliver thrombo-inflammatory signals to locations that are distant from the initial site of inflammation, infection, or thrombosis. Circulating EVs carry archetypic molecules from their parent cell including membrane proteins such as integrins, phosphatidylserine, adhesion proteins (eg. P-sel, PSGL-1), receptors (e.g. TF, TM, EPCR), and intracellular molecules such as cytokines, DAMPs, RNA and microRNAs. Consistent with their parent cells, EVs have complex biological effects on the vasculature. Platelets and megakaryocytes are the primary source of circulating EVs in healthy individuals, although most other vascular cells are also capable of producing EVs7. Platelet-EVs remain the predominant EVs in diseased states, where they may serve procoagulant functions by activating the tenase and prothrombinase complexes and by interacting with leukocytes via P-sel to trigger TF expression or NETosis (reviewed in Foley et al)7. EVs released by activated monocytes can carry a cargo of TF, phosphatidylserine and other signaling molecules that initiate inflammation and coagulation in remote locations197. Similar to platelet-EVs, EVs bearing PSGL-1 from monocytes can ligate P-selectin on platelets and the endothelium61. During the interaction of EVs with other cells, origin cell cargo such as TF may fuse with the plasma membrane of recipient cells and become available for activity131. A recent study suggests that perivascular cells may be an additional source of circulating TF-positive EVs following oxidative stress and may contribute to intravascular coagulation and lung inflammation198. Cancer cell-derived EVs expressing high levels of TF and PSGL-1 injected into mice associate with platelets and the endothelium, enhancing DVT formation. Similar transcellular communication through EVs can shuttle neutrophil-derived arachidonic acid into platelets for thromboxane production and endothelial activation7, 122. Consistent with this role for EVs, phosphatidylserine on the surface of EVs from platelets, monocytes, and neutrophils may activate endothelial cells and platelets to amplify thrombosis. Intriguingly, platelet-EVs may incorporate into bone marrow where they may have the opportunity to functionally reprogram this hematopoietic niche62. The diverse functional capabilities of EVs are a subject of intense investigation to better understand their relevance to disease and leverage these for diagnostic and therapeutic purposes.
Roles for adaptive immunity in VTE formation and resolution
The contributions of the innate immune system to the pathophysiology of VTE have been firmly established; however, the role of the adaptive immune system, specifically B and T cells, in VTE formation and resolution remains incompletely understood. The most well-characterized contribution of the adaptive immune system to VTE is in patients with autoantibody-driven thrombosis as discussed above. The endogenous and/or exogenous signals that trigger autoantibody production and the specific mechanisms by which they exert prothrombotic effects are unclear. While both B and T cells have been detected in murine DVT during the thrombus accretion and resolution, their function hasn’t been studied until recently. SCID mice, which lack functional B and T cells, develop DVT that are of similar size, cellular composition, and clot resolution as their wild-type counterparts, suggesting the presence of B and T cells is not required for VTE formation or resolution in a flow-restriction model199. In two mouse studies, depletion of T cells did not affect initial clot formation, but influenced clot resolution200, 201. However, one study found that the depletion of T cells impaired clot resolution while the other study demonstrated T cell depletion accelerated clot resolution200, 201. The divergent results between these studies could depend on the mouse strain, timing of intervention, and global T-cell versus effector-memory T cell depletion. Platelets can also present antigens to T-cells via major histocompatibility class-1 (MHC-1) expression, and platelet-T-cell interactions are associated with altered cytokine production in patients with COVID-19158. Additional research will be necessary to more clearly define the interactions between the adaptive and innate immune systems in VTE.
Clonal hematopoiesis, inflammation, and VTE
Over the lifespan of an individual, hematopoietic stem cells can accumulate somatic mutations that provide the cells with a competitive fitness advantage. This process, called clonal hematopoiesis of indeterminate potential (CHIP), leads to a disproportionate increase in the circulating number of the advantaged stem cell clones with age (reviewed in detail by Jaiswal and Ebert)202. CHIP is associated with an increase in the risk of developing hematopoietic malignancies and mortality secondary to cardiovascular events. The majority of individuals with CHIP have variants in either Tet2, Dnmt3a, Jak2, or Asxl1, which are believed to contribute to a proinflammatory vascular environment. Studies in animal models suggest an increase in IL-1β and other cytokines may contribute to the inflammatory milieu. Notably, the most frequent Jak2V617F variant in myeloproliferative malignancies is associated with a 12-fold increase in VTE risk, possibly due to excess NET formation203. It is unclear whether one of more mutations in Tet2, Dnmt3a, and/or Asxl1 are associated with venous thrombotic events204, 205. Although the implications of CHIP for VTE and other vascular disorders are not ready for translation to the clinic, the relationship between CHIP and vascular thromboinflammation warrants further investigation.
Coagulation and fibrinolytic pathways overlap with inflammation
While traditionally, TF was thought to be exclusively responsible for in vivo hemostasis, the contact pathway is now appreciated to have important roles in both thrombin generation and inflammation173, 206. Under inflammatory conditions, the contact system is initiated via activation of FXII, with loss of some negative regulatory mechanism. Physiologic activators of FXII during inflammation including RNA, DNA, activated platelets7, 108, 175, NETs57, HS207, and LPS, polyP or peptidoglycans on the surface of bacteria208. FXIIa can then trigger two distinct pathways: the extrinsic pathway via activation of FXI that culminates in thrombin generation; and the kallikrein-kinin pathway via generation of PK, which cleaves HK and culminates in liberation of bradykinin (BK) with activation of diverse potent proinflammatory pathways (reviewed by Foley and Renné)7, 209.
Exploration of the contact system has exposed the overly simplistic classification of factors as either pro-or anti-thrombotic, or pro- or anti-inflammatory. Contained within these binaries is the expectation that absence of a procoagulant factor will result in hypocoagulable state with bleeding and excess procoagulant factor will result in hypercoagulability. FXII exemplifies the complexities of factors with roles in both the coagulation and inflammatory systems. Inhibitors of FXII and FXIIa are protective against thromboembolic disease210, 211, while conversely, patients with gain-of-function mutations in the F12 gene212 do not have an increased risk of clotting, but have hereditary angioedema (HAE), a serious inflammatory disease due to excess generation of BK212. These observations generated excitement regarding the potential to target FXI and FXII in thromboinflammatory conditions. However, they also demonstrate the possibility for unexpected side effects of inhibiting factors that work at the interface of inflammation and coagulation.
Regardless of the mechanism of thrombus formation, breakdown of thrombi through fibrinolysis is essential to physiologic hemostasis, however, it can also contribute to inflammation. Plasmin, the central fibrinolytic protease, has been traditionally defined by its role in fibrin polymer degradation. It also has vital functions in degradation of the extracellular matrix, tissue remodeling, cell migration, and tuning of inflammation through cleavage of fibrin, cytokine induction and complement activation213. The primary negative regulator of the fibrinolytic system is plasminogen activator inhibitor-1 (PAI-1), which inhibits both tPA and uPA. High levels of PAI-1 are a risk factor for thrombosis and are observed in inflammatory diseases without a concomitant rise in tPA or uPA7. Conversely, individuals with lower levels of PAI due to a rare loss-of-function mutation have longer life spans and reduced incidence of thromboinflammatory diseases214.
Dysregulation of fibrinolysis can have thrombotic implications due to a hypofibrinolytic state, but also due to uncontrolled inflammation. The specific role of fibrinogen and plasminogen in inflammation have been reviewed in greater detail213, 215, 216 and we will briefly focus on plasminogen. The localization of the plasminogen activators, tPA and uPA, to different compartments support plasmin’s diverse functions. Additionally, plasminogen receptors on cells localize cell-based plasminogen activation through generation of local high concentrations of plasminogen217. For example, the plasminogen receptor, annexin A2, has increased cell surface expression in response to certain inflammatory stimuli.218 On endothelial cells, annexin A2 forms a tetramer complex with S100A10, serving as a co-receptor/chaperone for tPA and plasminogen and accelerating plasmin generation more than 60-fold. Conversely, plasmin activates macrophages through the annexin A2-S100A10 tetramer complex, which results in JAK1/TYK2 signaling, STAT3 activation, NFκB activation, and the release of proinflammatory cytokines. Thus, interactions between plasminogen and annexin A2 support both fibrinolysis and a proinflammatory response219. Decreased annexin A2 protein expression has been associated with VTE in patients with a positive family history but no identifiable thrombophilia220 and in patients with VTE compared to healthy controls220. Anti-A2 antibodies have been found in patients with cerebral venous thrombosis221, thrombotic APS222, and an early report suggests in COVID-19 as well223. Overall, these findings posit that annexin A2-mediated cell surface fibrinolysis may be an important mediator in hemostasis and thrombosis.
Plasmin and the complement system also activate and regulate each other. During inflammatory states, plasminogen can also be activated by components of the complement system.224 Conversely, plasminogen can directly bind C3, C3b, and C5, cleave some complement factors into inactive fragments, and act as a complement inhibitor via its lysine residues225. This inhibitor function has been coopted by pathogens to avoid effective host killing. For example, Moraxella catarrhalis expresses plasmin on its surface that degrades C3b and C5b226. The importance of the fibrinolytic system to the immune response has been demonstrated experimentally in both mice deficient in fibrinogen or plasminogen11, 227, 228. Notably, the phenotype associated with plasminogen deficiency is abrogated with concomitant fibrinogen deficiency, demonstrating the dependence of plasminogen on fibrinogen (likely through fibrinolysis) for many of its inflammatory functions227, 229. While components of the fibrinolytic system have clear roles in both fibrinolysis and inflammation, it remains uncertain if dysregulation of its inflammatory roles contributes to thrombosis.
Targeting Thromboinflammatory Pathways
The complex relationship between immune function and coagulation has evolved to stop bleeding, prevent infection, and promote wound healing. Leukocyte and platelets interact with activated endothelium and each other, initiating a diverse set of inflammatory and thrombotic pathways that are stimulus- and context-dependent. Without timely negative regulation, unregulated reciprocal activation of the immune and coagulation systems can result in catastrophic thrombotic and inflammatory damage to organs and tissues. The current paradigm for prevention and treatment of VTE with anticoagulants alone is not universally effective. Even aggressive pharmacologic fibrinolytic therapy and mechanical thrombectomy of VTE insufficiently address the underlying inflammatory milieu, tissue damage, and risk of recurrent thrombotic diatheses. A major limitation to the use of more potent antithrombotic agents is the concomitant risk of bleeding. As a more nuanced understanding of molecular mechanisms at the interface of coagulation and inflammatory systems is acquired, novel and perhaps more insightful applications of prior inflammatory targets can be tested for the prevention and treatment of VTE. Indeed, treatment of patients with elevated inflammatory biomarkers with a statin reduced the risk of VTE230. The ideal therapy to prevent or treat VTE will suppress excess inflammation and coagulation without disrupting hemostasis or triggering immune deficiency. As highlighted in this review, interventions targeting a number of inflammatory steps in murine models of DVT, including the inhibition of cell adhesion receptors, NETosis, platelet activation, and cytokine signaling, decrease thrombosis. As research discoveries shed new light on the nexus of inflammation and coagulation, there is renewed enthusiasm to identify and develop therapies targeting thromboinflammation that can be translated to the clinic for improved patient care.
ACKNOWLEDGEMENTS
The authors would like to thank our health care workers and researchers; patients and families affected by COVID-19; and acknowledge contributions from the community of investigators in thrombosis, hemostasis, and vascular biology. Figures 2 and 3 were illustrated by Erina He, National Institutes of Health.
Funding:
MEC and YK are supported by the Intramural Research Program of the NIH and NHLBI. YK has received additional support from the Lasker Foundation, A. Alfred Taubman Medical Research Institute, University of Michigan Frankel Cardiovascular Center COVID-19 Ignitor Award, and the Falk Catalyst Award. BET is supported by grant funding from the NHLBI (HL136784).
Non-standard Abbreviations and Acronyms
- ADP
adenosine diphosphate
- APS
antiphospholipid syndrome
- ATIII
antithrombin
- BK
bradykinin
- C3
complement factor 3
- CHIP
clonal hematopoiesis of indeterminate potential
- CLEC-2
cell-surface lectin-like receptor type-C2
- COVID-19
Coronavirus disease 2019
- CS
chondroitin sulfate
- CXCL
C-X-C Motif Chemokine Ligand
- CXCR
C-X-C Motif Chemokine Receptor
- DAMPs
damage-associated molecular patterns
- DVT
deep vein thrombosis
- E-sel
E-selectin
- EPCR
endothelial cell protein C receptor
- GM-CSF
Granulocyte-macrophage colony-stimulating factor
- GPIbα
glycoprotein Ibα
- HAE
hereditary angioedema
- HIF-1α
hypoxia-inducible factor alpha
- HIT
heparin-induced thrombocytopenia
- HMGB1
high mobility group box 1
- HNA
human neutrophil antigen
- HS
heparan sulfate
- ICAM-1
intracellular adhesion molecule-1
- IFN
interferon
- IREα
inositol-requiring enzyme 1α
- LDGs
low-density granulocytes
- mTORC1
mammalian target of rapamycin complex 1
- NETs
neutrophil extracellular traps
- NF-κB
nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells
- NLRP3
NOD-like receptor family pryrin domain containing
- NO
nitric oxide
- NOD
nucleotide oligomerization domain
- P-sel
P-selectin
- PAD4
protein arginine deiminase
- PAI-1
plasminogen activator inhibitor-1
- PAMPs
pathogen-associated molecular patterns
- PARs
protease-activated receptors
- PDI
protein disulfide isomerase
- PE
pulmonary embolism
- PF4
platelet factor 4
- polyP
inorganic polyphosphates
- PSGL-1
P-selectin glycoprotein ligand-1
- RAGE
receptor for advanced glycation end products
- ROS
reactive oxygen species
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- Slc44a1
Solute Carrier Family 44 Member 2
- SMC
smooth muscle cells
- TAFI
thrombin-activatable fibrinolysis inhibitor
- TF
tissue factor
- TFPI
tissue factor pathway inhibitor
- TLRs
toll-like receptors
- TM
thrombomodulin
- TNF-α
tissue necrosis factor-alpha
- tPA
tissue-type plasminogen activator
- TTP
thrombotic thrombocytopenic purpura
- uPA
urokinase plasminogen activator
- VCAM-1
venous cellular adhesion molecule-1
- VITT
vaccine-induced immune thrombotic thrombocytopenia
- VTE
venous thromboembolism
- VWF
Von Willebrand Factor
- WPB
Weibel-Palade bodies
Footnotes
Conflict of Interest: YK is an inventor on a pending patent to the University of Michigan on the use of biogases in vascular disease (US20180369278A1). The authors declare no other conflicts of interest.
REFERENCES
- 1.Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, Hylek EM, Kakkar A, Konstantinides SV, McCumber M, Ozaki Y, Wendelboe A, Weitz JI and Day ISCfWT. Thrombosis: a major contributor to global disease burden. Arterioscler Thromb Vasc Biol. 2014;34:2363–71. [DOI] [PubMed] [Google Scholar]
- 2.Martin KA, Molsberry R, Cuttica MJ, Desai KR, Schimmel DR and Khan SS. Time Trends in Pulmonary Embolism Mortality Rates in the United States, 1999 to 2018. J Am Heart Assoc. 2020;9:e016784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolberg AS, Rosendaal FR, Weitz JI, Jaffer IH, Agnelli G, Baglin T and Mackman N. Venous thrombosis. Nat Rev Dis Primers. 2015;1:15006. [DOI] [PubMed] [Google Scholar]
- 4.Kanthi Y and Piazza G. Great Debates in Vascular Medicine: Extended duration anticoagulation for unprovoked venous thromboembolism - Coming to consensus when the debate rages on. Vasc Med 2018;23:384–387. [DOI] [PubMed] [Google Scholar]
- 5.Zuo Y, Kanthi Y, Knight JS and Kim AHJ. The interplay between neutrophils, complement, and microthrombi in COVID-19. Best Pract Res Clin Rheumatol. 2021;35:101661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mackman N New insights into the mechanisms of venous thrombosis. J Clin Invest. 2012;122:2331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foley JH and Conway EM. Cross Talk Pathways Between Coagulation and Inflammation. Circulation research. 2016;118:1392–408. [DOI] [PubMed] [Google Scholar]
- 8.Bovill EG and van der Vliet A. Venous valvular stasis-associated hypoxia and thrombosis: what is the link? Annu Rev Physiol. 2011;73:527–45. [DOI] [PubMed] [Google Scholar]
- 9.Engelmann B and Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13:34–45. [DOI] [PubMed] [Google Scholar]
- 10.Iwanaga S The molecular basis of innate immunity in the horseshoe crab. Curr Opin Immunol. 2002;14:87–95. [DOI] [PubMed] [Google Scholar]
- 11.Sun H, Ringdahl U, Homeister JW, Fay WP, Engleberg NC, Yang AY, Rozek LS, Wang X, Sjobring U and Ginsburg D. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science. 2004;305:1283–6. [DOI] [PubMed] [Google Scholar]
- 12.Sun H, Xu Y, Sitkiewicz I, Ma Y, Wang X, Yestrepsky BD, Huang Y, Lapadatescu MC, Larsen MJ, Larsen SD, Musser JM and Ginsburg D. Inhibitor of streptokinase gene expression improves survival after group A streptococcus infection in mice. Proc Natl Acad Sci U S A. 2012;109:3469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, Hayashi C, Genco CA, Iafrati M and Freedman JE. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circulation research. 2009;104:346–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanthi YM, Sutton NR and Pinsky DJ. CD39: Interface between vascular thrombosis and inflammation. Curr Atheroscler Rep. 2014;16:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baek AE, Sutton NR, Petrovic-Djergovic D, Liao H, Ray JJ, Park J, Kanthi Y and Pinsky DJ. Ischemic Cerebroprotection Conferred by Myeloid Lineage-Restricted or Global CD39 Transgene Expression. Circulation. 2017;135:2389–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levin EG, Santell L and Osborn KG. The expression of endothelial tissue plasminogen activator in vivo: a function defined by vessel size and anatomic location. J Cell Sci. 1997;110 (Pt 2):139–48. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi K, Uwabe Y, Sawasaki Y, Kiguchi T, Nakamura H, Kashiwabara K, Yagyu H and Matsuoka T. Increased secretion of urokinase-type plasminogen activator by human lung microvascular endothelial cells. Am J Physiol. 1998;275:L47–54. [DOI] [PubMed] [Google Scholar]
- 18.Yau JW, Teoh H and Verma S. Endothelial cell control of thrombosis. BMC Cardiovasc Disord. 2015;15:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sobczak AIS, Pitt SJ and Stewart AJ. Glycosaminoglycan Neutralization in Coagulation Control. Arterioscler Thromb Vasc Biol. 2018;38:1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zindel J and Kubes P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu Rev Pathol. 2020;15:493–518. [DOI] [PubMed] [Google Scholar]
- 21.Kanthi Y, Hyman MC, Liao H, Baek AE, Visovatti SH, Sutton NR, Goonewardena SN, Neral MK, Jo H and Pinsky DJ. Flow-dependent expression of ectonucleotide tri(di)phosphohydrolase-1 and suppression of atherosclerosis. J Clin Invest. 2015;125:3027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nieuwdorp M, van Haeften TW, Gouverneur MC, Mooij HL, van Lieshout MH, Levi M, Meijers JC, Holleman F, Hoekstra JB, Vink H, Kastelein JJ and Stroes ES. Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. 2006;55:480–6. [DOI] [PubMed] [Google Scholar]
- 23.Hofmann-Kiefer KF, Kemming GI, Chappell D, Flondor M, Kisch-Wedel H, Hauser A, Pallivathukal S, Conzen P and Rehm M. Serum heparan sulfate levels are elevated in endotoxemia. Eur J Med Res. 2009;14:526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esmon CT. Protein C anticoagulant system--anti-inflammatory effects. Semin Immunopathol. 2012;34:127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonfanti R, Furie BC, Furie B and Wagner DD. PADGEM (GMP140) is a component of Weibel-Palade bodies of human endothelial cells. Blood. 1989;73:1109–12. [PubMed] [Google Scholar]
- 26.Wagner DD, Olmsted JB and Marder VJ. Immunolocalization of von Willebrand protein in Weibel-Palade bodies of human endothelial cells. J Cell Biol. 1982;95:355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schillemans M, Karampini E, Kat M and Bierings R. Exocytosis of Weibel-Palade bodies: how to unpack a vascular emergency kit. J Thromb Haemost. 2019;17:6–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rondaij MG, Bierings R, Kragt A, van Mourik JA and Voorberg J. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1002–7. [DOI] [PubMed] [Google Scholar]
- 29.Ponomaryov T, Payne H, Fabritz L, Wagner DD and Brill A. Mast Cells Granular Contents Are Crucial for Deep Vein Thrombosis in Mice. Circulation research. 2017;121:941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ay C, Simanek R, Vormittag R, Dunkler D, Alguel G, Koder S, Kornek G, Marosi C, Wagner O, Zielinski C and Pabinger I. High plasma levels of soluble P-selectin are predictive of venous thromboembolism in cancer patients: results from the Vienna Cancer and Thrombosis Study (CATS). Blood. 2008;112:2703–8. [DOI] [PubMed] [Google Scholar]
- 31.Ramacciotti E, Blackburn S, Hawley AE, Vandy F, Ballard-Lipka N, Stabler C, Baker N, Guire KE, Rectenwald JE, Henke PK, Myers DD Jr. and Wakefield TW. Evaluation of soluble P-selectin as a marker for the diagnosis of deep venous thrombosis. Clin Appl Thromb Hemost. 2011;17:425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diaz JA, Wrobleski SK, Alvarado CM, Hawley AE, Doornbos NK, Lester PA, Lowe SE, Gabriel JE, Roelofs KJ, Henke PK, Schaub RG, Wakefield TW and Myers DD Jr. P-selectin inhibition therapeutically promotes thrombus resolution and prevents vein wall fibrosis better than enoxaparin and an inhibitor to von Willebrand factor. Arterioscler Thromb Vasc Biol. 2015;35:829–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myers DD Jr., Rectenwald JE, Bedard PW, Kaila N, Shaw GD, Schaub RG, Farris DM, Hawley AE, Wrobleski SK, Henke PK and Wakefield TW. Decreased venous thrombosis with an oral inhibitor of P selectin. J Vasc Surg. 2005;42:329–36. [DOI] [PubMed] [Google Scholar]
- 34.Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, Guthrie TH, Knight-Madden J, Alvarez OA, Gordeuk VR, Gualandro S, Colella MP, Smith WR, Rollins SA, Stocker JW and Rother RP. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med. 2017;376:429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lowenstein CJ. Crizanlizumab for Treating COVID-19 Vasculopathy. (2020, July 9 – 2021, Jan 4).
- 36.Culmer DL, Dunbar ML, Hawley AE, Sood S, Sigler RE, Henke PK, Wakefield TW, Magnani JL and Myers DD Jr. E-selectin inhibition with GMI-1271 decreases venous thrombosis without profoundly affecting tail vein bleeding in a mouse model. Thromb Haemost. 2017;117:1171–1181. [DOI] [PubMed] [Google Scholar]
- 37.Devata S, Angelini DE, Blackburn S, Hawley A, Myers DD, Schaefer JK, Hemmer M, Magnani JL, Thackray HM, Wakefield TW and Sood SL. Use of GMI-1271, an E-selectin antagonist, in healthy subjects and in 2 patients with calf vein thrombosis. Res Pract Thromb Haemost. 2020;4:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Kollnberger M, Wakefield TW, Lammle B, Massberg S and Wagner DD. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117:1400–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petri B, Broermann A, Li H, Khandoga AG, Zarbock A, Krombach F, Goerge T, Schneider SW, Jones C, Nieswandt B, Wild MK and Vestweber D. von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116:4712–9. [DOI] [PubMed] [Google Scholar]
- 40.Michels A, Dwyer CN, Mewburn J, Nesbitt K, Kawecki C, Lenting P, Swystun LL and Lillicrap D. von Willebrand Factor Is a Critical Mediator of Deep Vein Thrombosis in a Mouse Model of Diet-Induced Obesity. Arterioscler Thromb Vasc Biol. 2020;40:2860–2874. [DOI] [PubMed] [Google Scholar]
- 41.Smith NL, Rice KM, Bovill EG, Cushman M, Bis JC, McKnight B, Lumley T, Glazer NL, van Hylckama Vlieg A, Tang W, Dehghan A, Strachan DP, O’Donnell CJ, Rotter JI, Heckbert SR, Psaty BM and Rosendaal FR. Genetic variation associated with plasma von Willebrand factor levels and the risk of incident venous thrombosis. Blood. 2011;117:6007–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klarin D, Busenkell E, Judy R, Lynch J, Levin M, Haessler J, Aragam K, Chaffin M, Haas M, Lindstrom S, Assimes TL, Huang J, Min Lee K, Shao Q, Huffman JE, Kabrhel C, Huang Y, Sun YV, Vujkovic M, Saleheen D, Miller DR, Reaven P, DuVall S, Boden WE, Pyarajan S, Reiner AP, Tregouet DA, Henke P, Kooperberg C, Gaziano JM, Concato J, Rader DJ, Cho K, Chang KM, Wilson PWF, Smith NL, O’Donnell CJ, Tsao PS, Kathiresan S, Obi A, Damrauer SM, Natarajan P, Consortium I and Veterans Affairs’ Million Veteran P. Genome-wide association analysis of venous thromboembolism identifies new risk loci and genetic overlap with arterial vascular disease. Nat Genet. 2019;51:1574–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, Baluha A, Bar N, Bona RD, Burns AJ, Dela Cruz CS, Dumont A, Halene S, Hwa J, Koff J, Menninger H, Neparidze N, Price C, Siner JM, Tormey C, Rinder HM, Chun HJ and Lee AI. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020;7:e575–e582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagashima S, Mendes MC, Camargo Martins AP, Borges NH, Godoy TM, Miggiolaro A, da Silva Deziderio F, Machado-Souza C and de Noronha L. Endothelial Dysfunction and Thrombosis in Patients With COVID-19-Brief Report. Arterioscler Thromb Vasc Biol. 2020;40:2404–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang M, Hao H, Leeper NJ, Zhu L and Early Career C. Thrombotic Regulation From the Endothelial Cell Perspectives. Arterioscler Thromb Vasc Biol. 2018;38:e90–e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miki I, Kusano A, Ohta S, Hanai N, Otoshi M, Masaki S, Sato S and Ohmori K. Histamine enhanced the TNF-alpha-induced expression of E-selectin and ICAM-1 on vascular endothelial cells. Cell Immunol. 1996;171:285–8. [DOI] [PubMed] [Google Scholar]
- 47.Shi H, Zuo Y, Gandhi AA, Sule G, Yalavarthi S, Gockman K, Madison JA, Wang J, Zuo M, Shi Y, Knight JS and Kanthi Y. Endothelial cell-activating antibodies in COVID-19. medRxiv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, Hohensinner P, Basilio J, Petzelbauer P, Assinger A and Schmid JA. Cell Type-Specific Roles of NF-kappaB Linking Inflammation and Thrombosis. Front Immunol. 2019;10:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JK, Kim SH, Lewis EC, Azam T, Reznikov LL and Dinarello CA. Differences in signaling pathways by IL-1beta and IL-18. Proc Natl Acad Sci U S A. 2004;101:8815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Banfi C, Brioschi M, Barbieri SS, Eligini S, Barcella S, Tremoli E, Colli S and Mussoni L. Mitochondrial reactive oxygen species: a common pathway for PAR1- and PAR2-mediated tissue factor induction in human endothelial cells. J Thromb Haemost. 2009;7:206–16. [DOI] [PubMed] [Google Scholar]
- 51.Lee MH, Perl DP, Nair G, Li W, Maric D, Murray H, Dodd SJ, Koretsky AP, Watts JA, Cheung V, Masliah E, Horkayne-Szakaly I, Jones R, Stram MN, Moncur J, Hefti M, Folkerth RD and Nath A. Microvascular Injury in the Brains of Patients with Covid-19. N Engl J Med. 2021;384:481–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Atkins GB, Jain MK and Hamik A. Endothelial differentiation: molecular mechanisms of specification and heterogeneity. Arterioscler Thromb Vasc Biol. 2011;31:1476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circulation research. 2007;100:158–73. [DOI] [PubMed] [Google Scholar]
- 54.Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D and Brown PO. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci U S A. 2003;100:10623–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brooks EG, Trotman W, Wadsworth MP, Taatjes DJ, Evans MF, Ittleman FP, Callas PW, Esmon CT and Bovill EG. Valves of the deep venous system: an overlooked risk factor. Blood. 2009;114:1276–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Welsh JD, Hoofnagle MH, Bamezai S, Oxendine M, Lim L, Hall JD, Yang J, Schultz S, Engel JD, Kume T, Oliver G, Jimenez JM and Kahn ML. Hemodynamic regulation of perivalvular endothelial gene expression prevents deep venous thrombosis. J Clin Invest. 2019;129:5489–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B and Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Myers D Jr., Farris D, Hawley A, Wrobleski S, Chapman A, Stoolman L, Knibbs R, Strieter R and Wakefield T Selectins influence thrombosis in a mouse model of experimental deep venous thrombosis. J Surg Res. 2002;108:212–21. [DOI] [PubMed] [Google Scholar]
- 59.Knight JS, Meng H, Coit P, Yalavarthi S, Sule G, Gandhi AA, Grenn RC, Mazza LF, Ali RA, Renauer P, Wren JD, Bockenstedt PL, Wang H, Eitzman DT and Sawalha AH. Activated signature of antiphospholipid syndrome neutrophils reveals potential therapeutic target. JCI Insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yago T, Liu Z, Ahamed J and McEver RP. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood. 2018;132:1426–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang JG, Williams JC, Davis BK, Jacobson K, Doerschuk CM, Ting JP and Mackman N. Monocytic microparticles activate endothelial cells in an IL-1beta-dependent manner. Blood. 2011;118:2366–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.French SL, Butov KR, Allaeys I, Canas J, Morad G, Davenport P, Laroche A, Trubina NM, Italiano JE, Moses MA, Sola-Visner M, Boilard E, Panteleev MA and Machlus KR. Platelet-derived extracellular vesicles infiltrate and modify the bone marrow during inflammation. Blood Adv. 2020;4:3011–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tans G, Rosing J, Thomassen MC, Heeb MJ, Zwaal RF and Griffin JH. Comparison of anticoagulant and procoagulant activities of stimulated platelets and platelet-derived microparticles. Blood. 1991;77:2641–8. [PubMed] [Google Scholar]
- 64.Anyanwu AC, Kanthi Y, Fukase K, Liao H, Mimura T, Desch KC, Gruca M, Kaskar S, Sheikh-Aden H, Chi L, Zhao R, Yadav V, Wakefield TW, Hyman MC and Pinsky DJ. Tuning the Thromboinflammatory Response to Venous Flow Interruption by the Ectonucleotidase CD39. Arterioscler Thromb Vasc Biol. 2019;39:e118–e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, Grover SP, Zhang X, Li L, Xiang B, Shi J, Li XA, Daugherty A, Smyth SS, Kirchhofer D, Shiroishi T, Shao F, Mackman N, Wei Y and Li Z. Inflammasome Activation Triggers Blood Clotting and Host Death through Pyroptosis. Immunity. 2019;50:1401–1411 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen G, Shaw MH, Kim YG and Nunez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. 2009;4:365–98. [DOI] [PubMed] [Google Scholar]
- 67.Brown GD, Willment JA and Whitehead L. C-type lectins in immunity and homeostasis. Nat Rev Immunol. 2018;18:374–389. [DOI] [PubMed] [Google Scholar]
- 68.Kurt-Jones EA, Mandell L, Whitney C, Padgett A, Gosselin K, Newburger PE and Finberg RW. Role of toll-like receptor 2 (TLR2) in neutrophil activation: GM-CSF enhances TLR2 expression and TLR2-mediated interleukin 8 responses in neutrophils. Blood. 2002;100:1860–8. [PubMed] [Google Scholar]
- 69.Yipp BG and Kubes P. NETosis: how vital is it? Blood. 2013;122:2784–94. [DOI] [PubMed] [Google Scholar]
- 70.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y and Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5. [DOI] [PubMed] [Google Scholar]
- 71.Meng H, Yalavarthi S, Kanthi Y, Mazza LF, Elfline MA, Luke CE, Pinsky DJ, Henke PK and Knight JS. In Vivo Role of Neutrophil Extracellular Traps in Antiphospholipid Antibody-Mediated Venous Thrombosis. Arthritis Rheumatol. 2017;69:655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cui BB, Tan CY, Schorn C, Tang HH, Liu Y and Zhao Y. Neutrophil extracellular traps in sterile inflammation: the story after dying? Autoimmunity. 2012;45:593–6. [DOI] [PubMed] [Google Scholar]
- 73.Sule G, Abuaita BH, Steffes PA, Fernandes AT, Estes SK, Dobry C, Pandian D, Gudjonsson JE, Kahlenberg JM, O’Riordan MX and Knight JS. Endoplasmic reticulum stress sensor IRE1alpha propels neutrophil hyperactivity in lupus. J Clin Invest. 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, Bhandari AA and Wagner DD. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10:136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Granger V, Faille D, Marani V, Noel B, Gallais Y, Szely N, Flament H, Pallardy M, Chollet-Martin S and de Chaisemartin L. Human blood monocytes are able to form extracellular traps. J Leukoc Biol. 2017;102:775–781. [DOI] [PubMed] [Google Scholar]
- 76.Ali RA, Gandhi AA, Meng H, Yalavarthi S, Vreede AP, Estes SK, Palmer OR, Bockenstedt PL, Pinsky DJ, Greve JM, Diaz JA, Kanthi Y and Knight JS. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commun. 2019;10:1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, Grone HJ, Brinkmann V and Jenne DE. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD and Lammle B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood. 2012;120:1157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perdomo J, Leung HHL, Ahmadi Z, Yan F, Chong JJH, Passam FH and Chong BH. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat Commun. 2019;10:1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gupta AK, Hasler P, Holzgreve W, Gebhardt S and Hahn S. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum Immunol. 2005;66:1146–54. [DOI] [PubMed] [Google Scholar]
- 81.Zuo Y, Zuo M, Yalavarthi S, Gockman K, Madison JA, Shi H, Woodard W, Lezak SP, Lugogo NL, Knight JS and Kanthi Y. Neutrophil extracellular traps and thrombosis in COVID-19. J Thromb Thrombolysis. 2021;51:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair C, Weber A, Barnes BJ, Egeblad M, Woods RJ, Kanthi Y and Knight JS. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Greinacher ASK, Wesche J, Handtke S, Palankar R, Aurich K, Lalk M, Methling K, Völker U, Hentschker C, Michalik S, Steil L, Schönborn L, Beer M, Franzke K, Rangaswamy C, Mailer RK, Thiele T, Kochanek S, Krutzke L, Siegerist F, Endlich N, Warkentin TE, Renné T. Understanding ChAdOx1 nCov-19 Vaccine-induced Immune Thrombotic Thrombocytopenia (VITT). Preprint available at Research Square. 2021. [Google Scholar]
- 84.Jawale CV, Ramani K, Li DD, Coleman BM, Oberoi RS, Kupul S, Lin L, Desai JV, Delgoffe GM, Lionakis MS, Bender FH, Prokopienko AJ, Nolin TD, Gaffen SL and Biswas PS. Restoring glucose uptake rescues neutrophil dysfunction and protects against systemic fungal infection in mouse models of kidney disease. Sci Transl Med. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, Mostyka M, Baxter-Stoltzfus A, Borczuk AC, Loda M, Cody MJ, Manne BK, Portier I, Harris ES, Petrey AC, Beswick EJ, Caulin AF, Iovino A, Abegglen LM, Weyrich AS, Rondina MT, Egeblad M, Schiffman JD and Yost CC. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136:1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, Hu J, Wang Y and Wagner DD. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A. 2013;110:8674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fuchs TA, Brill A and Wagner DD. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:1777–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, Dummer R, Simon HU and Yousefi S. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem. 2004;279:44123–32. [DOI] [PubMed] [Google Scholar]
- 89.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V and Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH and Kubes P. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–9. [DOI] [PubMed] [Google Scholar]
- 91.Zuo Y, Estes SK, Ali RA, Gandhi AA, Yalavarthi S, Shi H, Sule G, Gockman K, Madison JA, Zuo M, Yadav V, Wang J, Woodard W, Lezak SP, Lugogo NL, Smith SA, Morrissey JH, Kanthi Y and Knight JS. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci Transl Med. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Neubert E, Meyer D, Rocca F, Gunay G, Kwaczala-Tessmann A, Grandke J, Senger-Sander S, Geisler C, Egner A, Schon MP, Erpenbeck L and Kruss S. Chromatin swelling drives neutrophil extracellular trap release. Nat Commun. 2018;9:3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, Goldman RD, Wagner DD and Waterman CM. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci U S A. 2020;117:7326–7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou Y, An LL, Chaerkady R, Mittereder N, Clarke L, Cohen TS, Chen B, Hess S, Sims GP and Mustelin T. Evidence for a direct link between PAD4-mediated citrullination and the oxidative burst in human neutrophils. Sci Rep. 2018;8:15228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thiam HR, Wong SL, Wagner DD and Waterman CM. Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020;36:191–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB and Kaplan MJ. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22:146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, Bowden MG, Hussain M, Zhang K and Kubes P. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185:7413–25. [DOI] [PubMed] [Google Scholar]
- 98.Martinod K, Fuchs TA, Zitomersky NL, Wong SL, Demers M, Gallant M, Wang Y and Wagner DD. PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood. 2015;125:1948–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Martinod K, Witsch T, Erpenbeck L, Savchenko A, Hayashi H, Cherpokova D, Gallant M, Mauler M, Cifuni SM and Wagner DD. Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J Exp Med. 2017;214:439–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sollberger G, Tilley DO and Zychlinsky A. Neutrophil Extracellular Traps: The Biology of Chromatin Externalization. Dev Cell. 2018;44:542–553. [DOI] [PubMed] [Google Scholar]
- 101.Germain M, Chasman DI, de Haan H, Tang W, Lindstrom S, Weng LC, de Andrade M, de Visser MC, Wiggins KL, Suchon P, Saut N, Smadja DM, Le Gal G, van Hylckama Vlieg A, Di Narzo A, Hao K, Nelson CP, Rocanin-Arjo A, Folkersen L, Monajemi R, Rose LM, Brody JA, Slagboom E, Aissi D, Gagnon F, Deleuze JF, Deloukas P, Tzourio C, Dartigues JF, Berr C, Taylor KD, Civelek M, Eriksson P, Cardiogenics C, Psaty BM, Houwing-Duitermaat J, Goodall AH, Cambien F, Kraft P, Amouyel P, Samani NJ, Basu S, Ridker PM, Rosendaal FR, Kabrhel C, Folsom AR, Heit J, Reitsma PH, Tregouet DA, Smith NL and Morange PE. Meta-analysis of 65,734 individuals identifies TSPAN15 and SLC44A2 as two susceptibility loci for venous thromboembolism. Am J Hum Genet. 2015;96:532–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bayat B, Tjahjono Y, Berghofer H, Werth S, Deckmyn H, De Meyer SF, Sachs UJ and Santoso S. Choline Transporter-Like Protein-2: New von Willebrand Factor-Binding Partner Involved in Antibody-Mediated Neutrophil Activation and Transfusion-Related Acute Lung Injury. Arterioscler Thromb Vasc Biol. 2015;35:1616–22. [DOI] [PubMed] [Google Scholar]
- 103.Constantinescu-Bercu A, Grassi L, Frontini M, Salles C II, Woollard K and Crawley JT. Activated alphaIIbbeta3 on platelets mediates flow-dependent NETosis via SLC44A2. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tilburg J, Coenen DM, Zirka G, Dolleman S, van Oeveren-Rietdijk AM, Karel MFA, de Boer HC, Cosemans J, Versteeg HH, Morange PE, van Vlijmen BJM, Maracle CX and Thomas GM. SLC44A2 deficient mice have a reduced response in stenosis but not in hypercoagulability driven venous thrombosis. J Thromb Haemost. 2020;18:1714–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zirka G, Robert P, Tilburg J, Tishkova V, Maracle CX, Legendre P, van Vlijmen BJM, Alessi MC, Lenting PJ, Morange PE and Thomas GM. Impaired adhesion of neutrophils expressing Slc44a2/HNA-3b to VWF protects against NETosis under venous shear rate. Blood. 2021. [DOI] [PubMed] [Google Scholar]
- 106.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., Wrobleski SK, Wakefield TW, Hartwig JH and Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Konings J, Govers-Riemslag JW, Philippou H, Mutch NJ, Borissoff JI, Allan P, Mohan S, Tans G, Ten Cate H and Ariens RA. Factor XIIa regulates the structure of the fibrin clot independently of thrombin generation through direct interaction with fibrin. Blood. 2011;118:3942–51. [DOI] [PubMed] [Google Scholar]
- 108.Noubouossie DF, Whelihan MF, Yu YB, Sparkenbaugh E, Pawlinski R, Monroe DM and Key NS. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood. 2017;129:1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zuo Y, Yalavarthi S, Gockman K, Madison JA, Gudjonsson JE, Kahlenberg JM, Joseph McCune W, Bockenstedt PL, Karp DR and Knight JS. Anti-Neutrophil Extracellular Trap Antibodies and Impaired Neutrophil Extracellular Trap Degradation in Antiphospholipid Syndrome. Arthritis Rheumatol. 2020;72:2130–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Varju I, Longstaff C, Szabo L, Farkas AZ, Varga-Szabo VJ, Tanka-Salamon A, Machovich R and Kolev K. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb Haemost. 2015;113:1289–98. [DOI] [PubMed] [Google Scholar]
- 111.McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E and Jenne CN. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129:1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gardiner EE, De Luca M, McNally T, Michelson AD, Andrews RK and Berndt MC. Regulation of P-selectin binding to the neutrophil P-selectin counter-receptor P-selectin glycoprotein ligand-1 by neutrophil elastase and cathepsin G. Blood. 2001;98:1440–7. [DOI] [PubMed] [Google Scholar]
- 113.Nicklas JM, Gordon AE and Henke PK. Resolution of Deep Venous Thrombosis: Proposed Immune Paradigms. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ammollo CT, Semeraro F, Xu J, Esmon NL and Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J Thromb Haemost. 2011;9:1795–803. [DOI] [PubMed] [Google Scholar]
- 115.Fuchs TA, Bhandari AA and Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. 2011;118:3708–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zuo Y, Yalavarthi S, Navaz S, Hoy C, Shi H, Harbaugh A, Gockman K, Zuo M, Madison JA, Kanthi Y and Knight JS. Autoantibodies stabilize neutrophil extracellular traps in COVID-19. medRxiv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE and Zychlinsky A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107:9813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P and Pitzalis C. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Annals of the rheumatic diseases. 2016;75:1866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, Burk RD, Kunisaki Y, Jang JE, Scheiermann C, Merad M and Frenette PS. Neutrophil ageing is regulated by the microbiome. Nature. 2015;525:528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT and Kaplan MJ. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187:538–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ortmann W and Kolaczkowska E. Age is the work of art? Impact of neutrophil and organism age on neutrophil extracellular trap formation. Cell Tissue Res. 2018;371:473–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rossaint J, Kuhne K, Skupski J, Van Aken H, Looney MR, Hidalgo A and Zarbock A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat Commun. 2016;7:13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen B, Miller AL, Rebelatto M, Brewah Y, Rowe DC, Clarke L, Czapiga M, Rosenthal K, Imamichi T, Chen Y, Chang CS, Chowdhury PS, Naiman B, Wang Y, Yang D, Humbles AA, Herbst R and Sims GP. S100A9 induced inflammatory responses are mediated by distinct damage associated molecular patterns (DAMP) receptors in vitro and in vivo. PLoS One. 2015;10:e0115828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang Y, Gao H, Kessinger CW, Schmaier A, Jaffer FA and Simon DI. Myeloid-related protein-14 regulates deep vein thrombosis. JCI Insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shi H, Zuo Y, Yalavarthi S, Gockman K, Zuo M, Madison JA, Blair C, Woodward W, Lezak SP, Lugogo NL, Woods RJ, Lood C, Knight JS and Kanthi Y. Neutrophil calprotectin identifies severe pulmonary disease in COVID-19. J Leukoc Biol. 2021;109:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Silvin A, Chapuis N, Dunsmore G, Goubet AG, Dubuisson A, Derosa L, Almire C, Henon C, Kosmider O, Droin N, Rameau P, Catelain C, Alfaro A, Dussiau C, Friedrich C, Sourdeau E, Marin N, Szwebel TA, Cantin D, Mouthon L, Borderie D, Deloger M, Bredel D, Mouraud S, Drubay D, Andrieu M, Lhonneur AS, Saada V, Stoclin A, Willekens C, Pommeret F, Griscelli F, Ng LG, Zhang Z, Bost P, Amit I, Barlesi F, Marabelle A, Pene F, Gachot B, Andre F, Zitvogel L, Ginhoux F, Fontenay M and Solary E. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell. 2020;182:1401–1418 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Saffarzadeh M Neutrophil Extracellular Traps as a Drug Target to Counteract Chronic and Acute Inflammation. Curr Pharm Biotechnol. 2018;19:1196–1202. [DOI] [PubMed] [Google Scholar]
- 128.Chirivi RGS, van Rosmalen JWG, van der Linden M, Euler M, Schmets G, Bogatkevich G, Kambas K, Hahn J, Braster Q, Soehnlein O, Hoffmann MH, Es H and Raats JMH. Therapeutic ACPA inhibits NET formation: a potential therapy for neutrophil-mediated inflammatory diseases. Cell Mol Immunol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kanthi Y, Knight JS, Zuo Y and Pinsky DJ. New (re)purpose for an old drug: purinergic modulation may extinguish the COVID-19 thromboinflammatory firestorm. JCI Insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Strich JR, Ramos-Benitez MJ, Randazzo D, Stein SR, Babyak A, Davey RT, Suffredini AF, Childs RW and Chertow DS. Fostamatinib Inhibits Neutrophils Extracellular Traps Induced by COVID-19 Patient Plasma: A Potential Therapeutic. J Infect Dis. 2021;223:981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Grover SP and Mackman N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler Thromb Vasc Biol. 2018;38:709–725. [DOI] [PubMed] [Google Scholar]
- 132.Kimball AS, Obi AT, Luke CE, Dowling AR, Cai Q, Adili R, Jankowski H, Schaller M, Holinstadt M, Jaffer FA, Kunkel SL, Gallagher KA and Henke PK. Ly6CLo Monocyte/Macrophages are Essential for Thrombus Resolution in a Murine Model of Venous Thrombosis. Thromb Haemost. 2020;120:289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Laurance S, Bertin FR, Ebrahimian T, Kassim Y, Rys RN, Lehoux S, Lemarie CA and Blostein MD. Gas6 Promotes Inflammatory (CCR2(hi)CX3CR1(lo)) Monocyte Recruitment in Venous Thrombosis. Arterioscler Thromb Vasc Biol. 2017;37:1315–1322. [DOI] [PubMed] [Google Scholar]
- 134.Langer F, Spath B, Fischer C, Stolz M, Ayuk FA, Kroger N, Bokemeyer C and Ruf W. Rapid activation of monocyte tissue factor by antithymocyte globulin is dependent on complement and protein disulfide isomerase. Blood. 2013;121:2324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Al-Ani B, Hewett PW, Cudmore MJ, Fujisawa T, Saifeddine M, Williams H, Ramma W, Sissaoui S, Jayaraman PS, Ohba M, Ahmad S, Hollenberg MD and Ahmed A. Activation of proteinase-activated receptor 2 stimulates soluble vascular endothelial growth factor receptor 1 release via epidermal growth factor receptor transactivation in endothelial cells. Hypertension. 2010;55:689–97. [DOI] [PubMed] [Google Scholar]
- 136.Heuberger DM and Schuepbach RA. Protease-activated receptors (PARs): mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb J. 2019;17:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Komiyama Y, Pedersen AH and Kisiel W. Proteolytic activation of human factors IX and X by recombinant human factor VIIa: effects of calcium, phospholipids, and tissue factor. Biochemistry. 1990;29:9418–25. [DOI] [PubMed] [Google Scholar]
- 138.Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pao CRR, Righy C, Franco S, Souza TML, Kurtz P, Bozza FA and Bozza PT. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. 2020;136:1330–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rosell A, Havervall S, von Meijenfeldt F, Hisada Y, Aguilera K, Grover SP, Lisman T, Mackman N and Thalin C. Patients With COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That Is Associated With Severity and Mortality-Brief Report. Arterioscler Thromb Vasc Biol. 2021;41:878–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, Ntinopoulou M, Sertaridou E, Tsironidou V, Tsigalou C, Tektonidou M, Konstantinidis T, Papagoras C, Mitroulis I, Germanidis G, Lambris JD and Ritis K. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest. 2020;130:6151–6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yadav V, Chi L, Zhao R, Tourdot BE, Yalavarthi S, Jacobs BN, Banka A, Liao H, Koonse S, Anyanwu AC, Visovatti SH, Holinstat MA, Kahlenberg JM, Knight JS, Pinsky DJ and Kanthi Y. Ectonucleotidase tri(di)phosphohydrolase-1 (ENTPD-1) disrupts inflammasome/interleukin 1beta-driven venous thrombosis. J Clin Invest. 2019;129:2872–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Warnatsch A, Ioannou M, Wang Q and Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, Wu J, Wang Z, Liu Y, Chen F, Xiao X, Mackman N, Billiar TR, Han J and Lu B. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity. 2019;51:983–996 e6. [DOI] [PubMed] [Google Scholar]
- 144.Broz P and Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20. [DOI] [PubMed] [Google Scholar]
- 145.Karmakar M, Katsnelson MA, Dubyak GR and Pearlman E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP. Nat Commun. 2016;7:10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA and Weyrich AS. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol. 2001;154:485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vogel S, Arora T, Wang X, Mendelsohn L, Nichols J, Allen D, Shet AS, Combs CA, Quezado ZMN and Thein SL. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv. 2018;2:2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gupta N, Sahu A, Prabhakar A, Chatterjee T, Tyagi T, Kumari B, Khan N, Nair V, Bajaj N, Sharma M and Ashraf MZ. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc Natl Acad Sci U S A. 2017;114:4763–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Knight JS, Mazza LF, Yalavarthi S, Sule G, Ali RA, Hodgin JB, Kanthi Y and Pinsky DJ. Ectonucleotidase-Mediated Suppression of Lupus Autoimmunity and Vascular Dysfunction. Front Immunol. 2018;9:1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Moik F, Chan WE, Wiedemann S, Hoeller C, Tuchmann F, Aretin MB, Fuereder T, Zochbauer-Muller S, Preusser M, Pabinger I and Ay C. Incidence, risk factors, and outcomes of venous and arterial thromboembolism in immune checkpoint inhibitor therapy. Blood. 2021;137:1669–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Semple JW, Italiano JE Jr. and Freedman J Platelets and the immune continuum. Nat Rev Immunol. 2011;11:264–74. [DOI] [PubMed] [Google Scholar]
- 152.van der Meijden PEJ and Heemskerk JWM. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol. 2019;16:166–179. [DOI] [PubMed] [Google Scholar]
- 153.Koupenova M, Clancy L, Corkrey HA and Freedman JE. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circulation research. 2018;122:337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Morrell CN, Aggrey AA, Chapman LM and Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. 2014;123:2759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Portier I and Campbell RA. Role of Platelets in Detection and Regulation of Infection. Arterioscler Thromb Vasc Biol. 2021;41:70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Garshick MS, Tawil M, Barrett TJ, Salud-Gnilo CM, Eppler M, Lee A, Scher JU, Neimann AL, Jelic S, Mehta NN, Fisher EA, Krueger JG and Berger JS. Activated Platelets Induce Endothelial Cell Inflammatory Response in Psoriasis via COX-1. Arterioscler Thromb Vasc Biol. 2020;40:1340–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Assinger A, Schrottmaier WC, Salzmann M and Rayes J. Platelets in Sepsis: An Update on Experimental Models and Clinical Data. Front Immunol. 2019;10:1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, Petrey AC, Tolley ND, Guo L, Cody M, Weyrich AS, Yost CC, Rondina MT and Campbell RA. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136:1317–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Becattini C, Agnelli G, Schenone A, Eichinger S, Bucherini E, Silingardi M, Bianchi M, Moia M, Ageno W, Vandelli MR, Grandone E, Prandoni P and Investigators W. Aspirin for preventing the recurrence of venous thromboembolism. N Engl J Med. 2012;366:1959–67. [DOI] [PubMed] [Google Scholar]
- 160.Anderson DR, Dunbar M, Murnaghan J, Kahn SR, Gross P, Forsythe M, Pelet S, Fisher W, Belzile E, Dolan S, Crowther M, Bohm E, MacDonald SJ, Gofton W, Kim P, Zukor D, Pleasance S, Andreou P, Doucette S, Theriault C, Abianui A, Carrier M, Kovacs MJ, Rodger MA, Coyle D, Wells PS and Vendittoli PA. Aspirin or Rivaroxaban for VTE Prophylaxis after Hip or Knee Arthroplasty. N Engl J Med. 2018;378:699–707. [DOI] [PubMed] [Google Scholar]
- 161.Hamad OA, Mitroulis I, Fromell K, Kozarcanin H, Chavakis T, Ricklin D, Lambris JD, Ekdahl KN and Nilsson B. Contact activation of C3 enables tethering between activated platelets and polymorphonuclear leukocytes via CD11b/CD18. Thromb Haemost. 2015;114:1207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, Yao C, Rade J, Levy D, Wang JP, Finberg RW, Kurt-Jones EA and Freedman JE. The role of platelets in mediating a response to human influenza infection. Nat Commun. 2019;10:1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yang J, Zhou X, Fan X, Xiao M, Yang D, Liang B, Dai M, Shan L, Lu J, Lin Z, Liu R, Liu J, Wang L, Zhong M, Jiang Y and Bai X. mTORC1 promotes aging-related venous thrombosis in mice via elevation of platelet volume and activation. Blood. 2016;128:615–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dayal S, Wilson KM, Motto DG, Miller FJ Jr., Chauhan AK and Lentz SR. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation. 2013;127:1308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Payne H, Ponomaryov T, Watson SP and Brill A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood. 2017;129:2013–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Banerjee M, Huang Y, Joshi S, Popa GJ, Mendenhall MD, Wang QJ, Garvy BA, Myint T and Whiteheart SW. Platelets Endocytose Viral Particles and Are Activated via TLR (Toll-Like Receptor) Signaling. Arterioscler Thromb Vasc Biol. 2020;40:1635–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Maugeri N, Franchini S, Campana L, Baldini M, Ramirez GA, Sabbadini MG, Rovere-Querini P and Manfredi AA. Circulating platelets as a source of the damage-associated molecular pattern HMGB1 in patients with systemic sclerosis. Autoimmunity. 2012;45:584–7. [DOI] [PubMed] [Google Scholar]
- 168.Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, Schubert I, Hoseinpour P, Chandraratne S, von Bruhl ML, Gaertner F, Lorenz M, Agresti A, Coletti R, Antoine DJ, Heermann R, Jung K, Reese S, Laitinen I, Schwaiger M, Walch A, Sperandio M, Nawroth PP, Reinhardt C, Jackel S, Bianchi ME and Massberg S. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016;128:2435–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jin R, Yu S, Song Z, Zhu X, Wang C, Yan J, Wu F, Nanda A, Granger DN and Li G. Soluble CD40 ligand stimulates CD40-dependent activation of the beta2 integrin Mac-1 and protein kinase C zeda (PKCzeta) in neutrophils: implications for neutrophil-platelet interactions and neutrophil oxidative burst. PLoS One. 2013;8:e64631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, D’Atri LP, Gomez RM and Schattner M. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol. 2016;99:153–62. [DOI] [PubMed] [Google Scholar]
- 171.Smith SA, Choi SH, Davis-Harrison R, Huyck J, Boettcher J, Rienstra CM and Morrissey JH. Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood. 2010;116:4353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Roewe J, Stavrides G, Strueve M, Sharma A, Marini F, Mann A, Smith SA, Kaya Z, Strobl B, Mueller M, Reinhardt C, Morrissey JH and Bosmann M. Bacterial polyphosphates interfere with the innate host defense to infection. Nat Commun. 2020;11:4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH and Renne T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Morrissey JH, Choi SH and Smith SA. Polyphosphate: an ancient molecule that links platelets, coagulation, and inflammation. Blood. 2012;119:5972–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R and Morrissey JH. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci U S A. 2006;103:903–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mailer RKW, Hanel L, Allende M and Renne T. Polyphosphate as a Target for Interference With Inflammation and Thrombosis. Front Med (Lausanne). 2019;6:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wat JM, Foley JH, Krisinger MJ, Ocariza LM, Lei V, Wasney GA, Lameignere E, Strynadka NC, Smith SA, Morrissey JH and Conway EM. Polyphosphate suppresses complement via the terminal pathway. Blood. 2014;123:768–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, Kahr WH, Lindemann S, Seizer P, Yost CC, Zimmerman GA and Weyrich AS. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog. 2011;7:e1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Haberle H, Mehrlander M, Hammer S, Schulze H, Bitzer M, Malek N, Rath D, Bosmuller H, Nieswandt B, Gawaz M, Bakchoul T and Rosenberger P. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood. 2021;137:1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Krauel K, Potschke C, Weber C, Kessler W, Furll B, Ittermann T, Maier S, Hammerschmidt S, Broker BM and Greinacher A. Platelet factor 4 binds to bacteria, [corrected] inducing antibodies cross-reacting with the major antigen in heparin-induced thrombocytopenia. Blood. 2011;117:1370–8. [DOI] [PubMed] [Google Scholar]
- 181.Greinacher A, Holtfreter B, Krauel K, Gatke D, Weber C, Ittermann T, Hammerschmidt S and Kocher T. Association of natural anti-platelet factor 4/heparin antibodies with periodontal disease. Blood. 2011;118:1395–401. [DOI] [PubMed] [Google Scholar]
- 182.Zhu WZY, Mei Y, Wei J, Zhang Y, Topchyan P, Nguyen C, Janecke R, Kreuziger LB, White GC, Hari P, Aster R, Cui W, Jobe S, Graham MB, Wang D, Wen R. . SARS-CoV-2 receptor binding domain-specific antibodies activate platelets with features resembling the pathogenic antibodies in heparin-induced thrombocytopenia. Preprint available at Research Square. 2021. [Google Scholar]
- 183.Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA and Eichinger S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N Engl J Med. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Schultz NH, Sorvoll IH, Michelsen AE, Munthe LA, Lund-Johansen F, Ahlen MT, Wiedmann M, Aamodt AH, Skattor TH, Tjonnfjord GE and Holme PA. Thrombosis and Thrombocytopenia after ChAdOx1 nCoV-19 Vaccination. N Engl J Med. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kim H and Conway EM. Platelets and Complement Cross-Talk in Early Atherogenesis. Front Cardiovasc Med. 2019;6:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Subramaniam S, Jurk K, Hobohm L, Jackel S, Saffarzadeh M, Schwierczek K, Wenzel P, Langer F, Reinhardt C and Ruf W. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood. 2017;129:2291–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cai S and Davis AE 3rd. Complement regulatory protein C1 inhibitor binds to selectins and interferes with endothelial-leukocyte adhesion. J Immunol. 2003;171:4786–91. [DOI] [PubMed] [Google Scholar]
- 188.Kirschfink M and Nurnberger W. C1 inhibitor in anti-inflammatory therapy: from animal experiment to clinical application. Mol Immunol. 1999;36:225–32. [DOI] [PubMed] [Google Scholar]
- 189.Conway EM. Polyphosphates and Complement Activation. Front Med (Lausanne). 2019;6:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mnjoyan Z, Li J and Afshar-Kharghan V. Factor H binds to platelet integrin alphaIIbbeta3. Platelets. 2008;19:512–9. [DOI] [PubMed] [Google Scholar]
- 191.Gnatenko DV, Dunn JJ, McCorkle SR, Weissmann D, Perrotta PL and Bahou WF. Transcript profiling of human platelets using microarray and serial analysis of gene expression. Blood. 2003;101:2285–93. [DOI] [PubMed] [Google Scholar]
- 192.Luo S, Hu D, Wang M, Zipfel PF and Hu Y. Complement in Hemolysis- and Thrombosis- Related Diseases. Front Immunol. 2020;11:1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hillmen P, Szer J, Weitz I, Roth A, Hochsmann B, Panse J, Usuki K, Griffin M, Kiladjian JJ, de Castro C, Nishimori H, Tan L, Hamdani M, Deschatelets P, Francois C, Grossi F, Ajayi T, Risitano A and de la Tour RP. Pegcetacoplan versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med. 2021;384:1028–1037. [DOI] [PubMed] [Google Scholar]
- 194.Wang Y, Nanda V, Direnzo D, Ye J, Xiao S, Kojima Y, Howe KL, Jarr KU, Flores AM, Tsantilas P, Tsao N, Rao A, Newman AAC, Eberhard AV, Priest JR, Ruusalepp A, Pasterkamp G, Maegdefessel L, Miller CL, Lind L, Koplev S, Bjorkegren JLM, Owens GK, Ingelsson E, Weissman IL and Leeper NJ. Clonally expanding smooth muscle cells promote atherosclerosis by escaping efferocytosis and activating the complement cascade. Proc Natl Acad Sci U S A. 2020;117:15818–15826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Jarr KU, Nakamoto R, Doan BH, Kojima Y, Weissman IL, Advani RH, Iagaru A and Leeper NJ. Effect of CD47 Blockade on Vascular Inflammation. N Engl J Med. 2021;384:382–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Norgaard I, Nielsen SF and Nordestgaard BG. Complement C3 and High Risk of Venous Thromboembolism: 80517 Individuals from the Copenhagen General Population Study. Clin Chem. 2016;62:525–34. [DOI] [PubMed] [Google Scholar]
- 197.Zwicker JI, Trenor CC 3rd, Furie BC and Furie B Tissue factor-bearing microparticles and thrombus formation. Arterioscler Thromb Vasc Biol. 2011;31:728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ansari SA, Keshava S, Pendurthi UR and Rao LVM. Oxidative Stress Product, 4-Hydroxy-2-Nonenal, Induces the Release of Tissue Factor-Positive Microvesicles From Perivascular Cells Into Circulation. Arterioscler Thromb Vasc Biol. 2021;41:250–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Humphries J, Gossage JA, Modarai B, Burnand KG, Sisson TH, Murdoch C and Smith A. Monocyte urokinase-type plasminogen activator up-regulation reduces thrombus size in a model of venous thrombosis. J Vasc Surg. 2009;50:1127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Luther N, Shahneh F, Brahler M, Krebs F, Jackel S, Subramaniam S, Stanger C, Schonfelder T, Kleis-Fischer B, Reinhardt C, Probst HC, Wenzel P, Schafer K and Becker C. Innate Effector-Memory T-Cell Activation Regulates Post-Thrombotic Vein Wall Inflammation and Thrombus Resolution. Circ Res. 2016;119:1286–1295. [DOI] [PubMed] [Google Scholar]
- 201.Mukhopadhyay S, Gabre J, Chabasse C, Bromberg JS, Antalis TM and Sarkar R. Depletion of CD4 and CD8 Positive T Cells Impairs Venous Thrombus Resolution in Mice. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Jaiswal S and Ebert BL. Clonal hematopoiesis in human aging and disease. Science. 2019;366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, Silver AJ, Adams D, Castellano CA, Schneider RK, Padera RF, DeAngelo DJ, Wadleigh M, Steensma DP, Galinsky I, Stone RM, Genovese G, McCarroll SA, Iliadou B, Hultman C, Neuberg D, Mullally A, Wagner DD and Ebert BL. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Cerquozzi S, Barraco D, Lasho T, Finke C, Hanson CA, Ketterling RP, Pardanani A, Gangat N and Tefferi A. Risk factors for arterial versus venous thrombosis in polycythemia vera: a single center experience in 587 patients. Blood Cancer J. 2017;7:662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Segura-Diaz A, Stuckey R, Florido Y, Gonzalez-Martin JM, Lopez-Rodriguez JF, Sanchez-Sosa S, Gonzalez-Perez E, Saez Perdomo MN, Perera MDM, de la Iglesia S, Molero-Labarta T, Gomez-Casares MT and Bilbao-Sieyro C. Thrombotic Risk Detection in Patients with Polycythemia Vera: The Predictive Role of DNMT3A/TET2/ASXL1 Mutations. Cancers (Basel). 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Renne T, Pozgajova M, Gruner S, Schuh K, Pauer HU, Burfeind P, Gailani D and Nieswandt B. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202:271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Oschatz C, Maas C, Lecher B, Jansen T, Bjorkqvist J, Tradler T, Sedlmeier R, Burfeind P, Cichon S, Hammerschmidt S, Muller-Esterl W, Wuillemin WA, Nilsson G and Renne T. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity. 2011;34:258–68. [DOI] [PubMed] [Google Scholar]
- 208.Morrison DC and Cochrane CG. Direct evidence for Hageman factor (factor XII) activation by bacterial lipopolysaccharides (endotoxins). J Exp Med. 1974;140:797–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Renne T and Stavrou EX. Roles of Factor XII in Innate Immunity. Front Immunol. 2019;10:2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Matafonov A, Leung PY, Gailani AE, Grach SL, Puy C, Cheng Q, Sun MF, McCarty OJ, Tucker EI, Kataoka H, Renne T, Morrissey JH, Gruber A and Gailani D. Factor XII inhibition reduces thrombus formation in a primate thrombosis model. Blood. 2014;123:1739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Nickel KF, Long AT, Fuchs TA, Butler LM and Renne T. Factor XII as a Therapeutic Target in Thromboembolic and Inflammatory Diseases. Arterioscler Thromb Vasc Biol. 2017;37:13–20. [DOI] [PubMed] [Google Scholar]
- 212.Cichon S, Martin L, Hennies HC, Muller F, Van Driessche K, Karpushova A, Stevens W, Colombo R, Renne T, Drouet C, Bork K and Nothen MM. Increased activity of coagulation factor XII (Hageman factor) causes hereditary angioedema type III. Am J Hum Genet. 2006;79:1098–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Baker SK and Strickland S. A critical role for plasminogen in inflammation. J Exp Med. 2020;217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Khan SS, Shah SJ, Klyachko E, Baldridge AS, Eren M, Place AT, Aviv A, Puterman E, Lloyd-Jones DM, Heiman M, Miyata T, Gupta S, Shapiro AD and Vaughan DE. A null mutation in SERPINE1 protects against biological aging in humans. Sci Adv. 2017;3:eaao1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Gobel K, Eichler S, Wiendl H, Chavakis T, Kleinschnitz C and Meuth SG. The Coagulation Factors Fibrinogen, Thrombin, and Factor XII in Inflammatory Disorders-A Systematic Review. Front Immunol. 2018;9:1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Heissig B, Salama Y, Takahashi S, Osada T and Hattori K. The multifaceted role of plasminogen in inflammation. Cell Signal. 2020;75:109761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Plow EF, Doeuvre L and Das R. So many plasminogen receptors: why? J Biomed Biotechnol. 2012;2012:141806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Fang YT, Lin CF, Wang CY, Anderson R and Lin YS. Interferon-gamma stimulates p11-dependent surface expression of annexin A2 in lung epithelial cells to enhance phagocytosis. J Cell Physiol. 2012;227:2775–87. [DOI] [PubMed] [Google Scholar]
- 219.Madureira PA, Surette AP, Phipps KD, Taboski MA, Miller VA and Waisman DM. The role of the annexin A2 heterotetramer in vascular fibrinolysis. Blood. 2011;118:4789–97. [DOI] [PubMed] [Google Scholar]
- 220.Fassel H, Chen H, Ruisi M, Kumar N, DeSancho MT and Hajjar KA. Reduced Expression of Annexin A2 is Associated with Impaired Cell Surface Fibrinolysis and Venous Thromboembolism. Blood. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Cesarman-Maus G, Cantu-Brito C, Barinagarrementeria F, Villa R, Reyes E, Sanchez-Guerrero J, Hajjar KA and Latorre EG. Autoantibodies against the fibrinolytic receptor, annexin A2, in cerebral venous thrombosis. Stroke. 2011;42:501–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Cesarman-Maus G, Rios-Luna NP, Deora AB, Huang B, Villa R, Cravioto Mdel C, Alarcon-Segovia D, Sanchez-Guerrero J and Hajjar KA. Autoantibodies against the fibrinolytic receptor, annexin 2, in antiphospholipid syndrome. Blood. 2006;107:4375–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zuniga M, Gomes C, Carsons SE, Bender MT, Cotzia P, Miao QR, Lee DC and Rodriguez A. Autoimmunity to the Lung Protective Phospholipid-Binding Protein Annexin A2 Predicts Mortality Among Hospitalized COVID-19 Patients. medRxiv. 2021:2020.12.28.20248807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Jorg M and Binder BR. Kinetic analysis of plasminogen activation by purified plasma kallikrein. Thromb Res. 1985;39:323–31. [DOI] [PubMed] [Google Scholar]
- 225.Barthel D, Schindler S and Zipfel PF. Plasminogen is a complement inhibitor. J Biol Chem. 2012;287:18831–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Singh B, Al-Jubair T, Voraganti C, Andersson T, Mukherjee O, Su YC, Zipfel P and Riesbeck K. Moraxella catarrhalis Binds Plasminogen To Evade Host Innate Immunity. Infect Immun. 2015;83:3458–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Silva LM, Lum AG, Tran C, Shaw MW, Gao Z, Flick MJ, Moutsopoulos NM, Bugge TH and Mullins ES. Plasmin-mediated fibrinolysis enables macrophage migration in a murine model of inflammation. Blood. 2019;134:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Prasad JM, Gorkun OV, Raghu H, Thornton S, Mullins ES, Palumbo JS, Ko YP, Hook M, David T, Coughlin SR, Degen JL and Flick MJ. Mice expressing a mutant form of fibrinogen that cannot support fibrin formation exhibit compromised antimicrobial host defense. Blood. 2015;126:2047–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bugge TH, Kombrinck KW, Flick MJ, Daugherty CC, Danton MJ and Degen JL. Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency. Cell. 1996;87:709–19. [DOI] [PubMed] [Google Scholar]
- 230.Joseph P, Glynn R, Lonn E, Ramasundarahettige C, Eikelboom J, MacFadyen J, Ridker P and Yusuf S. Rosuvastatin for the prevention of venous thromboembolism: a pooled analysis of the HOPE-3 and JUPITER randomized controlled trials. Cardiovasc Res. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]