

Abstract
Background
The indoleamine 2,3-dioxygenase (IDO) pathway is a key counter-regulatory mechanism that, in cancer, is exploited by tumors to evade antitumor immunity. Indoximod is a small-molecule IDO pathway inhibitor that reverses the immunosuppressive effects of low tryptophan (Trp) and high kynurenine (Kyn) that result from IDO activity. In this study, indoximod was used in combination with a checkpoint inhibitor (CPI) pembrolizumab for the treatment for advanced melanoma.
Methods
Patients with advanced melanoma were enrolled in a single-arm phase II clinical trial evaluating the addition of indoximod to standard of care CPI approved for melanoma. Investigators administered their choice of CPI including pembrolizumab (P), nivolumab (N), or ipilimumab (I). Indoximod was administered continuously (1200 mg orally two times per day), with concurrent CPI dosed per US Food and Drug Administration (FDA)-approved label.
Results
Between July 2014 and July 2017, 131 patients were enrolled. (P) was used more frequently (n=114, 87%) per investigator’s choice. The efficacy evaluable population consisted of 89 patients from the phase II cohort with non-ocular melanoma who received indoximod combined with (P).
The objective response rate (ORR) for the evaluable population was 51% with confirmed complete response of 20% and disease control rate of 70%. Median progression-free survival was 12.4 months (95% CI 6.4 to 24.9). The ORR for Programmed Death-Ligand 1 (PD-L1)-positive patients was 70% compared with 46% for PD-L1-negative patients. The combination was well tolerated, and side effects were similar to what was expected from single agent (P).
Conclusion
In this study, the combination of indoximod and (P) was well tolerated and showed antitumor efficacy that is worth further evaluation in selected patients with advanced melanoma.
Keywords: melanoma, immunotherapy, indoleamine-pyrrole 2, 3, -Dioxygenase
Background
New approaches to metastatic melanoma (MM) treatment that rely on inhibition of immune checkpoints, such as the cytotoxic T-lymphocyte-associated antigen (CTLA-4) and the programmed death (PD-1) receptors, have generated improved antitumor activity over the last decade. Ipilimumab, an anti-CTLA-4 antibody, has an objective response rate (ORR) of 10%–20% in patients with MM,1 while pembrolizumab and nivolumab, both anti-PD-1 antibodies, have shown an ORR of approximately 43%.2 3 The combination of anti-PD-1 and anti-CTLA-4 inhibitors has improved ORR even further to approximately 58%; however, this was associated with significant grade 3 and 4 toxicities.3
Several preclinical and clinical studies suggest that inflammation induced by immunotherapy stimulates the upregulation of counter-regulatory mechanisms in the tumor microenvironment (TME), including the upregulation and activation of indoleamine 2,3-dioxygenase (IDO).4 5
The IDO pathway mediates immunosuppressive effects through the metabolism of tryptophan (Trp) to kynurenine (Kyn).6 Indoximod is an orally administered, small-molecule IDO pathway inhibitor that reverses the immunosuppressive effects of low Trp and high Kyn that result from IDO activity.7 Preclinical studies demonstrate that indoximod influences immune responses by altering the function of multiple immune cell types, including the enhancement of T cell proliferation, the preferential differentiation of CD4+ T cells into helper T cells over Tregs, reprogramming of IDO-activated Tregs into helper T cells and the downregulation of IDO expression in dendritic cells (DCs), all contribute to enhanced antitumor immune responses (online supplemental figure S1).7–12
jitc-2020-002057supp001.pdf (62.9KB, pdf)
Increasing evidence suggests that the IDO pathway activity can reinforce the immunosuppressive effects of checkpoints such as CTLA-4 and PD-1.5 13–15 Preclinical models further suggest that the inhibition of multiple checkpoints is necessary to optimize the antitumor efficacy of checkpoint inhibitor (CPI) immunotherapies.5 13–15 These studies provided the rationale for the present study where indoximod was administered in combination with CPI immunotherapy.
Here we report clinical and biomarker results for a phase II study of the concomitant administration of indoximod plus pembrolizumab for adult patients with unresectable, advanced or metastatic melanoma.
Methods
Study design and participants
After completion of phase Ib trial of indoximod with ipilimumab, the recommended phase II dose of indoximod used in this trial was determined to be 1200 mg two times per day.16 The initial phase II study design was a prospective single-arm trial combining ipilimumab (the standard of care at the time) with indoximod in adult patients with unresectable and metastatic melanoma. However, with the approval of anti-PD-1 antibodies as a standard first-line treatment, the protocol was amended to basket approach allowing investigator’s choice of standard doses of either of ipilimumab, nivolumab, or pembrolizumab in combination with indoximod 1200 mg two times per day, until disease progression or toxicity. At disease progression, the CPI could be changed from a PD-1 to a CTLA-4 inhibitor or vice versa while maintaining the combination with indoximod (online supplemental figure S2). Efficacy data would be determined and reported separately for each commercially available CPI combined with indoximod. Eligible patients were 18 years or older who had histologically confirmed unresectable stage III or IV cutaneous, mucosal or ocular melanoma. Patients must have had measurable disease, defined as lesions that can be measured per Response Evaluation Criteria in Solid Tumors (RECIST) 1.1.17 Other key eligibility criteria included Eastern Cooperative Oncology Group (ECOG) performance status of ≤2, Alanine Aminotransferase (ALT) and Aspartate Aminotransferase (AST) ≤3 times the upper limit of normal (ULN), serum bilirubin, amylase and lipase ≤1.5 ULN. Key exclusion criteria included active, uncontrolled brain metastases; a history of autoimmune disease; prior therapy with IDO inhibitor, immune CPI or immune stimulating agents in any prior line for metastatic disease (including, but not limited to, interleukin-2 (IL-2), interferons, CTLA-4 or PD1 antagonists, CD40 or CD137 agonist, or cancer therapeutic vaccines). Pregnant women, patients with HIV or autoimmune diseases were also excluded.
jitc-2020-002057supp002.pdf (66.7KB, pdf)
A portion of the patients enrolled had paired biopsies collected at baseline before enrolling on study and after three cycles of treatment, repeated on the same lesion for biomarker studies.
The original protocol and all amendments were approved by the relevant institutional review board. The study was conducted in accordance with the protocol, Good Clinical Practice guidelines, and the provisions of the Declaration of Helsinki. All patients provided written informed consent.
An external data and safety monitoring committee oversaw the study. All data were collected by investigators and associated site personnel, analyzed by statisticians employed by the sponsor, and interpreted by the authors, including those from the sponsor. All authors participated in reviewing and editing the manuscript, approved the submitted draft, had full access to the data used to write the manuscript and vouched for their accuracy, and attested that the study was conducted in accordance with the protocol.
Procedures and biomarkers
Patients were assessed according to RECIST 1.1,17 after the first 12 weeks of treatment and every 8 weeks thereafter. In addition to a baseline scan, confirmatory scans were obtained 4–6 weeks following initial documentation of an objective response (complete response (CR), partial response (PR)) or progressive disease (PD). The tumor assessment (TA) performed during screening was used as a baseline for efficacy assessments. CT/MRI of the chest, abdomen and pelvis was required at screening and at each TA, regardless of the location of known metastases. The same imaging modality was used for all TAs, unless contraindicated.
Total RNA was isolated from sections of tumor tissue from biopsies (pretreatment and on-treatment) followed by RNA sequencing (RNA-seq) analysis. The standard RNA-seq data analysis pipeline was applied for RNA-seq reads normalization transcript expression profiling, with subsequent analysis for identifying differentially expressed genes. Any gene with a proper cut-off of false discovery rate (FDR), after Benjamini-Hochberg correction for multiple testing, was deemed significantly differentially expressed in the test condition as compared with the control. More detailed methodology is described in the.
IDO1 expression was assessed in tumor biopsy samples using immunofluorescence staining. Expanded methodology is described in the.
PD-L1 staining and scoring for PD-L1 expression was conducted in pretreatment tumor biopsy samples using a validated assay for clone 22C3, as previously described. The Kaplan-Meier method was used to evaluate the relationship between progression-free survival (PFS) and baseline expression of IDO1 or PD-L1, in all patients with evaluable IDO1 and PD-L1 expression who received one or more doses of study treatment data.
Outcomes
The primary efficacy endpoint was best objective response rate (ORR) defined as the proportion of all treated patients whose best response at any time during the study following initiation of therapy was confirmed CR or PR.17
Secondary endpoint included PFS, defined as the time between the first dose of study therapy and the earliest date of progression or death. Patients who had neither progressed nor died were censored at the date of the last radiological evidence documenting absence of relapse. Additional efficacy endpoints included disease control rate (DCR), defined as the percentage of patients achieving CR+PR+stable disease (SD) at any time during the study following initiation of therapy. The duration of response (DOR) was defined as the time between the date that the criteria were first met for CR or PR or SD and the earliest date of progression or death. Overall survival (OS) was defined as the time between the first dose of study therapy and death. Patients who did not die were censored at the day they were last known to be alive.
Statistical analysis
Descriptive statistics were employed to evaluate the endpoint and analyze the data. Summary statistics for continuous variables included mean, SD, median and range. Categorical variables were presented as frequency counts and percentages including binomial CIs, and time-to-event variables were summarized by Kaplan-Meier estimates of median survival time and survival plots. The efficacy analysis was conducted on the efficacy evaluable population, and safety analysis was performed on the safety population. SAS V.9.4 was used for data analysis. The data were tabulated and analyzed with respect to patient enrollment and disposition, demographic and baseline.
Due to change in clinical practice and the approval of multiple anti-PD-1 antibodies as first-line therapy in this patient population, the protocol analysis plan called for separate statistical analysis of each treatment combination. Because pembrolizumab was by far the most frequently used in this study, the current analysis reports the treatment combination of pembrolizumab and indoximod in patients with advanced melanoma. For definition of objective response, as per protocol, patients were considered evaluable if they received at least one dose of study combination and had at least one post-treatment radiographic imaging assessment. Efficacy analyses were based on efficacy population who receive indoximod combined with pembrolizumab.
Role of the funding source
The study sponsor was involved in study design, data collection, data analyses, and writing of the report. All authors had full access to all data and approved the final content of this report. The corresponding author had final responsibility for the decision to submit for publication.
Results
Between July 2014 and July 2017, patients were enrolled at six academic centers in the USA. Among 140 patients screened for eligibility, 131 were enrolled. Nine were in the phase I dose escalation cohort, treated with ipilimumab/indoximod combination. Pembrolizumab was used more frequently (n=114, 87%) per investigator’s choice, and this is the population reported here. Of those, 18 patients had ocular melanoma; these were not included in the efficacy cohort due to different biology but were included in calculation of overall response. Seven patients came off study prior to first on-treatment imaging due to adverse events (AEs) or withdrawal of consent (table 1); these were not evaluable for efficacy, but were included in calculation of overall response, and in the safety cohort. Thus, the efficacy population consisted of 89 patients from the phase II cohort with non-ocular melanoma who received indoximod combined with pembrolizumab. Among these 89 patients, we obtained biopsies of 14 patients collected both pretreatment and while on treatment.
Table 1.
Participants in both study phases (N=131), safety (n=114) and efficacy (n=89) cohorts
| Study cohort | N |
| Phase I | 9 |
| Phase II | 122 |
| Indoximod+ipilimumab | 4 |
| Indoximod+nivolumab | 4 |
| Indoximod+pembrolizumab (safety cohort) | 114 |
| Efficacy cohort (non-ocular melanoma) | 89 |
| Ocular melanoma | 18 |
| Off study prior to first on-treatment imaging study | 7 |
| Adverse event | 5 |
| Withdrew | 2 |
At the data cut-off date of June 6, 2019, in the overall pembrolizumab and indoximod population (114 patients), ORR was 41.2% and DCR was 59.6%. The intention-to-treat (ITT) population included all patients treated with pemrolizumab and indoximod (n=96), excluding ocular melanoma due to different biology. The ORR in the ITT population was 47% and DCR was 65%. The efficacy population group (n=89) included all patients who were evaluable for efficacy. The median age of the efficacy population was 61.5 years (27–88 years). Fifty-six of patients were male (63%), and 85 patients (96%) were Caucasian. Four (4%) patients in this group had unresectable stage IIIB disease, 6 (7%) patients had stage IIIC, and 79 patients (89%) had stage IV disease at the time of study enrollment, of those 42% had stage M1c disease, 4 patients had mucosal melanoma and 2 patients had history of treated brain metastasis. All patients (n=89; 100%) had ECOG performance status of 0 or 1 (table 2). Fourteen patients (16%) had prior radiation therapy to non-target lesions, and 16 patients (18%) had prior systemic therapy, of those 4 patients had previous BRAF/MEK inhibitor, 1 had prior IL-2 and 11 had previous interferon alpha-2B. None of which was CPI. Four patients progressed while on prior treatment, 11 patients relapsed off therapy and 1 patient has no progression recorded. The median duration of follow-up at the time of data cut-off was 24.8 months (range 1.6–41.8) in the efficacy population, 21.1 months (1.1–49.4 months) for the overall population.
Table 2.
Demographics and baseline characteristics of non-ocular melanoma patients treated with indoximod+pembrolizumab (N=89; efficacy population)
| Characteristic | n (%) |
| Median age (range), years | 61.5 (27–88) |
| Male | 56 (63) |
| Caucasian | 85 (96) |
| Disease stage | |
| IIIB (unresectable) | 4 (4) |
| IIIC (unresectable) | 6 (7) |
| IV | 79 (89) |
| M1a | 25 (28) |
| M1b | 17 (19) |
| M1c | 37 (42) |
| LDH above ULN | 22 (25) |
| ECOG 0 or 1 | 89 (100) |
| Prior therapy | |
| Radiation | 14 (16) |
| Systemic* | 16 (18) |
*Includes BRAF and IL-1, but treatment with checkpoint inhibitor was not allowed per exclusion criteria.
BRAF, 8-rat proto-oncogene, serine/threonine kinase; ECOG, Eastern Cooperative Oncology Group; IL-1, interleukin-1; LDH, lactate dehydrogenase; ULN, upper limit of normal.
The ORR was 51% in the efficacy patient population according to investigator assessment and was confirmed using RECIST 1.1 criteria (table 3). A total of 18 patients (20%) had CR, and 27 patients (30%) had PR (figures 1 and 2). Seventeen patients (19%) had SD accounting for 70% of patients achieving DCR (CR+PR+SD), the median DOR was 33 months. Of note, in the subset of patients with prior radiation therapy (14 patients), the ORR was 64%, and 5 patients (36%) achieved CR. The overall median PFS was 12.4 months (95% CI 6.4 to 24.9) (table 3).
Table 3.
Efficacy results of non-ocular melanoma patients (n=89)
| Response status | As % of ITT population (N=96) | As % of efficacy population (N=89) who were evaluable for efficacy | ||
| Overall (N=89) | Prior systemic therapy (n=16)* | Prior radiation therapy (n=14) | ||
| PFS, median months (95% CI) | 12.4 (6.4 to 24.9) | |||
| ORR, n (%) | 45 (47) | 45 (51) | 10 (63) | 9 (64) |
| CR | 18 (19) | 18 (20) | 5 (31) | 5 (36) |
| PR | 27 (28) | 27 (30) | 5 (31) | 4 (29) |
| SD | 17 (18) | 17 (19) | 2 (13) | 3 (21) |
| DCR | 62 (65) | 62 (70) | 12 (75) | 12 (86) |
| PD | 27 (28) | 27 (30) | 4 (25) | 2 (14) |
| PD-L1 status known (n/N, %) | 56/89 (63) | |||
| Positive | 30/56 (54) | |||
| Negative | 26/56 (46) | |||
| ORR (CR+PR; n/N, %) | ||||
| PD-L1 positive | 21/30 (70) | |||
| PD-L1 negative | 12/26 (46) | |||
| DCR (CR+PR+SD; n/N, %) | ||||
| PD-L1 positive | 26/30 (87) | |||
| PD-L1 negative | 16/26 (62) | |||
*Includes BRAF inhibitors and IL-1.
BRAF, 8-rat proto-oncogene, serine/threonine kinase; CR, complete response; DCR, disease control rate; IL-1, interleukin-1; ITT, intention-to-treat; ORR, overall response rate; PD, progressive disease; PD-L1, Programmed Death-Ligand 1; PFS, progression-free survival; PR, partial response; SD, stable disease.
Figure 1.

(A) Change in tumor size per treatment cycle according to best overall response (N=85). Four patients progressed clinically prior to first on-study scan, not shown here, but counted towards disease progression. (B) Duration of response by patient (N=47). Two patients with target lesions response but had progression with non-target lesions, counted as PD. CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Figure 2.

Best response in target lesion size by patient relative to baseline (Response Evaluation Criteria in Solid Tumors (RECIST) 1.1. CR patients where best response in change in tumor size is not 100% have target lesions that are pathological lymph nodes <10 mm. SD or PD patients with a reduction in tumor volume of 30% or more due to either unequivocal non-target lesion progression or an unconfirmed response (N=85). Four patients progressed clinically prior to first on-study scan, not shown here, but counted towards disease progression. CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Among 56 patients with tumor tissue samples available for PD-L1 staining at the beginning of treatment, 30 (54%) were positive for PD-L1. The ORR for PD-L1-positive patients was 70% compared with 46% for PD-L1-negative patients. DCR was 87% and 62% in the PD-L1 positive and negative patient population, respectively (table 3).
The median time to response was 2.79 months (2.07, 20.68). Responses were achieved as early as 8 weeks. Many patients who achieved CR or PR had durable responses (figure 1A), with the median DOR being 30.4 months (19.1, 32.7) (figure 1B). The best response in target lesion size is shown in figure 2. At the time of data cut-off, responses were ongoing in 34 patients. Fifty-three patients (60%) were still alive, and the median OS was not reached.
The combination of indoximod and pembrolizumab was generally well tolerated with limited grade 3/4 treatment-related AEs (table 4/). The full recommended phase II dose of indoximod could be used in combination with pembrolizumab, with no adjustments required for toxicity. The most common (≥10% of patients) treatment-related AEs regardless of grade are shown in table 4. Twenty-four patients (21%) discontinued due to treatment-related AEs; most common causes leading to treatment discontinuation included rash, transaminitis or elevated lipase and amylase. Serious treatment-related AEs are shown in. No treatment-related death was reported.
Table 4.
Most common treatment-related adverse events in patients treated with indoximod+pembrolizumab (≥10% of patients, N=114)
| Adverse event | All grades, n (%) | Grade 3, n (%) |
| Fatigue | 71 (62.3) | 2 (1.8) |
| Rash | 46 (40.4) | 5 (4.3) |
| Pruritus | 40 (35.1) | 0 |
| Nausea | 32 (28.1) | 1 (0.9) |
| Diarrhea | 26 (22.8) | 0 |
| Arthralgia | 25 (21.9) | 1 (0.9) |
| Decreased appetite | 21 (18.4) | 0 |
| Headache | 21 (18.4) | 0 |
| Constipation | 19 (16.7) | 0 |
| Hypothyroidism | 15 (13.2) | 0 |
| Cough | 13 (11.4) | 1 (0.9) |
| Vomiting | 12 (10.5) | 0 |
A subset of patients had paired biopsies available. Expression of IDO within the TME was assessed by immunofluorescence staining and compared in baseline and on-treatment biopsies (figure 3A). Individual cell boundaries were assigned based on nuclear staining and image-analysis software, and the percentage of cells calculated that were positive for IDO, as described in the. IDO can be expressed by both tumor cells and host stromal/immune cells (macrophages, DCs, and others), and these two sites of expression might have different biologic significance. Therefore, to estimate the possible contribution of non-tumor stromal/immune cells, biopsies were co-stained with the proliferation marker Ki67. Since stromal/immune cells were non-proliferating, this allowed us to enrich for the ‘non-tumor cell’ component of the IDO expression. The total number of IDO-expressing cells (Ki67 positive or negative) was downregulated on treatment in responding patients, but unchanged in non-responding patients (figure 3A). When fractionated by expression of Ki67, the decrease in IDO expression was almost entirely contributed by the Ki67-negative fraction (enriched for stromal/immune cells). The Ki67+ fraction (tumor cells) showed little change in IDO expression with therapy.
Figure 3.
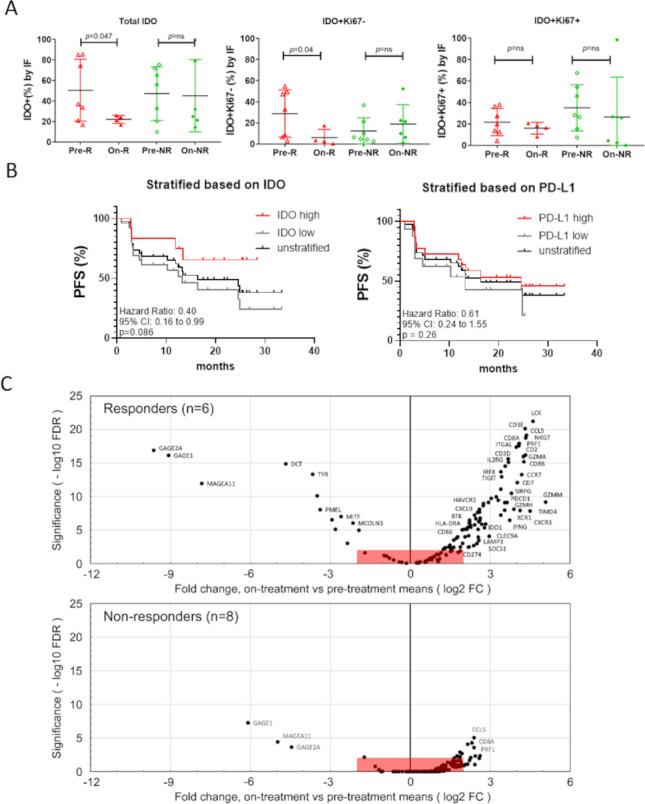
(A) Percentage of IDO1+ cells, IDO1+Ki67− cells and IDO1+Ki67+ cells in pretreatment (n=14) and on-treatment biopsies (n=10) of responders (R) and non-responders (NR) using immunofluorescence (IF) staining. Welch’s t-tests were used for statistical analyses. (B) Kaplan-Meier (log-rank test) curve of progression-free survival (PFS) based on IDO1 (stratified into 0%–20% or ≥20% groups as low vs high) and PD-L1 expression (stratified into 0%–1% or ≥1% groups as low vs high) at baseline. IDO1 measured by IF and PD-L1 measured by immunohistochemistry. The non-stratified survival curve has been included for reference (n=38). (C) RNA-seq data from paired pretreatment and on-treatment biopsies for n=14 patients, comprising 6 responders (CR/PR, upper plot) and 8 non-responders (SD/PD, lower plot). Volcano plots show log2 fold change (FC) (on-treatment mean/pretreatment mean) for each gene, plotted versus statistical significance (−log10 of the false discovery rate (FDR)-adjusted p value). All points outside red boxes represent a |log2 fold change|>2, and significance at an FDR<0.01. Plotted genes are from a curated list of 131 immune-related and melanoma-related genes (). CR, complete response; IDO, indoleamine 2,3-dioxygenase; PD, progressive disease; PD-L1, Programmed Death-Ligand 1; PR, partial response; SD, stable disease.
In 38 patients who were evaluable for both IDO and PD-L1 expression in pretreatment samples, PFS was stratified based on high IDO expression (>20% of cells expressing IDO, as defined by computer image analysis) or positive PD-L1 expression (>1% of cells positive for PD-L1 by immunohistochemistry) (figure 3B). In this small sample size, the differences did not reach significance, but further research workup is warranted to confirm whether baseline IDO expression could serve as a predictive biomarker for the treatment with indoximod in combination with pembrolizumab.
Whole transcriptome RNA sequencing (RNA-seq) was used to evaluate how the gene expression landscape was altered during indoximod plus pembrolizumab therapy in 14 patients with paired pretreatment and on-treatment tumor biopsies. The biopsy cohort comprised responders (CR/PR, n=6) and non-responders (SD/PD, n=8). Unsupervised clustering showed a clear cluster of inflammatory genes upregulated by treatment, primarily in the responder population (online supplemental figure S3). Volcano plots (figure 3C) show the change in the expression of a curated list of inflammation-related and melanoma-related genes (each dot represents a gene; the complete list with values is given in). Data are presented as fold change in the on-treatment samples as compared with pretreatment samples (positive values show an increase on treatment; negative values show a decrease). The statistical significance (FDR) for the change in each gene is shown on the vertical axis, with a cut-off for significance (red shading) of FDR<0.01. The responder group showed multiple genes that were significantly increased on-therapy; many of these genes were related either to activated T cells and Natural Killer (NK) cells (CD3, CD8, IFNγ, LCK, NKG7, perforin, and granzyme genes), or to antigen-presenting DCs (CLEC9A, XCR1, CD86, MHC class II, Tim-4). Downregulated genes (negative fold change) were mostly melanoma-related genes (MAGE and GAGE genes, and melanin pathway). In contrast, the non-responder patients showed fewer genes with statistically significant changes, and fold change was lower. Thus, overall, patients with objective clinical response showed greater evidence of inflammatory activation in their on-treatment biopsies.
jitc-2020-002057supp003.pdf (530.1KB, pdf)
Discussion
The combination of indoximod and pembrolizumab demonstrated encouraging safety and efficacy in patients with advanced melanoma, with nearly three-quarters of the efficacy population achieving disease control. Responses to the combination were deep and durable, with median PFS exceeding 1 year.
Even though the number of patients is low, the ORR seems to be higher in patients who received prior radiation therapy. This interesting observation merits future evaluation in clinical trials to address whether endo-vaccination caused by radiation might elicit a superior immune response in the context of therapy involving indoximod and PD-1 inhibitor.
The results of the current single-arm study do not allow us to conclude whether the addition of indoximod to CPI is clearly better than CPI alone. However, the median PFS of 12.4 months, and the CR rate of 20%, in the current study are encouraging and deserve vetting in a randomized, well-balanced phase II biomarker-driven study in carefully selected patient population.
The combination of indoximod and pembrolizumab was generally well tolerated with limited grade 3/4 treatment-related AEs. It is worth noting though that the treatment-related AEs that led to discontinuation of pembrolizumab and indoximod was 21%, higher from what was reported with pembrolizumab alone (10%).2
Although many IDO inhibitors remain in clinical development in other malignancies,18 the enthusiasm about the IDO pathway in melanoma was dampened after the failure of ECHO-301 clinical trial.19 That was the first phase III trial to evaluate the combination of an IDO1-selective enzyme inhibitor (epacadostat) with pembrolizumab in patients with MM. The combination of epacadostat plus pembrolizumab was not superior to pembrolizumab alone. The reason for the failure of that trial to achieve the primary endpoint is unknown. Questions have been raised whether the reduced dose of epacadostat (100 mg two times per day) used in this clinical trial was lower than ideal to achieve maximum pharmacodynamics effect.20 A combined analysis of multiple epacadostat clinical studies demonstrated that, when combined with CPI, doses of epacadostat <600 mg two times per day are unable to maintain suppression of plasma Kyn production.21 Higher doses of epacadostat are being tested in ongoing clinical trials. It is important to point out, however, that indoximod has a different mechanism of action from epacadostat and other available IDO inhibitors in different clinical trials (including navoximod, Linrodostat and KHK-2455). While epacadostat and others are direct enzymatic inhibitor of IDO1, indoximod is a tryptophan mimetic and works directly on immune cells to reverse IDO pathway–mediated suppression.
Importantly, indoximod was well tolerated and did not require any dose reduction from the planned recommended phase II dose (RP2D) when combined with pembrolizumab. In biopsies from indoximod-treated patients, those patients with clinical response showed evidence of greater immune activation by RNA-seq than patients who did not respond. Additionally, in on-treatment tumor biopsies, indoximod treatment was associated with downregulation of IDO protein expression by immunofluorescence in non-proliferating (presumptive host stromal) cells, suggesting efficacy of the treatment regimen against the IDO pathway. In this regard, it is important to emphasize that the reduction in IDO in the TME is not proposed as a direct effect of indoximod on the IDO protein. Rather, we interpret this as a therapy-induced change in the overall tumor immune microenvironment—from a milieu dominated by IDO to one with lower expression of IDO protein. It has recently been reported that the baseline DC pool in tumors (ie, the key antigen-presenting cells) were unexpectedly immunosuppressive, including extensive expression of IDO.22 Other immunosuppressive cells such as tumor-associated macrophages and myeloid-derived suppressor cells are also potential sources of IDO in tumors. We speculate that, in those patients who responded to therapy, treatment with indoximod plus PD-1 blockade elicited a change in the immune milieu that reduced the number of IDO-expressing cells. Of note, this reduction in IDO-expressing cells did not occur in the patients who did not respond to combination.
In other studies, indoximod shows encouraging clinical safety and activity signals in different types of cancers, utilizing different combinations with chemotherapy,23–26 chemoradiotherapy,27 vaccines28 and immune checkpoint therapy.13
Despite these encouraging clinical results, the concentration of indoximod drug achievable in plasma is barely within the active range observed in vitro for T cell activation.26 29 A prodrug of indoximod (NLG802), tested in the first phase I clinical trial, produced 6-fold increase in Cmax and 4.7-fold increase in Area Under the Curve (AUC) of the active indoximod moiety in plasma, compared with molar equivalent dosing of the parent indoximod drug.30 This substantially higher plasma concentration was well tolerated, and is not accompanied by any discernible increase in toxicity, which remained low.
Given the unique mechanism of action, the clinical activity, and the safety profile, we believe that the use of indoximod/NLG802 in combination therapies merits further evaluation in clinical trials, whether upfront in combination CPI or after CPI failure.
Acknowledgments
The authors would like to thank the patients and their families who enrolled on this clinical trial, along with the clinical research staff.
Footnotes
Twitter: @ZakhariaYousef
Contributors: YZ, OR, DM, RS, LLT, EPK, CJL, and NNV designed the study. YZ, RRM, OR, JD, MFS, KFG, and MM were study principal investigators. DM, RK, RP, CMS, JY, ELB, MM, GRR, RP, and SSS processed and analyzed correlative samples. YZ, GRR, EPK, and LLT wrote the manuscript. All authors reviewed and gave approval for the final manuscript.
Funding: This study was supported by NewLink Genetics.
Competing interests: YZ reports personal fees for Advisory Board: Amgen, Roche Diagnostics, Novartis, Janssen, Eisai, Exelixis, Castle Bioscience, Array, Bayer, Pfizer, Clovis, and EMD serono. Grant/research support from Institutional clinical trial support from NewLink Genetics, Pfizer, Exelixis, and Eisai. DSMC honorarium: Janssen Research and Development. Consultant honorarium: Pfizer, Novartis. RRM reports personal fees from Newlink Genetics, outside the submitted work. MFS and RK report personal fees from Illumina Inc, BMS, and Qiagen. DM reports grants from NewLink Genetics, during the conduct of the study; grants and personal fees from NewLink Genetics, outside the submitted work. In addition, DM has a patent US patent 8,232,313 licensed to NewLink Genetics, and a patent US patent 6,451,840 licensed to NewLink Genetics. MM reports personal fees from Blueprints Medicine, Immunocore, Amgen, Treiza, Array Biopharma, Biontech, and Novartis. LLT, CMS, EPK, CJL, NNV, JY, ELB, and GRR were employed by NewLink Genetics at the time of conducting the study. All remaining authors have declared no conflicts of interest.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Availability of data and material: All data relevant to the study are included in the article or uploaded as online supplemental information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics statements
Patient consent for publication
Not required.
Ethics approval
The study was done in accordance with both the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice guidelines and was approved by the institutional review board.
References
- 1.Hersh EM, O'Day SJ, Powderly J, et al. A phase II multicenter study of ipilimumab with or without dacarbazine in chemotherapy-naïve patients with advanced melanoma. Invest New Drugs 2011;29:489–98. 10.1007/s10637-009-9376-8 [DOI] [PubMed] [Google Scholar]
- 2.Robert C, Ribas A, Schachter J, et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol 2019;20:1239–51. 10.1016/S1470-2045(19)30388-2 [DOI] [PubMed] [Google Scholar]
- 3.Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373:23–34. 10.1056/NEJMoa1504030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holmgaard RB, Zamarin D, Munn DH, et al. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med 2013;210:1389–402. 10.1084/jem.20130066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spranger S, Koblish HK, Horton B, et al. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer 2014;2:3. 10.1186/2051-1426-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGaha TL, Huang L, Lemos H, et al. Amino acid catabolism: a pivotal regulator of innate and adaptive immunity. Immunol Rev 2012;249:135–57. 10.1111/j.1600-065X.2012.01149.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brincks EL, Adams J, Essmann M, et al. Indoximod modulates AhR-driven transcription of genes that control immune function. Proceedings of the AACR Annual Meeting; 2018 Apr 14-18, Chicago, IL, 2018. [Google Scholar]
- 8.Frumento G, Rotondo R, Tonetti M, et al. Tryptophan-Derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med 2002;196:459–68. 10.1084/jem.20020121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munn DH, Sharma MD, Hou D, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest 2004;114:280–90. 10.1172/JCI21583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma MD, Baban B, Chandler P, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest 2007;117:2570–82. 10.1172/JCI31911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma MD, Hou D-Y, Baban B, et al. Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity 2010;33:942–54. 10.1016/j.immuni.2010.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma MD, Hou D-Y, Liu Y, et al. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 2009;113:6102–11. 10.1182/blood-2008-12-195354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmgaard RB, Zamarin D, Li Y, et al. Tumor-Expressed IDO recruits and activates MDSCs in a Treg-Dependent manner. Cell Rep 2015;13:412–24. 10.1016/j.celrep.2015.08.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spranger S, Spaapen RM, Zha Y, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013;5:200ra116. 10.1126/scitranslmed.3006504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wainwright DA, Chang AL, Dey M, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res 2014;20:5290–301. 10.1158/1078-0432.CCR-14-0514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zakharia Y, Drabick J, Khleif S, et al. 514 results of phase 1B trial of the indoleamine 2,3-dioxygenase (IDO) pathway inhibitor Indoximod plus ipilimumab for the treatment of unresectable stage III or IV melanoma. Eur J Cancer 2015;51:S108. 10.1016/S0959-8049(16)30315-X [DOI] [Google Scholar]
- 17.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. 10.1016/j.ejca.2008.10.026 [DOI] [PubMed] [Google Scholar]
- 18.Sonpavde G, Necchi A, Gupta S, et al. ENERGIZE: a phase III study of neoadjuvant chemotherapy alone or with nivolumab with/without linrodostat mesylate for muscle-invasive bladder cancer. Future Oncol 2020;16:4359–68. 10.2217/fon-2019-0611 [DOI] [PubMed] [Google Scholar]
- 19.Long GV, Dummer R, Hamid O, et al. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (PTS) with unresectable or metastatic melanoma: results of the phase 3 ECHO-301/KEYNOTE-252 study. J Clin Oncol 2018;36:108. 10.1200/JCO.2018.36.15_suppl.108 [DOI] [Google Scholar]
- 20.Muller AJ, Manfredi MG, Zakharia Y, et al. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol 2019;41:41–8. 10.1007/s00281-018-0702-0 [DOI] [PubMed] [Google Scholar]
- 21.Smith MNR, Owens S, Gong X. Retrospective pooled analysis of Epacadostat clinical studies identifies doses required for maximal pharmacodynamic effect in anti-PD-1 combination studies SITC; 2020. [Google Scholar]
- 22.Maier B, Leader AM, Chen ST, et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 2020;580:257–62. 10.1038/s41586-020-2134-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bahary N, Wang-Gillam A, Haraldsdottir S, Sigurdis H, et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus gemcitabine / nab-paclitaxel for the treatment of patients with metastatic pancreas cancer. Journal of Clinical Oncology 2018;36:4015. 10.1200/JCO.2018.36.15_suppl.4015 [DOI] [Google Scholar]
- 24.Colman H, Mott F, Spira AI, et al. A phase 1b/2 study of the combination of the IDO pathway inhibitor indoximod and temozolomide for adult patients with temozolomide-refractory primary malignant brain tumors: safety analysis and preliminary efficacy of the phase 1B component. Journal of Clinical Oncology 2015;33:2070. 10.1200/jco.2015.33.15_suppl.2070 [DOI] [Google Scholar]
- 25.Emadi A, Duong VH, Pantin J, et al. Indoximod combined with standard induction chemotherapy is well tolerated and induces a high rate of complete remission with MRD-Negativity in patients with newly diagnosed AML: results from a phase 1 trial. 132. San Diego, CA: American Society of Hematology Annual Meeting, 2018: 332. 10.1182/blood-2018-99-117433 [DOI] [Google Scholar]
- 26.Hou D-Y, Muller AJ, Sharma MD, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res 2007;67:792–801. 10.1158/0008-5472.CAN-06-2925 [DOI] [PubMed] [Google Scholar]
- 27.Johnson TS, Aguilera D, Al-Basheer A. Abstract CT004: front-line therapy of DIPG using the IDO pathway inhibitor indoximod in combination with radiation and chemotherapy. Cancer Research 2018;78:CT004-CT. [Google Scholar]
- 28.Jha GG, Gupta S, Tagawa ST, et al. A phase II randomized, double-blind study of sipuleucel-T followed by IDO pathway inhibitor, indoximod, or placebo in the treatment of patients with metastatic castration resistant prostate cancer (mCRPC). J Clin Oncol 2017;35:3066. 10.1200/JCO.2017.35.15_suppl.3066 [DOI] [Google Scholar]
- 29.Soliman HH, Minton SE, Han HS, et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget 2016;7:22928–38. 10.18632/oncotarget.8216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.George T, Wang-Gillam A, Park H. A phase 1 clinical trial of NLG802, a prodrug of Indoximod with enhanced pharmacokinetic properties. Immuno-Oncology 2019 2nd World Congress, Barcelona, Spain, 2019. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2020-002057supp001.pdf (62.9KB, pdf)
jitc-2020-002057supp002.pdf (66.7KB, pdf)
jitc-2020-002057supp003.pdf (530.1KB, pdf)
Data Availability Statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Availability of data and material: All data relevant to the study are included in the article or uploaded as online supplemental information. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.