

Keywords: intestinal failure-associated liver disease, short bowel syndrome, small bowel resection
Abstract
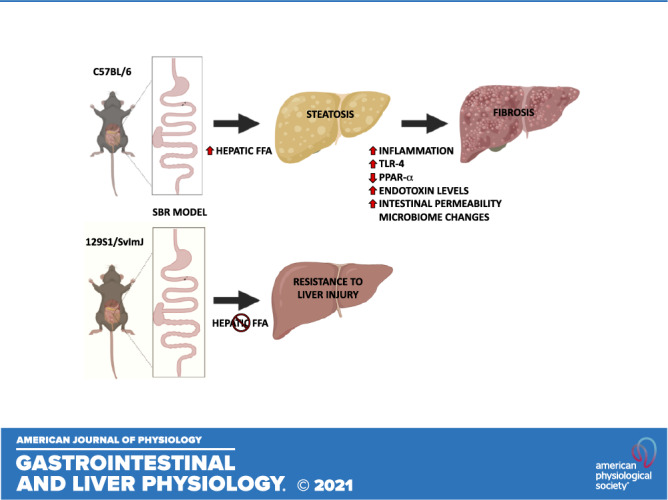
Intestinal failure-associated liver disease is a major morbidity associated with short bowel syndrome. We sought to determine if the obesity-resistant mouse strain (129S1/SvImJ) conferred protection from liver injury after small bowel resection (SBR). Using a parenteral nutrition-independent model of resection-associated liver injury, C57BL/6J and 129S1/SvImJ mice underwent a 50% proximal SBR or sham operation. At postoperative week 10, hepatic steatosis, fibrosis, and cholestasis were assessed. Hepatic and systemic inflammatory pathways were evaluated using oxidative markers and abundance of tissue macrophages. Potential mechanisms of endotoxin resistance were also explored. Serum lipid levels were elevated in all mouse lines. Hepatic triglyceride levels were no different between mouse strains, but there was an increased accumulation of free fatty acids in the C57BL/6J mice. Histological and serum markers of hepatic fibrosis, steatosis, and cholestasis were significantly elevated in resected C57BL/6J SBR mice as well as oxidative stress markers and macrophage recruitment in both the liver and visceral white fat in C57BL/6J mice compared with sham controls and the 129S1/SvImJ mouse line. Serum endotoxin levels were significantly elevated in C57BL/6J mice with significant elevation of hepatic TLR4 and reduction in PPARα expression levels. Despite high levels of serum lipids, 129S1/SvImJ mice did not develop liver inflammation, fibrosis, or cholestasis after SBR, unlike C57BL/6J mice. These data suggest that the accumulation of hepatic free fatty acids as well as increased endotoxin-driven inflammatory pathways through PPARα and TLR4 contribute to the liver injury seen in C57BL/6J mice with short bowel syndrome.
NEW & NOTEWORTHY Unlike C57BL/6 mice, the 129S1/SvImJ strain is resistant to liver inflammation and injury after small bowel resection. These disparate outcomes are likely due to the accumulation of hepatic free fatty acids as well as increased endotoxin-driven inflammatory pathways through PPARα and TLR4 in C57BL/6 mice with short bowel syndrome.
INTRODUCTION
Short bowel syndrome (SBS) occurs due to the surgical removal of a significant length of small intestine, with the resulting malabsorptive state leading to an approximate mortality rate of 25% (1). Morbidity persists among survivors with 25% requiring a small bowel transplantation and an overall five-year survival of 58% (2, 3). Intestinal failure-associated liver disease (IFALD), a common complication in patients with SBS, is the leading indication for transplantation (4).
The development of IFALD in patients with SBS is the most consistent negative prognostic factor for survival (5–7). Although initially thought to be due to prolonged intravenous nutrition, the mechanism to developing IFALD is multifactorial (8–10). Host factors, such as prematurity, as well as nutritional components, including plant sterols, have been implicated retrospectively (11, 12). Recent efforts to elucidate the mechanism behind liver injury have included multiple investigations directed toward concentrations of the proinflammatory effects of omega-6 polyunsaturated fatty acid in parenteral nutrition (PN) lipid emulsions, Kupffer cell activation due to increased intestinal permeability, and alterations in the microbiome (13, 14).
We have previously established a model for SBS with liver injury independent of parenteral nutrition (PN) by performing 50% proximal small bowel resections (SBR) in C57Bl/6J mice (15, 16). At 10 wk after SBR, C57Bl/6J mice developed steatosis and liver fibrosis (16). The appearance of steatosis appeared to be dependent on the higher fat content of our postoperative liquid diet (35% kcal fat vs. 13% kcal fat in standard rodent chow), however, the progression to fibrosis only occurred in mice undergoing SBR, thereby incriminating both dietary fat and intestinal resection as requirements for liver injury (16). In addition, we found that the profile of liver injury after SBR was ameliorated in global Toll-like receptor 4 (TLR4) knockout mice, suggesting a role for bacteria-derived endotoxin in this process (17). Here, we sought to explore the similarities of the resection-associated hepatic changes in our murine model to IFALD.
It has been previously demonstrated that C57BL/6J mice gain more weight at baseline than the 129S1/SvImJ mouse strain, a phenotype that is further exacerbated on a high-fat diet (18, 19). Another important feature of the 129S1/SvImJ mice is that they have diminished sensitivity to endotoxin when compared with C57Bl/6J (20, 21). Given these differences, the 129S1/SvImJ mice allowed for investigation of the role of steatosis and inflammation in the pathogenesis of resection-associated liver injury in an obesity and endotoxin-resistant mouse strain.
METHODS
Animals, Housing, and Operations
C57BL/6J and 129S1/SvImJ 6-wk-old mice were obtained from Jackson laboratories (Bar Harbor, ME). Mice were housed in a temperature-controlled, pathogen-free animal holding area on a 12-h light-dark cycle. After acclimation to the facility for 1 wk, they were redistributed by mouse line into different cages and were placed on liquid diet (PMI Micro-Stabilized Rodent Liquid Diet LD 101, TestDiet, St. Louis, MO) to synchronize baseline gut microbiota. Two weeks later, the 9-wk-old mice underwent either a sham control or 50% proximal small bowel resection (SBR), as previously described (22). Briefly, SBR involved a transection of the small intestine 12 cm proximal to the ileocecal junction and 1–2 cm distal to the ligament of Treitz, with removal of the intervening bowel. Intestinal continuity was restored with a handsewn end-to-end anastomosis using 9-0 monofilament suture. Sham control operations involved a single transection and re-anastomosis at 12 cm proximal to the ileocecal junction. Mice were provided with water ad libitum for the first 24 h and then restarted on liquid diet. They were co-housed both by mouse line and operation type. Protocols and experiments were approved by the Washington University in St. Louis Animal Studies Committee (Protocol 20170252) in accordance with the National Institute of Health Laboratory Animal Care and Use guidelines. Additional 9-wk-old C57BL/6J mice underwent operation and were harvested 12 wk postoperatively for serum lipid levels, hepatic mRNA expression levels, liver enzyme levels, and hepatic immunostaining.
Confirmation of Structural Adaptation
Intraoperative (IO) bowel from the distal end of the removed intestine at the time of resection as well as distal to the anastomosis at postoperative week 10–12 were fixed in 10% formalin and embedded in paraffin. As previously described, 5-µm thick longitudinal sections were stained with hematoxylin and eosin (H&E) to assess for structural adaptation via villus height and crypt (NIS elements AR 4; Nikon, Melville, NY) (23). Only mice that survived and adapted appropriately by villus elongation and/or crypt deepening (greater than 20%) were included in the analysis (24).
Average Daily Caloric Intake
Food intake was measured at postoperative weeks 4–5 and 9–10. Mice were either singly housed or cohoused within same operation type, with food intake measured daily for 4–5 days. Average daily food intake was converted to caloric intake.
Body Composition
Accretion of body fat mass and lean mass were measured preoperatively, on postoperative week 5, and on postoperative week 10 using a quantitative nuclear magnetic resonance instrument (Echo Medical Systems, Houston, TX) (25).
Hepatic Lipid Extraction and Analysis
As previously described, liver tissue was homogenized in PBS before protein concentration determination for control (26). Hepatic lipids were extracted from the homogenate using chloroform:methanol (2:1) and 0.1% sulfuric acid. The organic phase was then dried under nitrogen, resuspended using 2% Triton X-100, and reconstituted. Total hepatic triglyceride, cholesterol, and free fatty acid concentrations were determined using commercially available L-Type TG-H, Cholesterol E, and NEFA C kits, respectively (Wako Chemicals, Richmond, VA).
Serum Lipid, Alanine Aminotransferase, Aspartate Aminotransferase, Alkaline Phosphatase, Total Bilirubin, Direct Bilirubin, and Endotoxin Levels
Serum was obtained via submandibular vein sampling at postoperative weeks 5 and 10. Serum lipids and liver enzyme activity levels were measured using L-Type TG-H, Cholesterol E (Wako Chemicals, Richmond, VA), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) kits (Sigma-Aldrich, St. Louis, MO). Portal venous blood was used to measure serum alkaline phosphatase (ALP) (Abcam, Cambridge, MA) and total/direct bilirubin levels (Promocell, ThermoFisher, Berkely, MO). Serum levels of endotoxin were measured using a commercially available LAL chromogenic kit (Pierce, Thermo Fisher, Berkley, MO).
Hepatic Fibrosis and Cholestasis Staining and Quantification
After formalin fixation and paraffin embedding, liver tissues were stained with Sirius red as a marker of fibrosis and cytokeratin-19 (CK19; Proteintech; 10712-1-AP) as a marker of cholestasis (27). As previously described, the intensity of fibrotic and cholestatic staining was quantified using Image-J software (28). Briefly, 10 fields of each specimen were examined at ×20 magnification. All slides were analyzed in a blinded manner by the same investigator.
Hepatic and Visceral White Adipose Tissue RNA Isolation and PCR
Total RNA from the liver and visceral white fat was isolated using RNeasy Mini kits as per the manufacturer’s protocol (Qiagen, Germantown, MD). Visceral white adipose tissue was obtained from the perigonadal regions (29). qRT-PCR was conducted using the ABI StepOnePlus Real-Time PCR system with specific primers [NADPH oxidase 2 (NOX2): forward primer CGGAGAGTTTGGAAGAGCATAA, reverse primer GGTACTGGGCACTCCTTTATTT; tumor necrosis factor α (TNFα): forward primer AATGGCCTCCCTCTCATCAGTT, reverse primer CCACTTGGTGGTTTGCTACGA; glutathione synthetase (GSS): forward primer GCCTCCTACATCCTCATGGA, reverse primer CCACATGCTTGTTCATCACC; interleukin 1-β (IL-1β): forward primer AGAGCCCATCCTCTGTGACTCA, reverse primer TGCTTGGGATCCACACTCTCCA; IL-6: forward primer GAACAACGATGATGCACTTGC, reverse primer TCCAGGTAGCTATGGTACTCC; mitochondrial genome integrity 1 (MgI1): forward primer TGGCCTGAAGCTGACAAGTA, reverse primer AGGCCGATCCAACTAACCACAT; Applied Biosystems, Waltham, MA]. RNA was converted to cDNA using the qScript cDNA Synthesis Kit (Quanta Bio Beverly, MA). Real-time polymerase chain reaction (RT-PCR) was performed using the TaqMan Gene Expression Master Mix (Applied Biosystems, Foster City, CA, Cat. 4369016) and Applied Biosystems 7500 Fast Real-Time PCR to determine relative concentrations [Actb: Mm04394036_g1, peroxisome proliferator-activated receptor alpha (PPARα): Mm00440939_m1, TLR4: Mm00445274_m1; Applied Biosystems, Waltham, MA]. The relative mRNA levels were estimated from the equation 2–ΔCt (ΔCt = Cttarget gene − Ct18S rRNA). Fold changes in the mRNA level of genes were calculated with a control group level set at 1.
Immunofluorescent Staining for Macrophage Abundance in Liver and Visceral White Fat
Hepatic and white adipose tissue was fixed in 10% formalin and embedded in paraffin. Antigen retrieval was performed on deparaffinized slides in a pressurized chamber for 10 min (Diva Decloaking solution, Biocare Medical, Concord, CA). Sections were then incubated with blocking buffer (1% BSA, 5% donkey serum, and 0.03% Triton-X; Sigma Aldrich, St. Louis, MO). They were subsequently incubated overnight at 4°C with F4/80 antibody (Abcam, Cambridge, MA). Ten fields of each specimen were examined at ×200 magnification and counted for F4/80 positive cells. All slides were analyzed in a blinded fashion by the same investigator.
Statistical Analysis
Statistical analysis was performed using GraphPad-Prism 6 software (La Jolla, CA). Adaptation data were analyzed using an unpaired Student’s t test. Food intake, body composition, hepatic lipid content, mRNA expression levels, histology staining, and systemic endotoxin levels were analyzed using a two-way ANOVA with Tukey’s multiple comparison tests between groups. Serum triglyceride, cholesterol, and AST/ALT/ALP/bilirubin levels were analyzed using a two-way ANOVA with Tukey’s multiple comparisons between mouse lines. Unpaired Student’s t test was used to compare time periods between and within each mouse line. Graphs show the sample means with significance of Student’s t tests or Tukey’s multiple comparison tests. A P value of <0.05 was considered significant.
RESULTS
Structural Intestinal Adaptation and Body Composition
Villus height significantly increased in all mice after SBR regardless of strain, confirming appropriate structural intestinal adaptation (Fig. 1A) (22). The effect of SBR on average daily food intake did not differ among mouse lines at early (4 wk) or later (9 wk) timepoints after operation (Fig. 1B). Interestingly, C57BL/6J mice started off with higher lean mass but lower fat mass, even though they are more obesity prone than the 129S1/SvImJ mice. C57BL/6J sham controls and resected 129S1/SvImJ and sham mice followed a similar pattern of significantly decreased lean mass with corresponding increased fat mass (Fig. 1, D–F). However, resected C57BL/6J mice showed no large redistribution of mass comparatively (Fig. 1F). With this postoperative redistribution of lean and fat body composition, resected 129S1/SvImJ mice maintained their weight without significant difference between sham controls by 10 wk, in contrast to resected C57BL/6J mice, which had significantly less body mass than their sham controls (Fig. 1C).
Figure 1.
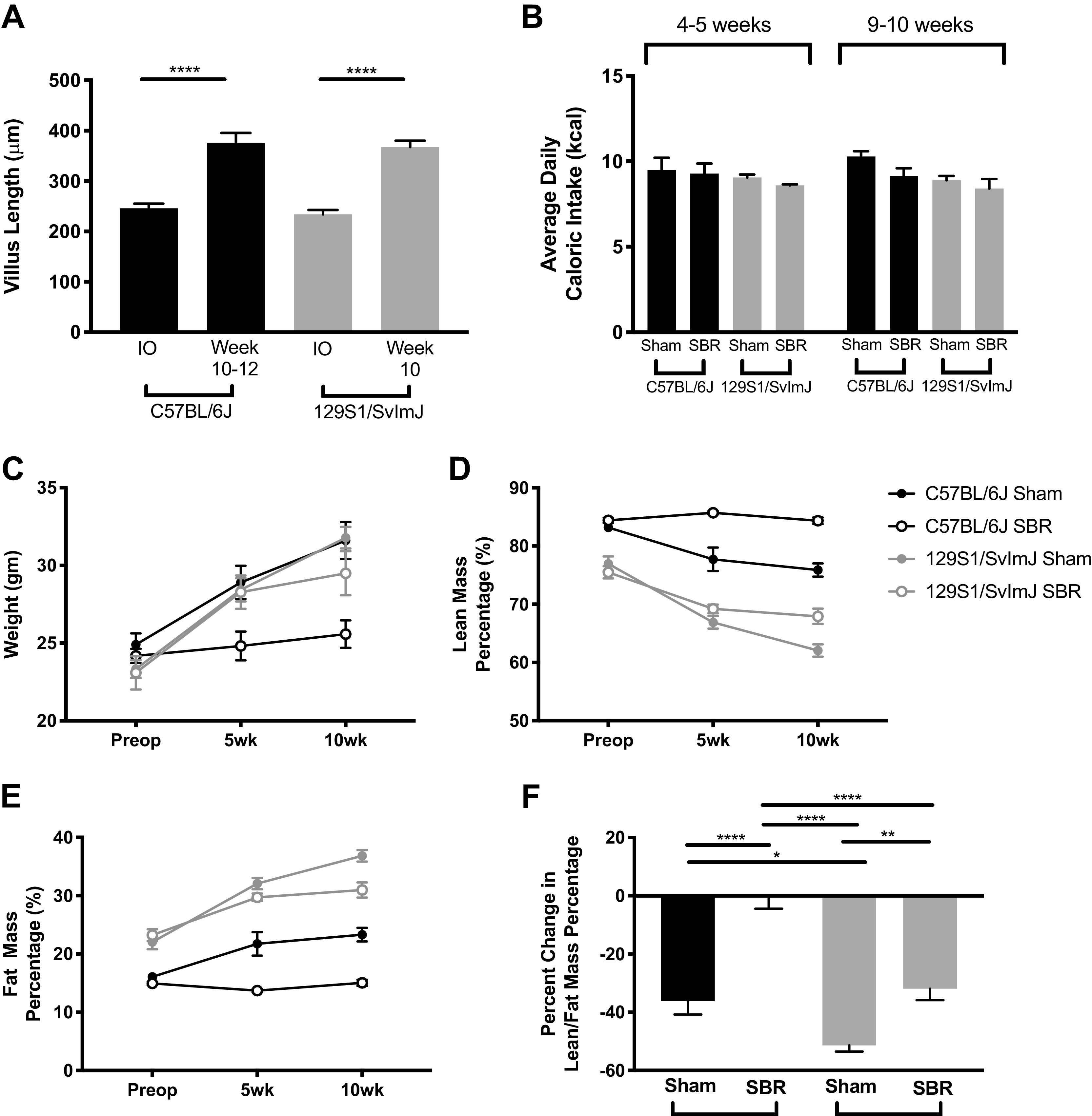
A: villus height increases in resected mice in C57BL/6J (n = 9) and 129S1/SvImJ (n = 5) mice. B: average daily caloric intake in C57BL/6J sham (n = 5), C57BL/6J small bowel resection (SBR) (n = 4), sham 129S1/SvImJ (n = 8), and 129S1/SvImJ SBR (n = 5) mice at 4 and 9 wk postoperatively. Weight (C), lean mass percentage (D), and fat mass percentage (E) changes in sham C57BL/6J (n = 5), C57BL/6J SBR (n = 5), sham 129S1/SvImJ (n = 8), and 129S1/SvImJ SBR (n = 5) mice preoperatively, at 5-wk postoperatively, and at 10-wk postoperatively with (F) percent change in lean mass percentage/fat mass percentage from baseline to 10 wk. ****P < 0.0001, **P < 0.005, and *P < 0.05.
Serum and Hepatic Lipid Concentrations
In general, SBR resulted in significantly reduced serum triglyceride levels in both strains of mice at the 10-wk time point. The most dramatic finding was a significantly higher serum triglyceride level in the C57BL/6J sham controls (Fig. 2A). In C57BL/6J mice, serum cholesterol levels were significantly decreased after SBR at 10 wk (P < 0.0005) and to a much greater extent than in the 129S1/SvImJ mice at this time point (Fig. 2B, P < 0.005). When normalized to hepatic protein, there was no difference in hepatic triglyceride levels between sham and resected mice in either mouse line (Fig. 2C). Hepatic cholesterol levels were significantly higher in sham 129S1/SvImJ mice and were significantly lower after SBR. In contrast, SBR did not affect hepatic cholesterol content in the C57Bl/6J mice. Hepatic free fatty acid levels significantly increased in both sham and resected B6 mice compared with their 129S1 equivalents (Fig. 2, D and E). As visualized by H&E staining, small bowel resected C57BL/6J mice demonstrated increased lipid droplets with micro/macrovesicular steatosis, dysregulated hepatocytes, and infiltration of small nucleus immune cells compared with their sham controls. In contrast, 129S1/SvImJ mice, both resected and sham controls, showed normal liver histology without steatosis, damaged hepatocytes, or enhanced small nucleus cells (Fig. 2F).
Figure 2.
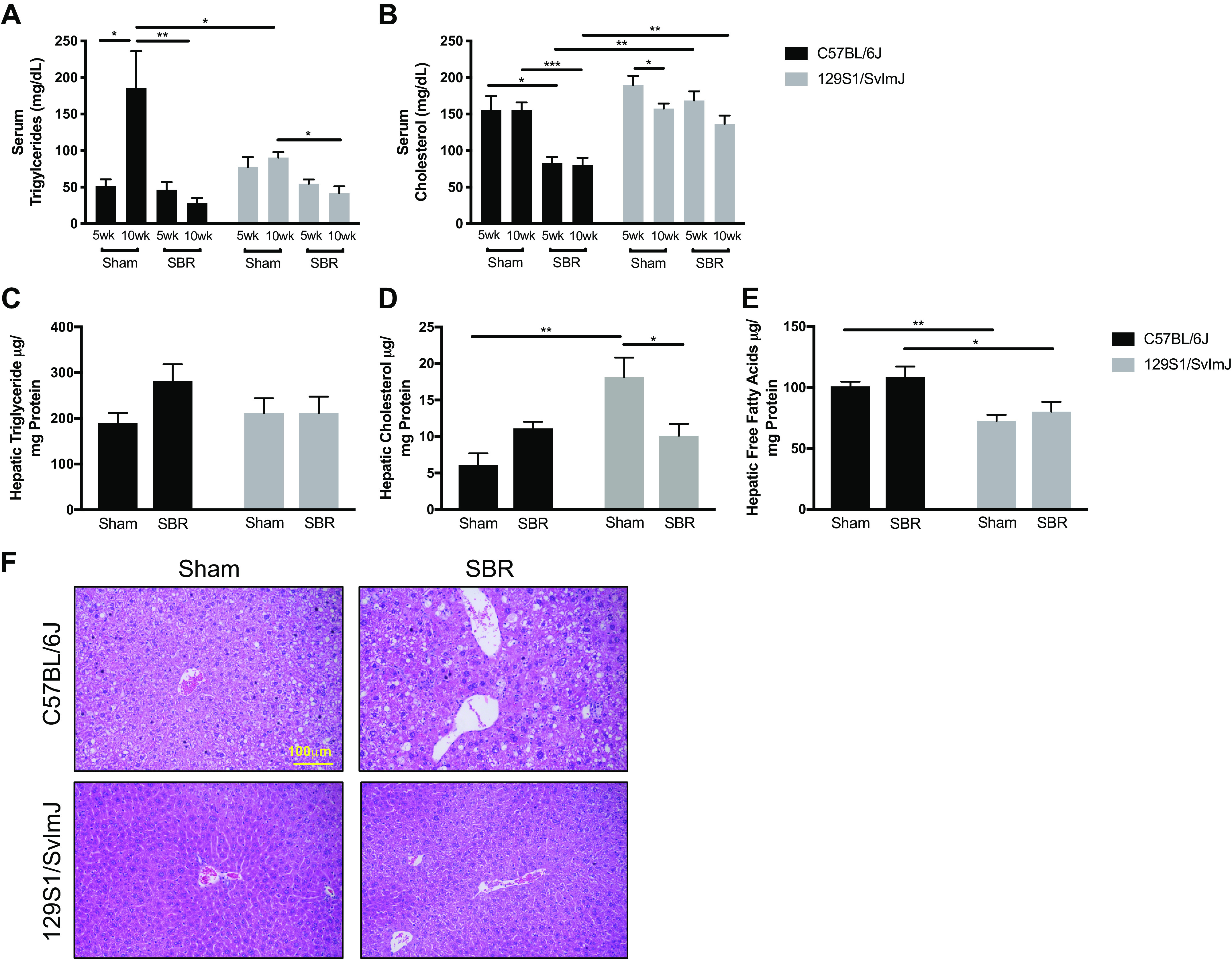
Serum triglyceride (A) and cholesterol (B) levels in C57BL/6J sham (n = 5, 5) and small bowel resection (SBR) (n = 5, 6) mice and in 129S1/SvImJ sham (n = 8, 8) and SBR (n = 5, 5) mice at postoperative weeks 5 and 10–12 (noted as 10 wk), respectively. Hepatic triglyceride (C), cholesterol (D), and free fatty acids (E) in sham C57BL/6J (n = 6) and SBR (n = 6) mice and in sham 129S1/SvImJ (n = 7) and SBR (n = 5) mice at 10-wk postoperatively. F: representative hematoxylin and eosin (H&E) images of livers in C57BL/6J and 129S1/SvImJ mice at 10-wk postoperatively showing increased lipid droplets with micro/macrovesicular steatosis, dysregulated hepatocytes, and infiltration of small nucleus immune cells in C57BL/6J SBR livers compared with their sham controls as well as both experimental groups in the 129S1/SvImJ line. ***P = 0.001, **P < 0.005, *P < 0.05.
Resection-Associated Liver Injury and Fibrosis is Mitigated in 129S1/SvImJ Mice
In C57BL/6J mice, serum ALT and AST levels in resected compared with sham mice were 69% and 293% higher at 5 wk and 134% and 123% higher at 10 wk, respectively (P < 0.0001, P < 0.0005). In marked contrast, 129S1/SvImJ ALT levels were unchanged between sham and SBR mice at both time points and the increase associated with SBR in AST levels was attenuated when compared with the C57Bl/6J group (Fig. 3, A and B). Sirius red staining of the liver qualitatively and quantitively showed increased perisinusoidal fibrosis in resected C57BL/6J mice after SBR. Fibrosis was not evident after SBR in the 129S1/SvImJ mice (Fig. 3, C and D).
Figure 3.
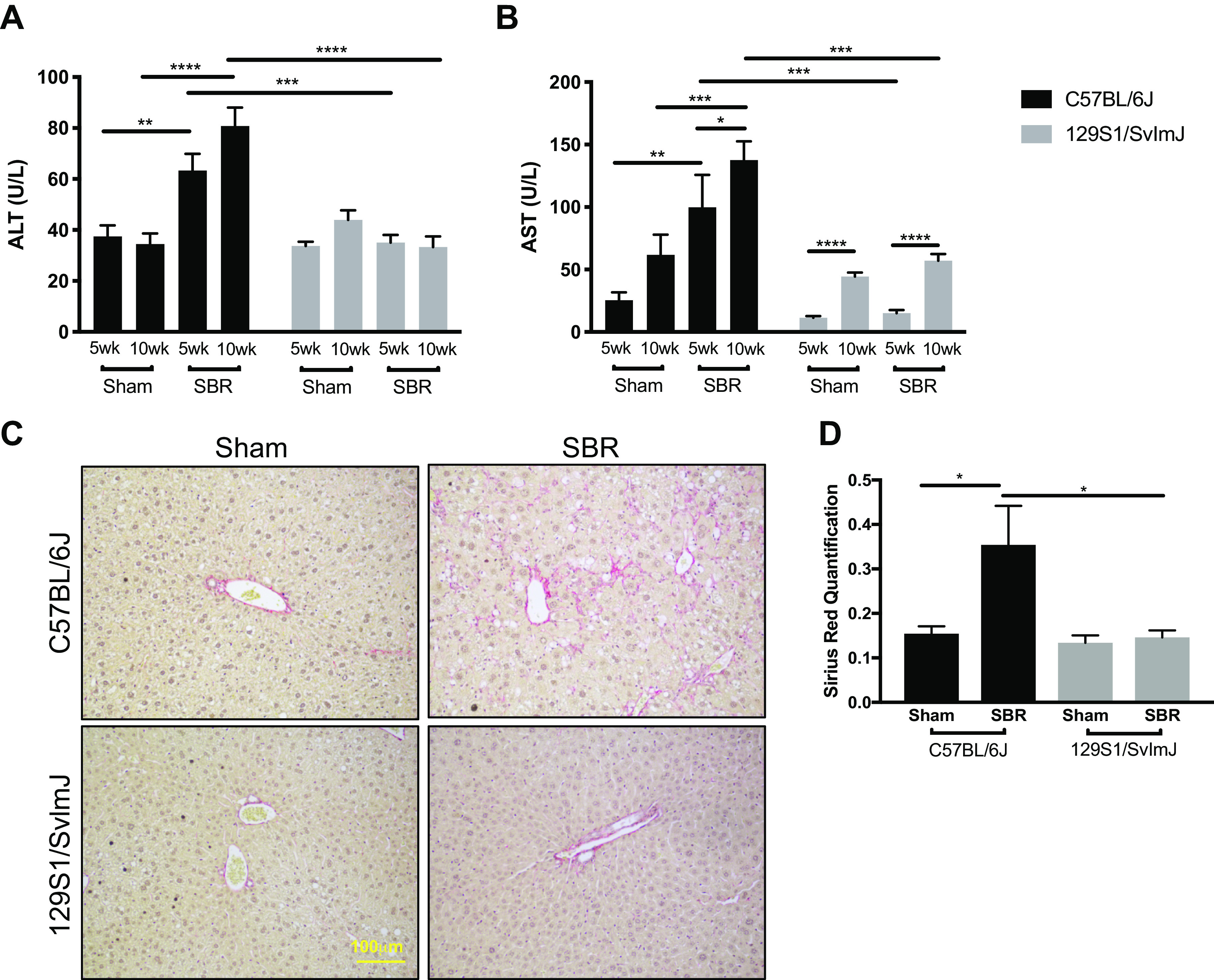
Serum alanine aminotransferase (ALT) (A) and aspartate aminotransferase (AST) (B) levels in C57BL/6J sham (n = 5, 5) and small bowel resection (SBR) (n = 5, 6) mice and in 129S1/SvImJ sham (n = 8, 8) and SBR (n = 5, 5) mice at postoperative weeks 5 and 10–12, respectively. C: representative images of Sirius red staining in livers of sham and SBR mice in C57BL/6J and 129S1/SvImJ lines, showing increased perisinusoidal fibrosis in the resected C57BL/6J livers compared with their controls and the two experimental groups of the 129S1/SvImJ mouse line. D: quantification of Sirius red staining for hepatic fibrosis in sham (n = 6, 8) and SBR (n = 6, 5) mice in C57BL/6J and 129S1/SvImJ lines, respectively. *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001.
Resection-Associated Cholestasis is Mitigated in 129S1/SvImJ Mice
In C57BL/6J mice, serum ALP levels were 194% higher in resected compared with sham mice at 10 wk (P < 0.0001). In addition, total and direct bilirubin levels were 95% and 145% higher in resected compared with sham C57BL/6J mice, respectively (P = 0.001, P < 0.0001). In marked contrast, 129S1/SvImJ ALP, total bilirubin, and direct bilirubin levels were unchanged between sham and SBR mice (Fig. 4, A–C). Hepatic staining for CK19 qualitatively and quantitively showed increased cholestasis with peri-portal ductular proliferations in resected C57BL/6J mice after SBR. Cholestasis was not evident after SBR in the 129S1/SvImJ mice (Fig. 4, C and D).
Figure 4.
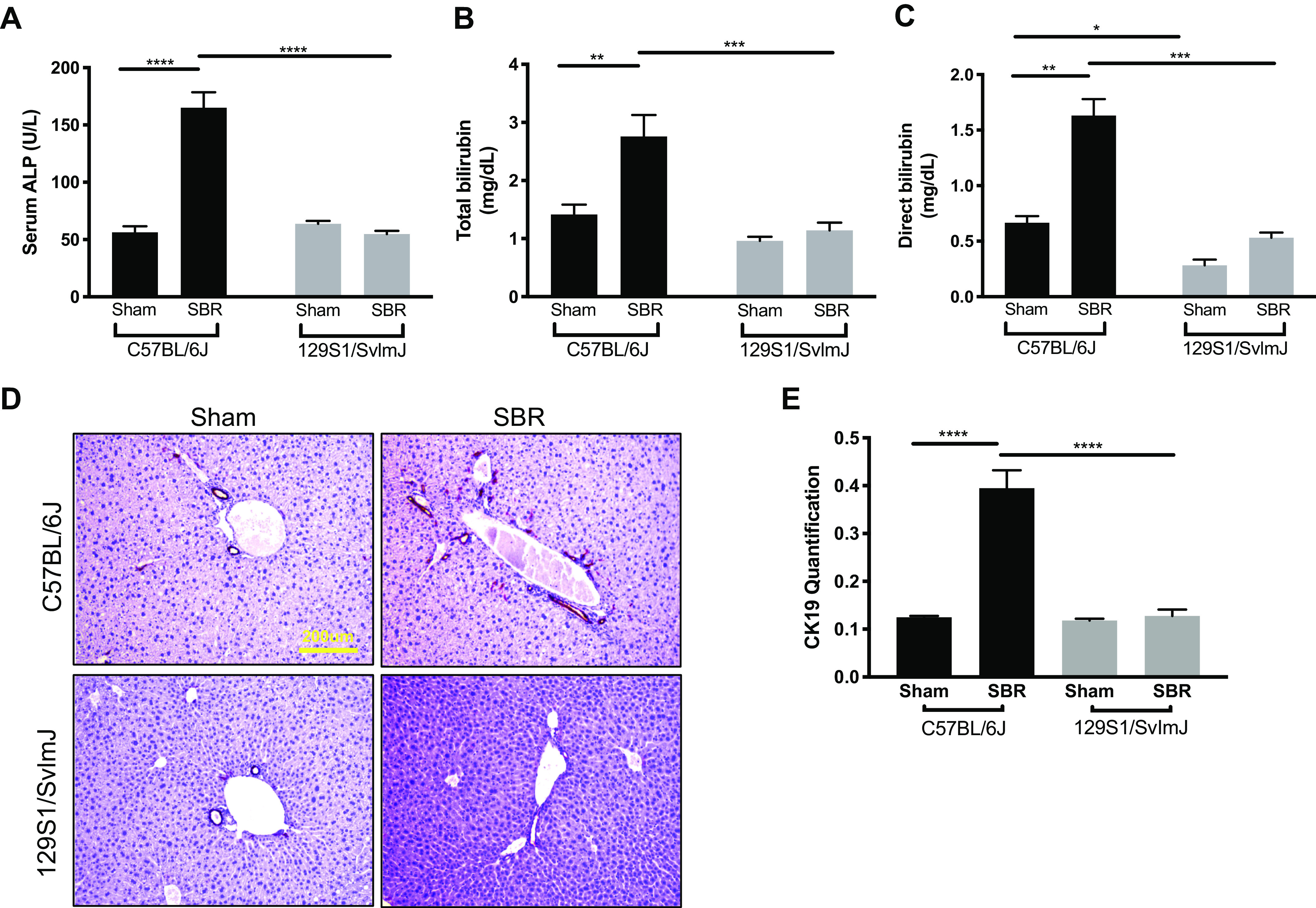
Serum alkaline phosphatase (ALP) (A), total bilirubin (B), and direct bilirubin (C) levels in C57BL/6J sham (n = 5) and small bowel resection (SBR) (n = 5) mice and in sham 129S1/SvImJ (n = 8) and SBR (n = 5) mice at postoperative week 10–12, respectively. D: representative images of CK19 staining in livers of sham and SBR mice in C57BL/6J and 129S1/SvImJ lines. E: quantification of CK19 staining for hepatic cholestasis in sham (n = 5, 5) and SBR (n = 5, 5) mice in C57BL/6J and 129S1/SvImJ lines, respectively. ****P < 0.0001, ***P < 0.001, **P < 0.005, *P < 0.05.
Anti-Inflammatory Effects of 129S1/SvImJ after Resection
To assess for oxidative stress in the liver, we measured the relative mRNA expression of NADPH oxidase 2 (NOX2), tumor necrosis factor α (TNFα), and glutathione synthetase (GSS). In C57BL/6J mice, there were significant 1.6-, 1.9-, and 2.5-fold increases in NOX2, TNFα, and GSS expression after resection. In contrast, no differences were detected between sham and SBR in the 129S1/SvImJ cohort (Fig. 5, A–C). Immunostaining for the macrophage marker F4/80 identified significantly greater numbers of macrophages in the liver after SBR in the C57B/l6J, but not in the 129S1/SvImJ mice (Fig. 5, D and E).
Figure 5.
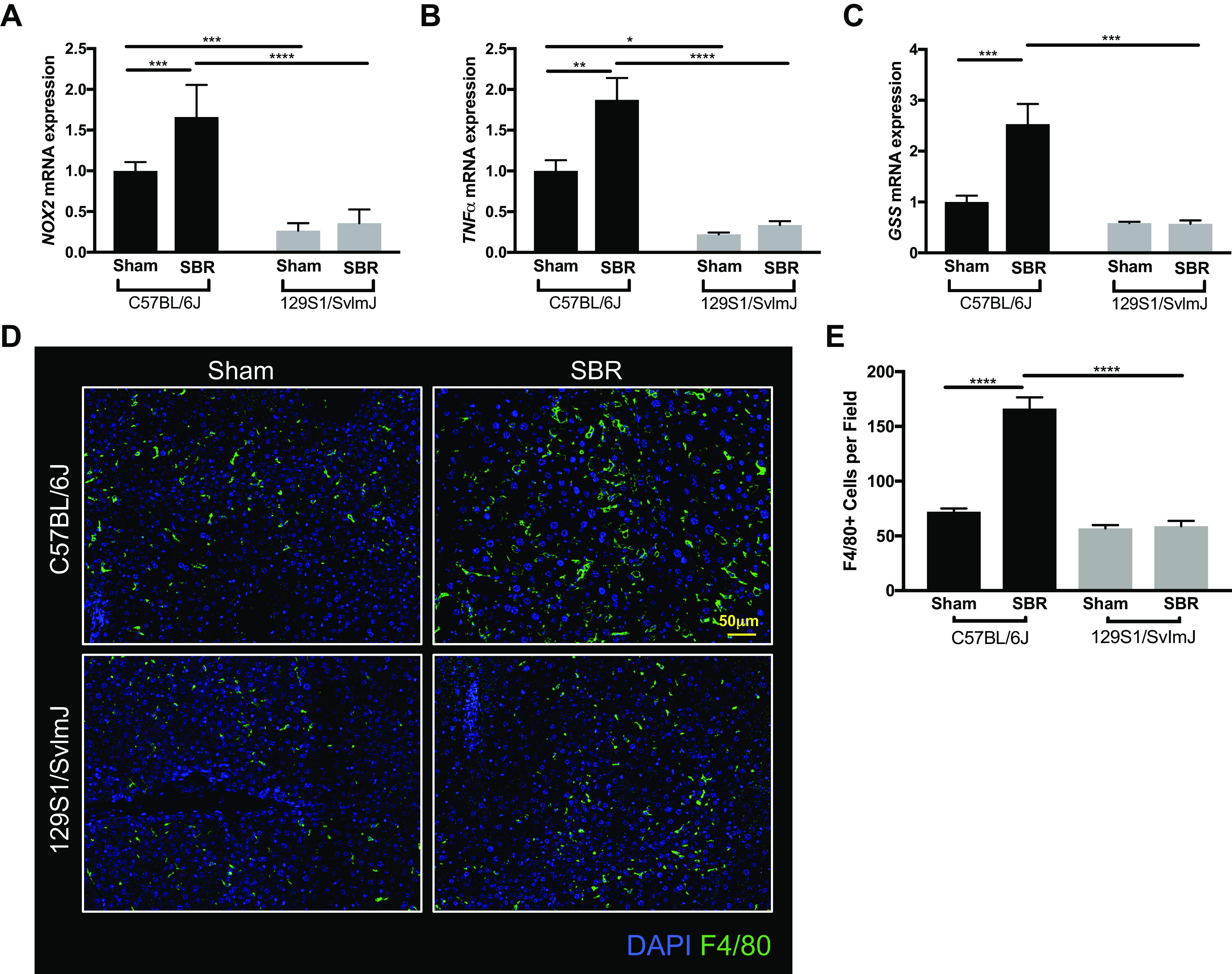
mRNA expression levels of NADPH oxidase 2 (NOX2) (A), tumor necrosis factor α (TNFα) (B), and glutathione synthetase (GSS) (C) in liver tissue from sham (n = 5, 7) and small bowel resection (SBR) (n = 6, 4) operated C57BL/6J and 129S1/SvImJ mice, respectively. D: representative images of F4/80-stained liver tissue. E: quantification of F4/80 staining for hepatic macrophages in sham (n = 3, 3) and SBR (n = 3, 3) mice in C57BL/6J and 129S1/SvImJ lines, respectively. ****P < 0.0001, ***P < 0.001, **P < 0.005, *P < 0.05.
Furthermore, we measured inflammatory markers in visceral white adipose tissue as a marker of systemic inflammation. Expression of proinflammatory TNFα and interleukin 1-β (IL-1β) mRNA were significantly elevated after SBR in the C57BL/6J line compared with sham controls, whereas expression of anti-inflammatory mitochondrial genome integrity 1 (MgI1) was significantly decreased (Fig. 6, A–C). Similar to the findings of oxidative stress in the liver, there were no changes identified in the 129S1/SvImJ mouse line after SBR. We also performed F4/80 staining to assess for macrophage presence in white adipose tissue, which was markedly increased in the resected C57BL/6J, but not in the 129S1/SvImJ group (Fig. 6, D and E; P < 0.0001).
Figure 6.
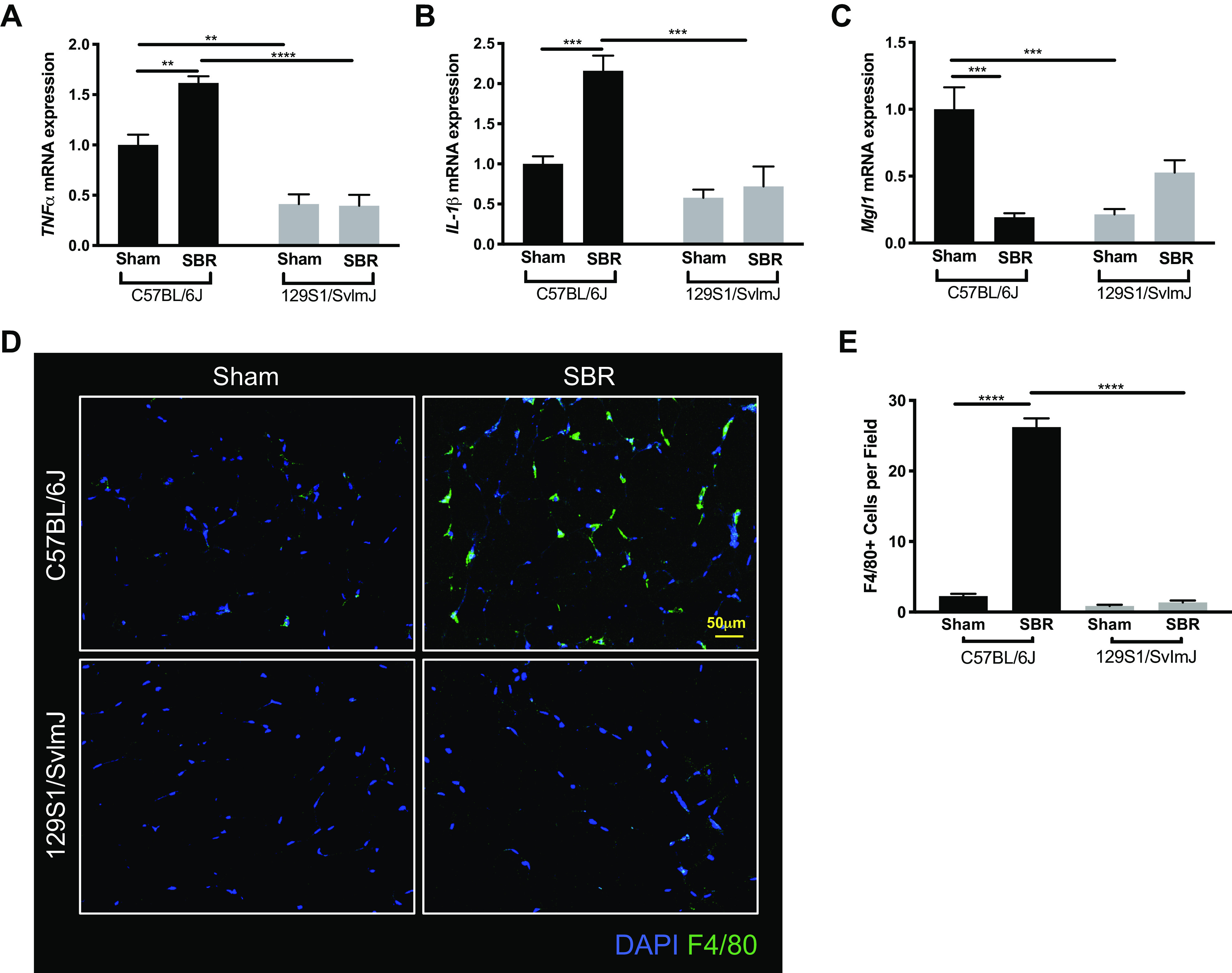
mRNA expression levels of tumor necrosis factor α (TNFα) (A), interleukin 1-β (IL-1β) (B), and mitochondrial genome integrity 1 (MgI1) (C) in white adipose tissue from sham (n = 5, 4) and small bowel resection (SBR) (n = 5, 4) operated C57BL/6J and 129S1/SvImJ mice, respectively. D: representative images of F4/80-stained white adipose tissue. E: quantification of F4/80 staining for visceral white adipose tissue macrophages in sham (n = 3, 3) and SBR (n = 3, 3) mice in C57BL/6J and 129S1/SvImJ lines, respectively. ****P < 0.0001, ***P < 0.001, **P < 0.005.
Mechanism of Liver Injury after Resection is Associated with Inflammation
To investigate possible mechanisms of IFALD development in our model, we measured the relative expression of peroxisome proliferator-activated receptor alpha (PPARα) and Toll-like receptor 4 (TLR4) as well as systemic endotoxin levels. In C57BL/6J SBR mice, hepatic expression of PPARα was significantly (twofold) decreased whereas TLR4 expression was significantly (twofold) elevated. These differences were not seen in the 129S1/SvImJ line (Fig. 7, A and B). Systemic endotoxin levels were significantly higher in C57BL/6J mice regardless of operation when compared with the 129S1/SvImJ line (Fig. 7C).
Figure 7.

Hepatic peroxisome proliferator-activated receptor α (PPARα) (A) and Toll-like receptor 4 (TLR4) (B) mRNA expression in sham C57BL/6J (n = 5) and small bowel resection (SBR) (n = 6) mice and in sham 129S1/SvImJ (n = 7) and SBR (n = 4) mice at 10–12 wk. C: serum endotoxin levels in sham C57BL/6J (n = 4) and SBR (n = 5) mice and in sham 129S1/SvImJ (n = 8) and SBR (n = 4) mice at 10–12 wk. **P < 0.005, *P < 0.05.
DISCUSSION
In this study, we demonstrate that intestinal resection-associated liver steatosis, with progression to injury, cholestasis, and fibrosis is prevented in 129S1/SvImJ mice. This murine strain is resistant to dietary-induced obesity and has a diminished sensitivity to endotoxin (20, 21). These data suggest that disrupted lipid metabolism, as well as sensitivity to endotoxin-driven inflammation play important roles in the pathogenesis of intestine resection-associated liver injury.
It is important to note that the correlation of this model to IFALD is limited by the fact that the mice do not truly have intestinal failure given the fact that there is only 50% proximal small bowel loss. In addition, parenteral nutrition is not provided. On the other hand, we have detected liver injury and fibrosis in the context of intestinal resection alone. This perturbation in the gut/liver axis without confounding contributions of parenteral nutrition is important. Since the majority of patients with short bowel syndrome receive nutrition by both intravenous and enteral routes, our model highlights the contribution of disordered intestinal function alone on this liver phenotype. We have previously studied the effects of a higher fat diet (35% kcal fat) in this resection model compared with standard chow, which showed that a diet high in a long-chain fatty acid (oleic acid) worsens steatosis and compounds liver injury (16). The hepatic steatosis in our model could be caused by the proinflammatory effects of oleic acid, which are well characterized in non-alcoholic fatty liver disease (NAFLD); however, it is important to note that average caloric intake was not different between experimental groups (30, 31).
The pathogenesis of IFALD is unique as patients with SBS are enterally starved without insulin resistance or obesity. This contrasts with NAFLD, which occurs in patients with normal intestinal function, insulin resistance, and is associated with the metabolic syndrome (32, 33). Moreover, IFALD is characterized by both steatosis and cholestasis, of which the latter is rare, both histologically and metabolically, in NAFLD (34, 35). We have shown that resected C57BL/6J mice develop a liver injury showing micro/macro vesicular steatosis, fibrosis, and cholestasis, resembling IFALD, which is absent in obesity-resistant 129S1/SvImJ mice as well as the C57BL/6J sham controls. Although perisinusoidal liver fibrosis is common in NAFLD, all other histological characteristics and biochemical markers resemble IFALD (which normally has a “jigsaw” pattern of fibrosis) (36). In addition, the presence of cholestasis, present in our model, is a hallmark of IFALD and is rarely observed in patients with NAFLD (37–39).
All mice except C57BL/6J SBR mice had a significant increase in body weight by postoperative week 10. Despite having the same average caloric intake across all mouse lines and operative interventions, resected C57BL/6J mice had no significant change in their body weight or phenotypic distribution of mass. However, both 129S1/SvImJ sham and resected mice showed similar trends to the sham C57BL/6J mice, showing a decrease in lean mass with a subsequent increase in fat mass after operation. Prior studies in C57BL/6J mice up to 3 wk postoperatively have shown a significant decrease in energy expenditure in resected mice compared with sham controls, which is most likely related to the significant removal of metabolically active intestinal tissue (25). This corresponds with the reduced serum triglyceride and cholesterol levels in resected C57BL/6J mice, postulating that dietary lipid malabsorption as well as other lipid metabolism pathways could alter serum lipid profiles. Interestingly, the obesity-resistant resected 129S1/SvImJ mice were able to gain weight similar to C57BL/6J control sham mice, demonstrating either an augmented adaptive capacity for nutrient absorption or a greater resistance to resection-induced inflammation. Given that structural features of intestinal adaptation were equivalent between the mouse lines, we believe that the preserved weight gain in 129S1/SvImJ mice was largely due to greater resistance to resection-induced inflammation in both the liver and peripheral tissues.
Similar to the pathogenesis of non-alcoholic steatohepatitis (NASH), we hypothesized that altered hepatic lipid metabolism, oxidative stress, and inflammation contribute to the hepatic injury after SBR (40–42). We, therefore, measured hepatic lipid levels after resection (43). 129S1/SvImJ mice were resistant to accumulation of hepatic free fatty acids, which are considered to be the most lipotoxic and a main driver of NASH (44, 45). Only the resected C57BL/6J mice develop heightened inflammatory markers with associated hepatitis and ultimately fibrosis and cholestasis. We propose that steatosis may therefore sensitize the liver to factors that produce inflammation in this model of resection-associated liver injury.
Hepatic free fatty acids are significantly elevated in the obesity-prone C57BL/6J mice compared with 129S1/SvImJ mice, regardless of operation (sham vs. resection). In the C57BL/6J mouse line, although the difference was not statistically significant, there was a trend toward greater free fatty acids in the resected mice compared with sham controls. Interestingly, the serum triglyceride and cholesterol levels were significantly higher in C57BL/6J sham controls compared with resected mice. The adipose tissue inflammation was higher in the C57BL/6J mice showing elevated transcriptions of inflammatory markers and numbers of F4/80+ macrophages. Inflamed adipose tissues released free fatty acids into circulation, which influenced accumulation of hepatic free fatty acid (46). We can speculate that increased free fatty acid-induced liver injuries following resection alter lipid metabolisms such as VLDL secretion, cholesterol handling, and/or triglyceride accumulation. Despite having a lower fat mass, the higher inflammation in adipose and liver tissues may result in the reduction in serum lipids. Therefore, despite the reduced fatty acid serum profile after resection, there is still an equal if not greater propensity toward steatosis and liver injury compared with sham controls. Given the role of lipids as structural, energy storage, and signaling molecules, there are multiple pathways by which dysregulated lipid signaling could contribute to liver injury. These include altered de novo lipogenesis with classes of free fatty acids promoting lipotoxicity, distorted membrane composition, mitochondrial dysfunction, and the generation of reactive oxygen species, as well as activation of endoplasmic reticulum stress and the unfolded protein response (47, 48).
Beyond steatosis, the resected C57BL/6J mice developed significant hepatitis, fibrosis, and cholestasis as revealed by the elevated liver enzyme levels and immunostaining. These elements were absent in the 129S1/SvImJ cohort. Because systemic inflammation has been correlated with the pathogenesis of NAFLD and NASH (49), the potential contribution of both hepatic and systemic inflammation to resection-associated liver injury were investigated in our model. The resected C57BL/6J mice showed an increase in hepatic expression levels of oxidative stress markers NOX2 and GSS as well as apoptotic marker TNFa. In addition, there was an increased predominance of Kupffer cells in the liver. This inflammation was also seen systemically in the resected C57BL/6J mice, confirmed by increased expression of the proinflammatory markers TNFα and IL-1β, decreased expression of anti-inflammatory marker MgI1, and a predominance in macrophages in visceral white adipose tissue. Despite the C57BL/6J mice having no significant change in their body weight, they had increased adipose tissue inflammation, which has been shown to be associated with obesity (50, 51). However, the obesity-resistant resected 129S1/SvImJ mice did not show liver injury or adipose tissue inflammation. Given that hepatic and systemic inflammatory effects were only seen in the C57BL/6J mouse line, we assert that steatosis is associated with a proinflammatory milieu promoting progression of liver injury and fibrosis.
To further determine why hepatic oxidative stress and macrophage recruitment did not develop in resected mice compared with shams in the obesity-resistant 129S1/SvImJ mice but did in the C57BL/6J mice, we explored two potential target pathways common to the development of inflammation-induced NAFLD: PPARα and TLR4. PPARα plays a critical role in fatty acid oxidation as well as having anti-inflammatory effect on the liver (52, 53). It has been shown that hepatic-specific PPARα knockout mice have an increase in hepatic free fatty acid accumulation and that murine treatment with a PPARα-agonist reverses steatohepatitis (54, 55). Although there was no difference in hepatic lipid composition in the C57BL/6J mice, hepatic PPARα expression was significantly reduced in the resected mice compared with sham controls. We posit that PPARα deficiency permits an enhanced inflammatory response to lipids present in the resected C57BL/6J mice by reducing the capacity for fatty acid oxidation. Hepatic PPARα has been shown to repress NFκB, IL-6 receptor, and endotoxin-induced inflammatory responses (56–58). In our model, systemic endotoxin levels were significantly higher in the C57BL/6J mouse line, with an elevated trend in the resected mice. Therefore, we suggest that a one mechanism for the repression of liver injury in resected 129S1/SvImJ mice could be due to resistance to endotoxin, through both PPARα as well as TLR4.
TLR4, the major effector of endotoxin, has been previously implicated in the development of NAFLD, as well as the liver injury associated in our mouse model (17, 59). Hepatic TLR4 expression was significantly increased in the resected C57BL/6J mice but not its sham controls or the resected 129S1/SvImJ mice. Therefore, we conclude C57BL/6J mice are more susceptible to endotoxin-driven hepatic injury in the setting of SBS via repression of the protective effects of PPARα and the stimulation of hepatic TLR4-driven inflammation. The source of increased endotoxin signaling is likely derived from alterations in the gut microbiota and/or increased intestinal permeability after intestinal resection. In human pediatric patients with IFALD, we have demonstrated a bloom of Gram-negative Proteobacteria in the feces that correlated with reduced remnant intestinal length, total parenteral nutrition (TPN) dependence, and diminished linear growth (33). Furthermore, we have recently confirmed increased gut permeability and disrupted expression of enterocyte tight junction proteins in our SBR model (60).
In conclusion, the 129J/SvImJ mouse line is resistant to the liver injury associated with SBS seen in the C57BL/6J mouse line. Although the 129J/SvImJ mice have elevated hepatic triglyceride and cholesterol levels, they have lower hepatic free fatty acid concentration. We believe that the resection-associated liver injury starts with an accumulation of hepatic free fatty acids despite intestinal malabsorption of fatty acids (as indicated by reduced serum levels) and stable fat mass percentages. The prolonged exposure to proinflammatory hepatic free fatty acids drives subsequent inflammation, fibrosis, and cholestasis in the resected mice, likely via endotoxin-mediated pathways of PPARα repression and TLR4 stimulation.
Preventative strategies for IFALD are limited and with moderate success; we believe that targeting these pathways with PPARα agonists or TLR4 inhibitors may offer clinically translatable treatment strategies. Increasing enteral caloric intake has been shown as an effective treatment for IFALD, with current recommendations being an enteral diet with a mixture of long and medium chain fatty acids (61–64). The impact of how these dietary fats could affect the development of IFALD is presently unknown. Our PN-independent resection model will potentially illuminate modifiable dietary interventions to prevent liver injury.
Conclusions
Unlike C57BL/6 mice, the 129S1/SvImJ strain is resistant to liver inflammation and injury after SBR. These disparate outcomes are likely due to the accumulation of hepatic free fatty acids as well as increased endotoxin-driven inflammatory pathways through PPARα and TLR4 in C57BL/6 mice with SBS.
GRANTS
This work was supported by The Digestive Diseases Research Core Center of the Washington University School of Medicine (NIH No. P30DK52574), the Children’s Surgical Sciences Research Institute of the St. Louis Children’s Hospital Foundation, NIH DP1DK109668 (to G. J. Randolph), the Immunology Training Grant NIH T32 AI007163 (to Y-H. Han, R. Sanguinetti Czepielewski), NIAID Primary Caregiver Award R37 AI049653 20S1 (to G. J. Randolph, E. J. Onufer), and the Department of Pediatrics Training Grant NIH T32 DK077653 (E. J. Onufer).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.J.O., J.O., R.S., and B.W.W. conceived and designed research; E.J.O., Y-H.H., C.C., S.S., and A. Sescleifer performed experiments; E.J.O. and Y-H.H. analyzed data; E.J.O., Y-H.H., R.S., and G.J.R. interpreted results of experiments; E.J.O., Y-H.H., and A. Steinberger prepared figures; E.J.O. drafted manuscript; E.J.O., Y-H.H., A.S., M.T., G.J.R., and B.W.W., edited and revised manuscript; E.J.O., Y-H.H., and B.W.W. approved final version of manuscript.
REFERENCES
- 1.Squires RH, Duggan C, Teitelbaum DH, Wales PW, Balint J, Venick R, Rhee S, Sudan D, Mercer D, Martinez JA, Carter BA, Soden J, Horslen S, Rudolph JA, Kocoshis S, Superina R, Lawlor S, Haller T, Kurs-Lasky M, Belle SH. Natural history of pediatric intestinal failure: initial report from the Pediatric Intestinal Failure Consortium. J Pediatr 161: 723–728.e2, 2012. doi: 10.1016/j.jpeds.2012.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grant D, Abu-Elmagd K, Mazariegos G, Vianna R, Langnas A, Mangus R, Farmer D, Lacaille F, Iyer K, Fishbein T; Intestinal Transplant Association. Intestinal transplant registry report: global activity and trends. Am J Transplant 15: 210–219, 2015. doi: 10.1111/ajt.12979. [DOI] [PubMed] [Google Scholar]
- 3.Kahn AB, Tulla KA, Tzvetanov IG. Indications of intestinal transplantation. Gastroenterol Clin North Am 48: 575–583, 2019. doi: 10.1016/j.gtc.2019.08.010. [DOI] [PubMed] [Google Scholar]
- 4.Kaufman SS, Atkinson JB, Bianchi A, Goulet OJ, Grant D, Langnas AN, McDiarmid SV, Mittal N, Reyes J, Tzakis AG; American Society of Transplantation. Indications for pediatric intestinal transplantation: a position paper of the American Society of Transplantation. Pediatr Transplant 5: 80–87, 2001. doi: 10.1034/j.1399-3046.2001.005002080.x. [DOI] [PubMed] [Google Scholar]
- 5.Bishay M, Pichler J, Horn V, Macdonald S, Ellmer M, Eaton S, Hill S, Pierro A. Intestinal failure-associated liver disease in surgical infants requiring long-term parenteral nutrition. J Pediatr Surg 47: 359–362, 2012. doi: 10.1016/j.jpedsurg.2011.11.032. [DOI] [PubMed] [Google Scholar]
- 6.Kelly DA. Liver complications of pediatric parenteral nutrition—epidemiology. Nutrition 14: 153–157, 1998. doi: 10.1016/s0899-9007(97)00232-3. [DOI] [PubMed] [Google Scholar]
- 7.Soden JS. Clinical assessment of the child with intestinal failure. Semin Pediatr Surg 19: 10–19, 2010. doi: 10.1053/j.sempedsurg.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Beath SV, Davies P, Papadopoulou A, Khan AR, Buick RG, Corkery JJ, Gornall P, Booth IW. Parenteral nutrition-related cholestasis in postsurgical neonates: multivariate analysis of risk factors. J Pediatr Surg 31: 604–606, 1996. doi: 10.1016/s0022-3468(96)90507-2. [DOI] [PubMed] [Google Scholar]
- 9.Courtney CM, Warner BW. Pediatric intestinal failure-associated liver disease. Curr Opin Pediatr 29: 363–370, 2017. doi: 10.1097/MOP.0000000000000484. [DOI] [PubMed] [Google Scholar]
- 10.Korpela K, Mutanen A, Salonen A, Savilahti E, de Vos WM, Pakarinen MP. Intestinal microbiota signatures associated with histological liver steatosis in pediatric-onset intestinal failure. JPEN J Parenter Enteral Nutr 41: 238–248, 2017. doi: 10.1177/0148607115584388. [DOI] [PubMed] [Google Scholar]
- 11.Rangel SJ, Calkins CM, Cowles RA, Barnhart DC, Huang EY, Abdullah F, Arca MJ, Teitelbaum DH; 2011 American Pediatric Surgical Association Outcomes and Clinical Trials Committee. Parenteral nutrition-associated cholestasis: an American Pediatric Surgical Association Outcomes and Clinical Trials Committee systematic review. J Pediatr Surg 47: 225–240, 2012. doi: 10.1016/j.jpedsurg.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Wales PW, Allen N, Worthington P, George D, Compher C, American Society for Parenteral and Enteral Nutrition, Teitelbaum D. A.S.P.E.N clinical guidelines: support of pediatric patients with intestinal failure at risk of parenteral nutrition-associated liver disease. JPEN J Parenter Enteral Nutr 38: 538–557, 2014. [DOI] [PubMed] [Google Scholar]
- 13.El Kasmi KC, Anderson AL, Devereaux MW, Fillon SA, Harris JK, Lovell MA, Finegold MJ, Sokol RJ. Toll-like receptor 4-dependent Kupffer cell activation and liver injury in a novel mouse model of parenteral nutrition and intestinal injury. Hepatology 55: 1518–1528, 2012. doi: 10.1002/hep.25500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Kasmi KC, Anderson AL, Devereaux MW, Vue PM, Zhang W, Setchell KD, Karpen SJ, Sokol RJ. Phytosterols promote liver injury and Kupffer cell activation in parenteral nutrition-associated liver disease. Sci Transl Med 5: 206ra137, 2013. doi: 10.1126/scitranslmed.3006898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barron L, Courtney C, Bao J, Onufer E, Panni RZ, Aladegbami B, Warner BW. Intestinal resection-associated metabolic syndrome. J Pediatr Surg 53: 1142–1147, 2018. doi: 10.1016/j.jpedsurg.2018.02.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onufer EJ, Han Y-H, Czepielewski RS, Courtney CM, Sutton S, Randolph GJ, Warner BW. Effects of high fat diet on liver injury after small bowel resection. J Pediatr Surg 55: 1099–1106, 2020. doi: 10.1016/j.jpedsurg.2020.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barron LK, Bao JW, Aladegbami BG, Colasanti JJ, Guo J, Erwin CR, Warner BW. Toll-like receptor 4 is critical for the development of resection-associated hepatic steatosis. J Pediatr Surg 52: 1014–1019, 2017. doi: 10.1016/j.jpedsurg.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Almind K, Kahn CR. Genetic determinants of energy expenditure and insulin resistance in diet-induced obesity in mice. Diabetes 53: 3274–3285, 2004. doi: 10.2337/diabetes.53.12.3274. [DOI] [PubMed] [Google Scholar]
- 19.Almind K, Manieri M, Sivitz WI, Cinti S, Kahn CR. Ectopic brown adipose tissue in muscle provides a mechanism for differences in risk of metabolic syndrome in mice. Proc Natl Acad Sci USA 104: 2366–2371, 2007. doi: 10.1073/pnas.0610416104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang IV, Alper S, Lackford B, Rutledge H, Warg LA, Burch LH, Schwartz DA. Novel regulators of the systemic response to lipopolysaccharide. Am J Respir Cell Mol Biol 45: 393–402, 2011. doi: 10.1165/rcmb.2010-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang IV, Wade CM, Kang HM, Alper S, Rutledge H, Lackford B, Eskin E, Daly MJ, Schwartz DA. Identification of novel genes that mediate innate immunity using inbred mice. Genetics 183: 1535–1544, 2009. doi: 10.1534/genetics.109.107540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helmrath MA, VanderKolk WE, Can G, Erwin CR, Warner BW. Intestinal adaptation following massive small bowel resection in the mouse. J. Am Coll Surg 183: 441–449, 1996. [PubMed] [Google Scholar]
- 23.Wakeman D, Guo J, Santos JA, Wandu WS, Schneider JE, McMellen ME, Leinicke JA, Erwin CR, Warner BW. p38 MAPK regulates Bax activity and apoptosis in enterocytes at baseline and after intestinal resection. Am J Physiol Gastrointest Liver Physiol 302: G997–G1005, 2012. doi: 10.1152/ajpgi.00485.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor JA, Martin CA, Nair R, Guo J, Erwin CR, Warner BW. Lessons learned: optimization of a murine small bowel resection model. J Pediatr Surg 43: 1018–1024, 2008. doi: 10.1016/j.jpedsurg.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tantemsapya N, Meinzner-Derr J, Erwin CR, Warner BW. Body composition and metabolic changes associated with massive intestinal resection in mice. J Pediatr Surg 43: 14–19, 2008. doi: 10.1016/j.jpedsurg.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 26.Newberry EP, Kennedy SM, Xie Y, Sternard BT, Luo J, Davidson NO. Diet-induced obesity and hepatic steatosis in L-Fabp−/− mice is abrogated with SF, but not PUFA, feeding and attenuated after cholesterol supplementation. Am J Physiol Gastrointest Liver Physiol 294: G307–G314, 2008. doi: 10.1152/ajpgi.00377.2007. [DOI] [PubMed] [Google Scholar]
- 27.Bateman AC, Hübscher SG. Cytokeratin expression as an aid to diagnosis in medical liver biopsies. Histopathology 56: 415–425, 2010. doi: 10.1111/j.1365-2559.2009.03391.x. [DOI] [PubMed] [Google Scholar]
- 28.Han Y-H, Shin K-O, Kim J-Y, Khadka DB, Kim H-J, Lee Y-M, Cho W-J, Cha J-Y, Lee B-J, Lee M-O. A maresin 1/RORα/12-lipoxygenase autoregulatory circuit prevents inflammation and progression of nonalcoholic steatohepatitis. J Clin Invest 129: 1684–1698, 2019. doi: 10.1172/JCI124219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez-Gurmaches J, Hung C-M, Guertin DA. Emerging complexities in adipocyte origins and identity. Trends Cell Biol 26: 313–326, 2016. doi: 10.1016/j.tcb.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n − 6/n − 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 106: 635–643, 2004. doi: 10.1042/CS20030326. [DOI] [PubMed] [Google Scholar]
- 31.Cui W, Chen SL, Hu K-Q. Quantification and mechanisms of oleic acid-induced steatosis in HepG2 cells. Am J Transl Res 2: 95–104, 2010. [PMC free article] [PubMed] [Google Scholar]
- 32.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 365: 1415–1428, 2005. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 33.Engelstad HJ, Barron L, Moen J, Wylie TN, Wylie K, Rubin DC, Davidson N, Cade WT, Warner BB, Warner BW. Remnant small bowel length in pediatric short bowel syndrome and the correlation with intestinal dysbiosis and linear growth. J Am Coll Surg 227: 439–449, 2018. doi: 10.1016/j.jamcollsurg.2018.07.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naini BV, Lassman CR. Total parenteral nutrition therapy and liver injury: a histopathologic study with clinical correlation. Hum Pathol 43: 826–833, 2012. doi: 10.1016/j.humpath.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 35.Stanko RT, Nathan G, Mendelow H, Adibi SA. Development of hepatic cholestasis and fibrosis in patients with massive loss of intestine supported by prolonged parenteral nutrition. Gastroenterology 92: 197–202, 1987. doi: 10.1016/0016-5085(87)90859-6. [DOI] [PubMed] [Google Scholar]
- 36.Buchman AL, Naini BV, Spilker B. The differentiation of intestinal-failure-associated liver disease from nonalcoholic fatty liver and nonalcoholic steatohepatitis. Semin Liver Dis 37: 33–44, 2017. doi: 10.1055/s-0036-1597771. [DOI] [PubMed] [Google Scholar]
- 37.Guglielmi FW, Regano N, Mazzuoli S, Fregnan S, Leogrande G, Guglielmi A, Merli M, Pironi L, Penco JM, Francavilla A. Cholestasis induced by total parenteral nutrition. Clin Liver Dis 12: 97–110, viii, 2008. [DOI] [PubMed] [Google Scholar]
- 38.Mullick FG, Moran CA, Ishak KG. Total parenteral nutrition: a histopathologic analysis of the liver changes in 20 children. Mod Pathol 7: 190–194, 1994. [PubMed] [Google Scholar]
- 39.Zambrano E, El-Hennawy M, Ehrenkranz RA, Zelterman D, Reyes-Múgica M. Total parenteral nutrition induced liver pathology: an autopsy series of 24 newborn cases. Pediatr Dev Pathol 7: 425–432, 2004. doi: 10.1007/s10024-001-0154-7. [DOI] [PubMed] [Google Scholar]
- 40.Farrell GC, Van Rooyen D, Gan L, Chitturi S. NASH is an inflammatory disorder: pathogenic, prognostic and therapeutic implications. Gut Liver 6: 149–171, 2012. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med 52: 59–69, 2012. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 42.Tessari P, Coracina A, Cosma A, Tiengo A. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis 19: 291–302, 2009. https://pubmed.ncbi.nlm.nih.gov/19359149/. [DOI] [PubMed] [Google Scholar]
- 43.Al-Shahwani NH, Sigalet DL. Pathophysiology, prevention, treatment, and outcomes of intestinal failure-associated liver disease. Pediatr Surg Int 33: 405–411, 2017. doi: 10.1007/s00383-016-4042-7. [DOI] [PubMed] [Google Scholar]
- 44.Larter CZ, Yeh MM, Haigh WG, Williams J, Brown S, Bell-Anderson KS, Lee SP, Farrell GC. Hepatic free fatty acids accumulate in experimental steatohepatitis: role of adaptive pathways. J Hepatol 48: 638–647, 2008. doi: 10.1016/j.jhep.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 45.Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, Patané G, Boggi U, Piro S, Anello M, Bergamini E, Mosca F, Di Mario U, Del Prato S, Marchetti P. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that β-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 51: 1437–1442, 2002. doi: 10.2337/diabetes.51.5.1437. [DOI] [PubMed] [Google Scholar]
- 46.Jacobi D, Stanya K, Lee C-H. Adipose tissue signaling by nuclear receptors in metabolic complications of obesity. Adipocyte 1: 4–12, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65: 1038–1048, 2016. doi: 10.1016/j.metabol.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 48.Hayes CN, Zhang P, Chayama K. The role of lipids in hepatocellular carcinoma. In: Hepatocellular Carcinoma, edited by Tirnitz-Parker JEE. Brisbane: Brisbane (AU): Codon Publications, 2019, p. 95–110. [PubMed] [Google Scholar]
- 49.Stojsavljević S, Palčić MG, Jukić LV, Duvnjak LS, Duvnjak M. Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol 20: 18070, 2014. doi: 10.3748/wjg.v20.i48.18070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, Shen Y, Czech MP, Aouadi M. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab 19: 162–171, 2014. doi: 10.1016/j.cmet.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reilly SM, Saltiel AR. Adapting to obesity with adipose tissue inflammation. Nat Rev Endocrinol 13: 633–643, 2017. doi: 10.1038/nrendo.2017.90. [DOI] [PubMed] [Google Scholar]
- 52.Duval C, Fruchart JC, Staels B. PPAR alpha, fibrates, lipid metabolism and inflammation. Arch Mal Coeur Vaiss 97: 665–672, 2004. [PubMed] [Google Scholar]
- 53.Vanden Berghe W, Vermeulen L, Delerive P, De Bosscher K, Staels B, Haegeman G. A paradigm for gene regulation: inflammation, NF-kappaB and PPAR. Adv Exp Med Biol 544: 181–196, 2003. doi: 10.1007/978-1-4419-9072-3_22. [DOI] [PubMed] [Google Scholar]
- 54.Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology 39: 1286–1296, 2004. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 55.Montagner A, Polizzi A, Fouché E, Ducheix S, Lippi Y, Lasserre F, Barquissau V, Régnier M, Lukowicz C, Benhamed F, Iroz A, Bertrand-Michel J, Al Saati T, Cano P, Mselli-Lakhal L, Mithieux G, Rajas F, Lagarrigue S, Pineau T, Loiseau N, Postic C, Langin D, Wahli W, Guillou H. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 65: 1202–1214, 2016. doi: 10.1136/gutjnl-2015-310798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bougarne N, Paumelle R, Caron S, Hennuyer N, Mansouri R, Gervois P, Staels B, Haegeman G, De Bosscher K. PPARα blocks glucocorticoid receptor α-mediated transactivation but cooperates with the activated glucocorticoid receptor α for transrepression on NF-κB. Proc Natl Acad Sci USA 106: 7397–7402, 2009. doi: 10.1073/pnas.0806742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gervois P, Vu-Dac N, Kleemann R, Kockx M, Dubois G, Laine B, Kosykh V, Fruchart J-C, Kooistra T, Staels B. Negative regulation of human fibrinogen gene expression by peroxisome proliferator-activated receptor α agonists via inhibition of CCAAT box/enhancer-binding protein β. J Biol Chem 276: 33471–33477, 2001. doi: 10.1074/jbc.M102839200. [DOI] [PubMed] [Google Scholar]
- 58.Pawlak M, Baugé E, Bourguet W, De Bosscher K, Lalloyer F, Tailleux A, Lebherz C, Lefebvre P, Staels B. The transrepressive activity of peroxisome proliferator-activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology 60: 1593–1606, 2014. doi: 10.1002/hep.27297. [DOI] [PubMed] [Google Scholar]
- 59.Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, Franchimont D, Louis H, Devière J, Le Moine O. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology 43: 989–1000, 2006. doi: 10.1002/hep.21138. [DOI] [PubMed] [Google Scholar]
- 60.Courtney CM, Onufer EJ, McDonald KG, Steinberger AE, Sescleifer AM, Seiler KM, Tecos ME, Newberry RD, Warner BW. Small bowel resection increases paracellular gut barrier permeability via alterations of tight junction complexes mediated by intestinal TLR4. J Surg Res 258: 73–81, 2021. doi: 10.1016/j.jss.2020.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi PM, Sun RC, Guo J, Erwin CR, Warner BW. High-fat diet enhances villus growth during the adaptation response to massive proximal small bowel resection. J Gastrointest Surg 18: 286–294, 2014. doi: 10.1007/s11605-013-2338-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chwals WJ. The metabolic response to surgery in neonates. Curr Opin Pediatr 6: 334–340, 1994. doi: 10.1097/00008480-199406000-00017. [DOI] [PubMed] [Google Scholar]
- 63.Kollman KA, Lien EL, Vanderhoof JA. Dietary lipids influence intestinal adaptation after massive bowel resection. J Pediatr Gastroenterol Nutr 28: 41–45, 1999. doi: 10.1097/00005176-199901000-00011. [DOI] [PubMed] [Google Scholar]
- 64.Park JH, Grandjean CJ, Hart MH, Baylor JM, Vanderhoof JA. Effects of dietary linoleic acid on mucosal adaptation after small bowel resection. Digestion 44: 57–65, 1989. doi: 10.1159/000199893. [DOI] [PubMed] [Google Scholar]