

Abstract
Background:
Complex regional pain syndrome type-1 (CRPS-1) is a severely disabling painful disease challenging to treat. This multicenter, randomized, double-blind placebo-controlled trial examined the efficacy of intramuscular (i.m.) neridronate in CRPS-1 patients.
Methods:
A total of 78 patients diagnosed with CRPS-1 (aged 59.5 ± 10.3, 66.7% female) were randomly assigned to 25 mg (i.m.) neridronate (N = 41) given once daily for 16 consecutive days or placebo control (N = 37). Efficacy was assessed after 30 days using a visual analogue scale (VAS) pain score and the number of patients achieving ⩾50% reduction in VAS score. Change in clinical signs and symptoms, quality of life (QoL) using Short Form Health Survey (SF-36) and the McGill Pain Questionnaire were also assessed.
Results:
After 30 days, VAS score decreased significantly to a greater extent in neridronate-treated patients versus placebo (31.9 ± 23.3 mm versus 52.3 ± 27.8 mm, p = 0.0003). Furthermore, the proportion of patients achieving a VAS reduction of ⩾50% was greater in the neridronate group (65.9% versus 29.7%, p = 0.0017). Clinical signs and symptoms were improved significantly in the neridronate group versus placebo for edema (72.5% versus 79.9%, p = 0.03), pain during motion (70% versus 83.3%, p = 0.0009), allodynia (20% versus 63.3%, p = 0.0004), and hyperalgesia (20% versus 56.7%, p = 0.0023). Whereas no difference was observed for QoL measures using the SF-36 questionnaire, three of the four pain variables using the McGill Pain Questionnaire improved significantly in the neridronate group. No serious drug-related adverse events were reported during the study.
Conclusion:
In patients with acute CRPS-1, i.m. injections of 25 mg neridronate were associated with clinically relevant benefit compared with placebo controls.
Trial registration:
EU Clinical Trials Register: https://www.clinicaltrialsregister.eu/ctr-search/search?query=2014-001156-28
Keywords: algodystrophy, bisphosphonates, complex regional pain syndrome type 1, neridronate, randomized clinical trial
Introduction
Complex regional pain syndrome type 1 (CRPS-1) is a disabling painful disease, the main characteristic of which is pain disproportionate to inciting events, together with other hallmarks of this disease including swelling, vasomotor instability, and abnormal sensory findings.1–3 As a result of pathogenic mechanisms that are not fully understood, a multitude of therapeutic interventions have been proposed, but a widely shared therapeutic algorithm has yet to be established.
Over the past three decades, several case reports, open studies and five randomized controlled trials (RCTs) have shown efficacy of bisphosphonate (BP) therapy in the treatment of CRPS-1.4–8 All these studies described positive results in controlling pain, local inflammation, functional disability, and improving the quality of life (QoL) of patients. The results of some meta-analyses confirm the clear benefit offered by BPs, mainly in patients with early disease.9,10 Among RCTs, the study on neridronate administered intravenously (i.v.) showed more convincing results for its design, such as the number of patients, the endpoints evaluated, and length of follow up. 8 From February 2015, i.v. neridronate was approved and licensed in Italy for the treatment of acute CRPS-1. Nevertheless, i.v. neridronate treatment can be given only in the hospital setting, in patients suffering from a severely disabling disease and therefore often unwilling or unable. To overcome these difficulties, an at-home treatment would be more convenient and less expensive. In Italy, neridronate is also licensed and used safely in 25 mg ampoules given intramuscularly (i.m.) for the treatment of Paget disease of bone and osteogenesis imperfecta.11,12
To offer a more suitable treatment to CRPS-1 patients sometimes not able to undergo i.v. treatment, the present study aimed to test whether the i.m. route would maintain the same efficacy of neridronate administered by i.v. infusion. 8
Methods
Patients
Patients were recruited from the outpatient services of 10 Italian Rheumatology centers. All patients included in the study fulfilled the International Association for the Study of Pain (IASP) diagnostic criteria for CRPS, 13 previously referred to as the “Budapest criteria”. 14 No patients showed any major nerve damage, suggestive of CRPS-2. To ensure a correct comparison, we maintained the same inclusion criteria employed in the i.v. neridronate study. 8 Patients were aged at least 18 years, disease duration was no longer than 4 months, spontaneous pain intensity in the affected limb was of at least 50 mm on a visual analogue scale (VAS) ranging from 0 (no pain) to 100 mm (maximum pain). 15 In all patients, bone scintigraphy was performed and only patients with an increased uptake in the late phase at the disease site were recruited. 16 Opioid analgesics, non-opioid analgesics, non-steroidal anti-inflammatory drugs (NSAIDs), anticonvulsants, antidepressant drugs, and other non-drug therapies prescribed for CRPS-1 were asked to be suspended when patients were enrolled (5–7 days before the first neridronate administration) and a rescue medication (acetaminophen 500 mg tablets) was provided and allowed throughout the study. Women of childbearing potential had to have a negative pregnancy test before entering the study. Prior treatment with BP and the presence of renal disease were exclusion criteria. The study was conducted under the provisions of the Declaration of Helsinki, and in accordance with the International Conference on Harmonization Consolidated Guideline on Good Clinical Practice. The study was approved by Milano B Ethics committee (28 October 2014). All patients provided written informed consent to participate in the study.
Study design
This was a multicenter, randomized, double-blind, placebo-controlled study to assess the efficacy and safety of 25 mg neridronate ampoules (2 ml), after repeated i.m. administrations in patients diagnosed with CRPS-1 [EudraCT Number: 2014-001156-28]. Patients assigned placebo received i.m. indistinguishable sham injections of saline (0.9% sodium chloride solution). The site recommended for i.m. injection was the ventrogluteal muscle of the hip by alternating right and left sides, preferably at the same hour each day. All patients received instructions from a nurse on how to correctly perform the injection. The study was performed in 10 centers, located across Italy from April 2015 to April 2019. Eligible patients were randomized (1:1 ratio, on the basis of a predetermined computer-generated randomization list) to receive either neridronate 25 mg i.m. or matched placebo i.m. daily day for 16 consecutive days. Neither patients nor investigators knew whether assignment would be to the placebo or neridronate group. At the end of the double-blind phase of the study (30 days from the start of treatment), the blind code was broken and patients treated with placebo during the double-blind phase of the study could be treated with neridronate at a dose of four 100 mg i.v. infusions (one every third day). The results of the double-blind phase of this trial are reported in the present manuscript.
Outcome measures
Outcome measures were assessed on the day of the first injection (day 1). Further assessments were obtained on day 8, at the end of treatment (day 16) and after 30 days.
The primary efficacy measure was the change in VAS pain score 30 days after the first administration of neridronate in the double-blind phase of the study. A decrease from the baseline value of at least 50% was considered clinically significant and qualified the patient as a responder. 16 Clinical assessment included: allodynia tested as pain to light stroking with a small brush (the end of a Q-Tip) and hyperalgesia defined as a stimulus evoked by a pinprick being perceived as more painful or lasting longer than the duration of the stimulus in the affected limb compared with the contralateral limb, both rated as a dichotomous variable (present/absent). 17 Local edema and pain at passive motion were also recorded: local edema scored as 0 = none, 1 = mild, 2 = moderate, 3 = severe was evaluated at ankle and midfoot level for the foot involvement, wrist, center of hand dorsum and finger for hand involvement; pain evoked by passive motion (ankle and finger joints for foot involvement and wrist and finger joints for hand involvement) was rated as 0 = none, 1 = mild, 2 = moderate, 3 = severe; both parameters were scored by the direct comparison with the contralateral unaffected limb. In each participating center the clinical evaluation was performed independently by two investigators unaware of the treatment and any side effects; in case of discordance, assessment was repeated by a third investigator and the score shared by at least two investigators was assigned.
To assess functional status, McGill Pain Questionnaire and 36-Item Short Form Health Survey (SF-36) questionnaire were evaluated.18,19 Both questionnaires were self-administered. All these outcome parameters were considered as secondary endpoints.
Safety
Physicians at the study sites reported adverse events (AEs) that were coded as preferred terms in the Medical Dictionary for Regulatory Activities (MedDRA) system. According to investigators’ judgment, a drug-related AE was defined as definitely, probably, or possibly related to study treatment. The percentage of AEs are expressed as the number of patients reporting an AE divided by the total number of patients in that treatment arm (expressed as a percentage).
All patients were informed about a possible acute-phase reaction (polyarthralgia and/or fever) 20 occurring after parenteral aminobisphosphonate administration and assumption of 500 mg acetaminophen tablets is recommended if these symptoms appeared.
Statistical analysis
The sample size calculation was based on the primary efficacy endpoint of the study, that is, the proportion of patients achieving a reduction in pain intensity ⩾50% from baseline to day 30. Sample size calculation was performed assuming a two-tailed probability of type I error equal to 0.05. A total sample size of 74 subjects, 37 in each treatment group, would have achieved an 80% power to detect a difference of 35% between the null hypothesis that both group proportions of responders were 30%, and the alternative hypothesis that the proportion of responders in the neridronate i.m. group was 65%. Assuming a drop-out rate or otherwise non-evaluable patients of about 20%, 90 patients were to be randomized to obtain the 74 evaluable patients. Statistical analysis was carried out according to the intention-to-treat principle, including all randomized patients who received at least one dose of the study medication. Missing data were replaced by LOCF (last observation carried forward). Data were evaluated for normal distribution using the Kolmogorov–Smirnov test. Baseline characteristics were compared with the use of Student’s t test for quantitative variables whereas the Fisher’s exact test was used to compare differences between binary variables. Changes in VAS scores were evaluated using an analysis of covariance (ANCOVA) model for repeated measures using the change from baseline as the dependent variable; treatment, visit, center, and the treatment-by-visit interaction as factors and baseline as covariates. In a separate ANCOVA model, differences in the cumulative dose of rescue medication were assessed: treatment group, gender, and center were considered as factors, while patient’s age and the VAS pain value at baseline were used as covariates. Differences between treatments were reported as least-square mean estimates together with associated two-sided 95% confidence intervals (CIs). The proportion of responders (VAS reduction ⩾50%) as well as dichotomous variables (allodynia and hyperalgesia) were compared with Fisher’s exact test while results were reported as risk difference together with associated two-sided 95% CIs. The results of the McGill Pain Questionnaire and SF-36 questionnaire were analysed using ANCOVA models using the change from baseline to day 30 as dependent variable, treatment and center as factors, and the baseline value as covariate. The comparison of clinical parameters evaluated by means of rating scales (edema and pain at passive motion) were performed using the Wilcoxon rank sum test. All statistical analyses were performed on changes in raw values, not transformed data (e.g., percentage change).
Statistical analysis was performed using SAS® Software (release 9.4) for Microsoft Windows. All tests were two-tailed and a p value of <0.05 was considered statistically significant.
Results
Study population
A total of 112 patients were screened and 78 were randomized to the assigned treatment group: 41 to neridronate and 37 to placebo. Patient disposition is summarised in Figure 1. A total of 34 patients were screening failures and were not randomized, mainly because the diagnostic criteria for research purposes could not be applied. 12 Overall, eight patients (one in the neridronate group and seven in the placebo group) discontinued the study after randomization during the double-blind phase of the study.
Figure 1.

Flowchart according to CONSORT statement for the report of randomized trials.
i.m., intramuscularly.
The two treatment groups were well matched for demographic clinical characteristics (Table 1). The majority of patients were female (66.7%) with mean age of 59.5 ± 10.3 years. The most frequent precipitating event was a trauma without fracture (contusion, sprain, or ligament tear). Fracture (all types/locations) was the second most frequent predisposing event. No difference was found between groups, either for surgery (elective) and for other/unknown predisposing events. Mean pain VAS score at baseline was 73.4 ± 12.5 mm in the neridronate group and 74.6 ± 11.2 mm in the placebo group.
Table 1.
Baseline demographic and clinical characteristics of CRPS-1 patients treated with neridronate or placebo.
| Characteristic | Neridronate (n = 41) | Placebo (n = 37) | p value |
|---|---|---|---|
| General | |||
| Female gender | 25 (61) | 27 (73) | 0.77 |
| Age, (years) | 59.3 ± 10.2 | 59.7 ± 10.5 | 0.87 |
| Caucasian | 40 (97.6) | 37 (100) | 1 |
| BMI, (kg/m²) | 26.7 ± 5.1 | 26.6 ± 4.4 | 0.93 |
| Disease characteristics | |||
| VAS pain score | 73.4 ± 12.5 | 74.6 ± 11.2 | 0.66 |
| Time since CRPS-1 diagnosis (weeks) | 4.8 ± 4.9 | 4.3 ± 5.5 | 0.67 |
| Precipitating event | |||
| Trauma | 22 (53.7) | 15 (40.5) | 0.24 |
| Fracture | 10 (24.4) | 10 (27.0) | 0.79 |
| Surgery | 4 (9.8) | 8 (21.6) | 0.14 |
| Other/unknown | 5 (12.1) | 4 (10.8) | 0.84 |
| Site | |||
| Hand | 12 (29.3) | 17 (45.9) | 0.16 |
| Foot | 29 (70.7) | 20 (54.1) | 0.16 |
| Comorbid diseases | |||
| At least 1 concomitant disease | 33 (80.5) | 26 (70.3) | 0.43 |
| Concomitant medication | |||
| At least 1 concomitant medication | 31 (75.6) | 25 (67.6) | 0.59 |
Data are presented as mean ± SD for continuous variables and number and % for dichotomous variables.
BMI, body mass index; CRPS-I, complex regional pain syndrome type-1; SD, standard deviation; VAS, visual analogue scale.
Pain VAS
Mean pain VAS score decreased progressively from baseline up to day 30 in both treatment groups (Figure 2a). The extent of the mean decrease from baseline was higher in the neridronate group than in the placebo group at any post-baseline time point. The mean [± standard deviation (SD)] change from baseline to day 30 in pain VAS score was −41.6 ± 22.7 mm in the neridronate group and −22.3 ± 27.1 mm in the placebo group. The adjusted mean changes from baseline at day 30 estimated by the ANCOVA model for repeated measures were −43.1 mm (95% CI: −50.3 mm to −35.8 mm) for neridronate and −23.9 mm (95% CI: −31.7 mm to −16.2 mm) for placebo groups, respectively. The adjusted mean difference between the neridronate and the placebo group was −19.12 (95% CI: −29.3 to −8.97; p = 0.0003) (Figure 2a).
Figure 2.
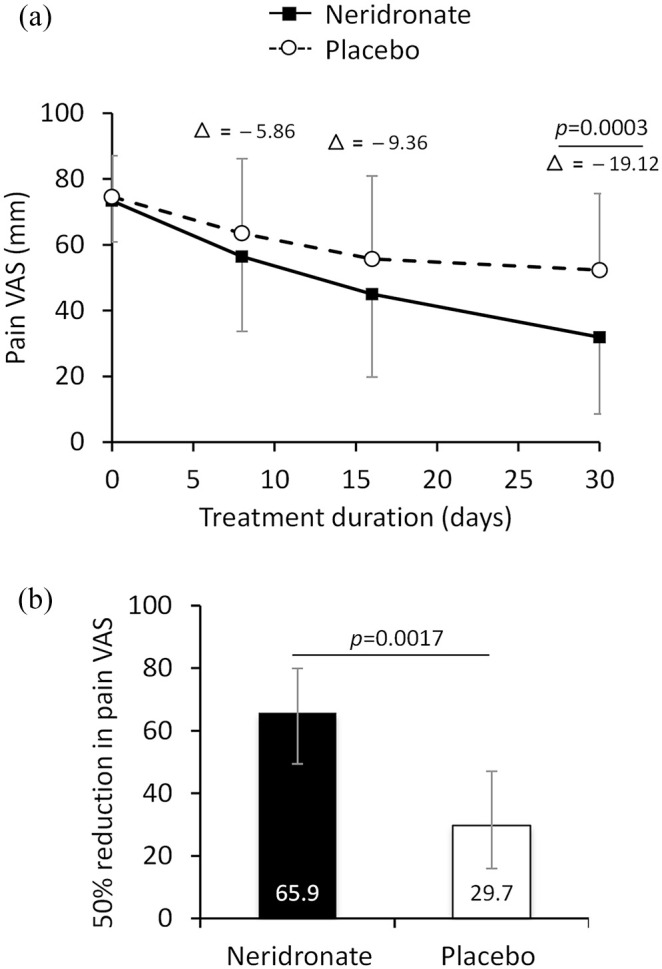
Change in VAS pain score in CRPS-I treated with neridronate or placebo. (a) Change in mean VAS pain score (mm) from baseline to day 30 in CRPS-1 patients treated with neridronate or placebo. Absolute mean differences (delta) between VAS scores for the two groups are shown for 8, 16, and 30 days. Data are presented as mean ± SD. (b) Proportion of patients achieving a ⩾50% reduction in pain VAS score from baseline to day 30 in the neridronate (N = 27/41) and placebo (11/37) group. Data are presented as % and 95% CIs.
p values denote level of statistical significance between groups.
CI, confidence interval; CRPS-I, complex regional pain syndrome type-1; SD, standard deviation; VAS, visual analogue scale.
A ⩾50% reduction in pain VAS at day 30 was seen in 27 patients (65.9%, 95% CI: 49.4%–79.9%) in the neridronate group versus 11 patients in the placebo group (29.7%, 95% CI: 15.9%–47%), with a treatment difference of 36.1% (95% CI: 14.01%–55.7%; p = 0.0017) (Figure 2b).
Clinical signs and symptoms
The number of patients experiencing edema and pain evoked by passive motion significantly decreased in the neridronate group compared with the placebo group. At day 30, the edema score fell from a baseline value of 1.93–0.9 in the neridronate group in comparison with a decrease from 1.86–1.3 in the placebo group (p = 0.03). Similarly, at day 30, pain on passive motion was absent in 12 patients (30.0%) in the neridronate group, and 5 patients (16.7%) in the placebo group (Figure 3b). At day 30, pain at passive motion score decreased from a baseline value of 2.17–0.85 in the neridronate group in comparison with a decrease from 2.16 to 1.5 in the placebo group (p = 0.009).
Figure 3.

Change in clinical signs and symptoms in CRPS-I treated with neridronate or placebo. Data are presented as the proportion of patients presenting with signs/symptoms such as edema, pain on motion, allodynia, or hyperalgesia. Edema and pain on passive motion are based on a 0–3 rating scale (none, mild, moderate, severe), while allodynia and hyperalgesia are dichotomous data (present/absent).
p values denote level of statistical significance between groups.
CRPS-I, complex regional pain syndrome type-1.
A marked improvement was seen for allodynia and hyperalgesia in the neridronate group compared with placebo. At baseline, allodynia was detected in 33 (80.5%) patients in the neridronate group and in 33 (89.2%) patients in the placebo group. At day 30, allodynia was present in 8 (20%) neridronate-treated patients and in 19 (63.3%) placebo patients, a 43.3% difference between groups (95% CI: 20.02%–63.1%, p = 0.0004) (Figure 3c). Hyperalgesia was present at baseline in all 41 patients in the neridronate group and in 35 (94.6%) in the placebo group. At day 30, hyperalgesia was detected in 8 (20%) patients in the neridronate group and in 17 (56.7%) patients in the placebo group, with a 36.7% difference between groups (95% CI: 12.98%–57.31%, p = 0.0023 (Figure 3d).
Short-form 36
Although the majority (7/10) of SF-36 scores for domains and components improved significantly to a greater extent from baseline to day 30 in the neridronate group compared with placebo, the differences between groups were not statistically significant. The domain pain and physical component scale showed the greatest improvement in the neridronate group compared with placebo (Table 2).
Table 2.
SF-36: treatment difference estimates from baseline to day 30 in CRPS-1 patients treated with neridronate or placebo.
| Domains | Difference between neridronate and placebo |
||
|---|---|---|---|
| Estimate | 95% CI | p value | |
| Physical functioning | 6.14 | −1.92, 14.21 | 0.13 |
| Role limitations due to physical health | −4.7 | −17.6, 8.2 | 0.47 |
| Role limitations due to emotional problems | −2.95 | −18.9, 13 | 0.71 |
| Energy/fatigue (vitality) | −0.016 | −6.45, 6.41 | 0.99 |
| Emotional well-being (mental health) | 0.46 | −7.1, 7.99 | 0.9 |
| Social functioning | −4.05 | −13.1, 5 | 0.37 |
| Pain | 5.13 | −2.8, 13 | 0.19 |
| General health | 4.8 | −1.58, 11.2 | 0.14 |
| Components | |||
| Physical component scale | 1.92 | −0.4, 4.2 | 0.1 |
| Mental component scale | −1.45 | −5.2, 2.3 | 0.45 |
Data are presented as least-square mean estimates with associated 95%CIs.
CI, confidence interval; CRPS-I, complex regional pain syndrome type-1; SF-36, 36-Item Short Form Health Survey.
McGill Pain Questionnaire Short-Form
Scores for pain measures (sensory, affective, present pain intensity, and VAS pain intensity) using the McGill Pain Short-Form (SF) questionnaire improved in patients treated with neridronate compared with placebo after 30 days, although affective components of pain were not statistically different between the two groups (Figure 4).
Figure 4.

Change in McGill Pain Questionnaire items in CRPS-I treated with neridronate or placebo from baseline to 30 days. Data are presented as mean ± SD for the following items: sensory, affective, present pain intensity and VAS.
p values denote level of statistical significance between groups.
CRPS-I, complex regional pain syndrome type-1; PPI, present pain intensity; SD, standard deviation; SF-MPQ, short-form McGill pain questionnaire; VAS, visual analogue scale.
Drug compliance and use of rescue medication
The mean number of i.m. injections was 15.6 ± 2.2 in the neridronate group and 15.2 ± 2.5 in the placebo (p = 0.45). Over the entire double-blind phase of the study, 35 patients (85.4%) in the neridronate group and 26 (70.3%; p = 0.18) in the placebo group received at least one dose of rescue medication. The mean cumulative dose over the entire study period in the neridronate group was 11.1 ± 12.82 g versus 11.0 ± 12.7 g in the placebo group, p = 0.87).
Safety
Neridronate was well tolerated and the safety results were in line with the known safety profile of this drug. A total of 114 AEs were reported in 26 patients (63.4%) in the neridronate group and 45 AEs were reported in 17 patients (45.9%) in the placebo group. AEs judged as treatment-related (TAEs) were reported in 18 patients (43.9%) in the neridronate group (74 TAEs) and in 9 (24.3%) in the placebo group (19 TAEs). No severe AEs occurred in either group. The most commonly reported TAEs in the neridronate group were symptoms of the acute-phase reaction or were due to local effects at the site of injection. These events were generally well tolerated by patients for the entire 16-day treatment phase, as only one patient in each group discontinued the study drug due to TAEs. None of the patients in the neridronate group discontinued the study due to AEs at the injection site. In both treatment groups, there were no clinically important changes from baseline to any post-baseline time point in safety laboratory parameters and in vital signs.
Discussion
The results of this randomized, double-blind, placebo-controlled study provide evidence that patients treated with neridronate given i.m. experienced significant benefits not seen in a control group treated with sham injections. These results have been achieved by an intention-to-treat analysis, irrespective of age, gender, site of disease, and precipitating event. These findings confirm and extend our previous study demonstrating that 400 mg neridronate course is effective in the treatment of acute CRPS-1, administered both i.v. and now via i.m. in the present study, with a closely similar temporal trend in the reduction of pain and overlapping efficacy profile. 8 Indeed, by comparing the results of this study with those obtained when the same drug amount was administered in 4 × 100 mg i.v. infusions over 10 days, 8 the rate of patients attaining the primary endpoint, i.e., reduction in pain greater than 50%, was not statistically different (p = 0.63). Of note, all clinical parameters exploring the local signs of disease gave consistent results.
With regard to QoL measures, three out of four pain measures significantly improved using the McGill Pain Questionnaire while results from the SF-36 questionnaire were less convincing. This may be due to an earlier evaluation (30 days in the present study versus 40 days for the i.v. study), as further improvement over time cannot be excluded.
The almost identical efficacy of i.m. and i.v. neridronate administration is likely to be true only if the same dosage of drug is given over a similar time frame and there is substantial bioequivalence between neridronate administered i.v. or i.m. To reach the same total amount of 400 mg of the i.v. schedule, the length of treatment had to be slightly prolonged, from 10 to 16 days, because the only neridronate formulation available and safely given via i.m. is the 25 mg ampoule.
It is worth observing that the assessment of change in pain was carried out 30 days from the start of the study, 10 days before the pain evaluation performed in the i.v. neridronate study, in which the pain measure was assessed 40 days after starting therapy. This difference does not necessarily mean a faster effect of the i.m. administration because, in the i.v. study, no assessment was performed at 30 days. For the present study, we selected this time frame to accomplish an evaluation on a possible earlier effect, as can be suggested by a significant pain reduction (p = 0.043) observed 10 days after starting i.v. treatment almost attaining statistical significance (p = 0.06) at the day of the last i.m. injection. 8
Furthermore, the changes observed in scores for edema and pain at passive motion were very similar to those observed in the neridronate i.v. study in which these improvements further increased over time. 8 We think that the real clinical meaning of our findings is that a small but significant improvement has been achieved only 14 days after the end of the treatment, therefore allowing an earlier rehabilitation program, when a more painful disease subsides and physical therapy can be more effective.
Besides the observed overlap in efficacy of neridronate regardless of the administration route, the choice to explore the added value of i.m. administration has been prompted by the needs of patients unwilling or unable to be treated with i.v. administration. The Italian National Health System requires that i.v. treatment is performed in patients only in a hospital setting, but without hospitalization. In this regard, sometimes severely disabled patients may need to go to the hospital up to four times, necessitating a more expensive treatment. The i.m. treatment can instead be administered at home, without the presence of a medical doctor.
As discussed elsewhere, 21 and confirmed by the results of an animal model study, 22 it is conceivable that many BPs can provide effectiveness for the treatment of acute CRPS-1 to some extent, as long as some important requirements are met. Firstly, patients should be treated as early as possible, when peripheral pathogenic mechanisms prevail. In a retrospective study on 194 CRPS-1 patients treated with BPs, 23 disease duration was predictive of responsiveness to treatment, with a progressive responder rate loss over time and an estimated absence of effect after 12–14 months from onset of disease. 24 Furthermore, among BPs, the drug used should have some specific pharmacokinetic and pharmacodynamic characteristics. The most suitable BP is likely to be a drug with an affinity to hydroxyapatite crystals that is not too high to avoid being captured throughout the entire skeleton (e.g., zoledronate), but high enough to ensure adequate uptake at the site of disease as bone scan frequently shows in the early phase of the disease.25,26 In this regard, neridronate shows an affinity profile lower than that of zoledronate, 27 but higher than that of clodronate or pamidronate,25,28 so likely allowing the appropriate local drug concentration. Moreover, when high local uptake has been reached, the drug should exert an inhibitory effect on the local production of inflammatory mediators by pro-apoptotic properties, 29 mainly on macrophage population, 30 reducing cytokine levels and counteracting pain and clinical signs of the disease. 22 Like all other aminosubstituted BPs, neridronate is able to exert a good level of activity in inhibiting local inflammation. 31 Finally, the last requirement is to employ a dosage large enough in a short period of time to avoid only a partial remission of the symptoms achieved when BPs have been used at an overall dosage that is too low,4,7 or administered for too long. 6 The same lesson can be learned also by the failure of some trials on neridronate in CRPS treatment performed elsewhere. When the drug has been employed at too low a dosage [ClinicalTrials.gov identifier: NCT02402530], 32 or when the patients have been inadequately selected because of too long disease duration [ClinicalTrials.gov identifier: NCT03560986], 33 poor efficacy can be expected. In these circumstances, the successful treatment of CRPS by oral administration would probably be difficult to achieve.
The safety profile of i.m. neridronate did not reveal any untoward effects. No serious drug-related AEs were reported during the study and no changes in biochemical analyses were found. As expected, the more frequent AE was an acute-phase reaction, such as a flu-like syndrome with polyarthralgia and/or fever never exceeding 38°C. This side effect disappeared within a few days after initial injections, without causing any study discontinuation. However, the similar assumption of rescue medication in both groups (despite the significant pain reduction in neridronate-treated patients) can be attributed to the greater amount of acetaminophen consumption in patients treated with neridronate to counteract symptoms of the acute-phase reaction. In fact, in the first (days 1–8) and second week (days 9–16) of treatment, 65.9% and 73.2% of patients received rescue medication in the neridronate group compared with 56.8% and 59.5% in the placebo group, respectively. More importantly, no difference was observed in the last 2 weeks (day 17–day 30) up to day 30 in the proportion of patients receiving rescue medication (56.1% in neridronate group versus 54.1% in placebo group), when the primary outcome measure was assessed.
A further concern could have been local tolerability. Patients treated with neridronate showed a significant greater incidence of site injection pain, decreasing progressively after 30 min. This side effect has been reported at day 8, but not on the day of the last injection. No local inflammation was induced and no patients treated with neridronate left the study due to local injection pain. With regard to other possible AEs shared by all BPs, the short treatment length shelters from fear of osteonecrosis of the jaw and atypical fractures. Neridronate treatment for CRPS-1 was licensed in Italy more than 5 years ago and, to date, no case of these AEs have been reported, despite hundreds of patients being treated.
Limitations
This study has some weaknesses. A limitation refers to one of the inclusion criteria employed. The short disease duration does not exclude patients who spontaneously recover as described in some reports,34,35 although none of the patients with early CRPS-1 recruited by using the same diagnostic criteria we employed were healed after 12 months. 36 A further limitation would be the potential for the study not to be fully blinded due to the acute phase reaction occurring only in some neridronate-treated patients. However, outcome measures included self-administered tools and, according to study design, investigators who assessed clinical measures were blinded to possible side effects reported by patients. Lastly, this study described the effects of a neridronate course given i.m. after 30 days, so a possible relapsing disease cannot be excluded.
Conclusion
This study demonstrated a robust, clinically significant improvement in patients treated with neridronate compared with placebo, further confirming that this treatment is a helpful therapeutic strategy in acute CRPS-1, irrespective of the route of administration. The efficacy of neridronate has been demonstrated when this extremely painful, disabling, and possibly long-standing disease is treated at an early stage with an effective molecule administered at the appropriate dosing regimen.
Acknowledgments
Additional investigators and significant contributors in the study were: Valerio Sansone, Department of Orthopedics, Istituto Ortopedico Galeazzi, Milano; Maria Grazia Benedetti, Department of Physiotherapy, Istituto Ortopedico Rizzoli, Bologna; Maria Teresa Mascia, Department of Medical Sciences, University of Modena and Marco Bulleri, Statistician, Pisa Italy.
Footnotes
Author contributions: MV: study concept and design, analysis and interpretation of data, drafting the manuscript.
VB, GI, and BF: data collection. DG, FZ, and CC: data collection, analysis and interpretation of data. FN: analysis and interpretation of data. MR: critical revision of the manuscript for important intellectual content. All authors edited and approved the report for final submission.
Conflict of interest statement: Massimo Varenna has received advisory board honoraria from UCB and Kyowa Kirin, consultancy fees from Amgen, and speaker fees from Eli-Lilly and Sandoz. Davide Gatti has received advisory board honoraria from Pfizer, consultancy fees from Abiogen and UCB, and speaker fees from Celgene, Eli-Lilly, and Neopharmed-Gentili. Giovanni Iolascon has received speaker fees from Eli-Lilly and UCB. Maurizio Rossini has received advisory board honoraria and consultancy fees from Abiogen, Celgene, Sanofi, Eli-Lilly, and UCB and speaker fees from Abiogen, Amgen, Abbvie, BMS, Celgene, Grunenthal, Eli-Lilly, MSD, Novartis, Pfizer, Sanofi, Sandoz, and UCB. Fabrizio Nannipieri is an employee of Abiogen Pharma S.p.A. All other authors have no conflict of interest to declare.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Abiogen Pharma S.p.A, Pisa, Italy.
ORCID iDs: Massimo Varenna  https://orcid.org/0000-0002-8644-0909
https://orcid.org/0000-0002-8644-0909
Chiara Crotti
https://orcid.org/0000-0001-7015-269X
Maurizio Rossini
https://orcid.org/0000-0001-9692-2293
Contributor Information
Massimo Varenna, Bone Diseases Unit, Department of Rheumatology, Gaetano Pini Institute, Via Pini, 9, Milan 20122, Italy.
Vania Braga, ULSS 9, Verona, Italy.
Davide Gatti, Rheumatology Unit, Department of Medicine, University of Verona, Verona, Veneto, Italy.
Giovanni Iolascon, Department of Medical and Surgical Specialties, University of Campania “Luigi Vanvitelli”, Naples, Italy.
Bruno Frediani, Unit of Rheumatology, University of Siena, Siena, Toscana, Italy.
Francesca Zucchi, Bone Diseases Unit, Department of Rheumatology, Gaetano Pini Institute, Milan, Italy.
Chiara Crotti, Bone Diseases Unit, Department of Rheumatology, Gaetano Pini Institute, Milan, Italy.
Fabrizio Nannipieri, Clinical Research, Abiogen Pharma, Pisa, Toscana, Italy.
Maurizio Rossini, Rheumatology Unit, Department of Medicine, University of Verona, Verona, Veneto, Italy.
References
- 1. Birklein F, Dimova V. Complex regional pain syndrome–up-to-date. Pain Rep 2017; 2: e624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dommerholt J. Complex regional pain syndrome—1: history, diagnostic criteria and etiology. J Bodyw Mov Ther 2004; 8: 167–177. [Google Scholar]
- 3. Bussa M, Guttilla D, Lucia M, et al. Complex regional pain syndrome type I: a comprehensive review. Acta Anaesthesiol Scand 2015; 59: 685–697. [DOI] [PubMed] [Google Scholar]
- 4. Adami S, Fossaluzza V, Gatti D, et al. Bisphosphonate therapy of reflex sympathetic dystrophy syndrome. Ann Rheum Dis 1997; 56: 201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Varenna M, Zucchi F, Ghiringhelli D, et al. Intravenous clodronate in the treatment of reflex sympathetic dystrophy syndrome. A randomized, double blind, placebo controlled study. J Rheumatol 2000; 27: 1477–1483. [PubMed] [Google Scholar]
- 6. Manicourt D-H, Brasseur J-P, Boutsen Y, et al. Role of alendronate in therapy for posttraumatic complex regional pain syndrome type I of the lower extremity. Arthritis Rheum 2004; 50: 3690–3697. [DOI] [PubMed] [Google Scholar]
- 7. Robinson JN, Sandom J, Chapman PT. Efficacy of pamidronate in complex regional pain syndrome type I. Pain Med 2004; 5: 276–280. [DOI] [PubMed] [Google Scholar]
- 8. Varenna M, Adami S, Rossini M, et al. Treatment of complex regional pain syndrome type I with neridronate: a randomized, double-blind, placebo-controlled study. Rheumatology (Oxford) 2013; 52: 534–542. [DOI] [PubMed] [Google Scholar]
- 9. Wertli MM, Kessels AGH, Perez RSGM, et al. Rational pain management in complex regional pain syndrome 1 (CRPS 1)—a network meta-analysis. Pain Med 2014; 15: 1575–1589. [DOI] [PubMed] [Google Scholar]
- 10. Chevreau M, Romand X, Gaudin P, et al. Bisphosphonates for treatment of complex regional pain syndrome type 1: a systematic literature review and meta-analysis of randomized controlled trials versus placebo. Joint Bone Spine 2017; 84: 393–399. [DOI] [PubMed] [Google Scholar]
- 11. Merlotti D, Rendina D, Gennari L, et al. Comparison of intravenous and intramuscular neridronate regimens for the treatment of Paget disease of bone. J Bone Miner Res 2011; 26: 512–518. [DOI] [PubMed] [Google Scholar]
- 12. Gatti D, Viapiana O, Idolazzi L, et al. Neridronic acid for the treatment of bone metabolic diseases. Expert Opin Drug Metab Toxicol 2009; 5: 1305–1311. [DOI] [PubMed] [Google Scholar]
- 13. Harden RN, Bruehl S, Stanton-Hicks M, et al. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007; 8: 326–331. [DOI] [PubMed] [Google Scholar]
- 14. Harden RN, Bruehl S, Perez RSGM, et al. Validation of proposed diagnostic criteria (the ‘budapest criteria’) for complex regional pain syndrome. Pain 2010; 150: 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huskisson EC. Measurement of pain. Lancet 1974; 2: 1127–1131. [DOI] [PubMed] [Google Scholar]
- 16. Forouzanfar T, Weber WEJ, Kemler M, et al. What is a meaningful pain reduction in patients with complex regional pain syndrome type 1? Clin J Pain 2003; 19: 281–285. [DOI] [PubMed] [Google Scholar]
- 17. Harden RN, Oaklander AL, Burton AW, et al. Complex regional pain syndrome: practical diagnostic and treatment guidelines, 4th edition. Pain Med 2013; 14: 180–229. [DOI] [PubMed] [Google Scholar]
- 18. Maiani G, Sanavio E. Semantics of pain in Italy: the Italian version of the McGill Pain Questionnaire. Pain 1985; 22: 399–405. [DOI] [PubMed] [Google Scholar]
- 19. Apolone G, Mosconi P. The Italian SF-36 Health Survey: translation, validation and norming. J Clin Epidemiol 1998; 51: 1025–1036. [DOI] [PubMed] [Google Scholar]
- 20. Adami S, Bhalla AK, Dorizzi R, et al. The acute-phase response after bisphosphonate administration. Calcif Tissue Int 1987; 41: 326–331. [DOI] [PubMed] [Google Scholar]
- 21. Varenna M. Bisphosphonates beyond their anti-osteoclastic properties. Rheumatology (Oxford) 2014; 53: 965–967. [DOI] [PubMed] [Google Scholar]
- 22. Wang L, Guo T-Z, Wei T, et al. Bisphosphonates inhibit pain, bone loss, and inflammation in a rat tibia fracture model of complex regional pain syndrome. Anesth Analg 2016; 123: 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Varenna M, Manara M, Rovelli F, et al. Predictors of responsiveness to bisphosphonate treatment in patients with complex regional pain syndrome type I: a retrospective chart analysis. Pain Med 2017; 18: 1131–1138. [DOI] [PubMed] [Google Scholar]
- 24. Varenna M, Crotti C. Bisphosphonates in the treatment of complex regional pain syndrome: is bone the main player at early stage of the disease? Rheumatol Int 2018; 38: 1959–1962. [DOI] [PubMed] [Google Scholar]
- 25. Nancollas GH, Tang R, Phipps RJ, et al. Novel insights into actions of bisphosphonates on bone: differences in interactions with hydroxyapatite. Bone 2006; 38: 617–627. [DOI] [PubMed] [Google Scholar]
- 26. Wertli MM, Brunner F, Steurer J, et al. Usefulness of bone scintigraphy for the diagnosis of complex regional pain syndrome 1: a systematic review and Bayesian meta-analysis. PLoS One 2017; 12: e0173688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fleisch H. Bisphosphonates: mechanisms of action. Endocr Rev 1998; 19: 80–100. [DOI] [PubMed] [Google Scholar]
- 28. Neves M, Gano L, Pereira N, et al. Synthesis, characterization and biodistribution of bisphosphonates Sm-153 complexes: correlation with molecular modeling interaction studies. Nucl Med Biol 2002; 29: 329–338. [DOI] [PubMed] [Google Scholar]
- 29. Cornish J, Bava U, Callon KE, et al. Bone-bound bisphosphonate inhibits growth of adjacent non-bone cells. Bone 2011; 49: 710–716. [DOI] [PubMed] [Google Scholar]
- 30. Rogers MJ, Chilton KM, Coxon FP, et al. Bisphosphonates induce apoptosis in mouse macrophage-like cells in vitro by a nitric oxide-independent mechanism. J Bone Miner Res 1996; 11: 1482–1491. [DOI] [PubMed] [Google Scholar]
- 31. Littlejohn G. Bisphosphonates for early complex regional pain syndrome. Nat Rev Rheumatol 2013; 9: 199–200. [DOI] [PubMed] [Google Scholar]
- 32. Grünenthal GmbH. A randomized, double-blind trial investigating the efficacy and safety of intravenous neridronic acid in subjects with Complex Regional Pain Syndrome type I (CRPS-I). Clinical Trial Registration NCT02402530, clinicaltrials.gov, https://clinicaltrials.gov/ct2/show/NCT02402530 (2018, accessed 19 May 2020).
- 33. Grünenthal GmbH. Placebo-controlled efficacy and safety trial of intravenous neridronic acid in subjects with Complex Regional Pain Syndrome (CRPS). Clinical Trial Registration NCT03560986, clinicaltrials.gov, https://clinicaltrials.gov/ct2/show/NCT03560986 (2019, accessed 19 May 2020).
- 34. Zyluk A. The natural history of post-traumatic reflex sympathetic dystrophy. J Hand Surg Br 1998; 23: 20–23. [DOI] [PubMed] [Google Scholar]
- 35. Sandroni P, Benrud-Larson LM, McClelland RL, et al. Complex regional pain syndrome type I: incidence and prevalence in Olmsted county, a population-based study. Pain 2003; 103: 199–207. [DOI] [PubMed] [Google Scholar]
- 36. Beerthuizen A, Stronks DL, Van’t Spijker A, et al. Demographic and medical parameters in the development of complex regional pain syndrome type 1 (CRPS1): prospective study on 596 patients with a fracture. Pain 2012; 153: 1187–1192. [DOI] [PubMed] [Google Scholar]