

Graphical abstract
Keywords: autophagy, LIR motif, phosphorylation, bio-layer interferometry, crystal structure
Highlights
-
•
Autophagic degradation of cellular material relies on LIR-ATG8 interactions.
-
•
Regulation of LIR-ATG8 interactions by phosphorylation is incompletely understood.
-
•
The Golgi protein SCOC binds to ATG8 proteins through a functional LIR domain.
-
•
SCOC LIR phosphorylation by ULK1-3, and TBK1 increases specifically LC3 binding.
-
•
New LIR phospho -regulation critical for ATG8 binding affinity and specificity.
Abstract
Autophagy is a highly conserved degradative pathway, essential for cellular homeostasis and implicated in diseases including cancer and neurodegeneration. Autophagy-related 8 (ATG8) proteins play a central role in autophagosome formation and selective delivery of cytoplasmic cargo to lysosomes by recruiting autophagy adaptors and receptors. The LC3-interacting region (LIR) docking site (LDS) of ATG8 proteins binds to LIR motifs present in autophagy adaptors and receptors. LIR-ATG8 interactions can be highly selective for specific mammalian ATG8 family members (LC3A-C, GABARAP, and GABARAPL1-2) and how this specificity is generated and regulated is incompletely understood.
We have identified a LIR motif in the Golgi protein SCOC (short coiled-coil protein) exhibiting strong binding to GABARAP, GABARAPL1, LC3A and LC3C. The residues within and surrounding the core LIR motif of the SCOC LIR domain were phosphorylated by autophagy-related kinases (ULK1-3, TBK1) increasing specifically LC3 family binding. More distant flanking residues also contributed to ATG8 binding. Loss of these residues was compensated by phosphorylation of serine residues immediately adjacent to the core LIR motif, indicating that the interactions of the flanking LIR regions with the LDS are important and highly dynamic.
Our comprehensive structural, biophysical and biochemical analyses support and provide novel mechanistic insights into how phosphorylation of LIR domain residues regulates the affinity and binding specificity of ATG8 proteins towards autophagy adaptors and receptors.
Introduction
Autophagy is a highly conserved recycling and stress survival pathway ensuring cellular health and homeostasis.1 During starvation, autophagy restores intracellular nutrients and molecular building blocks by sequestering and transferring cytosolic material in autophagosomes to lysosomes for degradation. Moreover, autophagy selectively removes toxic macromolecules, damaged organelles, and intracellular pathogens and therefore has been implicated in pathologies such as neurodegeneration, cancer, and infection.2, 3, 4, 5
The molecular machinery driving autophagosome formation comprises autophagy-related (ATG) proteins which are highly conserved from yeast to human. In mammals, autophagy is initiated by the UNC-51 like kinase (ULK) complex, which phosphorylates and activates the class III ATG14-Beclin1-VPS34-p150 phosphatidylinositol 3-phosphate (PI3P) kinase complex I.6, 7 This results in production of PI3P at autophagosome formation sites (omegasomes) on the endoplasmic reticulum (ER) and recruitment of PI3P-binding effectors, such as DFCP1 and WIPI proteins.8 WIPI2b further recruits the ATG12-5-16L1 complex9; which mediates conjugation of ATG8 proteins to phosphatidylethanolamine in the autophagic membrane.10, 11
Whereas yeast only has one ATG8 protein, mammalian cells have two ATG8 protein subfamilies, namely LC3s (LC3A, LC3B, LC3C) and GABARAPs (GABARAP, GABARAP-L1, GABARAP-L2),12 functioning in autophagosome formation and fusion with the lysosome.13, 14, 15, 16 During selective autophagy, ATG8 proteins mediate cargo recruitment by directly interacting with autophagy receptors, such as p62/SQSTM1.17 Moreover, interactions of ATG8 proteins with autophagy adaptors which are not degraded by autophagy regulate autophagosome formation (e.g. ULK1, PI3K class III complex, ATG2 and ATG4),18, 19, 20, 21, 22 fusion with the lysosome (e.g. PLEKHM1),23 lysosome or autolysosome biogenesis (e.g. STX16 and STX17),24 or autophagosome transport (e.g. FYCO1).25
Interaction with ATG8 proteins typically occurs via an ATG8-interacting motif (AIM), also known as LC3-interacting region (LIR) motif.5, 26, 27, 28 The consensus sequence of the canonical LIR motif is a small Θ0-X1-X2-Γ3 motif with Θ representing an aromatic residue (W/F/Y), Γ an aliphatic residue (L/V/I), and X any amino acid (aa). The side chains of the aromatic and aliphatic residues fit into hydrophobic pocket 1 (HP1) and 2 (HP2) of the ATG8 LIR docking site (LDS), respectively. Acidic and phosphorylated residues, often present in the N-terminal region directly preceding the core LIR motif, stabilize ATG8 binding through electrostatic interactions.26, 27, 28
Many LIR-containing proteins interact with ATG8 proteins in a highly selective manner, suggesting functional differences between LC3 and GABARAP subfamily proteins.5, 13, 14, 15, 28 Rogov et al. defined a GABARAP interaction motif (GIM), Θ0-[V/I]1-X2-Γ3, and showed that valine or isoleucine in position X1 promotes GABARAP binding.29 We recently showed that the binding preference of LIR motifs to GABARAPs is also regulated by the X2 residue within the core LIR motif as well as residues in the adjacent C-terminal region (positions X4-X10).30 ATG8-subfamily binding specificity is also determined by the residues of the LIR-docking site (LDS). In line with these findings, a recent study on plant ATG8 proteins revealed that an amino acid polymorphism in the N-terminal beta-strand comprising HP1 of the LDS determines the binding specificity of two potato ATG8 isoforms and contributes to their functional specialization.31
In addition, phosphorylation has been shown to modulate ATG8 binding specificity.20, 32, 33, 34, 35, 36, 37, 38 The LIR motif of the autophagy receptor optineurin (OPTN)32 interacts strongly with GABARAP and weakly with LC3B. Phosphorylation of OPTN serine S177 at position X−1, (the residue directly preceding the core LIR motif) by TANK-binding kinase1 (TBK1) activates LC3B binding and promotes selective autophagy of cytosolic Salmonella.32 Phosphorylation of residues N-terminal to the core LIR motif also positively regulates GABARAP binding to the VPS34 and Beclin1 LIR motifs.20 Conversely; phosphorylation of S13 (position X−5)37 and Y18; the aromatic core LIR motif residue (position Θ0)35; of the mitophagy receptor FUNDC1 prevent LC3 binding and inhibit mitophagy. Further analyses are required to fully understand the role of phosphorylation, the molecular determinants and constraints that confer and regulate selective ATG8 binding.
A genome wide siRNA screen identified the short coiled-coil protein (SCOC) as a novel positive regulator of starvation induced autophagy.39 SCOC is a small Golgi protein implicated in Golgi transport through interacting with ADP-ribosylation factor-like 1 (ARL1) via its coiled-coil domain.40 The SCOC coiled-coil domain also mediates a highly conserved interaction with the kinesin1-adaptor protein fasciculation and elongation protein zeta 1 (FEZ1)39, 41, 42, 43 and both proteins are required for axonal growth; transport and normal presynaptic organization.41, 44, 45 Interestingly; the Drosophila ULK1 homolog UNC-51 regulates axonal transport by binding and phosphorylating FEZ1 (UNC-76).46 The interaction between ULK1 and FEZ1 is also conserved in mammals and might be modulated by SCOC.39
To further understand the role of SCOC in autophagy, we identified a functional LIR motif in the unstructured region N-terminal of the coiled-coil domain of SCOC. The SCOC LIR motif exhibits a high binding preference for GABARAP, GABARAPL1 as well as LC3A and LC3C. The molecular details mediating SCOC LIR-ATG8 binding were investigated by X-ray crystallography and bio-layer interferometry (BLI) affinity measurements. Our data demonstrates a critical contribution of the flanking LIR domain residues, which are up to eight amino acid residues distant from the SCOC core LIR motif, to ATG8 protein binding. Moreover, we show that ULK kinases and TBK1 can phosphorylate SCOC serine/threonine residues (T15/S18SCOC) within and C-terminal to the core LIR motif to strongly increase the affinity to LC3 subfamily proteins. These data support and extend the notion of key phosphorylation sites that can regulate ATG8 binding affinity and specificity of LIR motifs.
Results
SCOC interacts with mammalian ATG8 proteins through a N-terminal LIR domain
SCOC is widely expressed in human tissues40, 41 and due to alternative splicing multiple isoforms have been reported. All SCOC isoforms are highly conserved in the C-terminal region comprising a coiled-coil domain; but vary in the N-terminal region (Figure 1A). To elucidate whether the diverse N-terminal regions confer functional differences we employed the T-REXTM HeLa cell line system to generate stable inducible HeLa cell lines expressing EGFP-tagged SCOC isoform 1 and 3 (Figure 1B). SCOC isoform 1 localizes primarily to the nucleus in interphase cells and possesses a potential nuclear localization signal (NLS) located in the N-terminal region (aa 1–29) (Figure 1A, B, predicted with cNLS Mapper tool.47 During cell division, a pronounced localization of SCOC isoform1 to the mitotic spindle (labelled by tubulin) was detected in prophase, metaphase, and anaphase cells (Supplementary Figure 1A). In contrast, SCOC isoform 3 distributed to both the nucleus and cytosol (Figure 1B), where it partially colocalized with the trans-Golgi network (TGN) marker TGN46 (Supplementary Figure 1B). Notably, the localisation of EGFP-SCOC isoform 3 to the Golgi and trans-Golgi network was less prominent than endogenous SCOC reported by immunofluorescence staining using an antibody recognising the conserved coiled-coil region (Figure 1A).39, 40 Endogenous SCOC partially colocalises with endogenous LC3 in EBSS-starved cells.39 In response to EBSS starvation; we detected partial colocalization of SCOC isoform 3-positive puncta with endogenous GABARAP (Figure 1B). Due to potential cross-reactivity of the anti-GABARAP antibody (Abgent) with other GABARAP subfamily proteins48; SCOC isoform 3 might also partially colocalize with GABARAPL1 and GABARAPL2.
Figure 1.

SCOC interacts with mammalian ATG8 proteins through a N-terminal LIR domain. A. Sequence alignment of SCOC isoform 1 (Q9UIL1), SCOC isoform 4 (Q9UIL1-4), SCOC isoform 3 (Q9UIL1-3), SCOC isoform 2 (Q9UIL1-2), SCOC isoform 5 (A0A0C4DGB0/protein accession AAK01707) using the Clustal Omega program of the UniProt database (www.uniprot.org). The highly conserved coiled-coil domain is highlighted in green. The LIR motif and the nuclear localisation signal sequence are indicated in red and blue, respectively. Residues conserved in all five isoforms are highlighted by asterisks (*) B. HeLa Flp-In T-Rex cells stably expressing EGFP-SCOC (isoform 1) or EGFP-SCOC (isoform 3) starved for 2 h in EBSS, fixed and labelled with anti-GABARAP and Hoechst for confocal microscopy. Expression of EGFP-SCOC constructs was induced with 0.5 μg/ml tetracycline overnight. Scale bars represent 10 μM. White boxes indicate position of insets and arrows indicate EGFP-SCOC (isoform 3)-GABARAP colocalization. C. Peptide array of 20-mer peptides covering full-length SCOC isoform 3 (each peptide shifted 3 amino acids relative to the previous) was incubated with GST-GABARAP and immunoblotted with anti-GST. The amino acid sequence for the GABARAP interacting peptides is shown with the interacting peptides depicted as black lines. D. GST pulldown assay of in vitro translated and [35S]methionine labelled wild-type (WT) and LIR mutant (F14A/I17A) GFP-SCOC (isoform 3) proteins binding to GST-ATG8 proteins. Autoradiograph (AR, upper panels) and representative coomassie stained immobilized GST fusion proteins (CBB, bottom panel) are shown. E. Affinities (Kd values) of SCOC (WT) LIR domain peptide to human ATG8 proteins determined by bio-layer interferometry (BLI). Color code indicates fold changes relative to GABARAP (data are average Kd values ± standard error calculated from non-linear regression curve fits, n = 2).
Bioinformatic analyses (iLIR49 identified a putative LIR motif (aa 13-FTNI-aa 18) in the non-conserved N-terminal region of both SCOC isoform 2 and 3 (varying by a single aa (alanine) at residue 48) (Figure 1A). This LIR motif was confirmed by probing a peptide array of 20-mer peptides, with each peptide shifted 3 amino acids C-terminally to cover full-length SCOC isoform 3, with GST-GABARAP (Figure 1C). In GST-pull down experiments SCOC isoform 3 interacted strongly with LC3A, LC3C, GABARAP and GABARAPL1, much weaker with GABARAPL2 and very little with LC3B (Figure 1D). Mutation of the LIR motif residues F14 and I17 to alanine (F14A/I17A) reduced the interaction with all ATG8 proteins and confirmed that this LIR motif is functional. However, these mutations did not abrogate colocalization of SCOC isoform 3 (F14A/I17A) with TGN46 and GABARAP (Supplementary Figure 1B) suggesting that the LIR motif is not essential for SCOC localization to the trans-Golgi network and GABARAP-positive puncta and this localization might occur through binding other proteins, such as Arl1 and FEZ1,39, 40 or dimerization with endogenous wild type SCOC.50
In line with the GST pull down experiments, biolayer-interferometry (BLI) affinity measurements showed that the SCOC LIR binds strongest to GABARAP (3.6 µM) and GABARAPL1 (2.3 µM), followed by LC3C (8.7 µM) and LC3A (27.8 µM), and weakest to GABARAPL2 (44.4 µM), and LC3B (270 µM) (Figure 1E, Supplementary Figure 2A). The binding affinities of the SCOC LIR motif, ranging from 2-300 µM are similar to Kd values reported for the autophagy adaptors PCM130 and the PI3K (class III) complex members, Beclin1, VPS34 and ATG14.20
To determine whether SCOC is functioning as an autophagy receptor and degraded by autophagy, we induced expression of EGFP-SCOC (isoform 1 and 3) in HeLa cells followed by 7 h EBSS starvation in the presence and absence of lysosomal or proteasomal inhibitors. Inhibition of proteasomal degradation using MG132 or epoxymycin, increased SCOC isoform 1 and 3 protein levels, whereas treatment with BafilomycinA1 (BafA1), which inhibits lysosomal degradation, did not have any effect in both fed and starved cells (Supplementary Figure 2B, 2C). Thus, both SCOC isoform1 and 3 are not turned over by autophagy, but by the proteasome. In summary, SCOC isoform 3 shows a high binding affinity to GABARAP, GABARAPL1, and LC3C and potentially is a novel autophagy adaptor protein.
Molecular determinants mediating SCOC LIR-GABARAP complex formation
The SCOC LIR motif and flanking regions were further characterised using mutational peptide array scans. Every position within the 23-mer SCOC LIR peptide was mutated to all alternative amino acids (aa) and binding of GST-tagged ATG8 proteins was analysed by immunoblotting with an anti-GST antibody. Based on our findings from GST-pull downs (Figure 1D) and BLI affinity measurements (Figure 1E), we focused on strong and medium interacting proteins and selected GST-GABARAP (Figure 2A), GST-LC3C (Figure 2B) and GST-LC3A (Supplementary Figure 2D). The mutational SCOC LIR peptide array scans confirmed a canonical LIR motif, as mutation of both F14 and I17 abrogated binding of all three ATG8 proteins. In line with previous reports,18, 30, 51 GABARAP binding was abolished by glycine or proline substitutions at any position of the core LIR (X0-X3), whereas LC3C and LC3A binding in addition was abolished by the presence of positively charged amino acids (K and R). Consistent with recent findings,30 substitution of N16 by a hydrophobic/aromatic aa (V, L, I, W, Y, F) strongly improved both GABARAP and LC3A/C binding further underscoring the stabilizing effect on ATG8 binding by these aa at the LIR motif position X2.30 Interestingly, L19 (in position X5) was also important for interaction. Only substitutions with the aromatic residues (Y, W, F) and the hydrophobic I gave significant binding, suggesting that residues C-terminal to the core LIR motif also contribute to ATG8 binding. N-terminal to the core LIR motif, substitution of D11 (position X−3) slightly reduced LC3A, LC3C and GABARAP binding. Moreover, substitutions of S12 and T13 by D/E improved LC3C (Figure 2B), LC3A (Supplementary Figure 2D) and GABARAP (Figure 2A) binding, further underscoring the positive regulation of LIR motif binding to ATG8 proteins by acidic residues and potential phosphorylation of S/T/Y residues in position X−1 and X−2.20, 27, 30, 32, 33, 34, 36
Figure 2.

Molecular determinants mediating SCOC LIR-GABARAP complex formation. A. and B. Mutational peptide array of 23-mer SCOC peptides covering the LIR motif incubated with GST-GABARAP (A.) or GST-LC3C (B.) and immunoblotted with anti-GST. Each amino acid position was substituted for every other amino acid. C. Structure of the SCOC LIR domain (aa 6–23) bound to GABARAP. The SCOC LIR sequence is shown in orange cartoon with interacting residues shown as sticks. GABARAP is displayed in white cartoon and transparent surface with hydrophobic pocket 1 and 2 coloured in pink and blue surfaces, respectively. The box with dashed lines indicates location of close-up view showing hydrophobic interactions of L19SCOC with the edge of HP2 (L55/L63GAB). D. Schematic overview of the interactions observed in the structure of GABARAP bound to the SCOC LIR domain. Orange boxes below the SCOC sequence display the residue position within the LIR domain. GABARAP residues are boxed and shown in black. Blue lines indicate hydrophobic interactions, green lines hydrogen bonds, and green double arrow salt bridges. Pink and blue boxes indicate canonical LIR motif interactions (hydrophobic contacts) with HP1 and HP2.
To further elucidate the molecular determinants mediating interaction of the SCOC LIR motif with GABARAP, we generated a chimera protein consisting of the SCOC6-23 LIR domain sequence N-terminally fused to GABARAP with a Gly-Ser linker. We solved the crystal structure of the SCOC6-23 LIR domain bound to GABARAP at a resolution of 1.25 Å (Figure 2C, Supplementary Figure 2E, Table 1). The SCOC LIR:GABARAP complex structure revealed a large surface of interactions between SCOC residues 6 to 19 and GABARAP. The SCOC core LIR motif displayed the canonical LIR interactions comprising the hydrophobic residues F14SCOC (Θ0) and I17SCOC (Γ3) deeply bound to HP1 and HP2, and three hydrogen bonds formed between the main chains of the SCOC LIR residues T15SCOC (X1) and I17SCOC (Γ3) and the main chains of the GABARAP residues K48GAB and L50GAB (Figure 2C). On top of these canonical LIR interactions additional specific contacts were observed between the SCOC core LIR motif as well as the adjacent N- and C-terminal regions and GABARAP (Figure 2C, D). In the region N-terminal to the SCOC core LIR motif we observed: a salt bridge between K6SCOC (X−8) and E8GAB, two hydrogen bonds between the backbone of E10SCOC (X−4) and the mainchain of D45/K47GAB, one hydrogen bond between the sidechain of D11SCOC (X−3) and the imidazole ring of H9GAB, and another hydrogen bond between the carbonyl of S12SCOC (X−2) and K48GAB sidechain. Within the core LIR motif, N16SCOC in position X2 was hydrogen bonded with the guanidinium of R28GAB. In the C-terminal region, hydrophobic interactions were observed between L19SCOC in position X5 and the edge of HP2 (L55/L63GAB).
Table 1.
Data collection and refinement statistics.
| SCOC:GABARAP | SCOC-2pS: GABARAPL1 | SCOC-2pT: GABARAPL1 | |
|---|---|---|---|
| PDB ID | 7AA8 | 7AA7 | 7AA9 |
| Resolution range | 39.92–1.25 (1.29–1.25) | 31.92–1.45 (1.47–1.45) | 59.94–1.72 (1.78–1.72) |
| Space group | P 32 2 1 | P 1 | P 32 |
| Unit cell | 52.82 52.82 81.72 90 90 120 | 28.36 37.17 62.48 81.75 89.60 67.82 | 90.94 90.94 92.40 90 90 120 |
| Total reflections | 289 346 (27 734) | 123 975 (6 189) | 349 139 (25 323) |
| Unique reflections | 37 098 (3 670) | 41 242 (2 051) | 90 238 (8 924) |
| Multiplicity | 7.8 (7.6) | 3.0 (3.0) | 3.9 (2.8) |
| Completeness (%) | 99.84 (99.78) | 99.5 (98.9) | 99.17 (96.98) |
| Mean I/sigma(I) | 15.85 (1.18) | 15.1 (5.0) | 7.55 (1.15) |
| R-merge | 0.04 (2.0) | 0.09 (0.6) | 0.09 (0.8) |
| R-meas | 0.04 (2.1) | 0.12 (0.92) | 0.11 (0.99) |
| R-pim | 0.01 (0.7) | 0.08 (0.60) | 0.05 (0.54) |
| CC1/2 | 0.99 (0.53) | 0.98 (0.46) | 0.99 (0.57) |
| Reflections used in refinement | 37 098 (3 671) | 41 159 (2 852) | 90 055 (8 825) |
| Reflections used for R-free | 1 828 (181) | 1 970 (150) | 4 406 (437) |
| R-work | 0.16 (0.34) | 0.15 (0.21) | 0.20 (0.35) |
| R-free | 0.19 (0.40) | 0.19 (0.29) | 0.25 (0.37) |
| Number of non-hydrogen atoms | 1 241 | 2 150 | 7 167 |
| Macromolecules | 1 100 | 1 967 | 6 424 |
| Solvent | 141 | 167 | 743 |
| RMS(bonds) | 0,008 | 0,01 | 0,002 |
| RMS(angles) | 0,96 | 1,12 | 0,46 |
| Ramachandran favored (%) | 99,24 | 98,27 | 97,32 |
| Ramachandran allowed (%) | 0,76 | 1,33 | 2,68 |
| Ramachandran outliers (%) | 0 | 0 | 0 |
| Average B-factor | 26,4 | 20,3 | 24,6 |
| Macromolecules | 24,9 | 19,5 | 23,8 |
| Solvent | 37,7 | 18 | 31,7 |
In summary, SCOC LIR domain binding to GABARAP is stabilized not only by core LIR residues but also by multiple interactions involving residues of the N- and C-terminal regions flanking the LIR motif, which range from residues in position X−8 to X+5. Several GABARAP residues interacting with the SCOC LIR motif are only conserved among GABARAP subfamily proteins (Supplementary Figure 2F).
ULK kinases and TBK1 phosphorylate SCOC in vitro
Phosphorylation of residues in position X−1,−2 have been shown to increase LIR binding.20, 32, 33, 36 Serine (S12SCOC) and threonine (T13SCOC) residues precede the SCOC core LIR motif, and moreover, within and directly after the core LIR motif (position X1 and X4) are serine and threonine residues (T15SCOC and S18SCOC), which could be modified by phosphorylation. In Drosophila and mammalian cells, UNC-51 and ULK1, respectively, bind and phosphorylate UNC-76/FEZ1 regulating axonal transport.39, 46 In line with our previous findings,39 GFP-Trap immunoprecipitation experiments showed that FEZ1-GFP but not GFP-SCOC (isoform 3) interacted with the endogenous ULK1 kinase complex members ULK1, ATG13 and FIP200 in both fed and starved HEK293A cells (Figure 3A, and 39). Moreover, we detected interaction of GFP-FEZ2 with the ULK1 kinase complex (Figure 3A). FEZ1 and FEZ2 share high sequence conservation between each other, and SCOC (isoform 3) interacted with both FEZ1 (39) and FEZ2 (Supplementary Figure 3A, 52).
Figure 3.

ULK kinases (ULK1-3) and TBK1 phosphorylate SCOC in vitro. A. GFP-Trap immunoprecipitation (IP) of GFP, GFP-SCOC (isoform 3), FEZ1-GFP or GFP-FEZ2 proteins from HEK293A cells and immunoblots with indicated antibodies. Cells were cultured in full medium (F) or Earle’s balanced salt solution (starvation medium (S)) for 2 h prior to lysis. B. Phosphorylation sites detected by mass spectrometry in GST-SCOC (isoform 3) after in vitro kinase assays using either recombinant TBK1, ULK1, ULK2 or ULK3. Posttranslational modification mapping was done using the Discoverer 2.4 software (ThermoFisher) and a threshold score (Xcorr) > 2.0. C. Western blot analysis of immunoprecipitated GFP, GFP-SCOC (isoform 3) WT, GFP-SCOC (isoform 3) 9A mutant (GFP-SCOC S26A, S27A, S36A, T60A, T72A, S77A, S106A, S108A, S109A), GFP-SCOC (isoform 3) 13A mutant (GFP-SCOC S12A, T13A, T15A, S18A, S26A, S27A, S36A, T60A, T72A, S77A, S106A, S108A, S109A), GFP-SCOC (isoform 3) 14A mutant (GFP-SCOC S12A, T13A, T15A, S18A, S26A, S27A, S36A, T60A, T72A, S77A, S92A, S106A, S108A, S109A) and GFP SCOC (isoform 3) 4A mutant (S12A, T13A, T15A, S18A) using Phos-tagTM SDS-Page. HEK293A cells expressing GFP, GFP-SCOC WT or mutants together with murine MYC-ULK1 wild type (WT) or MYC-ULK1 kinase inactive (KI) (MYC-ULK1-K46I) were starved in Earle’s balanced salt solution for 2 h prior to lysis and GFP-Trap IP. Half of the IP reaction was treated with λ-phosphatase prior to Phos-tagTM SDS-Page and Western blotting. Lower panel shows input analysed by SDS-PAGE and Western blotting.
As we have shown ULK1, FEZ1 and SCOC form a complex (39), we asked if SCOC is phosphorylated by ULK1 through its interaction with FEZ1 and FEZ2. We performed in-vitro kinase assays to determine whether recombinant ULK1 can directly phosphorylate SCOC. We also included ULK2 and ULK3 due to potential overlapping functions between these kinases.6, 50 Furthermore, we tested TBK1, since this kinase has been reported to phosphorylate the LIR domain of OPTN, enhancing OPTN binding to LC3 and clearance of intracellular Salmonella.32 Mass spectrometry analyses revealed that in vitro experiments all four kinases phosphorylated multiple sites of SCOC isoform 3 (Figure 3B, Supplementary Figure 3B). TBK1 was most active on SCOC isoform 3 with 14 different phosphorylation events. Four to five phosphorylation events were also detected for the three ULK kinases and the identified sites strongly overlapped (S18/S26/S108/S109), indicating similar substrate specificities between ULK1-3. Interestingly, four serine/threonine residues are located within or adjacent to the SCOC core LIR motif (S12/T13/T15/S18SCOC). TBK1 again displayed strongest activity on these SCOC LIR motif sites, since we detected phosphorylation of all four serine/threonine residues (S12/T13/T15/S18SCOC) as well as the highest intensity values for the corresponding N-terminal peptides. All three ULK kinases phosphorylated S18SCOC in position X+4 of the SCOC LIR motif with ULK1 and ULK2 exhibiting higher activities than ULK3.
To test whether ULK1 phosphorylates SCOC in cells, we expressed GFP-tagged SCOC WT together with MYC-tagged ULK1 WT or ULK1 kinase inactive (KI) in HEK293A cells. GFP-tagged proteins were immunoprecipitated using GFP-Trap and half of the IP reaction was subjected to λ-phosphatase treatment (generating non-phosphorylated GFP-SCOC). SCOC phosphorylation was visualised by Western blotting after Phos-tagTM SDS-Page. The Phos-tag reagent reduces electrophoretic mobility of phosphorylated proteins and shifts them to higher molecular weight.53 Overexpression of WT but not KI ULK1 induced higher molecular weight shifts of GFP-SCOC (Figure 3C), but no upward shift of GFP. Multiple bands were detected for GFP-SCOC, indicating ULK1 overexpression results in two differentially phosphorylated SCOC species. To determine whether ULK1 phosphorylates residues of the SCOC LIR domain, we mutated all the ULK phosphorylation sites in SCOC identified by mass spectrometry (Figure 3B) to alanine except for those located in the LIR motif and flanking region (S12/T13/T15/S18SCOC). Moreover, we also mutated the majority of sites that were identified for TBK1 (Figure 3C). One higher molecular weight band was still detected after expression of the SCOC 9A mutant (S26A, S27A, S36A, T60A, T72A, S77A, S106A, S108A, S109A). However, mutation of the LIR domain residues (referred to SCOC 13A mutant) as well as S92A (referred to SCOC 14A) did not abolish the higher molecular weight band, indicating that ULK1 does not phosphorylate the LIR domain residues and might phosphorylate other sites not detected by mass spectrometry. Alternatively, ULK1 overexpression might lead to activation of other kinases. Equally, mutation of the LIR domain residues S12A, T13A, T15A, S18A alone (referred to SCOC 4A) did not result in loss of the higher molecular weight bands compared to SCOC WT.
Thus, ULK (ULK1-3) kinases and TBK1 phosphorylated SCOC LIR domain residues in vitro, but in cells (under overexpression conditions) ULK1 does not phosphorylate the SCOC LIR domain.
SCOC LIR affinity and specificity is modulated by phosphorylation of residues within and adjacent to the core LIR motif
To determine whether phosphorylation alters binding of ATG8 proteins to the SCOC LIR motif, we performed a peptide array experiment using SCOC LIR domain peptide sequences carrying single as well as multiple phospho-mimetic mutations of S12ESCOC, T13ESCOC, T15ESCOC and S18ESCOC (Figure 4A). Single point mutations in the SCOC LIR peptide sequence affected mainly the binding of LC3A and LC3B. Mutation of S12SCOC and S18SCOC to glutamate enhanced both GST-LC3B and GST-LC3A binding. The single point mutations T13ESCOC and T15ESCOC also increased GST-LC3A binding to SCOC LIR peptides. The S12E/S18ESCOC or S12E/T13ESCOC double mutation of the SCOC LIR peptide improved both LC3 and GABARAP subfamily binding. The strongest increase in ATG8 binding was detected for the S12E/T13E/S18ESCOC, S12E/T15E/S18ESCOC triple mutation as well as mutation of all four residues (S12E/T13E/T15E/S18ESCOC), indicating additive effects when combined.
Figure 4.

Phosphorylation of the SCOC LIR domain modulates ATG8 binding affinity and specificity. A. Twenty four-mer peptide array of SCOC LIR peptides containing phospho-mimetic mutations incubated with indicated GST-ATG8 protein and immunoblotted with anti-GST. Each peptide is spotted in triplicates. Mutated residues are highlighted in red. B. Affinities (Kd values) of wild-type (WT) and phosphorylated SCOC LIR peptides determined by bio-layer interferometry (BLI). Colour code indicates fold changes relative to Kd value of SCOC WT LIR peptide binding to the corresponding ATG8 protein. Yellow circles indicate phosphorylation modification in SCOC LIR peptide. (data are average Kd values ± standard error calculated from non-linear regression curve fits, n = 2–3). C. GFP-Trap immunoprecipitation (IP) of indicated GFP-ATG8 proteins and immunoprecipitated MYC-tagged SCOC from HEK293A cells analysed by Western blots. To ensure equal amounts of tagged protein are present in each reaction, MYC-tagged SCOC (isoform 3) proteins and GFP-tagged ATG8 proteins were overexpressed separately and lysates were mixed prior to performing the IP experiments. D. and E. Quantification of WT and phospho-mimetic mutant, MYC-tagged SCOC (isoform 3) protein binding to GFP-LC3A (D.) and GFP-GABARAP (E.). Statistical analysis using one-way analysis of variance (ANOVA) test; mean ± s.d.; data from at least three independent experiments (n = 4 [GFP-LC3A]; n = 3 [GFP-GABARAP]); ****p ≤ 0.0001; ***p ≤ 0.001; *p ≤ 0.05.
To better understand how phosphorylation of these LIR residues alters the affinity of the SCOC LIR motif to individual ATG8 proteins, we next performed BLI affinity measurements using phosphorylated SCOC LIR peptides (Figure 4B). Consistent with previous findings on the LIR domain of NIX,33 phosphorylation of S12SCOC preceding the LIR motif (position X−2) strongly increased LC3 subfamily binding by six- to nine-fold. GABARAP subfamily binding was also enhanced two-fold for GABARAP/GABARAPL1 and three-fold for GABARAPL2. However, T13SCOC phosphorylation only slightly improved SCOC LIR binding (two-fold) to LC3A. Phosphorylation of T15SCOC and S18SCOC resulted in seven- to five-fold increase in LC3A and LC3B binding as well as two- to four-fold increase in LC3C binding, whereas no or less than 2-fold changes in affinity were observed for the GABARAPs. Double mutation of S12/T15SCOC and S12/S18SCOC had additive effects on LC3 subfamily binding and particularly, the KD values of LC3A and LC3B changed dramatically (LC3A: 40- and 17-fold, LC3B: 56- and 34-fold, respectively). A 21-fold increase in affinity was observed for LC3C, while the affinity of GABARAP proteins only improved three- to six-fold.
Consistently, phospho-mimetic mutation S18ESCOC increased MYC-SCOC binding to GFP-LC3A in GFP-Trap IP experiments (Figure 4C, 4D), but did not alter MYC-SCOC interaction with GFP-GABARAP (Figure 4C, 4E). MYC-SCOC S12E/S18E bound significantly more to both GFP-LC3A and GFP-GABARAP and the strongest binding was detected when phospho-mimetic mutations were introduced for all four potential phosphorylation sites (Figure 4C-E), further confirming additive effects of these sites.
In summary, analysis of potential phosphorylation sites in the SCOC LIR domain in position X−1, X−2, X1 and X4 revealed that not only phosphorylation of position X−2 preceding the core LIR motif, but also positions X1 and X4 (located within and C-terminal to the core LIR motif) selectively enhance LC3 subfamily binding and that these residues are critical in regulating binding specificity of the SCOC LIR motif. Interestingly, phosphorylation of S12/T15SCOC and S12/S18SCOC had a substantial effect on LC3 binding promoting it from very weak to strong binding suggestive of an important regulatory function in vivo.
Structure of the phosphorylated SCOC LIR domain in complex with GABARAPL1
Next, we tried to co-crystalise GABARAPL1, LC3C and LC3A with phosphorylated SCOC LIR peptides to solve the structures of phosphorylated SCOC LIR:ATG8 complexes. GABARAPL1 was used instead of GABARAP, since purified GABARAP aggregated during concentration. Our attempts to co-crystalise SCOC-2pS and SCOC-2pT peptides in complex with LC3A and LC3C, were also unsuccessful, but we were able to determine the crystal structure of a SCOC LIR domain peptide phosphorylated at S12SCOC and S18SCOC (referred to as SCOC-2pS hereafter) in complex with GABARAPL1 at a resolution of 1.45 Å (Figure 5A, Supplementary Figure 3C) as well as the structure of a SCOC LIR domain peptide phosphorylated at T13SCOC and T15SCOC (referred to as SCOC-2pT) in complex with GABARAPL1 at a resolution of 1.72 Å (Figure 5B, Supplementary Figure 3D).
Figure 5.

Structure of the phosphorylated SCOC LIR domain in complex with GABARAPL1. A. Structure of the phosphorylated SCOC pS12/S18 LIR domain peptide (aa 9–19) bound to GABARAPL1. The SCOC LIR peptide sequence is shown in light green cartoon with interacting residues depicted as sticks. GABARAPL1 is displayed in white cartoon and transparent surface with hydrophobic pocket 1 (pink) and 2 (blue). B. Structure of the phosphorylated SCOC pT13/T15 LIR domain peptide (aa 10–21) bound to GABARAPL1. The SCOC LIR sequence is shown in yellow cartoon with interacting residues depicted as sticks. C. Schematic overview of LIR interactions observed in the structures of GABARAP bound to SCOC WT LIR domain (orange), and GABARAPL1 bound to SCOC pS12/S18 (light green) and SCOC pT13/T15 (yellow) LIR domains. GABARAP/GABARAPL1 residues are boxed and shown in black. Blue lines indicate hydrophobic interactions, green lines hydrogen bonds, and green double arrow salt bridges. Pink and blue boxes indicate canonical LIR interactions (hydrophobic contacts) with HP1 and HP2. D. Superposition of the structures of GABARAP bound to SCOC WT (orange), and GABARAPL1 bound to SCOC pS12/S18 (light green) and SCOC pT13/T15 (yellow) LIR domains. Only the surface of GABARAP from the GABARAP:SCOC WT LIR complex structure is displayed in white transparency with HP1 and HP2 coloured in pink and blue, respectively.
In both structures SCOC formed the same canonical LIR interactions with GABARAPL1 as described previously for the SCOC WT LIR-GABARAP complex (Figure 2C). Furthermore, we observed multiple additional contacts in both structures: Hydrogen bonds were formed between the carbonyl of pS12SCOC-2pS/S12SCOC-2pT and K48GABL1, as well as between R28GABL1 and the carbonyl of I17SCOC-2pS or the sidechain of N16SCOC-2pT (Figure 5 A-C). Moreover, we detected some hydrophobic contacts between L19SCOC-2pS/2pT and the edge of HP2. In the SCOC-2pS LIR, the phosphoserine pS12 formed a large network of electrostatic interactions with H9GABL1, K48GABL1 and R47GABL1, while pS18 was not interacting with GABARAPL1. Similarly, in the SCOC-2pT LIR crystal structure, pT13 was not interacting with GABARAPL1, but pT15 in position X1 formed salt bridges with K46GABL1 and R67GABL1.
The leucine residue L19SCOC of SCOC (position X+5) also formed hydrophobic interactions with L55GAB/GABL1 and L63GAB/GABL1 of GABARAP in the SCOC WT-LIR:GABARAP complex (Figure 2C,D and Figure 5C). In mutational peptide array scans (Figure 2A, B and Supplementary Figure 2D) only substitutions of L19SCOC with aromatic aa (W, F, Y) or hydrophobic aa (I) resulted in substantial ATG8 binding, indicating that hydrophobic contacts mediated by the LIR residue in position X+5 were important for both GABARAP and LC3 subfamily binding.
Whereas more distant N-terminal residues (K6SCOC (X−8), E10SCOC (X−4) and D11SCOC (X−3)) contributed to binding of the SCOC WT LIR motif to GABARAP, no involvement of E10SCOC (X−4) and D11SCOC (X−3) (which were present in the phosphorylated peptides) was detected in the structures of the SCOC-2pS:GABARAPL1 and SCOC-2pT:GABARAPL1 complexes (Figure 5 C-D).
Distant flanking residues in the SCOC LIR motif contribute to ATG8 binding
To further refine the data and elucidate the importance of the flanking residues in regulating SCOC LIR binding, we performed BLI affinity measurements (Figure 6A). Removal of the N-terminal sequence (except for T13SCOC) resulted in a profound decrease (more than 140-fold) in ATG8 binding to the SCOC LIR motif (aa 13–25). Loss of the N-terminal lysine and glutamate residues in the SCOC LIR motif (aa 11–25) also strongly reduced interaction with LC3A (27-fold), LC3C (16-fold), GABARAP (37-fold) and GABARAPL1 (104-fold). Equivalently, truncation of the C-terminal flanking residues of the SCOC LIR motif (except for S18SCOC) resulted in a substantial decrease in affinity ranging from four-fold for GABARAP and GABARAPL1 to 10-fold and 22-fold for LC3A and LC3C, respectively. Loss of the C-terminal aspartate, isoleucine and histidine residues in the SCOC LIR motif (aa 6–20) lead to a two- to three-fold reduction binding to GABARAPs and LC3C and a more than seven-fold reduction in LC3A binding. Interestingly, both phosphorylation of S12SCOC and S18SCOC rescued ATG8 binding of the SCOC LIR motif, indicating that phosphorylation of these residues can compensate the loss of flanking residues and positively regulates SCOC LIR binding to ATG8 proteins.
Figure 6.

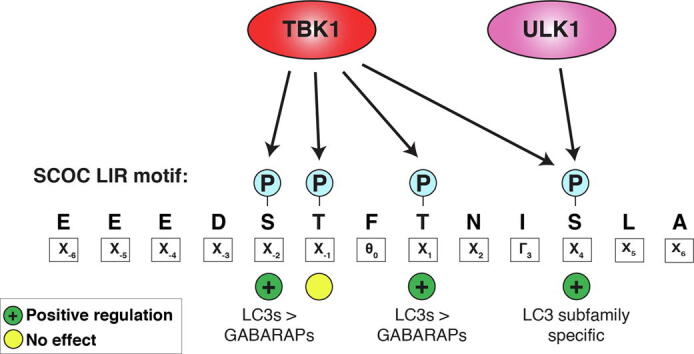
Contribution of the flanking regions in the SCOC LIR domain to ATG8 binding. A. Affinities (Kd values) of wild-type (WT), truncated and phosphorylated SCOC LIR domain peptides determined by bio-layer interferometry (BLI). Colour code indicates fold changes relative to Kd value of SCOC WT LIR peptide binding to the corresponding ATG8 protein. Blue circles indicate phosphorylation site in SCOC LIR sequence. Acidic residues are highlighted in red. (data are average Kd values ± standard error calculated from non-linear regression curve fits, n = 2). B. Schematic overview of regulatory phosphorylation sites identified in SCOC and other LIR motifs. Red circle with minus sign indicates negative regulation, green circle with plus sign positive regulation and yellow circle no regulation of ATG8 binding.
Discussion
Phosphorylation of LIR motifs of autophagy receptors and adaptors plays an important role in regulating selective autophagy pathways, e.g. mitophagy33, 34, 35, 36, 37, 38 and xenophagy.32 Our understanding of the underlying regulatory mechanisms, i.e. kinases mediating LIR motif phosphorylation as well as key phosphorylation sites in LIR motifs modulating ATG8 binding affinity and specificity, is still limited. Several studies have shown that phosphorylation of residues directly preceding the core LIR motif (in position X−1, X−2, and X−6) enhance ATG8 binding20, 32, 33, 34, 36, 38 (Figure 6B). Inhibition of LC3 binding has been shown for phosphorylation of the aromatic core LIR residue Y18FUNDC1 (Θ0)35 and the N-terminal residue S13FUNDC1 (X−5)37 of the mitophagy receptor FUNDC1. Whether LIR-ATG8 interactions are regulated by phosphorylation of other residues within or C-terminal to the core LIR motif still remains elusive.
In this study we identified a functional LIR motif in the small Golgi protein SCOC (isoform 3), which was phosphorylated by ULK kinases (ULK1-3) as well as TBK1 at serine/threonine residues immediately flanking or within the core LIR motif of SCOC (S12/T13/T15/S18SCOC). To determine and understand the regulatory effects of these phosphorylation sites on SCOC LIR binding affinity and specificity towards all six mammalian ATG8 proteins we carried out a comprehensive analysis involving high resolution crystal structures, peptide array and IP binding assays, as well as BLI affinity measurements.
Phosphorylation of S12SCOC in position X−2 of the SCOC LIR motif increased ATG8 binding (Figure 4A, B). Surprisingly, phosphorylation of T13SCOC had little or no effect on ATG8 binding affinities although phosphorylation of X-1 residues or substitution with D/E (mimicking phosphorylation) has been widely reported to positively regulate interaction with ATG8 proteins20, 32, 33, 34, 35, 36 (Figure 6B). In our crystal structure phosphorylated S12SCOC formed a large network of hydrogen bonds with H9GABL1, K48GABL1 and R47GABL1, whereas phosphorylated T13SCOC was solvent exposed and not interacting with LDS amino acids of GABARAPL1 (Figure 5A-C). This lack of interaction explains why phosphorylation of T13SCOC of the SCOC LIR domain did not enhance GABARAP subfamily binding (Figure 4B).
S12SCOC phosphorylation enhanced the affinity of the SCOC LIR motif to LC3s rather than GABARAPs (Figure 4B). Since the LDS of LC3 subfamily proteins has more basic residues at the edge of HP1 (R10/R11LC3A/B and R16/K17LC3C on helix α1), a larger network of electrostatic interactions with acidic or phosphorylated LIR residues in position X−2 to X−4 can be formed,27, 30, 54, 55, 56, 57 which promotes more binding to LC3s than GABARAPs.
Phosphorylation of T15SCOC at position X1 of the SCOC LIR motif also increased much stronger the binding affinities of LC3 subfamily proteins (especially LC3A and LC3B) than of GABARAPs (Figure 4B). In the WT SCOC LIR:GABARAP complex structure two hydrogen bonds were formed between the main chain residues of T15SCOC and K48GAB and L50GAB (Figure 2C, D). In the SCOC-2pT LIR:GABARAPL1 complex structure phosphorylated T15SCOC formed two additional salt bridges with K46GABL1 and R67GABL1 of GABARAPL1 (Figure 5B). Both K46GABL1 and R67GABL1 are conserved in both LC3 and GABARAP proteins (Supplementary Figure 2F). Additional electrostatic interactions therefore may also occur between phosphorylated T15SCOC and the corresponding residues (K49LC3A/B/K55LC3C and R70LC3A/B/R76LC3C) in LC3 subfamily proteins. Interestingly, in a compilation of 100 LIR sequences E is the residue most frequently found in position X1 followed by V and D.5 Several LIR-ATG8 complex structures of LIR motifs with E/D in position X1 have been solved. Salt bridges between E in position X1 and R67GAB/GABL1 are observed in the structures of the ATG4B LIR:GABARAPL1,22 VPS34 LIR:GABARAP, and ATG14-LIR:GABARAP20 complexes. Electrostatic interactions between E/D in position X1 and R70LC3B (corresponding to R67GAB of GABARAP) are also reported for the TECPR2 LIR:LC3B,58 FYCO1 LIR:LC3B,54, 55, 56 and RavZ LIR2:LC3B59 complex structures. Thus, acidic or phosphorylated residues in position X1 seem to generally stabilize LIR-ATG8 interactions. We currently do not understand why these additional electrostatic interactions significantly increase the affinity of the pT15 SCOC LIR motif to LC3s more than GABARAPs (Figure 4B) and this needs further investigation.
Growing evidence indicates that the region C-terminal to the core LIR motif is involved in ATG8 binding and can even regulate ATG8 binding specificity.30, 54, 55, 56, 60, 61 We recently demonstrated a critical role of the X4 LIR domain residue in regulating ATG8 binding specificity.30 The proline residue in position X4 specifically impairs binding of the ULK1 and FIP200 LIR motifs to LC3s, whereas mutation of this residue to D (P361DULK1 and P706DFIP200) improves it. The LIR domain of the centrosomal satellite protein PCM1 has an E (E1959PCM1) in position X4. Mutation of E1959PCM1 to T specifically decreases the affinity of the PCM1 LIR motif to LC3s, but does not alter the affinity to GABARAP proteins.30 In support of a role for X4 in generating ATG8 specificity, phosphorylation of S18SCOC in position X4 of the SCOC LIR domain also increased LC3 subfamily binding (Figure 4B, C). Notably, in the SCOC-2pS LIR:GABARAPL1 complex structure phosphorylated S18SCOC was solvent exposed and not interacting with GABARAPL1 (Figure 5A). Similarly, phosphorylation of the LIR motif of the cysteine protease ATG4B at position X4 (S392ATG4B), potentially implicated in LC3 delipidation50, does not contribute to GABARAPL1 binding.22 However, in the PCM1 LIR:GABARAP complex structure E1959PCM1 forms a salt bridge with R28GAB of GABARAP.30 Both GABARAP and LC3 subfamily proteins have a basic residue (R28GAB/GABL1/GABL2, K30LC3A/LC3B or K36LC3C) in the LDS that could potentially engage with the phosphorylated LIR residue in position X4. To further understand the binding mechanism of this region of the LIR domain, more structural data of C-terminally extended LIR motifs (with E/D/pS/T in position X4) in complex with LC3 proteins are needed.
Whereas electrostatic interactions between GABARAP and acidic residues in the N-terminal region of the SCOC LIR domain were observed, we did not detect interactions of acidic residues C-terminal (position X7 to X10) to the core LIR motif. Removal of C-terminal residues more distant than L19 (SCOC aa 20–27) reduced binding (Figure 6A), suggesting that these residues are important for ATG8 binding. However, electrostatic side chain and/or backbone interactions of these residues with the LDS seem to be highly dynamic and difficult to detect by structural analyses such as X-ray crystallography.
Notably, phosphorylation of S18SCOC rescued binding to GABARAPs and LC3s in the absence of the C-terminal SCOC LIR motif residues in position X7 to X11 (aa 21–25) (Figure 6A), suggesting that pS18SCOC can contribute or stabilize GABARAP and LC3 binding if interactions with more C-terminally located LIR motif residues are weak or weakened. Similarly, S12SCOC phosphorylation rescued ATG8 binding after deletion of aa 6–10 (positon X−4 to X−8) (Figure 6A). In the SCOC-2pS LIR:GABARAPL1 structure electrostatic interactions with the more distant SCOC LIR domain residues (E10SCOC (X−4) and D11SCOC (X−3)) were not detected and may not be required for LIR binding to GABARAPL1 due to additional interactions established by phosphorylated S12SCOC.
In summary, our data demonstrates that there is high plasticity and dynamics in the interactions that the LDS of ATG8 proteins forms with the extended N- and C-terminal regions flanking the core LIR motif. We identified novel regulatory phosphorylation sites within and C-terminal to the core LIR motif of SCOC. ATG8 binding affinity and specificity of other LIR motifs could potentially also be positively regulated by phosphorylation of these positions.
Lastly, autophagy-independent functions have been reported for ULK kinases,46, 62 GABARAP,63 SCOC and FEZ1.41, 44, 45, 64 SCOC is not degraded by autophagy (Supplementary Figure 2B and C) and therefore possibly functions as an autophagy adaptor protein and/or in autophagy-independent processes. Through its interaction with the kinesin1 adaptor protein FEZ139, 41, 42, 43 SCOC might be involved in microtubule-dependent transport of ATG8 proteins and autophagosomal membranes in neurons.
Phosphorylation of both S12SCOC and T15SCOC or S18SCOC resulted in a 34 to 56-fold increase in LC3B binding affinity with potential relevance in cells (Figure 4B). As the phos-tag gel assay to demonstrate ULK1 phosphorylation of the SCOC LIR domain in HEK293A cells (Figure 3C) did not confirm the in vitro kinase assay results (Figure 3B), further analyses are needed to determine whether this phosphoregulation of LC3B binding occurs in cells. Understanding the role of SCOC in neuronal autophagy and axonal transport will be an interesting direction for future research.
Methods
Antibodies
The following primary antibodies were used: HRP-conjugated anti-GST (GE Healthcare, RPN1236); anti-MYC (CRUK, 9E10; Abcam, ab9106); anti-GFP (CRUK, 3E1; Santa Cruz, sc-8334); anti-GABARAP (Abgent, AP1821a); anti-actin (Abcam, ab8227), anti-FLAG M2 (SIGMA, F1804), anti-TGN46 (Serotec, AHP500), anti-beta-tubulin (Abcam, ab6046), anti-ULK1 (Santa Cruz, sc-33182); anti-ATG1365; anti-FIP200 (Bethyl Labs, A301-536A-1); anti-FEZ2 (SIGMA, HPA0355978). Secondary antibodies for IF were: anti-rabbit IgG Alexa Fluor 555 and 647; anti-mouse IgG Alexa Fluor 555, 647; anti-sheep IgG Alexa Fluor 647 (all from Life Technologies). HRP-conjugated secondary antibodies used for WB were from GE Healthcare.
Plasmids
pDEST-EGFP, pDEST-EGFP-LC3A, pDEST-EGFP-GABARAP, pDEST15-ATG8 homologs (GST-tagged human ATG8 homologs), pENTRY-SCOC (isoform 3), pDEST-FLAG-SCOC (isoform 3) and pDEST-MYC-SCOC (isoform 3) were generated by the laboratory of Terje Johansen (UiT, The Arctic University of Norway, Tromsø).17, 18, 39, 66 pENTRY-SCOC F14A/I17A (isoform 3), pENTRY SCOC S18E, S12E/S18, and S12E/T13E/T15E/S18E (isoform 3) were generated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene). Gateway destination plasmids were made using Gateway LR recombination reactions (Invitrogen) and the pDEST-MYC vector (N-terminal MYC-tag) according to manufacturer’s instructions. STOP codons were introduced at the end of the SCOC (isoform 3) sequences using the QuickChange Multi Site-Directed Mutagenesis kit (Agilent). FEZ2 cDNA was amplified from a HeLa cDNA library and cloned into the pENTRY1A vector. pDEST-EGFP-FEZ2 was obtained from the pENTRY1A-FEZ2 by GATEWAY LR reaction.
Human ATG8 homologs in the pAL plasmid (containing N-terminal GST-tag followed by a 3C protease cleavage site)30 were used to recombinantly express ATG8 proteins for BLI affinity measurements. For crystallization, the SCOC wild type LIR sequence (aa 6–23) were N-terminally fused with a Gly-Ser linker to full-length GABARAP (GST-GABARAP (pAL)) using NcoI and BamHI sites. The QuickChange Multi Site-Directed Mutagenesis Kit (Agilent) was used to introduce the S113StopGABARAP mutation and remove the last five aa from the C-terminus of GABARAP.
The pEGFPN2-FEZ1 expression plasmid67 was kindly provided by Caroline Whitehouse (King’s College, UK). N-terminal MYC-tagged wild type and kinase-dead (K46I) murine ULK1 expression plasmids (pRK5)68 as well as pEGFPC1-SCOC39 expression plasmids have been described previously. pEGFPC1-SCOC 9A mutant (SCOC isoform 3 S26A, S27A, S36A, T60A, T72A, S77A, S106A, S108A, S109A), pEGFPC1-SCOC 13A mutant (SCOC isoform 3 S12A, T13A, T15A, S18A, S26A, S27A, S36A, T60A, T72A, S77A, S106A, S108A, S109A), pEGFPC1-SCOC 14A mutant (SCOC isoform 3 S12A, T13A, T15A, S18A, S26A, S27A, S36A, T60A, T72A, S77A, S92A, S106A, S108A, S109A) and pEGFPC1-SCOC 4A mutant (SCOC isoform 3 S12A, T13A, T15A, S18A) expression plasmid were generated using QuickChange Multi Site-Directed Mutagenesis Kit (Agilent) and/or Q5 Site-Directed Mutagenesis Kit (New England BioLabs).
All plasmid constructs generated in this study were verified by DNA sequencing.
Cell culture and generation of stable cell lines
HEK293A cells (provided by Cell Services of the Francis Crick Institute) were grown in a humidified incubator at 37 °C in 10% CO2 in full medium (Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and 4 mM l-glutamine). Lipofectamine 2000 (Life Technologies) was used for transient transfection of cells according to the manufacturer’s instructions. DNA plasmids were used at a concentration of 1 mg/ml of transfection mix. Cells were harvested after 24 h.
HeLa Flp-In T-Rex cell lines expressing GFP-SCOC (isoform 1), GFP-SCOC (isoform 3), GFP-SCOC F14A/I17A (isoform 3) from a tetracycline inducible CMV promoter were made using the FlpIn T-Rex system (Thermofisher, R71407). pDest-EGFP-Flp-In SCOC iso1, pDest-EGFP-Flp-In SCOC iso3 and pDest-EGFP-Flp-In SCOC iso3 LIRm constructs were established by GATEWAY cloning into the pDEST-EGFP-FlpIn vector.18 The HeLa-based Flp-In T-Rex host cell line (Invitrogen) contains a single integrated FRT site for insertion of selected constructs. To generate stable cell lines, Flp-In plasmids carrying the selected GFP-tagged constructs were co-transfected with pOG44 encoding the Flp-In recombinase in the ratio of 1:3. Cells were selected by treatment with 200 µg/ml Hygromycin B (Invitrogen, #10687010) and 7,5 µg/ml Blasticidine (Gibco, A1113903). Expression from the CMV-TetO2 promoter was induced by adding 0.5 μg/ml of tetracycline (Sigma, T7660) for 16 to 24 hours. HeLa Flp-In T-Rex cell lines were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 4 mM l-glutamine, 1% streptomycin-penicillin, 100 µg/ml Hygromycin B, and 3.5 µg/ml Blasticitidine. Cells were treated as indicated with 100 nM Bafilomycin A1, 10 µM MG132, or 1 µM epoxomicin in full medium or Earle’s balanced salt solution (EBSS).
Western blotting
Cells were lysed in ice-cold TNTE buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% w/v Triton X-100, 10% v/v glycerol, 5 mM EDTA) containing EDTA-free Complete Protease Inhibitor cocktail (Roche) and PhosSTOP (Roche). Lysates were cleared by centrifugation and resolved on NuPAGE Bis-Tris 4–12% gels (Life Technologies) followed by transfer onto a PVDF membrane (Millipore). After incubation with primary and secondary antibodies, the blots were developed by chemiluminescence using Immobilon Classico Western HRP substrate (Merck Millipore) or enhanced chemiluminescence reagents (GE Healthcare). Densitometry was performed with ImageJ software. For western blotting of weak signal antibodies, primary antibody was diluted with SignalBoost Immunoreaction Enhancer Kit (Merck Millipore, 407207) and blots were developed with Luminata Crescendo Western HRP substrate (Merck Millipore).
GST pulldown assay
GST- and GST-tagged proteins were expressed in E. coli LE392 and BL21(DE3), respectively, purified and immobilized on Glutathione Sepharose 4 Fast Flow beads (GE Healthcare). One μg of the appropriate DNA constructs were used to produce full reactions (50 μl) of 35S-labeled proteins following the TNT T7 Quick Coupled Transcription/Translation system (Promega). For GST pulldown assays 10 μl of each in vitro translated protein diluted in 200 μl of NET-N buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40 (v/) containing cOmplete Mini EDTA-free protease inhibitor cocktail (Roche Applied Science)). The binding assay (GST pulldown), gel electrophoresis and visualization of binding by autoradiography was performed as described by Johansen et al. 69.
Immunoprecipitation
For GFP-Trap IP experiments (Figure 4C), cells were lysed in ice-cold TNTE buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% w/v Triton X-100, 10% v/v glycerol, 1x Complete protease inhibitor (Roche), 1x PhosSTOP (Roche)). Lysates were cleared by centrifugation at 16,000xg, 4 °C for 15 min. To ensure that equal amounts of overexpressed GFP- and MYC-tagged proteins were present in each reaction, equal volumes of GFP-tagged protein extracts were mixed with equal volumes of MYC-tagged protein extracts. Lysates were precleared with control agarose beads (ChromoTek) for 1 hour at 4 °C. GFP-tagged proteins were immunoprecipitated using GFP-TRAP beads (ChromoTek) overnight at 4 °C. Beads were washed 3 times with TNTE (w/o PhosSTOP) and bound protein was eluted with 2x Laemmli buffer at 100 °C for 10 min before resolving by SDS-PAGE (4%–12% Bis-Tris NuPAGE gels, Life Technologies) and western blotting. GFP-Trap IP (Figure 3A) and FLAG IP (Supplementary Figure 3A) experiments were performed with slight modifications. Cells were lysed in TNTE buffer A (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% w/v Triton X-100, 10% v/v glycerol, 1x Complete protease inhibitor (Roche), 1x PhosSTOP (Roche)), cell lysates were not precleared with control agarose beads and IP reactions were washed with TNTE buffer B (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.1% w/v Triton X-100, 10% v/v glycerol, 1x Complete protease inhibitor (Roche), 1x PhosSTOP (Roche)). FLAG-tagged SCOC was immunocaptured using Anti-FLAG-M2 affinity agarose gel (SIGMA, A2220).
In vitro kinase assay and mass spectrometry
For in vitro kinase assay, 50 ng of recombinant kinases [ULK1 (SignalChem, #U01-11G), ULK2 (Merck Millipore, #14-772); ULK3 (Merck Millipore, #14-755); TBK1 (Merck Millipore, #14-628)], 1 µg of GST tagged SCOC isoform 3 and 60 mM ATP and combined in 30 μl of kinase buffer (35.5 mM Tris pH7.5, 10 mM MgCl2, 0.5 mM EGTA pH 8.0, 0.1 mM CaCl2) supplemented with cOmplete Mini EDTA-free protease inhibitor cocktail tablets (Roche Applied Science, #11836170001) and phosphatase inhibitor cocktail (Merck Millipore, #524625) and incubated for 20 minutes at 30 °C. The reaction was stopped by addition of 6xSDS-loading buffer and samples separated by SDS-PAGE. The gels were stained and SCOC band cut out for protease digestion and mass spectrometric analysis. In-gel chymotrypsin digestion was performed before analysis by high-performance liquid chromatography–tandem mass spectrometry (HPLC-MS/MS). Gel pieces were subjected to in-gel reduction, alkylation, and digestion using 6 ng/μl chymotrypsin (V1062; Promega) or Trypsin (V5111; Promega). OMIX C18 tips (Varian) were used for sample cleanup and concentration. Peptide mixtures containing 0.1% formic acid were loaded onto a ThermoFisher Scientific EASY-nLC1200 system. Samples were injected to a trap column (Acclaim PepMap 75 μm × 2 cm, C18, 3 μm, 100 Å; ThermoFisher) for desalting before elution to the separation column (EASY-Spray column, C18, 2 μm, 100 Å, 50 μm, 50 cm; ThermoFisher). Peptides were fractionated using a 4–40% gradient of increasing amounts of 80% Acetonitrile in water over 60 min at a flow rate of 300 nl/min. The mobile phases contained 0.1% formic acid. Separated peptides were analyzed using an Orbitrap Fusion Lumos mass spectrometer. The mass spectrometer was operated in a data-dependent mode with the precursor scan in the orbitrap over the range m/z 350–1500. The most intense ions were selected for ETD or CID fragmentation using 3 sec between each master scan. Dynamic exclusion was set to 8 s. The Orbitrap AGC target was set to 4E5 with maximum injection time 50 ms. The MS2 scans in the Ion Trap was set to 1E4 with dynamic injection time. Precursor ions with charge 3+ in the m/z range 350–650 and 4+ or 5+ ions in the m/z range 350–900 was fragmented with ETD. All ions with 6+ or higher were also fragmented using ETD. The rest of the precursor ions were fragmented using CID. Protein identification and PTM mapping was done using the Proteome Discoverer 2.4 software (ThermoFisher) using the ptmRS module (>75%). Peak lists generated in Proteome Discoverer was searched against the UniProt Homo sapiens proteome (april 2019; 73,645 sequences) using the built in Sequest HT search engine. Search parameters were:
| - Enzyme: | Chymotrypsin (Full); |
| - Max missed cleavage: | 2 |
| - Precursor mass tolerance: | 10 ppm |
| - Fragment mass tolerance: | 0.6 Da |
| - Fixed modifications: | Carbamidomethyl (C) |
| - Dynamic modifications: | Oxidation (M), Phospho (ST), Acetyl (protein N-term), Met-loss (protein N-term), Met-loss + Acetyl (protein N-term) |
| - Threshold score | Xcorr > 2.0 |
| - #peptides | >2 |
Lambda-phosphatase treatment and Phos-tagTM-SDS Page
To determine SCOC phosphorylation in vivo (Fig. 3C), myc-ULK1 WT or myc-ULK1 KI were co-expressed along with pEGFPC1, pEGFPC1-SCOC or pEGFPC1-SCOC 9A in HEK293A cells. 24 h after transfection, cells were starved for 2 h with EBSS. Cells were then harvested and washed with cold PBS, and lysed in TNTE buffer (w/o 10% v/v glycerol). The cell lysates were immunoprecipitated using GFP-TRAP (ChromoTek) as decribed above. Prior to lambda-phosphatase treatment, GFP-TRAP beads were washed 3 times with TNTE buffer (w/o 10% v/v glycerol, EDTA and PhosSTOP) and split into two equal portions for λ-phosphatase treatment and control. Lambda phosphatase was added into each sample with phosphatase reaction buffer and MnCl2 following manufacturer’s instructions (New England BioLabs). Sodium orthovanadate (Na3VO4) was added into each sample at the final concentration of 100 μM, as a control. All reactions were incubated at 30 °C for 30 min. 2x Laemmli buffer was added into all the samples to stop reaction and then boiled at 100 °C for 5 min. Samples were separated by 10% SDS-PAGE with 25 μM Phos binding reagent (Phosbind) acrylamide (APExBIO, F4002) and analysed by immunoblotting.
Peptide array and GST overlay assay
GST or GST-ATG8 proteins were expressed (from GST-ATG8 (pAL) plasmids) in E. coli BL21 (DE3) plysS cells (Agilent) in LB medium supplemented with 50 µg/ml kanamycin. Expression was induced by addition of 0.5 mM IPTG at OD600 = 0.6 and cells were incubated at 25 °C overnight or at 37 °C for 5 h. Harvested cells were lysed in 50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 0.1% Triton X-100, 0.4 mM AEBSF and 15 µg/ml Benzamidine. Fusion protein was batch-adsorbed onto Glutathione-Sepharose 4B beads (GE Healthcare). After five washes with wash buffer (50 mM Tris, pH 8.0, 250 mM NaCl, 0.4 mM AEBSF and 15 µg/ml benzamidine) fusion proteins were eluted in 50 mM Tris pH 8.0, 2 mM L-glutathione reduced, 0.4 mM AEBSF and 15 µg/ml benzamidine.
A MultiPep or ResPep SL automated synthesizer (INTAVIS Bioanalytical Instruments AG, Germany) was used for SPOT synthesis of peptide arrays on cellulose membranes.70 After blocking membranes in TBST with 5% nonfat dry milk, peptide interactions with GST or GST fusion proteins were tested by overlaying the membranes with either 1 µg/ml (mutational peptide array scan) or 2 µg/ml of recombinant protein (all other peptide arrays) for 2 h at room temperature. Membranes were washed in TBST, and bound proteins were detected with HRP-conjugated anti-GST antibody (1:5000, GE Healthcare, RPN1236).
Protein expression and purification for crystallization
GST-GABARAPL1 full-length (pAL)30 or GST-SCOC6-23LIR-GABARAP(S113Stop) (pAL) were expressed in E. coli Rosetta (DE3) pLysS at 25 °C overnight. Bacteria were harvested by centrifugation and resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 0.1% TX-100, 0.5 mM TCEP, 0.5 mM AEBSF and 15 ug/ml benzamidine). The fusion proteins were batch-adsorbed onto a glutathione-Sepharose affinity matrix and recovered by cleavage with 3C protease at 4 °C overnight in 50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.5 mM TCEP. The protein was then purified by size exclusion chromatography using a Superdex 75 column equilibrated and run in 25 mM Tris-HCl, pH 8.0, 150 mM NaCl and 0.5 mM TCEP. Peptides were synthesized by the Francis CRICK Institute peptide chemistry science technology platform.
Crystallisation and data processing
GABARAPL1:SCOC LIR peptide complexes were prepared by mixing purified full length GABARAPL1 and SCOC-2pS peptide (residues 9–19, EED-pS-TFTNI-pS-L) or SCOC-2pT peptide (residues 10–21, EDS-pT-F-pT-NISLAD) at a 1:3 molar ratio. The complexes were dialysed overnight in 25 mM Tris-HCl, pH 8.0, 150 mM NaCl and 0.5 mM TCEP buffer, using a 500–1000 Da MWCO dialysis tubing for both complex. The GABARAPL1:SCOC LIR peptide complexes and the SCOC6-23LIR-GABARAP chimera protein were all crystallized at 20 °C using the sitting-drop vapour diffusion method with a protein concentration of 10–20 mg/ml. The initial crystallization trial was performed using Qiagen (JCSG core 1–4, AMSO4), Molecular dimension (PACT, Wizard 1–4), Jena Bioscience (PiPEG). In all cases the drop included 0.5 μl of protein and 0.5 μl of mother liquor. For SCOC6-23LIR-GABARAP crystals grew in 50 mM Bicine pH 8.4, 30% PEG 1500. For SCOC-2pS:GABARAPL1 and SCOC-2pT:GABARAPL1 crystals grew in 0.1 M MES pH 6.5, 3.5 M AMSO4, 1% MPD and 0.1 M Tris pH 8.0, 20% PEG 1500, respectively. Crystals were flash-frozen in liquid nitrogen, and X-ray data sets were collected at 100 K on I03, I02 and I04-1 beamline (mx9826-49, mx9826-55 and mx9826-41) of the Diamond Light Source Synchrotron (Oxford, UK). Data collection and refinement statistics are summarized in Table 1. The data sets were indexed and scaled with xia2.71 Molecular replacement was achieved by using the atomic coordinates of the peptide-free GABARAP (PDB code: 1GNU) and GABARAPL1 (PDB code: 2R2Q) in PHASER.72 Refinement was carried out using Phenix.73 Model building was carried out in COOT.74 Model validation used PROCHECK75; and figures were prepared using the graphics program PYMOL (http://www.pymol.org).
Bio-layer interferometry assay
Bio-layer interferometry (BLI) is an optical analytical technique for measuring kinetics of interactions in real-time. The biosensor tip surface immobilized with a ligand is incubated with an analyte in solution, resulting in an increase in optical thickness at the biosensor tip and a wavelength shift, which is a direct measure of the change in thickness. Bio-layer interferometry analyses of ATG8s binding to immobilized biotinylated LIR peptides were performed using an Octet Red 96 (ForteBio). 50 μg/ml of biotinylated LIR peptide was immobilized on streptavidin coated biosensor (SA, ForteBio) and the typical immobilization levels were above 0.3 nm. Ligands-loaded SA biosensors were then incubated with different concentrations of ATG8. All binding experiments were performed in solid-black 96-well plates containing 200 μl of solution (25 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM TCEP, 0.1% Tween20, 1 mg/ml BSA) in each well at 25 °C with an agitation speed of 1000 rpm. Each measurement was repeated 2 to 3 times. Dissociation constants for LIR-ATG8 interactions were determined from plotting the increase in BLI response as a function of the protein concentration and fitting using non-linear regression of ForteBio 7.1 data analysis and GraphPad Prism 7 softwares.
The following peptides were synthesized using FMOC solid phase peptide chemistry by the Francis CRICK Institute peptide chemistry science technology platform:
| SCOC WT (aa 6–25): | Biotin-Linker-KEEEEDSTFTNISLADDIDH-Amide |
| SCOC pS12: | Biotin-Linker-KEEEED-pS12-TFTNISLADDIDH-Amide |
| SCOC pT13: | Biotin-Linker-KEEEEDS-pT13-FTNISLADDIDH-Amide |
| SCOC pT15: | Biotin-Linker-KEEEEDSTF-pT15-NISLADDIDH-Amide |
| SCOC pS18: | Biotin-Linker-KEEEEDSTFTNI-pS18-LADDIDH-Amide |
| SCOC pS12/pT15: | Biotin-Linker-KEEEED-pS12-TF-pT15-NISLADDIDH-Amide |
| SCOC aa 13–25: | Biotin-Linker-TFTNISLADDIDH-Amide |
| SCOC aa 11–25: | Biotin-Linker-DSTFTNISLADDIDH-Amide |
| SCOC aa 11–25/pS12: | Biotin-Linker-D-pS12-TFTNISLADDIDH-Amide |
| SCOC-aa 6–18: | Biotin-Linker-KEEEEDSTFTNIS-Amide |
| SCOC aa 6–20: | Biotin-Linker-KEEEEDSTFTNISLA-Amide |
| SCOC aa 6–20/pS18: | Biotin-Linker-KEEEEDSTFTNI-pS18-LA-Amide |
Immunostaining and confocal microscopy
Cells were grown on coverslips, fixed with 3% paraformaldehyde in PBS for 20 min before permeabilization with methanol at room temperature for 5 min. Coverslips were then blocked in 5% BSA (Roche) in PBS for 20 min. Coverslips were incubated with primary antibody in 1% BSA in PBS 1 hour at room temperature. Coverslips were washed and incubated with secondary antibody in 1% BSA for 1 hour. After final washing with PBS and water, coverslips were mounted in mowiol. Images were acquired using a Zeiss LSM 710 confocal microscope (x63 oil-immersion lens) and Zeiss ZEN imaging software.
Accession numbers and data availability
The UniProt database numbers (https://www.uniprot.org/) of the SCOC isoforms are: SCOC isoform 1 (Q9UIL1), SCOC isoform 4 (Q9UIL1-4), SCOC isoform 3 (Q9UIL1-3), SCOC isoform 2 (Q9UIL1-2), SCOC isoform 5 (A0A0C4DGB0/protein accession AAK01707). Atomic coordinates and crystallographic structure factors have been deposited in the Protein Data Bank under accession codes 7AA7, 7AA8 and 7AA9. All other data that support the findings of this study are available from the corresponding authors upon request.
Acknowledgments
Acknowledgement
We thank Caroline Whitehouse (King’s College London, UK) for the pEGFPN2-FEZ1 expression plasmid, the Francis Crick Fermentation Science Technology Platform (STP) for protein expression, the Francis Crick Structural Biology (STP) for technical support, the Tromsø University Proteomics Platform for assistance with mass spectrometry-based proteomic analyses, and Diamond Light Source synchrotron for access to beamlines I03, I02 and I04-1 beamline (mx9826-49, mx9826-55 and mx9826-41). M.W., S.M., W.Z., M.R., H.B.J.J., R.L., D.J., N. O’R., and S.A.T were supported by The Francis Crick Institute which receives its core funding from Cancer Research UK (FC001187, FC001999), the UK Medical Research Council (FC001187, FC001999). This research was funded in whole, or in part, by the Wellcome Trust (FC001187, FC001999). For the purpose of Open Access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. The work in T.J.’s laboratory was funded by grants from the FRIBIOMED (grant number 214448) and the TOPPFORSK (grant number 249884) programs of the Research Council of Norway, and the Norwegian Cancer Society (grant number 71043-PR-2006-0320). M.W. had been supported by a European Union Marie Curie fellowship IEF-330396.
Author contributions
M.W., W.Z., M.R., E.S., A.J., H.B.J.J and H.L.O. performed cell- and biochemical studies. S.M., M.W., W.Z. and R.L. performed structural studies. Y.P.A. and JA.B. performed mass spectrometry analyses. D.J. and N.O’R synthesized peptides and peptide arrays. M.W., S.M., S.A.T., E.S. and T.J. wrote the manuscript. All authors discussed the results and commented on the manuscript. M.W., S.M., S.A.T. and T.J. supervised the work. M.W. coordinated the project.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Edited by Martens Sascha
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2021.166987.
Contributor Information
Martina Wirth, Email: m.b.wirth@lumc.nl.
Terje Johansen, Email: terje.johansen@uit.no.
Sharon A. Tooze, Email: sharon.tooze@crick.ac.uk.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Lamb C.A., Yoshimori T., Tooze S.A. The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013;14:759–774. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- 2.Stolz A., Ernst A., Dikic I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 3.Johansen T., Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. https://www.ncbi.nlm.nih.gov/pubmed/21189453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levine B., Kroemer G. Biological functions of autophagy genes: A disease perspective. Cell. 2019;176:11–42. doi: 10.1016/J.CELL.2018.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johansen T., Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J. Mol. Biol. 2019 doi: 10.1016/J.JMB.2019.07.016. [DOI] [PubMed] [Google Scholar]
- 6.Wirth M., Joachim J., Tooze S.A. Autophagosome formation–the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin. Cancer Biol. 2013;23:301–309. doi: 10.1016/j.semcancer.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Mercer T.J., Gubas A., Tooze S.A. A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 2018;293:5386–5395. doi: 10.1074/jbc.R117.810366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karanasios E., Walker S.A., Okkenhaug H., Manifava M., Hummel E., Zimmermann H., Ahmed Q., Domart M.C., Collinson L., Ktistakis N.T. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat. Commun. 2016;7:12420. doi: 10.1038/ncomms12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dooley H.C., Razi M., Polson H.E., Girardin S.E., Wilson M.I., Tooze S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell. 2014;55:238–252. doi: 10.1016/j.molcel.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mizushima N., Sugita H., Yoshimori T., Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998;273:33889–33892. doi: 10.1074/jbc.273.51.33889. [DOI] [PubMed] [Google Scholar]
- 11.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slobodkin M.R., Elazar Z. The Atg8 family: multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 2013;55:51–64. doi: 10.1042/bse0550051. [DOI] [PubMed] [Google Scholar]
- 13.Weidberg H., Shvets E., Shpilka T., Shimron F., Shinder V., Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–1802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen T.N., Padman B.S., Usher J., Oorschot V., Ramm G., Lazarou M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016;215:857–874. doi: 10.1083/jcb.201607039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pontano Vaites L., Paulo J.A., Huttlin E.L., Harper J.W. Systematic analysis of human cells lacking ATG8 proteins uncovers roles for GABARAPs and the CCZ1/MON1 regulator C18orf8/RMC1 in macro and selective autophagic flux. Mol. Cell. Biol. 2017 doi: 10.1128/MCB.00392-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuboyama K., Koyama-Honda I., Sakamaki Y., Koike M., Morishita H., Mizushima N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036–1041. doi: 10.1126/science.aaf6136. [DOI] [PubMed] [Google Scholar]
- 17.Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.A., Outzen H., Overvatn A., Bjorkoy G., Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 18.Alemu E.A., Lamark T., Torgersen K.M., Birgisdottir A.B., Larsen K.B., Jain A., Olsvik H., Overvatn A., Kirkin V., Johansen T., Øvervatn A., Kirkin V., Johansen T. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J. Biol. Chem. 2012;287:39275–39290. doi: 10.1074/jbc.M112.378109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kraft C., Kijanska M., Kalie E., Siergiejuk E., Lee S.S., Semplicio G., Stoffel I., Brezovich A., Verma M., Hansmann I., Ammerer G., Hofmann K., Tooze S., Peter M. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J. 2012;31:3691–3703. doi: 10.1038/emboj.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Birgisdottir Å.B., Mouilleron S., Bhujabal Z., Wirth M., Sjøttem E., Evjen G., Zhang W., Lee R., O’Reilly N., Tooze S.A., Lamark T., Johansen T. Members of the autophagy class III phosphatidylinositol 3-kinase complex I interact with GABARAP and GABARAPL1 via LIR motifs. Autophagy. 2019;15:1333–1355. doi: 10.1080/15548627.2019.1581009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bozic M., van den Bekerom L., Milne B.A., Goodman N., Roberston L., Prescott A.R., Macartney T.J., Dawe N., McEwan D.G. A conserved ATG2-GABARAP family interaction is critical for phagophore formation. EMBO Rep. 2020;21 doi: 10.15252/embr.201948412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skytte Rasmussen M., Mouilleron S., Kumar Shrestha B., Wirth M., Lee R., Bowitz Larsen K., Abudu Princely Y., O’Reilly N., Sjøttem E., Tooze S.A., Lamark T., Johansen T. ATG4B contains a C-terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8. Autophagy. 2017;13:834–853. doi: 10.1080/15548627.2017.1287651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McEwan D.G., Popovic D., Gubas A., Terawaki S., Suzuki H., Stadel D., Coxon F.P., Miranda de Stegmann D., Bhogaraju S., Maddi K., Kirchof A., Gatti E., Helfrich M.H., Wakatsuki S., Behrends C., Pierre P., Dikic I. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell. 2015;57:39–54. doi: 10.1016/j.molcel.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 24.Gu Y., Princely Abudu Y., Kumar S., Bissa B., Choi S.W., Jia J., Lazarou M., Eskelinen E., Johansen T., Deretic V. Mammalian Atg8 proteins regulate lysosome and autolysosome biogenesis through <scp>SNARE</scp> s. EMBO J. 2019;38 doi: 10.15252/embj.2019101994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pankiv S., Alemu E.A., Brech A., Bruun J.A., Lamark T., Overvatn A., Bjorkoy G., Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010;188:253–269. doi: 10.1083/jcb.200907015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noda N.N., Ohsumi Y., Inagaki F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett. 2010;584:1379–1385. doi: 10.1016/j.febslet.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 27.Birgisdottir A.B., Lamark T., Johansen T. The LIR motif - crucial for selective autophagy. J. Cell Sci. 2013;126:3237–3247. doi: 10.1242/jcs.126128. [DOI] [PubMed] [Google Scholar]
- 28.Wild P., McEwan D.G., Dikic I. The LC3 interactome at a glance. J. Cell Sci. 2014;127:3–9. doi: 10.1242/jcs.140426. [DOI] [PubMed] [Google Scholar]
- 29.Rogov V.V., Stolz A., Ravichandran A.C., Rios-Szwed D.O., Suzuki H., Kniss A., Lohr F., Wakatsuki S., Dotsch V., Dikic I., Dobson R.C., McEwan D.G. Structural and functional analysis of the GABARAP interaction motif (GIM) EMBO Rep. 2017;18:1382–1396. doi: 10.15252/embr.201643587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wirth M., Zhang W., Razi M., Nyoni L., Joshi D., O’Reilly N., Johansen T., Tooze S.A., Mouilleron S. Molecular determinants regulating selective binding of autophagy adapters and receptors to ATG8 proteins. Nat. Commun. 2019;10:2055. doi: 10.1038/s41467-019-10059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zess E.K., Jensen C., Cruz-Mireles N., De la Concepcion J.C., Sklenar J., Stephani M., Imre R., Roitinger E., Hughes R., Belhaj K., Mechtler K., Menke F.L.H., Bozkurt T., Banfield M.J., Kamoun S., Maqbool A., Dagdas Y.F. N-terminal β-strand underpins biochemical specialization of an ATG8 isoform. PLoS Biol. 2019;17 doi: 10.1371/journal.pbio.3000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.P. Wild, H. Farhan, D.G. McEwan, S. Wagner, V. V. Rogov, N.R. Brady, B. Richter, J. Korac, O. Waidmann, C. Choudhary, V. Dötsch, D. Bumann, I. Dikic, Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth, Science (80-.). 333 (2011) 228–233. https://doi.org/10.1126/SCIENCE.1205405. [DOI] [PMC free article] [PubMed]
- 33.Rogov V.V., Suzuki H., Marinković M., Lang V., Kato R., Kawasaki M., Buljubašić M., Šprung M., Rogova N., Wakatsuki S., Hamacher-Brady A., Dötsch V., Dikic I., Brady N.R., Novak I. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017;7:1131. doi: 10.1038/s41598-017-01258-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Y., Massen S., Terenzio M., Lang V., Chen-Lindner S., Eils R., Novak I., Dikic I., Hamacher-Brady A., Brady N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013;288:1099–1113. doi: 10.1074/jbc.M112.399345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu L., Feng D., Chen G., Chen M., Zheng Q., Song P., Ma Q., Zhu C., Wang R., Qi W., Huang L., Xue P., Li B., Wang X., Jin H., Wang J., Yang F., Liu P., Zhu Y., Sui S., Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 36.Wu W., Tian W., Hu Z., Chen G., Huang L., Li W., Zhang X., Xue P., Zhou C., Liu L., Zhu Y., Zhang X., Li L., Zhang L., Sui S., Zhao B., Feng D. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15:566–575. doi: 10.1002/embr.201438501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen G., Han Z., Feng D., Chen Y., Chen L., Wu H., Huang L., Zhou C., Cai X., Fu C., Duan L., Wang X., Liu L., Liu X., Shen Y., Zhu Y., Chen Q. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell. 2014;54:362–377. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 38.Di Rita A., Peschiaroli A., D’Acunzo P., Strobbe D., Hu Z., Gruber J., Nygaard M., Lambrughi M., Melino G., Papaleo E., Dengjel J., El Alaoui S., Campanella M., Dötsch V., Rogov V.V., Strappazzon F., Cecconi F. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-05722-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKnight N.C., Jefferies H.B., Alemu E.A., Saunders R.E., Howell M., Johansen T., Tooze S.A. Genome-wide siRNA screen reveals amino acid starvation-induced autophagy requires SCOC and WAC. EMBO J. 2012;31:1931–1946. doi: 10.1038/emboj.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Valkenburgh H., Shern J.F., Sharer J.D., Zhu X., Kahn R.A. ADP-ribosylation factors (ARFs) and ARF-like 1 (ARL1) have both specific and shared effectors: characterizing ARL1-binding proteins. J. Biol. Chem. 2001;276:22826–22837. doi: 10.1074/jbc.M102359200. [DOI] [PubMed] [Google Scholar]
- 41.Su C.-W., Tharin S., Jin Y., Wightman B., Spector M., Meili D., Tsung N., Rhiner C., Bourikas D., Stoeckli E., Garriga G., Horvitz H.R., Hengartner M.O. The short coiled-coil domain-containing protein UNC-69 cooperates with UNC-76 to regulate axonal outgrowth and normal presynaptic organization in Caenorhabditis elegans. J. Biol. 2006;5:9. doi: 10.1186/jbiol39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blasius T.L., Cai D., Jih G.T., Toret C.P., Verhey K.J. Two binding partners cooperate to activate the molecular motor Kinesin-1. J. Cell Biol. 2007;176:11–17. doi: 10.1083/jcb.200605099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alborghetti M.R., Furlan A.da S., da Silva J.C., Sforça M.L., Honorato R.V., Granato D.C., dos Santos Migueleti D.L., Neves J.L., de Oliveira P.S.L., Paes-Leme A.F., Zeri A.C.de M., de Torriani I.C.L., Kobarg J. Structural analysis of intermolecular interactions in the kinesin adaptor complex fasciculation and elongation protein zeta 1/ short coiled-coil protein (FEZ1/SCOCO) PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0076602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Butkevich E., Härtig W., Nikolov M., Erck C., Grosche J., Urlaub H., Schmidt C.F., Klopfenstein D.R., Chua J.J.E. Phosphorylation of FEZ1 by microtubule affinity regulating kinases regulates its function in presynaptic protein trafficking. Sci. Rep. 2016;6:26965. doi: 10.1038/srep26965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chua J.J.E., Butkevich E., Worseck J.M., Kittelmann M., Grønborg M., Behrmann E., Stelzl U., Pavlos N.J., Lalowski M.M., Eimer S., Wanker E.E., Klopfenstein D.R., Jahn R. Phosphorylation-regulated axonal dependent transport of syntaxin 1 is mediated by a Kinesin-1 adapter. Proc. Natl. Acad. Sci. U. S. A. 2012;109:5862–5867. doi: 10.1073/pnas.1113819109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toda H., Mochizuki H., Flores R., Josowitz R., Krasieva T.B., LaMorte V.J., Suzuki E., Gindhart J.G., Furukubo-Tokunaga K., Tomoda T. UNC-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes Dev. 2008;22:3292–3307. doi: 10.1101/gad.1734608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kosugi S., Hasebe M., Tomita M., Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. U. S. A. 2009;106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simons I.M., Mohrlüder J., Feederle R., Kremmer E., Zobel T., Dobner J., Bleffert N., Hoffmann S., Willbold D. The highly GABARAP specific rat monoclonal antibody 8H5 visualizes GABARAP in immunofluorescence imaging at endogenous levels. Sci. Rep. 2019;9:1–11. doi: 10.1038/s41598-018-36717-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalvari I., Tsompanis S., Mulakkal N.C., Osgood R., Johansen T., Nezis I.P., Promponas V.J. iLIR: A web resource for prediction of Atg8-family interacting proteins. Autophagy. 2014;10:913–925. doi: 10.4161/auto.28260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alers S., Löffler A.S., Wesselborg S., Stork B. The incredible ULKs. Cell Commun. Signal. 2012;10:1–14. doi: 10.1186/1478-811X-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joachim J., Razi M., Judith D., Wirth M., Calamita E., Encheva V., Dynlacht B.D., Snijders A.P., O’Reilly N., Jefferies H.B.J., Tooze S.A. Centriolar satellites control GABARAP ubiquitination and GABARAP-mediated autophagy. Curr. Biol. 2017;27:2123–2136 e7. doi: 10.1016/j.cub.2017.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alborghetti M.R., Furlan A.S., Kobarg J. FEZ2 has acquired additional protein interaction partners relative to FEZ1: Functional and evolutionary implications. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0017426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kinoshita E., Kinoshita-Kikuta E., Takiyama K., Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics. 2006;5:749–757. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- 54.Olsvik H.L., Lamark T., Takagi K., Larsen K.B., Evjen G., Overvatn A., Mizushima T., Johansen T. FYCO1 contains a C-terminally extended, LC3A/B-preferring LC3-interacting region (LIR) motif required for efficient maturation of autophagosomes during basal autophagy. J. Biol. Chem. 2015;290:29361–29374. doi: 10.1074/jbc.M115.686915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng X., Wang Y., Gong Y., Li F., Guo Y., Hu S., Liu J., Pan L. Structural basis of FYCO1 and MAP1LC3A interaction reveals a novel binding mode for Atg8-family proteins. Autophagy. 2016;12:1330–1339. doi: 10.1080/15548627.2016.1185590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sakurai S., Tomita T., Shimizu T., Ohto U. The crystal structure of mouse LC3B in complex with the FYCO1 LIR reveals the importance of the flanking region of the LIR motif. Acta Crystallogr. F Struct. Biol. Commun. 2017;73:130–137. doi: 10.1107/S2053230X17001911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ichimura Y., Kumanomidou T., Sou Y.S., Mizushima T., Ezaki J., Ueno T., Kominami E., Yamane T., Tanaka K., Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 58.Stadel D., Millarte V., Tillmann K.D., Huber J., Tamin-Yecheskel B.C., Akutsu M., Demishtein A., Ben-Zeev B., Anikster Y., Perez F., Dötsch V., Elazar Z., Rogov V., Farhan H., Behrends C. TECPR2 cooperates with LC3C to regulate COPII-dependent ER export. Mol. Cell. 2015;60:89–104. doi: 10.1016/j.molcel.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 59.Yang A., Pantoom S., Wu Y.W. Elucidation of the anti-autophagy mechanism of the legionella effector ravz using semisynthetic LC3 proteins. Elife. 2017;6 doi: 10.7554/eLife.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lystad A.H., Ichimura Y., Takagi K., Yang Y., Pankiv S., Kanegae Y., Kageyama S., Suzuki M., Saito I., Mizushima T., Komatsu M., Simonsen A. Structural determinants in GABARAP required for the selective binding and recruitment of ALFY to LC3B-positive structures. EMBO Rep. 2014;15:557–565. doi: 10.1002/embr.201338003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li J., Zhu R., Chen K., Zheng H., Zhao H., Yuan C., Zhang H., Wang C., Zhang M. Potent and specific Atg8-targeting autophagy inhibitory peptides from giant ankyrins. Nat. Chem. Biol. 2018;14:778–787. doi: 10.1038/s41589-018-0082-8. [DOI] [PubMed] [Google Scholar]
- 62.Wang B., Iyengar R., Li-Harms X., Joo J.H., Wright C., Lavado A., Horner L., Yang M., Guan J.L., Frase S., Green D.R., Cao X., Kundu M. The autophagy-inducing kinases, ULK1 and ULK2, regulate axon guidance in the developing mouse forebrain via a noncanonical pathway. Autophagy. 2018;14:796–811. doi: 10.1080/15548627.2017.1386820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kittler J.T., Rostaing P., Schiavo G., Fritschy J.M., Olsen R., Triller A., Moss S.J. The subcellular distribution of GABARAP and its ability to interact with NSF suggest a role for this protein in the intracellular transport of GABAA receptors. Mol. Cell. Neurosci. 2001;18:13–25. doi: 10.1006/mcne.2001.1005. [DOI] [PubMed] [Google Scholar]
- 64.Teixeira M.B., Alborghetti M.R., Kobarg J. Fasciculation and elongation zeta proteins 1 and 2: From structural flexibility to functional diversity. World J. Biol. Chem. 2019;10:28–43. doi: 10.4331/wjbc.v10.i2.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan E.Y., Longatti A., McKnight N.C., Tooze S.A. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol. Cell. Biol. 2009;29:157–171. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bhujabal Z., Birgisdottir Å.B., Sjøttem E., Brenne H.B., Øvervatn A., Habisov S., Kirkin V., Lamark T., Johansen T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017;18:947–961. doi: 10.15252/embr.201643147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Whitehouse C., Chambers J., Howe K., Cobourne M., Sharpe P., Solomon E. NBR1 interacts with fasciculation and elongation protein zeta-1 (FEZ1) and calcium and integrin binding protein (CIB) and shows developmentally restricted expression in the neural tube. Eur. J. Biochem. 2002;269:538–545. doi: 10.1046/j.0014-2956.2001.02681.x. [DOI] [PubMed] [Google Scholar]
- 68.Chan E.Y.W., Kir S., Tooze S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007;282:25464–25474. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 69.Johansen T., Birgisdottir Å.B., Huber J., Kniss A., Dötsch V., Kirkin V., Rogov V.V. Methods for studying interactions between Atg8/LC3/GABARAP and LIR-containing proteins. Methods Enzymol. 2017;587:143–169. doi: 10.1016/BS.MIE.2016.10.023. [DOI] [PubMed] [Google Scholar]
- 70.Frank R. The SPOT-synthesis technique: Synthetic peptide arrays on membrane supports—principles and applications. J. Immunol. Methods. 2002;267:13–26. doi: 10.1016/S0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- 71.Winter G., Lobley C.M., Prince S.M. Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr. 2013;69:1260–1273. doi: 10.1107/S0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., McCoy A.J., Moriarty N.W., Oeffner R., Read R.J., Richardson D.C., Richardson J.S., Terwilliger T.C., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vaguine A.A., Richelle J., Wodak S.J. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 1999;55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.