

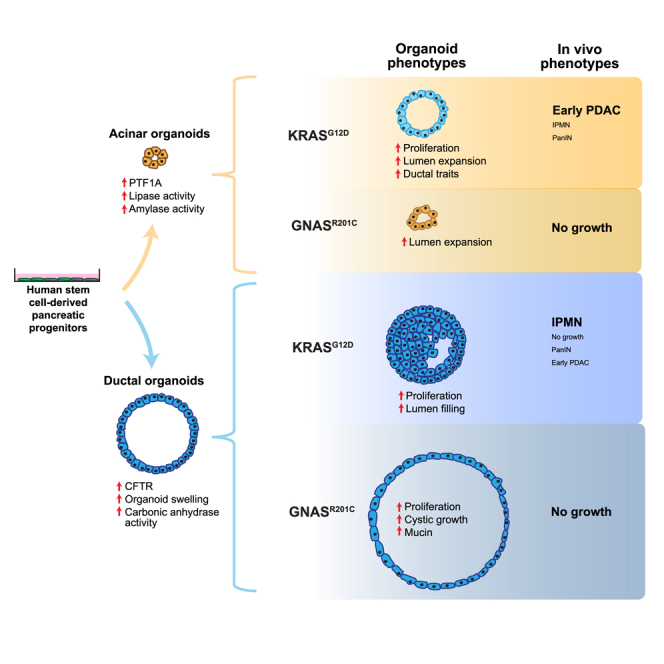
Summary
The exocrine pancreas, consisting of ducts and acini, is the site of origin of pancreatitis and pancreatic ductal adenocarcinoma (PDAC). Our understanding of the genesis and progression of human pancreatic diseases, including PDAC, is limited because of challenges in maintaining human acinar and ductal cells in culture. Here we report induction of human pluripotent stem cells toward pancreatic ductal and acinar organoids that recapitulate properties of the neonatal exocrine pancreas. Expression of the PDAC-associated oncogene GNASR201C induces cystic growth more effectively in ductal than acinar organoids, whereas KRASG12D is more effective in modeling cancer in vivo when expressed in acinar compared with ductal organoids. KRASG12D, but not GNASR201C, induces acinar-to-ductal metaplasia-like changes in culture and in vivo. We develop a renewable source of ductal and acinar organoids for modeling exocrine development and diseases and demonstrate lineage tropism and plasticity for oncogene action in the human pancreas.
Keywords: organoid, pancreatic cancer, KRAS, GNAS, acini, cancer precursor, pluripotent stem cell, plasticity, exocrine pancreas, lineage specification
Graphical abstract
Highlights
-
•
Generation and long-term maintenance of human acinar organoids from stem cells
-
•
GNASR201C induces cystic growth more effectively in ductal than in acinar organoids
-
•
KRASG12D induces cancerous lesions more often from acinar versus ductal organoids
-
•
KRASG12D in acinar organoids induces an acinar-to-ductal metaplasia-like phenotype
Huang et al. describe conditions for generation of pancreatic ductal and acinar organoids from human stem cells. Expression of KRASG12D or GNASR201C induced cell-lineage-specific phenotypes and cell-state plasticity in culture and in vivo, identifying a renewable source of ductal and acinar organoids for modeling development and diseases of the exocrine pancreas.
Introduction
The pancreas is composed of endocrine and exocrine compartments. Although the endocrine pancreas harbors islets of Langerhans, the exocrine pancreas, which makes up over 90% of the total pancreas volume, contains the ductal (~5%) and acinar (~85%) epithelia. The most common type of pancreatic cancer, pancreatic ductal adenocarcinoma (PDAC), is thought to arise from the exocrine compartment (Kleeff et al., 2016). Precursor lesions of pancreatic cancer can be classified into four main subtypes based on their clinical pathology: pancreatic intraepithelial neoplasia (PanIN), intraductal papillary mucinous neoplasms (IPMNs), intratubular papillary neoplasms (ITPNs), and mucinous neoplasms (MCNs) (Cooper et al., 2013; Hruban et al., 2000, 2007). Genomic studies reveal that KRAS mutations are observed in more than 80% of PanINs and associated with invasive PDAC (Almoguera et al., 1988; Kanda et al., 2012; Maitra and Hruban, 2008), identifying KRAS as the dominant driver gene in PDAC. Mutation in GNAS involving codon 201 is observed frequently in IPMN lesions, either by itself or in combination with mutant KRAS (Amato et al., 2014; Fukayama et al., 1986; Wu et al., 2011), and adjacent invasive PDAC, identifying GNAS as a driver in IPMN-derived PDAC. It is unclear how different precancerous lesions affect PDAC development and how the cellular origins of PDAC affect development of precancerous lesions and clinical prognosis.
Among the pancreatic cancer-associated mutations in KRAS, G12D is the most frequent (Waters and Der, 2018). In mouse models, multiple early studies using expression of mutant KRasG12D alone or in combination with loss of the tumor suppressors Tp53 or Cdkn2a under control of the Pdx1 promoter resulted in development of PanIN-like lesions that progressed to PDAC (Aguirre et al., 2003; Bardeesy et al., 2006; Hingorani et al., 2003). Pdx1 is expressed in progenitors of all pancreatic epithelia; hence, these studies do not provide direct insights into the cells of origin for PDAC. Expression of KRasG12D under multiple acinar cell-specific promoters (Mist1, Pftf1a) supports an acinar cell of origin for PDAC (De La O et al., 2008; Gidekel Friedlander et al., 2009; Habbe et al., 2008; Tuveson et al., 2006), where acinar cells undergo ductal metaplasia early during the tumorigenic process. Compelling evidence generated by Sanders, Kopp, and Behrens provided additional insights (Ferreira et al., 2017; Kopp et al., 2012; Lee et al., 2019). Expression of KRasG12D under acinar cell-specific Ptf1a-Cre induced PanINs more readily than under duct-selective Sox9-Cre (Kopp et al., 2012), suggesting that acinar cells are more sensitive to KRAS-induced PanIN initiation. Combining KRasG12D expression with loss of Tp53 or Fbw7 under control of acinus-specific elastase or Ptf1a-Cre induces PanIN lesions that progress to PDAC (Ferreira et al., 2017; Lee et al., 2019); however, the same gene combinations under control of ductal cell-specific Sox9 or Krt19-Cre results in PanIN-independent development of PDAC that was more aggressive, suggesting a distinct evolutionary path for ductal epithelium-initiated cancers (Ferreira et al., 2017; Lee et al., 2019). The existence of different paths for acinus-initiated and duct-initiated PDAC tumorigenesis highlights the importance of cells of origin during initiation and progression of PDAC.
IPMN is associated anatomically with ducts in the human pancreas; however, studies with duct-specific expression of GNAS mutants have not yet been reported. Expression of a PDAC-associated mutant of Gnas, GnasR201C, and KrasG12D under control of the Ptf1a promoter is sufficient to induce dilation of pancreatic ducts and loss of acini, representing the IPMN phenotype (Ideno et al., 2018; Patra et al., 2018; Taki et al., 2016). Although it is not known whether duct-specific expression of a GNAS mutant is sufficient to induce IPMN lesions, Pten loss or Lkb1 inactivation with KRas mutant expression in ductal epithelia drives IPMN-like lesions (Collet et al., 2020; Kopp et al., 2018), suggesting that IPMN-like lesions can be derived from ductal and acinar lesions in mice.
Modeling cancer initiation and progression in a human cell context will provide new biological insights that can be exploited for identification of biomarkers and treatments for PDAC. Ductal exocrine cells from the adult pancreas have been maintained successfully in culture (Boj et al., 2015; Huch et al., 2013; Seino et al., 2018) and engineered to express PDAC-associated mutations in KRAS, CDKN2A, SMAD4, and TP53 to induce cell proliferation in vitro and form PanIN-like lesions in vivo without progression to PDAC (Lee et al., 2017). Efforts to maintain human acinar cells in culture and use them to model cancer initiation has been challenging because acinar cell cultures are short lived or undergo trans-differentiation by a process referred to as acinar-to-ductal metaplasia (ADM) to assume a duct-like state (De Lisle and Logsdon, 1990; Rooman et al., 2000).
Human pluripotent stem cells (hPSCs) are a powerful platform to generate multiple cell types in culture (Sharma et al., 2020). We and others have reported the ability to induce hPSC-derived pancreatic progenitor (PP) cells toward the exocrine lineage in culture, supporting formation of ductal and acinar structures when transplanted into the mouse pancreas (Hohwieler et al., 2017; Huang et al., 2015; Simsek et al., 2016). Conditions to generate populations of pancreatic ductal or acinar cells in vitro are challenging. Although murine embryonic stem cells can be induced to differentiate into acinar cells (Rovira et al., 2008), whether such strategies can be used to generate human pancreatic acinar cells is not known. This study reports our ability to generate pancreatic duct-like or acinus-like organoid structures from human stem cell-derived PP cells. The two lineages of organoids exhibit distinct morphologies, express lineage-enriched markers, and have lineage-specific enzyme activities. Using these organoids, we demonstrate that GNASR201C and KRASG12D exert lineage-specific effects in culture and in vivo.
Results
Commitment of human PP cells toward acinar and ductal lineages
PDX1-positive PP cells obtained with an established protocol (Pagliuca et al., 2014) were used to generate ductal and acinar lineage-committed organoids in Matrigel-based cultures. To design the culture media, we focused on pathways with established roles in exocrine pancreas specification. For example, in the mouse embryonic pancreas, β-catenin was required for specification of pancreatic acinar cells, whereas it was largely dispensable for ductal and endocrine cells (Keefe et al., 2012). Continuous activation of the Notch signaling pathway promotes duct differentiation and suppresses acinar cell fate (Esni et al., 2004; Murtaugh et al., 2003; Shih et al., 2012). In addition, histone deacetylase (HDAC) inhibition suppresses the acinar fate and promotes ductal specification (Haumaitre et al., 2008). We methodically screened multiple combinations of factors and small molecules and optimized a protocol to induce differentiation of PP cells toward ductal or acinar lineage-specified organoids (see Figure 1A for a schematic). Specification of duct-like organoids was promoted by activation of the fibroblast growth factor (FGF), epidermal growth factor (EGF), non-canonical WNT family, and retinoic acid pathways and inhibition of the HDAC, canonical WNT family, and ALK5 pathways. On the other hand, specification of acinus-like organoids was promoted by activation of the canonical WNT, FGF, and cortisol pathways and inhibition of the hedgehog, NOTCH family, bone morphogenetic protein (BMP), and ALK5 pathways. Under these culture conditions, acinar and ductal organoids formed with 6.57% ± 0.23% and 1.7% – 0.24% efficiency (n = 3), respectively. Cells that did not develop into organoids died within 2 days in culture.
Figure 1.

Commitment of human PP cells toward the acinar and ductal lineages
(A) Schematic of the duct-like and acinus-like organoid induction protocols.
(B) Phase-contrast images of organoids during 16-day culture (n = 3, independent cultures). Scale bar, 50 μm.
(C) Changes in total area and form factor (circularity index) of organoids during morphogenesis (n > 150, n = 3 independent cultures). Box-and-whisker plot, range 5%–95%; center lines indicate median values; gray dots represent individual measurements. ∗∗∗p < 0.001.
(D) Immunostaining for collagen IV (red), DAPI (blue), and ZO1 (green). Scale bar, 50 μm.
(E) TEM images of acinar and ductal organoids. Red arrow, electron-dense vesicles; N, nucleus; L, lumen. DU (blue), duct-like organoid; AC (orange), acinus-like organoid.
The acinar organoids were small (~20–105 μm in diameter on day 16) with no visible lumen. In comparison, ductal organoids were large (~50–220 μm in diameter on day 16) and cystic with visible hollow lumens (Figure 1B). Morphologically, both types of organoids increased their sizes during 3D growth and became spherical, as indicated by increases in organoid areas and form factor (a form factor of 1.0 is a perfect circle) (Figure 1C). Lumen expansion of ductal organoids started from day 8 in culture, and the majority of these organoids had a visible lumen by day 12. Both types of organoids were polarized, as indicated by the apical localization of the tight junction protein ZO1 and basal localization of the basement membrane protein collagen IV (Figure 1D). We also investigated subcellular features of organoids by transmission electron microscopy (TEM) (Figure 1E). TEM analysis showed that acinus-like organoids were compact with a small lumen, whereas duct-like organoids were large with a dilated lumen (Figure 1E). Cells in acinar organoids had many secretory vesicles located at the apical side (red arrow, Figure 1E); these vesicles were 0.5–1.0 μm in diameter and slightly electron dense. Although these vesicles did not resemble the apically localized zymogen granules present in mature human acinar cells (Lugea et al., 2017), they were similar to precursors of zymogen granules observed in 12- to 14-week-old human fetal pancreata (Laitio et al., 1974), suggesting that our acinus-like organoids resembled fetal and not mature pancreatic acini. PP cells generated from two independent human induced pluripotent stem cells (iPSCs) (see Figure S1A for details) also formed acinar and ductal organoids with morphological features comparable with organoids obtained from human embryonic stem cell (hESC)-derived PP cells (Figures S1B and S1C), demonstrating that the condition we developed was effective in supporting differentiation of PP cells generated from hESC and iPSC lines.
Single-cell analysis of hESC-derived duct-like and acinus-like organoids
We used single-nucleus RNA (snRNA) sequencing as an unbiased assessment of the molecular differences between the two organoid lineages. Samples were multiplexed in the 10X Chromium system to obtain single-nucleus transcriptomes from PP cells and day 8 acinar and ductal organoids (McGinnis et al., 2019). Uniform manifold approximation and projection (UMAP) visualization of the data revealed three clusters of cells; each was enriched for cells from one of the three samples, and the replicates clustered together (Figure 2A). Of the common genes shared by two cell clusters, ductal organoids and progenitor cells exhibited the greatest overlap (Figure 2B), suggesting that acinar organoids were more divergent from progenitor cells. To assess the unique characteristics of each cell type, we recomputed markers to identify genes specifically enriched in each cluster (Table S1). Some classical exocrine lineage markers, including SOX9, HNF1B, CEL, PNLIP, CTRB1, and CTRC, showed trends in average expression across culture types that were in line with organoid differentiation (Figure S2A) but were not statistically different between cell groups. It is possible that use of nuclear RNA, because of technical challenges we experienced during isolation of cellular RNA from acinar organoids, contributed to the low signal.
Figure 2.

Single-cell analysis of hESC-derived duct-like and acinus-like organoids and expression of lineage markers and functional readouts
(A) Uniform manifold approximation and projection (UMAP) of single-nucleus transcriptomes of cells in pancreatic progenitors (PPs; pink), DUs (blue) and ACs (orange). Shown are results from two independent cultures and sequencing.
(B) Venn diagram of cell type markers identified by pairwise comparison using adjusted p ≤ 0.05 and average log(fold-change) > 0. The color scheme is the same as in (A).
(C) Comparison of Pearson correlation coefficients for expression of marker genes (1,012 genes; adjusted (adj.) p < 0.05) between organoid culture and human neonatal acinar and ductal cells. Marker genes were derived by identifying differentially expressed genes (p < 0.05) in cells under different culture conditions. Wilcoxon rank-sum test was used.
(D) Immunoblot analysis for acinar and ductal markers (n = 3 independent cultures). The numbers under the blots represent normalized signals of protein bands.
(E and F) Morphological changes of day 8 DUs (blue) and ACs (orange) in response to 10 μM forskolin treatment (2-h incubation). Scale bar, 50 μm.
(G) Measurements of organoid size changes induced by forskolin treatment. Box-and-whisker plot, range 5%–95%; center lines indicate median values; gray dots represent individual measurements (n > 200, combination of three independent cultures).
(H–J) Measurement of enzyme activities in DU and AC lysates (n = 3 independent cultures). y axis, enzyme activity in arbitrary units.
Error bars in bar charts represent standard deviation; gray dots represent individual measurements. ∗∗∗p < 0.001; ∗, 0.01 < p < 0.05; N.S., not significant, p > 0.05.
The top 20 differentially upregulated or downregulated genes detected in our snRNA analysis (Table S2) were not widely studied markers for ductal and acinar cells of the pancreas. To evaluate their localization in the human pancreas, we checked expression patterns in the Human Protein Atlas (Uhlén et al., 2015; Table S3). CALN1, CRID2, DLG2, and LRP1B, enriched in acinar organoids, showed localization patterns consistent with acinar cells in the human pancreas (Figures S2B–S2E), and SLC4A4, ANXA4, C8orf34, and MAGI1, enriched in duct-like organoids, showed localization patterns consistent with ductal cells in the human pancreas (Figures S2F–S2I), further supporting the lineage commitment of hESC-derived acinus-like and duct-like organoids. The ratio of RBPJL/RBPJ, an indicator of acinar specification (Masui et al., 2010), was also highest in acinus-like organoids (Figure S2A). These results suggest that ductal organoids and acinar organoids adopt distinct differentiation paths.
To contextualize the relative differentiation of our organoids compared with human acinar and ductal cells, we downloaded one published snRNA sequencing dataset of human pancreas tissue (Tosti et al., 2021). Using the average expression profiles of marker genes derived from our organoids and progenitors, we calculated Pearson correlation coefficients for our acinar and ductal organoids and the acinar and ductal cells from the neonatal dataset (Figure 2C). The trends in these correlations are consistent with the notion that, by day 8, duct-like and acinus-like organoid cultures were induced toward more duct- or acinus-like fates, respectively. Given the correlations with the neonatal exocrine pancreas and the low representation of genes expressed in adult ductal and acinar cells, our stem cell-derived organoids are likely to represent more neonatal states; hence, we refer to them as “acinus-like” and “duct-like” organoid structures.
Expression of acinar and ductal lineage markers and functional readouts
Next we took a candidate approach to monitor the expression of recognized ductal and acinar markers in our acinus-like and duct-like structures. Protein expression of the pancreatic ductal markers SOX9 and carbonic anhydrase II (CAII) and the pancreatic acinar markers PTF1A and chymotrypsin C (CTRC) (Figure 2D) demonstrated that the duct-like organoids expressed high levels of SOX9 and CAII, which were undetectable or very low in acinus-like organoids. In contrast, acinus-like organoids expressed PTF1A and CTRC at higher levels compared with duct-like organoids.
To assess functional differentiation, the organoids were treated with 10 μM forskolin for 2 h to monitor changes in swelling. Duct-like but not acinus-like organoids showed a significant expansion of the luminal space (Figures 2E–2G). Forskolin is known to induce luminal expansion in a cystic fibrosis transmembrane conductance regulator (CFTR)-dependent manner and loss of CFTR expression or cells with inactive mutants of CFTR fail to respond to forskolin treatment (Dekkers et al., 2013). The differential response to forskolin treatment by our duct-like and acinus-like organoids demonstrates functional CFTR in duct-like organoids and further supports ductal lineage commitment. In addition, carbonic anhydrase activity was four times higher in duct-like organoids compared with acinus-like organoids (Figure 2H). In contrast, amylase and lipase activities (associated with acinar cells) were more elevated in acinus-like organoids compared with duct-like organoids, demonstrating lineage-specific functions (Figures 2I and 2J). Acinus-like organoids derived from iPSCs also had lower expression of PDX1, CFTR, and NKX6.1 compared with duct-like organoids (Figure S3A). PTF1A expression in acinus-like organoids derived from iPSC line 11 was also higher than in duct-like organoids (Figure S3A).
Temporal changes in marker expression during duct-like and acinus-like lineage specification
To better understand the timeline of lineage commitment, we monitored the expression of ductal (SOX9, HNF1B, and CAII) and acinar (PTF1A, RBPJL, and CPA1) markers over time (Figure 3). HNF1B and SOX9 expression was high in PP cells and remained high during morphogenesis in duct-like organoids (Figures 3A, 3B, and 3E), whereas they decreased over time in acinus-like organoids (Figures 3A, 3B, and 3E). CAII RNA and protein and CFTR RNA expression increased from day 8 and was upregulated significantly by day12 during ductal differentiation but not during acinar differentiation (Figures 3C, 3D, and 3F).
Figure 3.

Temporal changes in marker expression during duct-like and acinus-like lineage specification
(A–D) RNA expression of pancreatic ductal markers SOX9 (A), HNF1B (B), CAII (C), and CFTR (D) in DUs and ACs during 16-day culture. Floating column charts represent RNA measurements from quantitative PCR, hinges represent maximal and minimal values, central lines indicate mean values, and dots represent individual measurements.
(E and F) Expression of SOX9 (E) and CAII (F) in organoids, detected by immunofluorescence. Numbers embedded in the images indicate the average percentage and standard deviation of marker-positive organoids. Scale bars, 50 μm.
(G–I) RNA expression of the pancreatic acinar markers PTF1A (G), RBPJL (H), and CPA1 (I) in DUs and ACs during 16-day culture. Floating column charts: representation as in (A).
(J and K) Expression of PTF1A (J) and CPA1 (K) in organoids, detected by immunofluorescence. Numbers embedded in images indicate the average percentage and standard deviation of marker-positive organoids. Scale bars, 50 μm.
All results represent the sum of three independent cultures.
Expression of PTF1A RNA and proteins in acinus-like organoids increased beginning on day 8 and remained high, whereas the levels decreased progressively during ductal differentiation (Figures 3G and 3J). RBPJL expression is associated with the acinar cell type (Masui et al., 2010). RBPJL RNA levels were high in acinar cultures starting on day 8 but undetectable in duct-like organoids (Figure 3H). However, CPA1, the acinar cell enzyme, was expressed in acinus-like and duct-like organoids from day 8 (Figures 3I and 3K). RNA expression of the PP marker PDX1 remained constant in duct-like organoids and was suppressed in acinus-like organoids (Figure S3B). Detectable expression of CPA1 in our duct-like organoids is consistent with findings that embryonic ducts are plastic and can give rise to acinar cells (Reichert and Rustgi, 2011). The above results demonstrate that our culture conditions support commitment of PPs toward the acinus-like or duct-like lineage.
The GNAS mutant shows tropism for ductal-lineage epithelia
GNAS mutations occur frequently in cystic lesions of the pancreas (Ohtsuka et al., 2019). It is not known whether GNAS shows lineage tropism in human pancreatic cells. We engineered PP cells with doxycycline-inducible wild-type GNAS (GNASWT) or GNASR201C (a mutation observed frequently in IPMN) and induced gene expression on day 8 of the differentiation protocol (Figure 4A). Expression of GNASR201C promoted an increase in general phosphorylation of PKA substrates and phosphorylation of the specific PKA substrate vasodilator stimulated phosphoprotein (VASP) (Figure 4A). GNASR201C was significantly better than GNASWT in its ability to induce lumen expansion in duct-like organoids compared with acinus-like organoids (Figure 4B). Duct-like organoids expanded by about 8-fold in response to GNASR201C expression, whereas acinar organoids showed only a 2.9-fold increase (Figure 4C). Overexpression of GNASWT induced an ~1.5-fold expansion in duct-like organoids and no change in acinus-like organoids (Figure 4C). More than 40% of duct-like organoids expressing GNASR201C were larger than 99% of the control organoids, whereas only 25% of acinus-like organoids expressing GNASR201C were larger than 99% of the control organoids (Figure 4D), demonstrating that duct-like organoids were more sensitive than acinus-like organoids to GNASR201C expression.
Figure 4.

The GNAS mutant shows tropism for ductal-lineage epithelia
(A) Immunoblot analysis for GNAS expression and phosphorylation of GNAS downstream effectors in DUs and ACs (n = 3, independent cultures).
(B) Phase-contrast images of DUs and ACs expressing wild-type (WT) GNAS and GNASR201C. (-)DOX, without doxycline treatment; (+)DOX, with doxycycline treatment. Scale bar, 100 μm.
(C) Change in size, measured as the total area of organoids expressing GNASWT or GNASR201C. Box-and-whisker plot, range 5%–95%; central lines, median values (n > 100, combination of three independent cultures).
(D) Percentage of organoids surpassing the 99th percentile of control organoid (no transgene expression) size (n = 3 independent cultures).
(E) Organoid cell number changes induced by GNASWT and GNASR201C as detected by CellTiter. Bar charts represent the mean and standard deviation of measurements from three independent cultures; gray dots represent individual measurements.
(F) Immunofluorescent staining of polarity proteins in organoids without (−DOX) and with (+DOX) GNASR201C expression. ZO1, red; collagen IV, green; DAPI, blue. Scale bar, 100 μm.
(G) Expression of SOX9 in organoids without (−DOX) and with (+DOX) GNASR201C expression. SOX9, red; DAPI, blue.
(H) AB (pH 2.5) staining of organoids without (−DOX) and with (+DOX) GNASR201C expression. Scale bar, 100 μm.
(I) Expression of E-cadherin (green) and MUC2 (red) in DUs without (−DOX) and with (+DOX) GNASR201C expression. Scale bar, 50 μm.
(J) Percentages of DUs expressing MUC2 (top chart) or MUC5AC (bottom chart). Bar charts represent the mean and standard deviation of measurements from three independent cultures; gray dots represent individual measurements.
(K and L) RNA expression of lineage markers in DUs (K) and ACs (L) altered by GNASR01C expression. Floating column charts represent RNA measurements from quantitative PCR (n = 3 technical repeats), hinges represent maximal and minimal values, central lines indicate mean values, and dots represent individual measurements. ∗∗∗p < 0.001; ∗∗, 0.001 < p < 0.01; ∗, 0. 01 < p < 0.05; N.S., p > 0.05.
GNASR021C but not GNASWT expression increased cell numbers in organoid cultures (Figure 4E), with no detectable effect on localization of the cell polarity proteins ZO1 (an apical protein) and collagen IV (a basement membrane protein) (Figure 4F) or SOX9 in duct-like and acinus-like organoids (Figure 4G). To investigate whether GNAS induces changes in mucin overexpression, a hallmark of premalignant pancreas lesions, we stained structures with Alcian blue (an acidic mucin dye). More than 30% of duct-like organoids expressing GNASR201C stained positive for Alcian blue (Figure 4H), with no detectable staining of acinus-like organoids. To better understand the patterns of mucin expression, we monitored changes in MUC2 and MUC5AC because these mucins are expressed in less than 4% of normal pancreatic cells (Kim et al., 2002) but are detected routinely in precursor lesions. Interestingly, MUC2 expression was observed in clusters of cells in about 60% of duct-like organoids expressing GNASR201C (Figures 4I and 4J) but not in organoids expressing GNASWT (Figure S4A). No MUC2 expression was detected in acinus-like organoids under any condition (Figures S4B and S4C). Neither duct-like nor acinus-like organoids expressed MUC5AC (Figures 4J, S4B, and S4C). These observations support our conclusion that human ductal epithelium is more sensitive to GNASR201C-induced changes than acinus-like cells.
Next we analyzed changes in expression of markers of PP cells (NKX6.1, PDX1), ductal (SOX9, CFTR), and acinar (PTF1A) epithelia (Figures 4K and 4L). Interestingly, the expression levels of PDX1 and NKX6.1 decreased in response to GNASR201C expression in duct-like epithelia (Figure 4K). In acinar organoids, NKX6.1 levels decreased to undetectable levels, and PTF1A levels increased (Figure 4L). CFTR was upregulated weakly by GNASR201C in both lineages (Figure S4D).
Next we transplanted GNASR201C-expressing duct-like organoids into the mouse pancreas to investigate whether GNASR201C expression was sufficient to induce precancerous lesions in vivo. Organoids were disassociated and injected into the pancreas of 6- to 8-week-old non-obese diabetic (NOD) CRISPR Prkdc Il2r Gamma (NCG) mice (N = 12). We did not observe any outgrowth that stained positive for human KRT19 or class I major histocompatibility complex (HLA) or human nuclear antigen. However, two of eight mice sacrificed after 8 weeks and one of four mice sacrificed after 14 weeks showed abnormal regions representing proliferative glands and metaplasia, likely because of injection-induced reactions (Figures S4E and S4F). The presence of the injection-site reaction in the pancreas validates the injection technique and supports the conclusion that GNASR201C expression is not sufficient to support survival and growth of duct-like cells in vivo.
KRASG12D induces ADM-like changes in acinus-like organoids but a progenitor phenotype in duct-like organoids
Mutations of KRAS are the most common genomic changes in precancerous lesions of PDAC (Buscail et al., 2020). KRAS is also overexpressed (Figure S5A) or amplified in human PDAC tumors (Mueller et al., 2018). To understand how oncogenic KRAS affects our human pancreatic acinus-like and duct-like lineages, we expressed KRASG12D in PP cells in a doxycycline-inducible manner. We titrated the virus to allow ectopic expression of KRASG12D within 2- to 4-fold of endogenous levels (Figures S5B and S5C) because a higher level of KRAS overexpression was not tolerated by these organoids (Figure S5D). In acinus-like and duct-like organoids, KRASG12D expression induced a modest increase in phosphorylation of mitogen-activated protein kinase (p-ERK) (Figures S5B and S5C), consistent with previous studies (Gillies et al., 2020; Tuveson et al., 2004). Expression of KRASG12D was induced on day 8 after cells were committed to a duct-like or acinus-like lineage, and the phenotypes were analyzed 16 days later. Consistent with previous reports (Serrano et al., 1997), expression of KRASG12D induced a modest increase in expression of nuclear p16INK4A (Figures S5E and S5F), a senescence-associated marker. However, in duct-like and acinus-like organoids, KRASG12D increased cell proliferation, as monitored by Ki67 positivity (Figures 5A and 5B) and total cell numbers (Figure 5C), suggesting that the level of expression of KRASG12D is pro-proliferative and not high enough to induce senescent arrest. Morphologically, KRASG12D induced a 4-fold increase in the number of structures with lumens filled by multilayering of cells, as monitored in phase-contrast and DAPI-stained images (Figures 5D and 5E). In contrast, expression of KRASG12D induced a 5-fold increase in organoids with a visible lumen (Figures 5D and 5E) in acinar lineage-committed organoids. KRASG12D induced no obvious increase in the overall size of duct-like organoids but a significant change in acinus-like organoids (Figure 5F). These observations demonstrate that, although KRASG12D is equally competent in inducing cell proliferation in both lineages, it differentially affects the morphology of acinus-like and duct-like organoids.
Figure 5.

KRASG12D induces ADM-like changes in ACs but a progenitor phenotype in DUs
(A) Expression of the cell proliferation marker Ki67 (red) in DUs (blue) and ACs (orange) with and without KRASG12D expression. Scale bar, 50 μm.
(B) Quantification of Ki67-positive organoids with and without KRASG12D expression. Box-and-whisker plot, range 5%–95%; center lines indicate median values; gray dots represent individual measurements.
(C) Organoid cell number changes induced by KRASG12D expression. Bar charts represent the mean and standard deviation of measurements from three independent cultures; gray dots represent individual measurements.
(D and E) Quantification of organoid morphological changes induced by KRASG12D expression. Bar chart representation as in (C).
(F) Organoid size changes induced by KRASG12D expression. Box-and-whisker plot representation as in (B).
(G and H) Expression of pancreatic lineage markers in DUs (G) and ACs (H) altered by KRASG12D expression. Floating column charts represent RNA measurements from quantitative PCR, hinges represent maximal and minimal values, central lines indicate mean values, and dots represent individual measurements. ∗∗∗p < 0.001; ∗∗, 0.001 < p < 0.01; ∗, 0. 01 < p < 0.05; N.S., p > 0.05.
All results represent the sum of three independent cultures.
To better understand the morphological changes in acinus-like organoids in response to expression of KRASG12D, we assessed changes in expression of markers of PP (NKX6.1, PDX1), ductal (SOX9, CFTR), and acinar (PTF1A, CPA1) epithelial cells. In duct-like organoids, KRASG12D upregulated PDX1, NKX6.1, and SOX9 and downregulated and CFTR, suggesting change toward a progenitor state (Figure 5G). Interestingly, in acinus-like organoids, KRASG12D downregulated genes associated with acinar epithelia, PTF1A and CPA1, and upregulated SOX9 expression (Figure 5H), suggesting trans-differentiation of an acinus-like into a duct-like state, recapitulating phenotypes of ADM.
Transforming growth factor β (TGF-β) augments KRASG12D-induced phenotypes in acinus-like and duct-like organoids
KRASG12D cooperates with cerulein-induced acute pancreatitis to initiate tumorigenesis in mouse models of PDAC (Guerra et al., 2007). Because TGF-β levels increase during acute pancreatitis (Riesle et al., 1997), and TGF-β induces trans-differentiation of exocrine pancreatic epithelial cells in culture (Handler et al., 2018; Liu et al., 2016), we investigated whether TGF-β cooperates with KRASG12D in duct-like and acinus-like organoids. TGF-β treatment did not alter expression of KRASG12D in organoids (Figure S5B). In the absence of KRASG12D expression, TGF-β induced cell death (Figure S6A) in acinus-like and duct-like organoids, whereas in KRASG12D expressing cells, TGF-β induced cell proliferation in ductal and acinar organoids by 1.7-fold and 2.4-fold, respectively (compare Figure 6A with Figures 5A and 5B), as well as suppression of KRASG12D-induced p16INK4A expression (compare Figures S6B and S7C with Figures S5E and S5F). Morphologically, TGF-β treatment of KRASG12D expressing duct-like organoids induced a significant increase in the percentage of organoids with filled lumens (compare Figures 6B and 6C with Figure 5E). TGF-β induced a shrinkage in the size of duct-like organoids (compare Figure 6D with Figure 5F), likely because of compaction of cells and collapsing lumens (Figures 6B and 5D). In acinus-like organoids, TGF-β-induced an increase in organoid area and in the number of lumen-containing structures (compare Figures 6B–6D with Figures 5D and 5F).
Figure 6.

TGF-β augments KRASG12D-induced phenotypes in ACs and DUs
(A) Expression of the cell proliferation marker Ki67 (red) in DUs (blue) and ACs (orange) with KRASG12D expression and TGF-β treatment. Quantification of Ki67-positive organoids represented by box-and-whisker plots: range, 5%–95%; center lines indicate median values; gray dots represent individual measurement.
(B and C) Organoid morphological changes induced by KRASG12D expression and TGF-β treatment. Bar charts represent the mean and standard deviation of measurements from three independent cultures; gray dots represent individual measurements.
(D) Organoid size changes induced by KRASG12D expression and TGF-β treatment. Box-and-whisker plot representation as in (A).
(E and F) Expression of pancreatic lineage markers in DUs (E) and ACs (F) altered by KRASG12D expression and TGF-β treatment. Floating column charts represent RNA measurements from quantitative PCR (n = 3), hinges represent maximal and minimal values, central lines indicate mean values, and dots represent individual measurements.
(G–J) Detection of KRT7 (G), AB (H), MUC2 (I), and MUC5AC (J) in organoids under different experimental conditions. Bar chart representation as in (C).
Scale bars in main images, 50 μm; scale bars in insets, 25 μm. ∗∗∗p < 0.001; ∗∗, 0.001 < p < 0.01; ∗, 0. 01 < p < 0.05; N.S., p > 0.05. All results represent the sum of three independent cultures.
In duct-like organoids, TGF-β further upregulated NKX6.1 expression and suppressed GATA4 and PTF1A expression compared with levels observed in the absence of TGF-β (Figures 6E and 5G). In acinus-like organoids, KRASG12D and TGF-β upregulated PDX1, NKX6.1, PTF1A, and CPA1 (Figures 6F and 5H), suggesting reversion to a progenitor state. Prolonged stimulation of transformed epithelial cells with TGF-β is known to induce epithelial-to-mesenchymal transition (EMT) (Xu et al., 2009). Phase-contrast and H&E morphology analysis of KRASG12D-expressing organoids showed that TGF-β did not induce invasive behavior typically associated with EMT (Figure 6H). Although we cannot rule out acquisition of partial EMT states (Aiello et al., 2018), TGF-β-treated organoids showed an increase in the ductal epithelial marker KRT7 (Figure 6G), suggesting that the cells maintain their epithelial state under TGF-β stimulation conditions.
We studied the effect of KRASG12D and TGF-β on presentation of acidic mucins using Alcian blue (AB) staining. AB signals were increased by KRASG12D and enhanced further by TGF-β (Figure 6H). This effect was more dramatic in duct-like organoids than in acinus-like organoids. Among the mucins, expression of MUC5AC is associated with PanINs and all stages of pancreatic cancer (Kim et al., 2002), and a low frequency of MUC2 expression is associated with cystic lesions. Control organoids did not express either marker (Figures 6I and 6J), whereas MUC2 was detected in KRASG12D-expressing duct-like organoids with low frequency (~2.5%), and this was lost in TGF-β-treated organoids. MUC2 expression was not detected in acinus-like organoids (Figure 6I), regardless of treatment conditions. MUC5AC was rarely expressed in duct-like organoids with KRASG12D expression (Figure 6J) but expressed strongly in ~30% of KRASG12D-expressing organoids, suggesting a PanIN-like change.
KRASG12D is more effective in inducing acinus-like organoids to form early pancreatic-cancer-like lesions in vivo
To investigate in vivo phenotypes, duct-like or acinus-like organoids that were grown in the presence or absence of KRASG12D induction were dissociated and injected into the pancreata of 6- to 8-week old NCG mice at a rate of 500,000 cells per mouse. Immediately after surgery, mice were fed regular food (for organoids cultures without doxycycline (-DOX) ; without KRASG12Dexpression) or DOX-containing food (for organoids from the +DOX cultures; with KRASG12Dexpression) and monitored by palpation twice a week. All mice were sacrificed 8 weeks after transplantation.
We did not observe any transplant growth from control organoids (-DOX, N = 10; Figure S7A). In the presence of DOX, 67% of mice (7 of 10) transplanted with duct-like organoids expressing KRASG12D exhibited a wide spectrum of growth patterns, and three had no growth. Four had lesions with large, convoluted, dilated ductal structures lined with epithelium showing mild to moderate dysplastic changes, analogous to IPMN in humans (Figure 7A), and one mouse had a slightly dilated pancreatic duct, analogous to PanIN, and lesions with a convoluted conglomeration of duct structures in myoxid stroma lined with mucinous epithelium showing moderate to severe dysplastic change, analogous to early adenocarcinoma (Figure 7A). In contrast, 100% of mice (10 of 10) transplanted with acinus-like organoids expressing KRASG12D showed diverse patterns of growth in vivo. Seven mice had lesions exhibiting mild to moderate dysplastic changes, containing occasional goblet cells and focal cribriform, which were analogous to early adenocarcinoma in humans (Figure 7B). Two had lesions with dilated duct structures, analogous to IPMN (Figure 7B), and one mouse had a slightly dilated duct lined by normal serous epithelium, analogous to human PanIN (Figure 7B). Cells in lesions formed by duct-like and acinus-like organoids were positive for human cytokeratin 19, confirming that they were epithelial cells derived from human cells (Figures 7C, 7D, S7C, and S7D). Lesions with more aggressive histological phenotypes (Figures 7A and 7B) also had more frequent expression of Ki67 compared with those resembling precursor lesions (Figures 7C and 7D). In regions adjacent to normal mouse pancreas tissue, we observed histological phenotypes analogous to pancreatitis (Figures 7A and 7B, yellow arrows, and S7B). These adjacent regions, however, were negative for human cell markers and likely originated from mouse pancreatic cells.
Figure 7.

ACs expressing KRASG12D were more effective than DUs in inducing formation of early pancreatic-cancer-like lesions in vivo
(A and B) Histology of lesions grown from KRASG12D-expressing DUs (A, N = 9) or ACs (B, N = 10) transplanted into the mouse pancreas. Tissues were stained with nuclear red. Scale bars, 100 μm.
(C and D) Expression of human KRT19 (red), Ki67 (green), KRT7 (teal), MUC5AC (yellow), and SOX9 (purple) in lesions grown from KRASG12D-expressing DUs (C) or ACs (D). Scale bars, 100 μm.
Because KRASG12D expression altered cell differentiation of organoids in vitro, we also investigated cell differentiation in lesions grown in vivo. Lesions grown from duct-like organoids had strong KRT7 and SOX9 expression (Figures 7C and S7C). Interestingly, lesions developed from acinus-like organoids also had strong KRT7 and SOX9 expression (Figures 7D and S7D), suggesting that the acinus-like cells with KRASG12D expression underwent trans-differentiation in vivo and shifted to a ductal fate. The lesions strongly expressed MUC5AC in a mosaic pattern (Figures 7C, 7D, S7C, and S7D), similar to what we observed in human PDAC samples (Figures S7E and S7F). These results demonstrate that both types of organoids could grow in vivo, recapitulating different stages of pancreatic tumorigenesis, with acinus-like organoids generating more aggressive lesions.
Discussion
We report the feasibility of generating pancreatic duct-like and acinus-like organoids resembling fetal or neonatal states from PP cells derived from hPSCs. Using these lineage-committed organoids, we demonstrate that KRASG12D and GNASR201C have lineage-specific effects in culture and in vivo. Our results are consistent with Breunig et al (Breunig et al., 2021) and together identify the pancreatic progenitor cell derived exocrine organoid platform as an opportunity to understand mechanisms regulating lineage commitment in the human exocrine pancreas and to model pancreatic cancer initiation and progression in culture.
Although acinus-like and ductal-like cells did not express all markers associated with adult pancreatic exocrine cells, the expression pattern we observed is consistent with human fetal exocrine pancreatic acinar cells, where CPA1 expression is detected first, and elastase production is a later event (Fukayama et al., 1986; Jennings et al., 2013). Our observations of the expression patterns of pancreatic enzymes were also consistent with the morphology of secretory vesicles in acinar organoids, which resemble precursors but not the mature form of zymogen granules. About 30% of duct-like organoids expressed the mature duct marker CAII, consistent with the early stages of lineage specification. Consistent with these correlations, single-nucleus gene expression analysis identified similarities between our acinus-like and duct-like organoids and previously reported neonatal acinar and ductal signatures. We recognize that further optimization of culture conditions will be needed to induce generation of maturate of human acinar and ductal cells, which can aid in developing better models of PDAC. The results presented here, however, will help define conditions to induce ductal or acinar lineage commitment and identify a platform for understanding the mechanisms that regulate lineage commitment in the human pancreas.
We find that activating mutations in KRAS and GNAS show lineage tropism. KRAS was more effective in acinar cells, whereas GNAS was more effective in ductal epithelia. Although KRASG12D was competent in inducing proliferation in acinar and ductal lineages in culture, acinar cells were more effective than ductal cells in promoting formation of cancerous lesions in vivo. Consistent with the notion that IPMN originates from ductal epithelia (Ren et al., 2019), ductal acini responded strongly to GNASR201C expression by changes in morphology and expression of mucin (MUC2) associated with IPMN neoplastic lesions. Expression of GNASR201C in acinar cells failed to induce expression of MUC2, which is consistent with a previous study where acinus-specific expression of GnasR201C in mouse models was not sufficient to initiate PDAC (Patra et al., 2018). Understanding the mechanisms that regulate this lineage tropism and engineering genetic cooperation events to model initiation and progression of PDAC will provide important insights into events regulating initiation of pancreatic cancer.
We demonstrate that hPSCs can be induced to differentiate into ductal and acinar organoids representing the exocrine pancreas. In particular, our results identify a renewable source for generating human exocrine pancreas organoids for studying exocrine development and disease modeling.
Limitations of study
The organoids generated with our protocols represented fetal or neonatal exocrine pancreatic ducts and acini and did not represent adult lineages. We also lack data demonstrating the functions and growth of normal organoids in vivo. Further optimization of conditions may be needed to generate mature acinar and ductal organoids in culture that can grow in vivo. In addition, our single-nucleus analysis data did not have large cell numbers for ductal organoids, although the quality of the data was excellent. We analyzed cells 8 days after differentiation because day 8 was used for oncogene activation studies. It is possible that snRNA analysis of day 16 organoids would add strength to our data. Last, our studies of oncogene-induced cell state changes were performed in the cell population. Performing single-cell analysis will likely provide more insights into the biology of pancreatic cancer initiation. Although addressing the limitations can help increase the strength of our conclusions, the results presented already provide direct support for all conclusions drawn from this study.
STAR★Methods
Key resources table
| REAGENTS or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SOX9 | Millipore | AB5535 |
| CAII | Santa Cruz | sc-133111 |
| PTF1A | Christopher Wright Lab (Vanderbilt) | N/A |
| PTF1A | BD Biosciences | s25-763 |
| CTRC | Sino Biological | 11456-T24 |
| CPA1 | Sino Biological | 10504-RP02 |
| MUC2 | Santa Cruz | sc7314 |
| MUC5AC | Santa Cruz | sc33667 |
| phospho-ERK | Cell Signaling | 4370 |
| KRAS | Cell Signaling | 3339 |
| ERK | Cell Signaling | 9107 |
| KRT19 | Abcam | ab9221 |
| Ki67 | Cell Signaling | 9027 |
| KRT7 | DAKO | M7018 |
| P16 | Santa Cruz | sc-1661 |
| Goat anti-mouse IgG | Invitrogen | A-21424 |
| Goat anti-rabbit IgG | Invitrogen | A-11034 |
| Oligonucleotides | ||
| TBP (PCR primers) | IDT | Hs.PT.39a.22214825 |
| PDX1 (PCR primers) | IDT | Hs.PT.58.2295117 |
| NXK6-1 (PCR primers) | IDT | Hs.PT.58.25073618 |
| GATA4 (PCR primers) | IDT | Hs.PT.58.259457 |
| SOX9 (PCR primers) | IDT | Hs.PT.58.38984663 |
| CFTR (PCR primers) | IDT | Hs.PT.58.3365414 |
| PTF1A (PCR primers) | IDT | Hs.PT.58.45382933.g |
| CPA1 (PCR primers) | IDT | Hs.PT.58.3138313 |
| CTRC (PCR primers) | IDT | Hs.PT.58.15671909 |
| HNF1B (PCR primers) | IDT | Hs.PT.58.25568705 |
| RBBPJL (PCR primers) | IDT | Hs.PT.58.15167632.gs |
| Critical commercial assays | ||
| Amylase Activity Colorimetric Assay Kit | Biovision | K711 |
| Lipase Activity Fluorometric Assay Kit III | Biovision | K724 |
| Anhydrase (CA) Activity Assay Kit | Biovision | K472 |
| Alcian Blue (pH 2.5) Stain Kit | Vector Laboratories | H3501 |
| Deposited data | ||
| Single nuclei transcriptome | GEO repository | GEO: GSE169008 |
| Experimental models: cell lines | ||
| HUES8 | Douglas Metlon Lab (Harvard University) | CVCL_B207 |
| iPSC #11 | Douglas Metlon Lab (Harvard University) | N/A |
| iPSC #13 | Douglas Metlon Lab (Harvard University) | N/A |
| Experimental models: organisms | ||
| NCG mice | Charles River | 572 |
| Recombinant DNA | ||
| pINDUCER21 (ORF-EG) | Addgene | #46948 |
| Biological samples | ||
| PDAC patient tissue #1 (paraffin slide) | BIDMC | N/A |
| PDAC patient tissue #2 (paraffin slide) | BIDMC | N/A |
| Software and algorithms | ||
| Analysis Code | Github | https://github.com/dannyconrad/PancreasOrganoidMULTIseq |
| Chemicals, peptides, and recombinant proteins | ||
| Forskolin | Selleckchem | S2449 |
| BSA (fatty acid-free) | Proliant Biologicals | 68700 |
| PBS | Invitrogen | 14155063 |
| DEMEM | Invitrogen | 10564045 |
| TrypLE | Invitrogen | 12605010 |
| Matrigel | Corning | 354263 |
| Cell Recovery Solution | Corning | 354253 |
| Trizol | Invitrogen | 15596018 |
| Direct-zol | Zymo Research | R2080 |
| Superscript IV | Invitrogen | 11756500 |
| STEMxyme I | Worthington Biochemical | STZ1 |
| Doxycycline diet | Envigo | TD01306 |
| TGF | Peprotech | 100-21 |
| FGF10 | Peprotech | 100-26 |
| FGF2 | Peprotech | 100-18C |
| EGF | Peprotech | AF-100-15 |
| Doxycycline (powder) | Tocris | 4090 |
| A83-01 | Tocris | 2939 |
| Y27632 | Tocris | 1245 |
| SKL2001 | Selleckchem | S8320 |
| WNT1 | Peprotech | 120-17 |
| Foxy5 | Tocris | 5461 |
| Dexamethasone | Tocris | 1126 |
| LDN193189 | Tocris | 6053 |
| CD3254 | Tocris | 3302 |
| BMS961 | Tocris | 3410 |
| DBZ | Tocris | 4489 |
| HPI-1 | Tocris | 3839 |
| XMU-MP-1 | Selleckchem | S8334 |
| CHIR99021 | Tocris | 4953 |
| Ascorbic acid | Sigma | A4544 |
| EPZ011989 | Selleckchem | S7805 |
| IWP2 | Tocris | 3533 |
| IQ1 | Tocris | 4713 |
| iCRT-14 | Tocris | 4299 |
| CPTH2 | Cayman Chemical | 12086 |
| SB939 | Selleckchem | S1515 |
| WT161 | Selleckchem | S8495 |
| XAV939 | Tocris | 3748 |
| FGF1 | Peprotech | 100-17A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the lead contact, Senthil K Muthuswamy (smuthusw@bidmc.harvard.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
The single nucleus transcriptome data generated in this study will be available at GEO repository with accession number GEO: GSE169008. R language code for analysis will be available at https://github.com/dannyconrad/PancreasOrganoidMULTIseq.
Experimental model and subject details
In vivo animal models
Animal experiments performed in this study followed protocols approved by the institutional animal care and use committee at Beth Israel Deaconess Medical Center (Boston, USA). Male immunodeficient NOD CRISPR Prkdc Il2r Gamma (NCG) mice of 6-8 week old were purchased from Charles River (catalog #572). Results for each experimental group are summary of two to three independent operations using independent organoid cultures.
Pluripotent stem cell lines
Human embryonic stem line HUES8 was generated at Harvard University and has a normal 46 XY karyotype (NIH registration number 0021; RRID: CVCL_B207). Induced pluripotent stem cell lines 11 and 13 were generated by the Melton laboratory at Harvard University. Donor for line 11 is a 26 year old healthy male. Donor for line 13 is a 27 year old healthy female. pINDUCER21 (Addgene #46948) was used for transgene expression.
Method details
Induction of organoids
Pancreatic progenitor cells were derived from Hues-8 cells by Dr. Douglas Melton’s group, following a previously published protocol (Leite et al., 2020; Pagliuca et al., 2014). The PP1 cell aggregates at stage S4+2d induction was collected and dissociated with TrypLE (Invitrogen) into single cells. Digested cells were centrifuged at 1500 rpm for 5 min then resuspended in differentiation media supplemented with 5% Matrigel, at final cell density of 50,000/ml. Cell suspensions were seeded in culture plates or chamber slides precoated with 100% Matrigel. Media were replaced every four days, all supplemented with 5% Matrigel. For ductal cell induction, the basic medium contains DMEM, 1% Pen-Strep, 1% B27, 10 ng/ml FGF1, 50 μg/ml ascorbic acid, 1 μM A83-01, 10 ng/ml FGF10, 1 ng/ml EGF, 100 nM BMS961. Stage I duct medium (day 0-4) contains basic medium, supplemented with 15 ng FGF10, 10 ng/ml FGF2, 5 ng/ml EGF, 100 nM EPZ011989, 1 μg/ml Foxy5, 3 μM IWP2, 25 nM IQ1, 50 nM iCRT-14, 10 μM Y27632, 1 μM CPTH2, 50 nM SB939. Stage II (day 4-8) duct medium contains basic medium, supplemented with 1 μg/ml Foxy5, 3 μM IWP2, 25 nM IQ1, 50 nM iCRT-14, 50 nM WT161, 5 μM Y27632, 5 μM CPTH2. Stage III (day 8-12) duct medium contains basic medium, supplemented with 3μM IWP2, 25 nM IQ1, 50 nM iCRT-14, 5 μM CPTH2, 400 nM BMS961, 0.1 μM XAV939. Stage IV (day 12-) duct medium contains basic medium, supplemented with 3 μM IWP2, 5 nM IQ1, 10 nM iCRT-14, 0.5 μM CPTH2. For acinar induction, the basic medium contains DMEM, 1% Pen-Strep, 1% B27, 10 ng/ml FGF1, 50 μg/ml ascorbic acid, 1 μM A83-01, 10 ng/ml WNT1. Stage I acinar (day 0-4) medium contains basic medium, supplemented with 10 μM Y27632, 3μM SKL2001, 15 ng/ml WNT1, 50 nM dexamethasone, 5 ng/ml FGF2, 1 ng/EGF, 0.3 μM CHIR, 5 ng/ml FGF10, 0.1 μM HPI-1, 50 nM XMU-MP-1. Stage II (day 4-8) acinar medium contains basic medium, supplemented with 3 μM SKL2001, 200 nM dexamethasone, 0.1 μM HPI-1, LDN193189 0.1 μM, 5 nM CD3254, 50 nM XMU-MP-1. Stage III (day8-12) acinar medium contains basic medium, supplemented 3 μM SKL2001, 200 nM dexamethasone, 0.1 μM LDN193189, 1 μM DBZ. Stage IV (day12-) acinar medium contains basic medium, 0.3 μM SKL2001, 25 nM dexamethasone, 0.1 μM DBZ. For TGFβ treatment experiments, A83-01 were removed from medium from day 8.
Phase contrast imaging and analysis
For phase contrast imaging, organoids were seeded in Falcon culture chamber slide and images were taken every two days using 10X objective. For each time point, three independent cultures were imaged with at least 100 organoids imaged per culture. For analysis of phase contrast images, CellProfiler program (McQuin et al., 2018) was used to measure the Area and Form Factor. Form Factor is defined as 4∗π∗Area/Perimeter2.
Immunofluorescence
For direct immunostaining, organoids were seeded in Falcon culture chamber slides. At the time of analysis, organoids in chambers were washed once with PBS then fixed with 4% PFA for 40 minutes. After fixation, organoids were washed three times with PBS-glycine (100mM glycine) with 10 minute incubation each wash. Organoids then were permeabilized by 0.5% Triton X-100 of 5 minute incubation. After permeabilization, organoids were blocked using blocking buffer (PBS, 100 mM glycine, 0.1% BSA, 1% goat serum, 0.05% Triton X-100) for one hour. For primary antibody staining, antibodies were diluted with the blocking buffer and organoids were incubated with the dilution for 2-4 hours at room temperature then washed three times with blocking buffer (10 minutes incubation per wash). For secondary antibody staining, goat anti-mouse IgG or goat anti-rabbit IgG antibodies from Invitrogen were diluted at 1:400 in blocking buffer then incubated organoids with the dilution for 1.5 hour. After the secondary staining, organoids were washed with PBS three times and stained with DAPI for 5 minute. Slides were mounted with DAKO fluorescence mounting media. Alcian blue staining were performed using the Alcian Blue (pH2.5) Stain Kit (#H3501, Vector Laboratories). Imaging of slides were conducted under Zeiss LSM880 confocal system, using 20X objective or a Keyence BZ microscope. Image analyses were performed using CellProfiler.
For preparation of paraffin blocks, organoids in chamber slides were fixed with 4% PFA for 2 hours then embedded with Histogel in cryomold. Histogel capsules were transferred into cassette and processed at BIDMC histology core using standard paraffin embedding protocols. For staining of slides, paraffine sections of 5 μm thick were incubated with xylene 2x5 minutes then rehydrated in 100%, 95%, 75% ethanol and H2O for 10 minute per incubation. Antigen retrieval were done by incubating slides with citrus buffer at high pressure heating for 15 minutes. After this, slides were washed twice with PBS then blocked with blocking buffer for one hour. Primary staining and secondary staining were done by incubating slides with antibodies for one hour at room temperature, with three washes in between using blocking buffer. After secondary staining, slides were washed with PBS three times and stained with DAPI for 5 minutes. Slides were mounted with DAKO fluorescence mounting media. Slides were imaged using Zeiss LSM or Keyence Vision System. Antibodies used: SOX9, 1:200, #AB5535, Millipore ; CAII, 1:100, #sc-133111, Santa Cruz; PTF1A, 1:100, #s25-763, BD ; CTRC, 1:100, #11456-T24, Sino Biological; CPA1,1:100, #10504-RP02, Sino Biological; Muc2, 1:100, #sc7314, Santa Cruz; Muc5AC, 1:100, #sc33667, Santa Cruz; phosphor-ERK, 1:200, #4370, Cell Signaling; KRT19, 1:200, #ab9221, Abcam;
Western blotting
For western blotting, organoids were seeded in 6 well plates. At the time of analysis, culture media were aspirated then 1ml ice-cold PBS was added into one well. Pipette several times to physically disrupt the Matrigel and transfer the slurry into 15ml conical tube. Centrifuge tubes at 3500 rpm x 5minutes. Aspirate the supernatant carefully to not disrupt Matrigel layers. Add 1ml of ice-cold Cell Recovery Solution (Corning) and pipette several times then incubated tubes on ice for 1 hour. After the incubation, centrifuge tubes at 3500rpm x 5 minutes and aspirate the supernatant carefully not to disrupt cell pellets. Add 1ml of PBS to wash the pellet then centrifuge tubes at 3500rpm x 5 minutes and aspirate the supernatant carefully not to disrupt cell pellets. Pellets were lysed with RIPA buffer supplemented with protease and phosphatase inhibitors (Roche). Protein concentration of cell lysates were determined by Bradford assay and lysates were subject to standard procedures of western blotting. Antibodies use: Sox9, 1:500, #AB5535, Millipore; CAII, 1:500, #sc-133111, Santa Cruz; PTF1A, 1:500, a gift from Dr. Christopher Wright’s lab (Vanderbilt University); CTRC, 1:500, #11456-T24, Sino Biological.
Enzyme function analysis
Organoids were seeded in 6 well plate. At the time of analysis, culture media were aspirated then 1ml ice-cold PBS was added into one well. Pipette several times to physically disrupt the Matrigel and transfer the slurry into 15ml conical tube. Centrifuge tubes at 3500 rpm x 5minutes. Aspirate the supernatant carefully to not disrupt Matrigel layers. Add 1ml of ice-cold Cell Recovery Solution (Corning) and pipette several times then incubated tubes on ice for 1 hour. After the incubation, centrifuge tubes at 3500rpm x 5 minutes and aspirate the supernatant carefully not to disrupt cell pellets. Add 1ml of PBS to wash the pellet then centrifuge tubes at 3500rpm x 5 minutes and aspirate the supernatant carefully not to disrupt cell pellets. Pellets were lysed with enzyme analysis buffer (150 mM NaCl, 10 mM Tris, 1% Triton X-100, pH = 7.2) with no protease inhibitors supplemented. The protein concentration of cell lysates was determined by Bradford assay. Enzyme activities were analyzed using Lipase Activity Fluorometric Assay Kit (#K724, Biovision) and Carbonic Anhydrase Activity Assay Kit (#K472, Biovision), following the instructions by the manufacturer.
RNA extraction and qPCR analysis
Organoids were seeded in 12 well plates. At the time of analysis, culture media were aspirated and 0.5 mL of ice-cold Cell Recovery Solution (Corning) was added into one well. Pipette several times to physically disrupt the Matrigel and transfer the slurry into 1.5ml microcentrifuge tube and incubate on ice. After 30 minutes, centrifuge the tubes at 8000rpm x 5 minutes. Aspirate supernatant then add 250 ul Trizol (Invitrogen). Lyze pellets for 15 minutes at room temperature. Purification of total RNA was done using Direct-Zol RNA kits (ZymoResearch). cDNA was synthesized using SuperScript IV First-Strand Master kit (Invitrogen). Gene expression was quantitated by quantitative PCR using PrimeTime qPCR Probe Assays kit.
Virus production and infection
Mutant genes were cloned into pINDUCER21 (Addgene #46948) and lentivirues produced were concentrated by ultracentrifugaton at 25,000rpm for 2.5 hours at 4 degrees. Viruses were resuspended in PBS. 6-well plate was coated with Matrigel by incubating with 5% Matrigel for 1 hour then aspirate the solution to air dry. 500,000 pancreatic progenitor cells with culture media were seeded in one well and allowed to grow overnight. On second day, lentiviruses were mixed with culture media supplemented with 4ug/ml polybrene and 2ml solution was added into each well. To improve infection rate, plates were centrifuged at 3500rpm for 30 mins. The plates were placed in incubator overnight. On third day, cells were digested with Accutase (Sigma), collected and seeded in 3D. For induction of transgene, doxycycline was added into culture media at final concentration of 1ug/ml.
Mouse transplantation
Day 16 organoid cultures with or without KRasG12D expression were digested with STEMxyme I (1mg/ml) for 1.5 hours then dissociated with Accutase (Sigma). Dissociated cells were counted and resuspended in 90% Matrigel with a final density of 10 million cells/ml. Orthotopic mouse pancreas transplantation was conducted following the protocol approved by the institutional animal care and use committee at BIDMC. 6-8 week old male NCG mice (#572, Charles River) were purchased for this study. Mice were anesthetized by isoflurane and a 1.5 cm incision was cut at the left upper abdominal flank. The pancreas was carefully pulled out, and 50 ul of cell suspension was injected at the tail of the pancreas using a 29G needle. After injection, the pancreas was gently placed back into the peritoneal cavity, and the wound was sutured. Meloxicam was used as analgesics. Mice were fed with regular food or diets with doxycycline (#TD01306, Envigo). Eight weeks after injection, mice were sacrificed following approved protocol, and the pancreas was collected then processed as paraffin blocks.
For mammary fat pad transplantation, experiments were performed following the protocol approved by the institutional animal care and use committee at BIDMC. 6-8 week old female NCG mice (#572, Charles River) were purchased for this study. At the time of operation, Mice were anesthetized by isoflurane and a 1.5 cm incision was cut on the skin near No.9 and No.4 glands. A 29G needle was used to inject 50ul of cell/Matrigel suspension into mammary fat pad, at density of 500,000 cells/gland. After injection, the wound was sutured and Meloxicam was used as analgesics. Mice were fed with regular food or diets with doxycycline (#TD01306, Envigo). Eight weeks after injection, mice were sacrificed following approved protocol and pancreas was collected then processed as paraffin blocks.
Single nuclei RNA sequencing and analysis
Isolation of single nuclei from organoids were performed based on a published protocol (Tosti et al., 2021). 2ml of citric acid-based buffer (sucrose 0.25 M, citric acid 25 mM, MgCl2 3mM) were added to one well of 6-well plate with organoid culture. Slurries were collected then centrifuged at 800 g for 3 minutes at 4 degrees. Supernatant was discarded and cell pellets were incubated with 2 mL of citric acid-Triton buffer (sucrose 0.25 M, citric acid 25 mM, MgCl2 3mM, 0.05% Triton X-100) for 3 minutes on ice then centrifuged at 800 g for 3 minutes at 4 degrees. Supernatant was discarded and pellets were resuspended with 2 mL of citric acid-based buffer (sucrose 0.25 M, citric acid 25 mM, MgCl2 3mM) then pass through a 35 μm cell strainer. The flow-through was centrifuged at 800 g for 3 minutes at 4 degrees. The supernatant was discarded and pellets were resuspended with 0.5 mL of storage buffer (KCl 25 mM, MgCl2 3mM, Tris 25 mM, SuperaseIn 1 U/μL, cOmplete protease inhibitor (4693159001, sigma), 40% Glycerol). Nuclei in storage buffer were transferred to 1.5 mL microcentrifuge tubes and froze at −80 degrees.
Frozen nuclei pellets were thawed in 50 μL 10% PBS-BSA, pelleted (800 g, 3 min, 4°C), and washed with 1 mL 2% PBS-BSA. Nuclei were counted, pelleted again, and resuspended at 5x10ˆ6 nuclei/mL in cold PBS. ~200k nuclei were aliquoted from each sample for labeling with MULTI-seq barcodes (McGinnis et al., 2019). After labeling, nuclei were washed once with 1 mL 2% PBS-BSA, pelleted (600 g, 4 min, 4°C), resuspended in 200μL 2% PBS-BSA, and then pooled. The pooled sample was counted, and cDNA libraries were generated using two lanes of a 10X Genomics Chromium Next GEM Single Cell 3′ v3.1 reagent kit, targeting 10k nuclei per lane. MULTI-seq barcode libraries were generated according to the MULTI-seq protocol (https://github.com/chris-mcginnis-ucsf/MULTI-seq). The quality of all libraries was assessed using the High Sensitivity DNA kit for the Agilent 2100 Bioanalyzer. cDNA and barcode libraries were multiplexed and submitted to the UCSF Center for Advanced Technology for sequencing on a NovaSeq 6000 SP flow cell, yielding 518M reads.
cDNA reads from both libraries were aligned to the GRCh38-3.0.0 reference transcriptome with Cell Ranger v5.0 (https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest), using the option “–include-introns” to enable detection of unspliced mRNAs. Using the Seurat v3.2 R package (Stuart et al., 2019), filtered gene expression matrices from each library were loaded and then integrated into a single merged dataset. MULTI-seq barcode libraries were loaded and parsed with the deMULTIplex v1.0 R package (https://github.com/chris-mcginnis-ucsf/MULTI-seq) to assign each nucleus to its sample of origin. Doublets, unclassifiable nuclei, and samples with insufficient nuclei recovery were removed from the dataset, yielding a final dataset of 4,450 nuclei. From these we captured an average of 959 UMIs and 780 genes per nucleus. Mitochondrial genes were removed from the dataset to mitigate biases arising from differences in nuclei isolation between samples. Expression data were normalized with SCTransform (Hafemeister and Satija, 2019). The first 32 principal components were used for UMAP embeddings and to calculate the k nearest neighbors per cell, followed by cluster identification using the Louvain algorithm.
Using the sample classifications derived from MULTI-seq barcodes, Clusters 1 & 3 were identified as Pancreatic Progenitor nuclei, Cluster 2 as Day 8 Ductal Organoid nuclei, and Clusters 0 and 4 as Day 8 Acinar Organoid nuclei. Using these new groupings, marker genes were calculated using FindAllMarkers with a logFC threshold of 0, and only those with an adjusted p value < 0.05 were kept for downstream analysis. The marker heatmap was produced using Seurat’s DoHeatmap function by downsampling all three cell types to the same number and using only marker genes overexpressed by 20%, i.e., avg_logFC > log(1.2). Pearson correlation coefficients were calculated pairwise between acinar organoids, ductal organoids, and progenitors using the average expression of all genes in the dataset computed by Seurat’s AverageExpression function.
As a reference point against which to compare our organoids, single-nucleus RNA-seq data of the human neonatal pancreas was downloaded from http://singlecell.charite.de/pancreas/ (Tosti et al., 2021) and filtered to contain only the acinar and ductal cells. Using markers derived from organoids and progenitors (adjusted p value ≤ 0.05), we calculated average expression profiles for each cell type in both datasets and computed Pearson correlation coefficients pairwise between them.
Quantification and statistical analysis
For data regarding RNA expression quantitated by qPCR, enzyme activities, protein marker expression detected by immunofluorescent staining, Alcian blue staining, cell number changes, percentages of organoids with lumen changes, t test was used for testing the statistical significance and p = 0.05 was used as the threshold. For data regarding organoid size and form factor changes, Ki67 expression, p16INK4A expression, Mann-Whitney test was used for testing the statistical significance and p = 0.05 was used as the threshold. Details for chart representation and experimental replication can found in figure legend.
Single nuclei sequencing results were analyzed using the Seurat package in R language. Statistical significance of differences in gene expression was tested using the Wilcoxon Rank Sum test and p = 0.05 was used as the threshold. Details can be found in experimental procedures.
Acknowledgments
We are grateful to members of the Melton laboratory, including Elise Engquist, Yi Yu, Kaleigh Biles, Jingping Zhang, and Kyle Boulanger, for technical support with stem cell culture and in vitro differentiation. We thank Dr. Anirban Maitra (MD Anderson) for providing wild-type and mutant GNAS cDNAs. We thank Dr. Christopher Wright (Vanderbilt University) for providing the PTF1A antibody. In addition, we thank members of the Muthuswamy laboratory for critical input throughout the development of this project. Institutional startup funds and UO1 (CA224193 to S.K.M.), an F32 fellowship (F32GM115201 to R.D.), seed grant from the Hirschberg Foundation for Pancreatic Cancer Research (to L.H.), and R01 from NIGMS (R01GM135462 to Z.G.) were used for completion of this study.
Author contributions
L.H. and S.K.M. conceived and designed the study, interpreted the results, and wrote the manuscript. L.H. developed the medium conditions for generation of acinar and ductal organoids and performed KRas experiments. R.D. performed all experiments using GNAS, analyzed the data, and wrote the corresponding results. C.M.L. and L.B.M. conducted bioinformatics analyses of TGCA pancreatic cancer cohorts. R.G. analyzed the histopathology of pancreatic transplants. D.N.C. and Z.G. performed single-nucleus gene expression studies, analyzed the data, and wrote the corresponding sections. N.C.L. generated PP lines from iPSCs.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Published: April 28, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stem.2021.03.022.
Supplemental information
Wilcoxon Rank Sum test was used to calculate the significance of differences between cell clusters, and p = 0.05 was used as the threshold. Column titles: avg_logFC, natural log values of average fold changes; pct1, gene expression in the cluster of interests; pct2, gene expression in the rest of cells.
Genes were ranked based on fold changes. The top 20 most upregulated and downregulated genes were selected.
Expression of top 20 organoid marker genes in normal human pancreas. Images obtained from Human Protein Atlas (HPA). Column titles: avg_logFC, natural log values of average fold changes; pct1, gene expression in the cluster of interests; pct2, gene expression in the rest of cells. Label for subcellular localization: not reported, data not available in HPA; Exo > Endo, proteins enriched in the exocrine pancreas; Exo = Endo, proteins expressed at similar levels across pancreas; Exo < Endo, proteins enriched in the endocrine pancreas.
References
- Aguirre A.J., Bardeesy N., Sinha M., Lopez L., Tuveson D.A., Horner J., Redston M.S., DePinho R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello N.M., Maddipati R., Norgard R.J., Balli D., Li J., Yuan S., Yamazoe T., Black T., Sahmoud A., Furth E.E. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell. 2018;45:681–695.e4. doi: 10.1016/j.devcel.2018.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almoguera C., Shibata D., Forrester K., Martin J., Arnheim N., Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- Amato E., Molin M.D., Mafficini A., Yu J., Malleo G., Rusev B., Fassan M., Antonello D., Sadakari Y., Castelli P. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J. Pathol. 2014;233:217–227. doi: 10.1002/path.4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardeesy N., Aguirre A.J., Chu G.C., Cheng K.H., Lopez L.V., Hezel A.F., Feng B., Brennan C., Weissleder R., Mahmood U. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boj S.F., Hwang C.I., Baker L.A., Chio I.I., Engle D.D., Corbo V., Jager M., Ponz-Sarvise M., Tiriac H., Spector M.S. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breunig M., Merkle J., Wagner M., Melzer M.K., Barth T.F.E., Engleitner T., Krumm J., Wiedenmann S., Cohrs C.M., Perkhofer L. Modeling plasticity and dysplasia of pancreatic ductal organoids derived from human pluripotent stem cells. Cell Stem Cell. 2021;28 doi: 10.1016/j.stem.2021.03.005. Published online April 28, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscail L., Bournet B., Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020;17:153–168. doi: 10.1038/s41575-019-0245-4. [DOI] [PubMed] [Google Scholar]
- Collet L., Ghurburrun E., Meyers N., Assi M., Pirlot B., Leclercq I.A., Couvelard A., Komuta M., Cros J., Demetter P. Kras and Lkb1 mutations synergistically induce intraductal papillary mucinous neoplasm derived from pancreatic duct cells. Gut. 2020;69:704–714. doi: 10.1136/gutjnl-2018-318059. [DOI] [PubMed] [Google Scholar]
- Cooper C.L., O’Toole S.A., Kench J.G. Classification, morphology and molecular pathology of premalignant lesions of the pancreas. Pathology. 2013;45:286–304. doi: 10.1097/PAT.0b013e32835f2205. [DOI] [PubMed] [Google Scholar]
- De La O J.P., Emerson L.L., Goodman J.L., Froebe S.C., Illum B.E., Curtis A.B., Murtaugh L.C. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. USA. 2008;105:18907–18912. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lisle R.C., Logsdon C.D. Pancreatic acinar cells in culture: expression of acinar and ductal antigens in a growth-related manner. Eur. J. Cell Biol. 1990;51:64–75. [PubMed] [Google Scholar]
- Dekkers J.F., Wiegerinck C.L., de Jonge H.R., Bronsveld I., Janssens H.M., de Winter-de Groot K.M., Brandsma A.M., de Jong N.W., Bijvelds M.J., Scholte B.J. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013;19:939–945. doi: 10.1038/nm.3201. [DOI] [PubMed] [Google Scholar]
- Esni F., Ghosh B., Biankin A.V., Lin J.W., Albert M.A., Yu X., MacDonald R.J., Civin C.I., Real F.X., Pack M.A. Notch inhibits Ptf1 function and acinar cell differentiation in developing mouse and zebrafish pancreas. Development. 2004;131:4213–4224. doi: 10.1242/dev.01280. [DOI] [PubMed] [Google Scholar]
- Ferreira R.M.M., Sancho R., Messal H.A., Nye E., Spencer-Dene B., Stone R.K., Stamp G., Rosewell I., Quaglia A., Behrens A. Duct- and Acinar-Derived Pancreatic Ductal Adenocarcinomas Show Distinct Tumor Progression and Marker Expression. Cell Rep. 2017;21:966–978. doi: 10.1016/j.celrep.2017.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukayama M., Ogawa M., Hayashi Y., Koike M. Development of human pancreas. Immunohistochemical study of fetal pancreatic secretory proteins. Differentiation. 1986;31:127–133. doi: 10.1111/j.1432-0436.1986.tb00393.x. [DOI] [PubMed] [Google Scholar]
- Gidekel Friedlander S.Y., Chu G.C., Snyder E.L., Girnius N., Dibelius G., Crowley D., Vasile E., DePinho R.A., Jacks T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16:379–389. doi: 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies T.E., Pargett M., Silva J.M., Teragawa C.K., McCormick F., Albeck J.G. Oncogenic mutant RAS signaling activity is rescaled by the ERK/MAPK pathway. Mol. Syst. Biol. 2020;16:e9518. doi: 10.15252/msb.20209518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C., Schuhmacher A.J., Cañamero M., Grippo P.J., Verdaguer L., Pérez-Gallego L., Dubus P., Sandgren E.P., Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Habbe N., Shi G., Meguid R.A., Fendrich V., Esni F., Chen H., Feldmann G., Stoffers D.A., Konieczny S.F., Leach S.D., Maitra A. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc. Natl. Acad. Sci. USA. 2008;105:18913–18918. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler J., Cullis J., Avanzi A., Vucic E.A., Bar-Sagi D. Pre-neoplastic pancreas cells enter a partially mesenchymal state following transient TGF-β exposure. Oncogene. 2018;37:4334–4342. doi: 10.1038/s41388-018-0264-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haumaitre C., Lenoir O., Scharfmann R. Histone deacetylase inhibitors modify pancreatic cell fate determination and amplify endocrine progenitors. Mol. Cell. Biol. 2008;28:6373–6383. doi: 10.1128/MCB.00413-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani S.R., Petricoin E.F., Maitra A., Rajapakse V., King C., Jacobetz M.A., Ross S., Conrads T.P., Veenstra T.D., Hitt B.A. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- Hohwieler M., Illing A., Hermann P.C., Mayer T., Stockmann M., Perkhofer L., Eiseler T., Antony J.S., Müller M., Renz S. Human pluripotent stem cell-derived acinar/ductal organoids generate human pancreas upon orthotopic transplantation and allow disease modelling. Gut. 2017;66:473–486. doi: 10.1136/gutjnl-2016-312423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruban R.H., Goggins M., Parsons J., Kern S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000;6:2969–2972. [PubMed] [Google Scholar]
- Hruban R.H., Maitra A., Kern S.E., Goggins M. Precursors to pancreatic cancer. Gastroenterol. Clin. North Am. 2007;36:831–849, vi. doi: 10.1016/j.gtc.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Holtzinger A., Jagan I., BeGora M., Lohse I., Ngai N., Nostro C., Wang R., Muthuswamy L.B., Crawford H.C. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015;21:1364–1371. doi: 10.1038/nm.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huch M., Bonfanti P., Boj S.F., Sato T., Loomans C.J., van de Wetering M., Sojoodi M., Li V.S., Schuijers J., Gracanin A. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013;32:2708–2721. doi: 10.1038/emboj.2013.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideno N., Yamaguchi H., Ghosh B., Gupta S., Okumura T., Steffen D.J., Fisher C.G., Wood L.D., Singhi A.D., Nakamura M. GNAS(R201C) Induces Pancreatic Cystic Neoplasms in Mice That Express Activated KRAS by Inhibiting YAP1 Signaling. Gastroenterology. 2018;155:1593–1607.e12. doi: 10.1053/j.gastro.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings R.E., Berry A.A., Kirkwood-Wilson R., Roberts N.A., Hearn T., Salisbury R.J., Blaylock J., Piper Hanley K., Hanley N.A. Development of the human pancreas from foregut to endocrine commitment. Diabetes. 2013;62:3514–3522. doi: 10.2337/db12-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda M., Matthaei H., Wu J., Hong S.M., Yu J., Borges M., Hruban R.H., Maitra A., Kinzler K., Vogelstein B., Goggins M. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730–733.e9. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe M.D., Wang H., De La O J.P., Khan A., Firpo M.A., Murtaugh L.C. β-catenin is selectively required for the expansion and regeneration of mature pancreatic acinar cells in mice. Dis. Model. Mech. 2012;5:503–514. doi: 10.1242/dmm.007799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G.E., Bae H.I., Park H.U., Kuan S.F., Crawley S.C., Ho J.J., Kim Y.S. Aberrant expression of MUC5AC and MUC6 gastric mucins and sialyl Tn antigen in intraepithelial neoplasms of the pancreas. Gastroenterology. 2002;123:1052–1060. doi: 10.1053/gast.2002.36018. [DOI] [PubMed] [Google Scholar]
- Kleeff J., Korc M., Apte M., La Vecchia C., Johnson C.D., Biankin A.V., Neale R.E., Tempero M., Tuveson D.A., Hruban R.H., Neoptolemos J.P. Pancreatic cancer. Nat. Rev. Dis. Primers. 2016;2:16022. doi: 10.1038/nrdp.2016.22. [DOI] [PubMed] [Google Scholar]
- Kopp J.L., Dubois C.L., Schaeffer D.F., Samani A., Taghizadeh F., Cowan R.W., Rhim A.D., Stiles B.L., Valasek M., Sander M. Loss of Pten and Activation of Kras Synergistically Induce Formation of Intraductal Papillary Mucinous Neoplasia From Pancreatic Ductal Cells in Mice. Gastroenterology. 2018;154:1509–1523.e5. doi: 10.1053/j.gastro.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp J.L., von Figura G., Mayes E., Liu F., Dubois C.L., Morris J.P., IV, Pan F.C., Akiyama H., Wright C.V.E., Jensen K., Hebrok M., Sander M. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(6):737–750. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitio M., Lev R., Orlic D. The developing human fetal pancreas: an ultrastructural and histochemical study with special reference to exocrine cells. J. Anat. 1974;117:619–634. [PMC free article] [PubMed] [Google Scholar]
- Lee J., Snyder E.R., Liu Y., Gu X., Wang J., Flowers B.M., Kim Y.J., Park S., Szot G.L., Hruban R.H. Reconstituting development of pancreatic intraepithelial neoplasia from primary human pancreas duct cells. Nat. Commun. 2017;8:14686. doi: 10.1038/ncomms14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.Y.L., Dubois C.L., Sarai K., Zarei S., Schaeffer D.F., Sander M., Kopp J.L. Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut. 2019;68:487–498. doi: 10.1136/gutjnl-2017-314426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite N.C., Sintov E., Meissner T.B., Brehm M.A., Greiner D.L., Harlan D.M., Melton D.A. Modeling Type 1 Diabetes In Vitro Using Human Pluripotent Stem Cells. Cell Rep. 2020;32:107894. doi: 10.1016/j.celrep.2020.107894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Akanuma N., Liu C., Naji A., Halff G.A., Washburn W.K., Sun L., Wang P. TGF-β1 promotes acinar to ductal metaplasia of human pancreatic acinar cells. Sci. Rep. 2016;6:30904. doi: 10.1038/srep30904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugea A., Waldron R.T., Mareninova O.A., Shalbueva N., Deng N., Su H.Y., Thomas D.D., Jones E.K., Messenger S.W., Yang J. Human Pancreatic Acinar Cells: Proteomic Characterization, Physiologic Responses, and Organellar Disorders in ex Vivo Pancreatitis. Am. J. Pathol. 2017;187:2726–2743. doi: 10.1016/j.ajpath.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra A., Hruban R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008;3:157–188. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui T., Swift G.H., Deering T., Shen C., Coats W.S., Long Q., Elsässer H.P., Magnuson M.A., MacDonald R.J. Replacement of Rbpj with Rbpjl in the PTF1 complex controls the final maturation of pancreatic acinar cells. Gastroenterology. 2010;139:270–280. doi: 10.1053/j.gastro.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis C.S., Patterson D.M., Winkler J., Conrad D.N., Hein M.Y., Srivastava V., Hu J.L., Murrow L.M., Weissman J.S., Werb Z. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods. 2019;16:619–626. doi: 10.1038/s41592-019-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C., Goodman A., Chernyshev V., Kamentsky L., Cimini B.A., Karhohs K.W., Doan M., Ding L., Rafelski S.M., Thirstrup D. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018;16:e2005970. doi: 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S., Engleitner T., Maresch R., Zukowska M., Lange S., Kaltenbacher T., Konukiewitz B., Öllinger R., Zwiebel M., Strong A. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature. 2018;554:62–68. doi: 10.1038/nature25459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh L.C., Stanger B.Z., Kwan K.M., Melton D.A. Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl. Acad. Sci. USA. 2003;100:14920–14925. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka T., Tomosugi T., Kimura R., Nakamura S., Miyasaka Y., Nakata K., Mori Y., Morita M., Torata N., Shindo K. Clinical assessment of the GNAS mutation status in patients with intraductal papillary mucinous neoplasm of the pancreas. Surg. Today. 2019;49:887–893. doi: 10.1007/s00595-019-01797-7. [DOI] [PubMed] [Google Scholar]
- Pagliuca F.W., Millman J.R., Gürtler M., Segel M., Van Dervort A., Ryu J.H., Peterson Q.P., Greiner D., Melton D.A. Generation of functional human pancreatic β cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra K.C., Kato Y., Mizukami Y., Widholz S., Boukhali M., Revenco I., Grossman E.A., Ji F., Sadreyev R.I., Liss A.S. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat. Cell Biol. 2018;20:811–822. doi: 10.1038/s41556-018-0122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert M., Rustgi A.K. Pancreatic ductal cells in development, regeneration, and neoplasia. J. Clin. Invest. 2011;121:4572–4578. doi: 10.1172/JCI57131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B., Liu X., Suriawinata A.A. Pancreatic Ductal Adenocarcinoma and Its Precursor Lesions: Histopathology, Cytopathology, and Molecular Pathology. Am. J. Pathol. 2019;189:9–21. doi: 10.1016/j.ajpath.2018.10.004. [DOI] [PubMed] [Google Scholar]
- Riesle E., Friess H., Zhao L., Wagner M., Uhl W., Baczako K., Gold L.I., Korc M., Büchler M.W. Increased expression of transforming growth factor beta s after acute oedematous pancreatitis in rats suggests a role in pancreatic repair. Gut. 1997;40:73–79. doi: 10.1136/gut.40.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooman I., Heremans Y., Heimberg H., Bouwens L. Modulation of rat pancreatic acinoductal transdifferentiation and expression of PDX-1 in vitro. Diabetologia. 2000;43:907–914. doi: 10.1007/s001250051468. [DOI] [PubMed] [Google Scholar]
- Rovira M., Delaspre F., Massumi M., Serra S.A., Valverde M.A., Lloreta J., Dufresne M., Payre B., Konieczny S.F., Savatier P. Murine embryonic stem cell-derived pancreatic acinar cells recapitulate features of early pancreatic differentiation. Gastroenterology. 2008;135:1301–1310. doi: 10.1053/j.gastro.2008.06.049. 1310.e1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino T., Kawasaki S., Shimokawa M., Tamagawa H., Toshimitsu K., Fujii M., Ohta Y., Matano M., Nanki K., Kawasaki K. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell. 2018;22:454–467.e6. doi: 10.1016/j.stem.2017.12.009. [DOI] [PubMed] [Google Scholar]
- Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sharma A., Sances S., Workman M.J., Svendsen C.N. Multi-lineage Human iPSC-Derived Platforms for Disease Modeling and Drug Discovery. Cell Stem Cell. 2020;26:309–329. doi: 10.1016/j.stem.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih H.P., Kopp J.L., Sandhu M., Dubois C.L., Seymour P.A., Grapin-Botton A., Sander M. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development. 2012;139:2488–2499. doi: 10.1242/dev.078634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek S., Zhou T., Robinson C.L., Tsai S.Y., Crespo M., Amin S., Lin X., Hon J., Evans T., Chen S. Modeling Cystic Fibrosis Using Pluripotent Stem Cell-Derived Human Pancreatic Ductal Epithelial Cells. Stem Cells Transl. Med. 2016;5:572–579. doi: 10.5966/sctm.2015-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taki K., Ohmuraya M., Tanji E., Komatsu H., Hashimoto D., Semba K., Araki K., Kawaguchi Y., Baba H., Furukawa T. GNAS(R201H) and Kras(G12D) cooperate to promote murine pancreatic tumorigenesis recapitulating human intraductal papillary mucinous neoplasm. Oncogene. 2016;35:2407–2412. doi: 10.1038/onc.2015.294. [DOI] [PubMed] [Google Scholar]
- Tosti L., Hang Y., Debnath O., Tiesmeyer S., Trefzer T., Steiger K., Ten F.W., Lukassen S., Ballke S., Kuhl A.A. Single-Nucleus and In Situ RNA-Sequencing Reveal Cell Topographies in the Human Pancreas. Gastroenterology. 2021;160:1330–1334.e11. doi: 10.1053/j.gastro.2020.11.010. [DOI] [PubMed] [Google Scholar]
- Tuveson D.A., Shaw A.T., Willis N.A., Silver D.P., Jackson E.L., Chang S., Mercer K.L., Grochow R., Hock H., Crowley D. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- Tuveson D.A., Zhu L., Gopinathan A., Willis N.A., Kachatrian L., Grochow R., Pin C.L., Mitin N.Y., Taparowsky E.J., Gimotty P.A. Mist1-KrasG12D knock-in mice develop mixed differentiation metastatic exocrine pancreatic carcinoma and hepatocellular carcinoma. Cancer Res. 2006;66:242–247. doi: 10.1158/0008-5472.CAN-05-2305. [DOI] [PubMed] [Google Scholar]
- Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Waters A.M., Der C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018;8:a031435. doi: 10.1101/cshperspect.a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Matthaei H., Maitra A., Dal Molin M., Wood L.D., Eshleman J.R., Goggins M., Canto M.I., Schulick R.D., Edil B.H. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci. Transl. Med. 2011;3:92ra66. doi: 10.1126/scitranslmed.3002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Lamouille S., Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Wilcoxon Rank Sum test was used to calculate the significance of differences between cell clusters, and p = 0.05 was used as the threshold. Column titles: avg_logFC, natural log values of average fold changes; pct1, gene expression in the cluster of interests; pct2, gene expression in the rest of cells.
Genes were ranked based on fold changes. The top 20 most upregulated and downregulated genes were selected.
Expression of top 20 organoid marker genes in normal human pancreas. Images obtained from Human Protein Atlas (HPA). Column titles: avg_logFC, natural log values of average fold changes; pct1, gene expression in the cluster of interests; pct2, gene expression in the rest of cells. Label for subcellular localization: not reported, data not available in HPA; Exo > Endo, proteins enriched in the exocrine pancreas; Exo = Endo, proteins expressed at similar levels across pancreas; Exo < Endo, proteins enriched in the endocrine pancreas.
Data Availability Statement
The single nucleus transcriptome data generated in this study will be available at GEO repository with accession number GEO: GSE169008. R language code for analysis will be available at https://github.com/dannyconrad/PancreasOrganoidMULTIseq.