

Abstract
BACKGROUND
Preclinical studies suggest that bb2121, a chimeric antigen receptor (CAR) T-cell therapy that targets B-cell maturation antigen (BCMA), has potential for the treatment of multiple myeloma.
METHODS
In this phase 1 study involving patients with relapsed or refractory multiple myeloma, we administered bb2121 as a single infusion at doses of 50×106, 150×106, 450×106, or 800×106 CAR-positive (CAR+) T cells in the dose-escalation phase and 150×106 to 450×106 CAR+ T cells in the expansion phase. Patients had received at least three previous lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or were refractory to both drug classes. The primary end point was safety.
RESULTS
Results for the first 33 consecutive patients who received a bb2121 infusion are reported. The data-cutoff date was 6.2 months after the last infusion date. Hematologic toxic effects were the most common events of grade 3 or higher, including neutropenia (in 85% of the patients), leukopenia (in 58%), anemia (in 45%), and thrombocytopenia (in 45%). A total of 25 patients (76%) had cytokine release syndrome, which was of grade 1 or 2 in 23 patients (70%) and grade 3 in 2 patients (6%). Neurologic toxic effects occurred in 14 patients (42%) and were of grade 1 or 2 in 13 patients (39%). One patient (3%) had a reversible grade 4 neurologic toxic effect. The objective response rate was 85%, including 15 patients (45%) with complete responses. Six of the 15 patients who had a complete response have had a relapse. The median progression-free survival was 11.8 months (95% confidence interval, 6.2 to 17.8). All 16 patients who had a response (partial response or better) and who could be evaluated for minimal residual disease (MRD) had MRD-negative status (≤10−4 nucleated cells). CAR T-cell expansion was associated with responses, and CAR T cells persisted up to 1 year after the infusion.
CONCLUSIONS
We report the initial toxicity profile of a BCMA-directed cellular immunotherapy for patients with relapsed or refractory multiple myeloma. Antitumor activity was documented. (Funded by Bluebird Bio and Celgene; CRB-401 ClinicalTrials.gov number, NCT02658929.)
Multiple myeloma remains an incurable plasma-cell cancer. Advancements in treatment, including the introduction of immunomodulatory drugs, proteasome inhibitors, and monoclonal antibodies, have prolonged survival.1–5 However, almost all patients eventually have a relapse, with worse survival outcomes seen in patients with a high-risk cytogenetic profile or treatment-refractory disease.6–9 Chimeric antigen receptor (CAR) T-cell therapy has emerged as a novel treatment that has the potential for long-term disease control in some hematologic cancers,10–12 with anti-CD19 CAR T-cell therapies showing efficacy in patients with leukemia or lymphoma.13–22 B-cell maturation antigen (BCMA) is a member of the tumor necrosis factor superfamily of proteins that is primarily expressed by malignant and normal plasma cells and some mature B cells, making it a potential target for multiple myeloma.23–27 We produced bb2121 by transducing autologous T cells with a lentiviral vector encoding a second-generation CAR incorporating an anti-BCMA single-chain variable fragment, a CD137 (4–1BB) costimulatory motif, and a CD3-zeta signaling domain.28
In preclinical studies, bb2121 showed low antigen-independent signaling and potent in vitro killing of myeloma tumor cells across a range of BCMA expression levels, even in the presence of soluble BCMA.28 In addition, bb2121 showed rapid, sustained elimination of tumors and 100% survival after single-dose administration in a mouse model of human multiple myeloma.28 On the basis of these findings, a phase 1 clinical study (CRB-401) of bb2121 involving patients with relapsed or refractory multiple myeloma was initiated. We report initial results from this ongoing study.
METHODS
STUDY DESIGN AND PATIENTS
This open-label, phase 1 study was conducted at multiple centers in the United States and consisted of two parts: a dose-escalation phase and a dose-expansion phase. Eligibility criteria included an age of 18 years or older; an Eastern Cooperative Oncology Group performance-status score of 0 or 1 (on a scale of 0 to 5, with higher scores indicating greater disability); measurable disease, defined by a concentration of monoclonal protein (M protein) in serum of at least 0.5 g per deciliter or in urine of at least 200 mg per 24 hours, serum free light chains (involved free light chain concentration of ≥10 mg per deciliter with abnormal ratio), or more than 30% bone marrow plasma cells; at least three previous lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or disease refractory to both drug classes; and adequate organ function. Additional eligibility criteria for the dose-escalation phase included BCMA expression on 50% or more of marrow plasma cells on immunohistochemical assay. Additional eligibility criteria for the dose-expansion phase included previous exposure to daratumumab and refractoriness to the most recent line of therapy per International Myeloma Working Group (IMWG) criteria (Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org); patients in the expansion cohort included in this report could have less than 50% tumor BCMA expression. A full description of the study design and eligibility criteria are provided in the study protocol (available at NEJM.org).
The bb2121 was manufactured (by Celgene) from autologous peripheral-blood mononuclear cells, stimulated with antibodies to CD3 and CD28, transduced with a lentiviral vector containing the anti-BCMA CAR, and expanded over a period of 10 days as described previously.28 No minimum absolute lymphocyte count was required in patients to proceed to apheresis, which targeted a collection of at least 2.5×109 mononuclear cells by the processing of approximately twice the patient’s total blood volume. Bridging therapy during manufacturing was allowed but was stopped at least 14 days before the start of lymphodepletion (Table S2 in the Supplementary Appendix).
Patients received lymphodepletion with fludarabine (30 mg per square meter of body-surface area per day) and cyclophosphamide (300 mg per square meter per day) on days –5, –4, and –3, followed by an infusion of bb2121 on day 0. Doses of 50×106, 150×106, 450×106, or 800×106 total CAR-positive (CAR+) T cells (with an allowance of ±20%) were tested in the dose-escalation phase and 150×106 to 450×106 total CAR+ T cells in the expansion phase. Patients were followed until disease progression. Thereafter, all patients who provided written informed consent were transferred to a long-term follow-up study of up to 15 years according to Food and Drug Administration (FDA) guidance.29
STUDY OVERSIGHT
The study was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation guidelines for Good Clinical Practice. The protocol was approved by local or independent institutional review boards at each study center. Written informed consent was obtained from each patient. All the authors had access to the data and interpreted the results. The authors affirm the accuracy and completeness of the data and the adherence of the study to the protocol. Assistance in the preparation of an earlier draft of the manuscript was provided by a medical writer and paid for by Celgene. All drafts were critically reviewed and revised by the authors.
END POINTS AND STUDY PROCEDURES
The primary end point was safety. Adverse events occurring during the first 8 weeks after the infusion and from 8 weeks through 6 months are reported separately. Severity was graded according to National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03. Cytokine release syndrome was defined and graded according to published criteria.30 Neurologic toxic effects were graded according to the highest grade of any individual event during the first 90 days after the infusion. Secondary end points were response rate and duration. Clinical response and disease progression were assessed according to IMWG Uniform Response Criteria for Multiple Myeloma.2 Exploratory end points included evaluation of minimal residual disease (MRD) by next-generation sequencing with the use of a minimum cutoff of 10−4 nucleated cells (clonoSEQ, Adaptive Biotechnologies) at specified time points, independent of response status; overall survival and progression-free survival; measurement of select cytokines and chemokines; quantification of bb2121 in blood and circulating serum BCMA; measurement of tumor BCMA expression as described previously28; replication-competent lentivirus testing; and immunogenicity assessment. (Additional information is provided in the Methods section in the Supplementary Appendix.)
STATISTICAL ANALYSIS
The sample size was based on clinical considerations and a standard dose-escalation design. Descriptive statistics include means with standard deviations or medians with minimum and maximum for continuous variables, and counts and percentages for categorical variables. Missing data were not imputed unless otherwise specified. Exact methods (Clopper–Pearson 95% confidence intervals) were used for categorical variables. Duration of response, progression-free survival, time to recovery from grade 3 or 4 cytopenia, and associated 95% confidence intervals were estimated with the use of Kaplan–Meier methods. Censoring of data for progression-free survival and response duration was based on FDA censoring rules.31 Given the exploratory nature of this study, no adjustments for multiple comparisons were made. Analyses were performed with SAS software, version 9.4.
RESULTS
PATIENTS
Between January 31, 2016, and April 30, 2018, a total of 36 consecutive patients were enrolled in the study and underwent leukapheresis. The manufacturing of bb2121 was successful for 100% of the patients. Three patients underwent leukapheresis but discontinued the study owing to disease progression before bb2121 infusion (Fig. S1 in the Supplementary Appendix). The results that are presented are based on the 33 patients who received bb2121. The median age was 60 years (range, 37 to 75), and the median time since diagnosis was 5 years (range, 1 to 36) (Table 1, and Table S3 in the Supplementary Appendix). A total of 67% of the patients had stage II or III disease, 27% had extramedullary disease, and 45% had a high-risk cytogenetic profile, defined by the presence of del(17p), t(4;14), or t(14;16).
Table 1.
Baseline Characteristics of the Safety Population.*
| Characteristic | Dose-Escalation Cohort (N = 21) | Expansion Cohort (N = 12) | Total (N = 33) |
|---|---|---|---|
| Median age (range) ― yr | 57 (37–74) | 64 (46–75) | 60 (37–75) |
| Male sex ― no. (%) | 13 (62) | 8 (67) | 21 (64) |
| Median time since diagnosis (range) ― yr† | 4 (1–16) | 6 (1–36) | 5 (1–36) |
| High tumor burden ― no.(%)‡ | 11 (52) | 5 (42) | 16 (48) |
| Extramedullary disease ― no. (%) | 4 (19) | 5 (42) | 9 (27) |
| Tumor BCMA expression ≥50% ― no. (%)§ | 21 (100) | 2 (17) | 23 (70) |
| ECOG performance-status score ― no. (%)¶ | |||
| 0 | 8 (38) | 2 (17) | 10 (30) |
| 1 | 11 (52) | 10 (83) | 21 (64) |
| 2 | 2 (10) | 0 | 2 (6) |
| High-risk cytogenetic profile ― no. (%)‖ | 8 (38) | 7 (58) | 15 (45) |
| Bridging therapy ― no. (%)** | 7 (33) | 7 (58) | 14 (42) |
| Progressive disease during most recent line of therapy — no. (%) | 11 (52) | 10 (83) | 21 (64) |
| Median no. of previous antimyeloma regimens (range) | 7 (3–14) | 8 (3–23) | 7 (3–23) |
| Previous autologous stem-cell transplantation ― no. (%) | 21 (100) | 11 (92) | 32 (97) |
| Previous therapies ― no. (%) | |||
| Bortezomib | |||
| Exposed | 21 (100) | 12 (100) | 33 (100) |
| Refractory | 13 (62) | 7 (58) | 20 (61) |
| Carfilzomib | |||
| Exposed | 19 (90) | 11 (92) | 30 (91) |
| Refractory | 12 (57) | 7 (58) | 19 (58) |
| Lenalidomide | |||
| Exposed | 21 (100) | 12 (100) | 33 (100) |
| Refractory | 17 (81) | 7 (58) | 24 (73) |
| Pomalidomide | |||
| Exposed | 19 (90) | 12 (100) | 31 (94) |
| Refractory | 14 (67) | 12 (100) | 26 (79) |
| Daratumumab | |||
| Exposed | 15 (71) | 12 (100) | 27 (82) |
| Refractory | 9 (43) | 9 (75) | 18 (55) |
BCMA denotes B-cell maturation antigen.
Shown is the time between the initial diagnosis and screening for the study.
Tumor burden was determined by the investigator, with a high burden defined as at least 50% CD138-positive cells by central laboratory analysis (first preference) or by local analysis of bone marrow plasma cells (second preference). In the absence of both, tumor burden was determined by the safety review committee.
Data are for tumor BCMA expression at screening.
Eastern Cooperative Oncology Group (ECOG) performance-status scores range from 0 to 5, with higher scores indicating greater disability; a score of 5 indicates death.
The cytogenetic risk profile was reported by investigators on the basis of local assessment of bone marrow obtained at screening. High risk was defined by the presence of the following abnormalities: del(17p), t(4;14), or t(14;16).
Bridging therapy was administered after leukapheresis and before lymphodepletion.
The median number of previous regimens was 7 (range, 3 to 14) among patients in the dose-escalation cohort and 8 (range, 3 to 23) among patients in the expansion cohort (Table 1). All but 1 patient received previous autologous stem-cell transplantation. All the patients had previously received both bortezomib and lenalidomide, and 79% were exposed to bortezomib, carfilzomib, lenalidomide, pomalidomide, and daratumumab. A total of 26 patients (79%) were refractory to both a proteasome inhibitor and an immunomodulatory agent; 6 patients (18%) were refractory to bortezomib, carfilzomib, lenalidomide, pomalidomide, and daratumumab (Table S3 in the Supplementary Appendix). A total of 14 patients (42%) received bridging therapy during the manufacturing window, and dexamethasone, daratumumab, bortezomib, and bendamustine were the most commonly used agents (Table S2 in the Supplementary Appendix). All treated patients still had measurable disease at baseline assessments performed after the completion of bridging therapy and before the start of lymphodepletion.
SAFETY
All 33 patients had adverse events, with 32 (97%) having events of grade 3 or higher (Table 2, and Tables S4 and S5 in the Supplementary Appendix). Hematologic toxic effects were the most common events of grade 3 or higher, including neutropenia (in 85% of the patients), leukopenia (in 58%), anemia (in 45%), and thrombocytopenia (in 45%); these are expected toxic effects of lymphodepleting chemotherapy. Among patients who had cytopenia of grade 3 or higher, 97% recovered to an absolute neutrophil count of at least 1000 cells per cubic millimeter and 65% to a platelet count of at least 50,000 per cubic millimeter by month 1; however, delayed recovery from cytopenia was observed (Fig. S2 in the Supplementary Appendix).32,33 The median time from infusion to recovery of an absolute neutrophil count of at least 1000 per cubic millimeter was 1.3 weeks (95% confidence interval [CI], 1.0 to 1.4). Recovery to a platelet count of at least 50,000 per cubic millimeter occurred in a median of 2.0 weeks (95% CI, 1.4 to 8.4). Nonhematologic events of grade 3 or higher were uncommon.
Table 2.
Adverse Events, Cytokine Release Syndrome, and Neurologic Toxic Effects.
| Variable | Total (N = 33) | ||
|---|---|---|---|
| Any Grade | Grade 3 | Grade 4 | |
| number of patients (percent) | |||
| Adverse event* | |||
| Any | 33 (100) | 4 (12)† | 28 (85) |
| Hematologic | |||
| Neutropenia | 28 (85) | 2 (6) | 26 (79) |
| Leukopenia | 20 (61) | 6 (18) | 13 (39) |
| Anemia | 19 (58) | 15 (45) | 0 |
| Thrombocytopenia | 19 (58) | 5 (15) | 10 (30) |
| Lymphopenia | 6 (18) | 3 (9) | 3 (9) |
| Gastrointestinal | |||
| Constipation | 9 (27) | 0 | 0 |
| Nausea | 7 (21) | 0 | 0 |
| Diarrhea | 7 (21) | 0 | 0 |
| Vomiting | 6 (18) | 0 | 0 |
| Other | |||
| Fatigue | 14 (42) | 1 (3) | 0 |
| Headache | 10 (30) | 0 | 0 |
| Hypocalcemia | 9 (27) | 0 | 0 |
| Pyrexia | 8 (24) | 1 (3) | 0 |
| Hypokalemia | 8 (24) | 0 | 0 |
| Hypophosphatemia | 7 (21) | 3 (9) | 0 |
| Peripheral edema | 6 (18) | 1 (3) | 0 |
| Hyperglycemia | 6 (18) | 1 (3) | 0 |
| Hypoalbuminemia | 6 (18) | 0 | 0 |
| Cough | 6 (18) | 0 | 0 |
| Dizziness | 6 (18) | 0 | 0 |
| Upper respiratory tract infection | 5 (15) | 0 | 0 |
| Sinus tachycardia | 5 (15) | 0 | 0 |
| Hypotension | 5 (15) | 2 (6) | 0 |
| Hyponatremia | 5 (15) | 2 (6) | 0 |
| Cytokine release syndrome‡ | 25 (76) | 2 (6) | 0 |
| Neurologic toxic effect § | 14 (42) | 0 | 1 (3) |
Shown are adverse events not designated as symptoms of cytokine release syndrome that occurred in 15% or more of the safety population during the first 8 weeks.
One patient who had a grade 3 event during the first 8 weeks later had a grade 5 event (cardiorespiratory arrest).
The clustered term included the preferred term. Cytokine release syndrome was graded uniformly according to the criteria in Lee et al.30
Data are for events occurring in the first 90 days and including the following preferred terms: bradyphrenia, brain edema, confusional state, dizziness, hallucination, insomnia, lethargy, memory impairment, neurotoxicity, nystagmus, somnolence, and tremor.
A total of 25 patients (76%) had cytokine release syndrome, which was of grade 1 or 2 in 23 patients (70%) and grade 3 in 2 patients (6%); there were no cases of cytokine release syndrome of grade 4 or higher (Tables S6 and S7 in the Supplementary Appendix). Cytokine release syndrome occurred early, with a median time to onset of 2 days (range, 1 to 25) and a median duration of 5 days (range, 1 to 32). A total of 7 patients received tocilizumab and 4 received glucocorticoids. The proportion of patients who had cytokine release syndrome correlated with the dose, with a higher percentage of patients who received more than 150×106 CAR+ T cells than those who received 150×106 or fewer being affected. A higher incidence of cytokine release syndrome was also associated with a higher peak level of serum C-reactive protein, a higher peak level of tumor necrosis factor α, and higher baseline levels of ferritin, tumor-associated serum free light chains, and serum BCMA (Figs. S3 through S5 in the Supplementary Appendix). In addition, peak CAR T-cell expansion was higher in patients who had cytokine release syndrome than in those who did not. Overall, CAR T-cell expansion did not appear to be negatively affected by tocilizumab or glucocorticoid use (Fig. S6 in the Supplementary Appendix).
Neurologic toxic effects occurred in 14 patients (42%) and were of grade 1 or 2 in 13 patients (39%) (Table 2, and Table S8 in the Supplementary Appendix). One patient (3%) with a high tumor burden had a grade 4 neurologic toxic effect starting 11 days after the infusion; this effect resolved within 1 month. One patient reported a grade 3 headache during a cytokine release syndrome event in the absence of other signs of neurotoxicity and did not warrant the use of glucocorticoids. Infection developed in 14 patients (42%), 2 of whom had grade 3 events (anal abscess and parvovirus infection); no grade 4 infections occurred.
EFFICACY
The objective response rate was 85% (95% CI, 68.1 to 94.9), with 45% of the patients having a complete response (9%) or stringent complete response (36%) (Table 3). A dose-dependent effect on the frequency and duration of response was observed (Table 3 and Fig. 1). Very good partial responses or better were observed only with doses of at least 150×106 CAR+ T cells. Although subgroup analyses were limited by small sample sizes, the occurrence of a partial response or better was not significantly influenced by baseline serum or tumor BCMA levels or previous treatment exposure but trended lower in patients with a high-risk cytogenetic profile, those who did not have cytokine release syndrome, those who received 150×106 or fewer CAR+ T cells, and those with less in vivo CAR T-cell expansion (Figs. 1 and 2A, and Table S9 and Fig. S7 in the Supplementary Appendix). In addition, response rates of 74% or higher were observed among patients who had progressive disease during their most recent line of therapy, those who had received daratumumab as part of their most recent line, those who did not receive bridging therapy, and those who had extramedullary disease (plasmacytomas) at baseline (Table S9 in the Supplementary Appendix). Among patients who received 450×106 CAR+ T cells, the percentage who had a response was similar in those with tumor BCMA expression of less than 50% and those with tumor BCMA expression of 50% or more on marrow plasma cells at screening (100% and 91%, respectively).
Table 3.
Tumor Response According to Dose of Chimeric Antigen Receptor–Positive (CAR+) T Cells.*
| Variable | 50×106 CAR+ T Cells (N = 3) | 150×106 CAR+ Tells (N = 8) | 450×106 CAR+ T Cells | 800×106 CAR+ T Cells (N = 3) | 150×106–800×106 CAR+ T Cells (N = 30) | 50×106–800×106 CAR+ T Cells (N = 33) | |
|---|---|---|---|---|---|---|---|
| <50% BCMA (N = 8)† | ≥50% BCMA (N = 11)† | ||||||
| Objective response‡ | |||||||
| No. of patients with a response | 1 | 6 | 8 | 10 | 3 | 27 | 28 |
| Rate ― % (95% CI) | 33 | 75 | 100 | 91 | 100 | 90 | 85 |
| (1–91) | (35–97) | (63–100) | (59–100) | (29–100) | (74–98) | (68–95) | |
| Best overall response ― no. (%) | |||||||
| Stringent complete response | 0 | 5 (63) | 3 (38) | 4 (36) | 0 | 12 (40) | 12 (36) |
| Complete response | 0 | 0 | 0 | 1 (9) | 2 (67) | 3 (10) | 3 (9) |
| Very good partial response | 0 | 0 | 4 (50) | 4 (36) | 1 (33) | 9 (30) | 9 (27) |
| Partial response | 1 (33) | 1 (12) | 1 (12) | 1 (9) | 0 | 3 (10) | 4 (12) |
| Stable disease | 2 (67) | 1 (12) | 0 | 1 (9) | 0 | 2 (7) | 4 (12) |
| Progressive disease | 0 | 1 (12) | 0 | 0 | 0 | 1 (3) | 1 (3) |
| Median duration of response (95% CI) — mo | 1.9 (NE–NE) | NE | 7.7 (5.3–14.8) | 12.9 (10.9–12.9) | 10.9 (7.2–NE) | 10.9 (7.2–NE) | |
| Negativity for MRD § | |||||||
| No. of patients with a response who could be evaluated for MRD | 0 | 4 | 11 | 1 | 16 | 16 | |
| Rate ― % | 0 | 100 | 100 | 100 | 100 | 100 | |
All responses were confirmed and assessed according to the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma (details on the criteria for disease response are provided in the Supplementary Appendix). Percentages may not total 100 because of rounding. MRD denotes minimal residual disease, and NE could not be estimated.
Data are for BCMA expression on bone marrow plasma cells at screening.
An objective response was defined as a partial response or better.
Negativity for MRD was defined by a sensitivity level of at least 10−4 nucleated cells on the next-generation sequencing assay clonoSEQ (Adaptive Biotechnologies); 15 patients were MRD-negative at 10−5 (indeterminate, 1 patient), and 3 were MRD-negative at 10−6 (indeterminate, 10 patients). A total of 12 of the 16 patients who had a response and were MRD-negative had at least two MRD-negative assessments. Reasons that MRD could not be evaluated were an inability to detect the malignant clone in the baseline bone marrow aspirate, which made follow-up evaluation impossible (in 7 patients), failure of the MRD result in quality-control testing (in 2 patients), and nonreceipt of the bone marrow aspirate sample (in 6 patients).
Figure 1. Response to bb2121 Infusion.
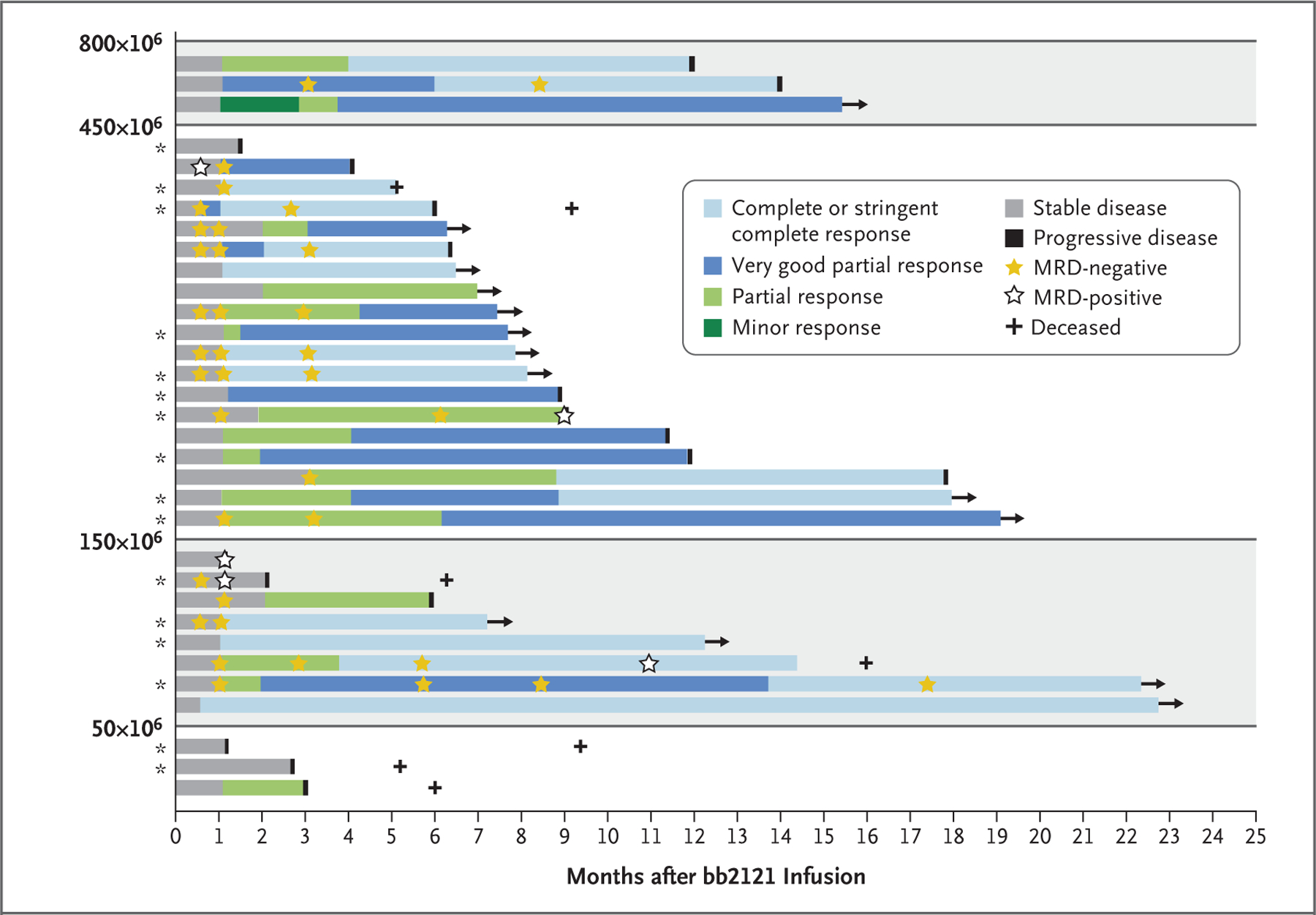
Shown are the best responses among individual patients according to dose (50×106 to 800×106) of chimeric antigen receptor–positive (CAR+) T cells. All responses were confirmed and assessed according to the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma (details on the criteria for disease response are provided in the Supplementary Appendix). Asterisks indicate patients with a high tumor burden (≥50% bone marrow plasma cells). MRD denotes minimal residual disease.
Figure 2. Objective Response Rate and Progression-free Survival.
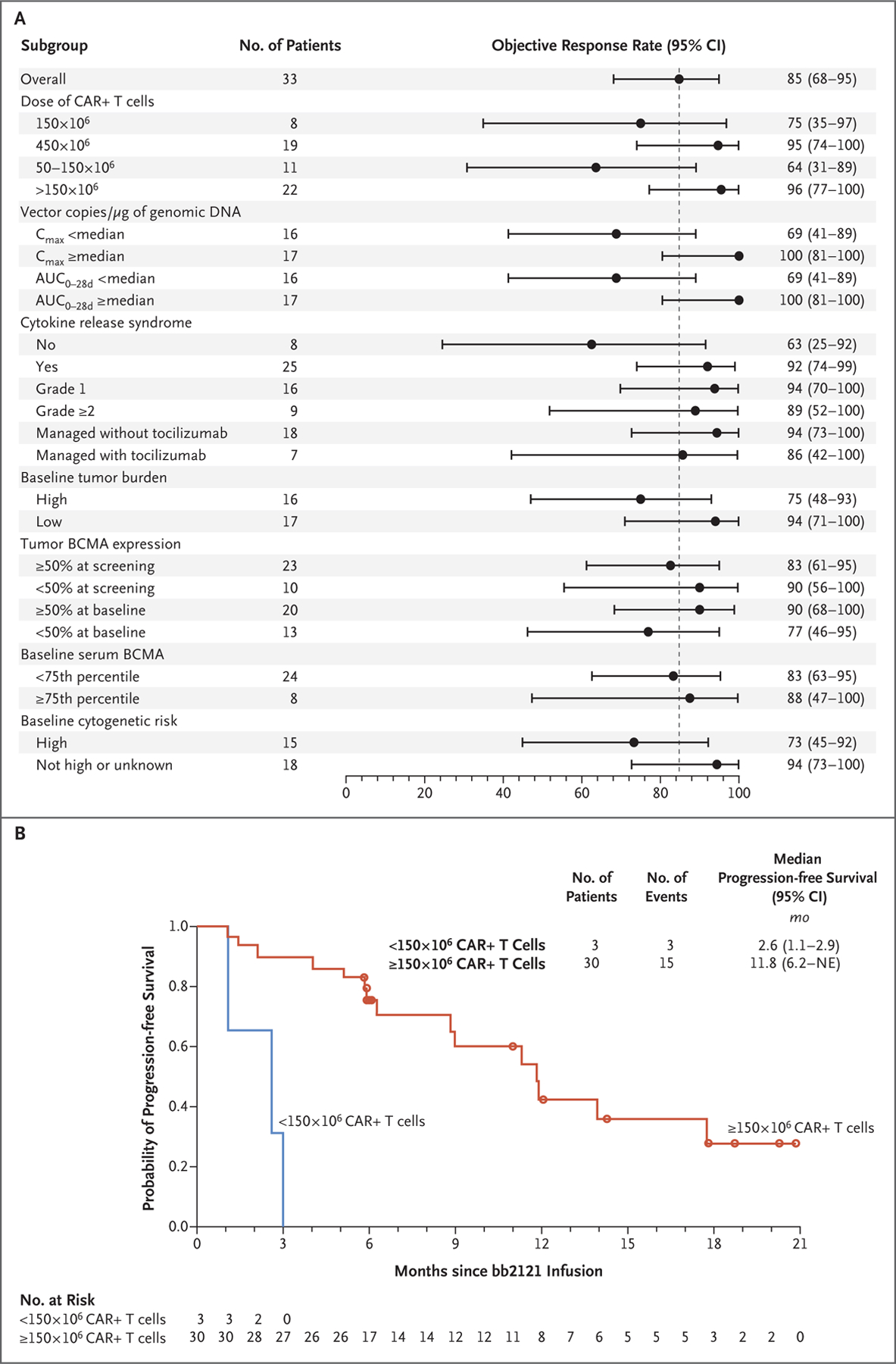
Panel A shows the rate of objective response (confirmed partial response or better) according to characteristics at baseline and during treatment. One patient who received 205×106 CAR+ T cells is included in the 450×106 dose group. Tumor burden was determined by the investigator, with a high burden defined as at least 50% CD138-positive cells by central laboratory analysis (first preference) or by local analysis of bone marrow plasma cells (second preference). In the absence of both, tumor burden was determined by the safety review committee. The cytogenetic risk profile was reported by investigators on the basis of local assessment of bone marrow obtained at screening. High risk was defined by the presence of the following abnormalities: del(17p), t(4;14), or t(14;16). AUC0–28d denotes area under the curve during the first 28 days after the infusion, BCMA B-cell maturation antigen, and Cmax maximum concentration. Panel B shows the rate of progression-free survival among patients who received less than 150×106 CAR+ T cells and those who received at least 150×106 CAR+ T cells. NE denotes could not be estimated.
Responses occurred early, with a median time to first partial response or better of 1.0 month (range, 0.5 to 3.0). There was a general trend for slower decreases in serum M protein levels, which led to a prolonged deepening of response over time in some patients. Two patients had clearance of serum M protein as late as month 9. Nearly complete decreases (>90%) from baseline in tumor-associated serum free light chains and serum BCMA were observed within 1 month in most patients who received at least 150×106 CAR+ T cells, a finding consistent with shorter serum half-lives for these markers than for intact immunoglobulin (Fig. S8 in the Supplementary Appendix). Furthermore, rapid bone marrow clearance of plasma cells (on immunohistochemical assay for BCMA and CD138) was observed at doses of at least 150×106 CAR+ T cells as early as day 14 (Fig. S9A in the Supplementary Appendix). Tumor responses in sites of extramedullary disease were also seen by month 1 in many patients (Fig. S9B in the Supplementary Appendix). The median duration of response was 10.9 months (range, 7.2 to could not be estimated) (Table 3).
A total of 18 patients could be evaluated for MRD status in the bone marrow, including 16 who had a response (partial response or better) and 2 who did not have a response (less than a partial response). All 16 patients who had a response were MRD-negative at 10−4 nucleated cells or better; 15 of 16 (94%) were MRD-negative at 10−5, and 3 were MRD-negative at 10−6 (Table 3). All 16 MRD-negative patients had MRD negativity at the first valid assessment (13 patients at month 1 and 3 patients at month 3), and 12 had at least two consecutive negative assessments. Two patients who did not have a response and who could be evaluated for MRD were MRD-positive at month 1. Early MRD negativity occurred in patients before the best response according to IMWG criteria. In fact, early MRD negativity was observed in the context of stable disease with later evolution to complete response due to decreases in serum M protein levels over time (Fig. 1).
The median duration of follow-up after bb2121 infusion was 11.3 months (range, 6.2 to 22.8). A total of 17 patients (52%) had disease progression, including 12 who had a response (among whom 6 had a complete response and 6 had an MRD-negative response); 14 patients had ongoing responses (Fig. 1). The median progression-free survival was 11.8 months (95% CI, 6.2 to 17.8) (Fig. 2B). One patient died from cardiopulmonary arrest that was considered by the investigators to be unrelated to study treatment, and another discontinued the study after starting chemotherapy for myelodysplastic syndrome.
EXPANSION AND PERSISTENCE
The final bb2121 CAR+ T-cell product was composed of a variable proportion of CAR+ CD4 and CD8 T cells, with a median of 85% (range, 42 to 98) CAR+ CD4 T cells and 13% (range, 2 to 47) CAR+ CD8 T cells. In vivo expansion of bb2121 CAR T cells was observed, with overlapping peak blood concentrations at all dose levels of more than 50×106 CAR+ T cells (Fig. 3A). Both CAR+ CD4 T cells and CD8 T cells expanded in vivo; a correlation was observed between the CAR+ CD4:CD8 T-cell ratio in the final product and that observed at peak expansion (r = 0.45, P = 0.02) (Fig. S10 in the Supplementary Appendix). Persistence was durable, with 96%, 86%, 57%, and 20% of the patients having detectable CAR T cells at 1, 3, 6, and 12 months, respectively (Table S10 in the Supplementary Appendix). Blood CAR T-cell levels were higher in patients who had a response (partial response or better) than in those who did not have a response, as measured by maximum vector transgene copies per microgram of genomic DNA and area under the curve during the first 28 days after the infusion (P = 0.006 and P = 0.007, respectively) (Fig. 3B, and Fig. S11 in the Supplementary Appendix).
Figure 3. Correlation of CAR T-Cell Expansion with Dose and Response.
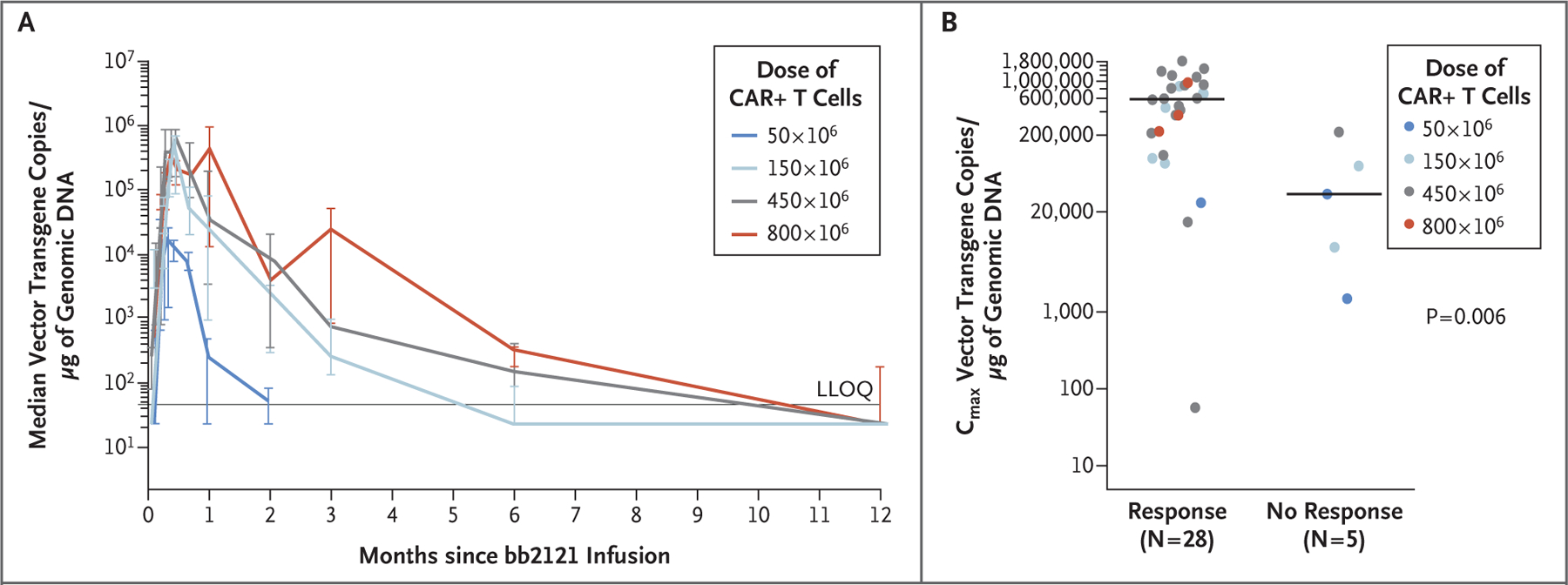
Panel A shows cellular kinetics as measured by median vector transgene copies per microgram of genomic DNA in CD3-enriched peripheral blood, according to dose group. Patients with a postbaseline vector transgene copy value were included. One patient received 205×106 CAR+ T cells instead of the planned 450×106 and was included in the 450×106 dose group. I bars indicate 95% confidence intervals. LLOQ denotes lower limit of quantitation. Panel B shows the correlation between peak vector transgene copies per microgram of genomic DNA after infusion and the occurrence of tumor response in patients with at least 1 month of cellular kinetics data (33 patients). Horizontal lines indicate the medians. Circles indicate individual patients according to dose. The P value is based on a two-sided Wilcoxon rank-sum test.
DISCUSSION
In this phase 1 study of bb2121 anti-BCMA CAR T cells involving patients with heavily pretreated relapsed or refractory multiple myeloma, 85% of the patients had a clinical response lasting a median of 10.9 months without any ongoing myeloma therapy. Complete responses were observed across all doses from 150×106 to 800×106 CAR+ T cells.
Response appeared to be independent of tumor BCMA expression, with similarly high response rates observed at tumor BCMA expression levels of less than 50% and 50% or more in patients who received 450×106 CAR+ T cells. High response rates (≥74%) were also observed in patients with a high-risk cytogenetic profile, progressive disease during their most recent line of therapy, or extramedullary disease at baseline and in the absence of bridging therapy. Results of subgroup analyses and correlative studies are preliminary given the small sample size in this phase 1 study and require confirmation in future studies. Clearance of bone marrow plasma cells was rapid, generally occurring within 1 month. This was associated with a rapid time to initial response and a near complete decline in serum BCMA and tumor-associated serum free light chains. The time to best response according to IMWG criteria often lagged owing to slower clearance of serum M protein. The mechanism for slower clearance of whole immunoglobulin M protein is unknown but may be due to the long circulating half-life of immunoglobulin molecules or possibly slow clearance from tissue reservoirs, including extramedullary plasmacytomas. Although potentially confounding, the fludarabine and cyclophosphamide lymphodepleting chemotherapy is unlikely to have contributed substantially to the responses observed given the lack of responses reported in this study and a previous study of anti-BCMA CAR T-cell therapy involving patients with myeloma who received the same fludarabine–cyclophosphamide regimen but who were treated at sub-therapeutic doses of CAR T cells.26
The median progression-free survival was 11.8 months, with 40% of the patients free of progression at 12 months in this heavily pretreated population. Although these results are preliminary and the sample size is small, the reported activity with this single-dose therapy compares favorably with other salvage therapies for relapsed or refractory myeloma that are administered in repeated doses until progression. In one study, patients who received pomalidomide plus dexamethasone had a response rate of 31% and a median progression-free survival of 4 months.34 In a similar population, treatment with single-agent daratumumab resulted in a response rate of 29%, including 3% with a stringent complete response, and a median progression-free survival of 3.7 months.35 Modest activity has been observed with selinexor plus dexamethasone in patients who had previously received daratumumab, with a response rate of 21%, no complete responses, and a median progression-free survival of 2.3 months.36,37 Emerging therapies for relapsed or refractory myeloma have had some encouraging results. In a phase 1 study of a BCMA-directed antibody–drug conjugate in which 67% of the patients had at least five previous lines of therapy, a response rate of 60% and a median progression-free survival of 7.9 months were observed.38
For treatment of multiple myeloma, BCMA has been identified as an important CAR target. In a recent study of another anti-BCMA CAR T-cell therapy involving 16 patients with refractory myeloma (median, 9.5 previous treatments), the median event-free survival was 7.1 months.25 Among patients with myeloma who were less heavily pretreated (median, three previous regimens; most had not undergone stem-cell transplantation or previously received daratumumab) and who were treated with a different anti-BCMA CAR T-cell therapy in China, the response rate was 88%, with a complete response rate of 68%.39 However, the patient populations are not directly comparable. Whether bb2121 has the potential to induce long-term durable remissions, as observed with CD19 CAR T cells, will require longer follow-up of ongoing patients who had a response (6 patients continued to have a response of at least a very good partial response with ≥12 months of follow-up) as well as further exploration in less heavily pretreated patients.
Although this study had a large number of patients who could not be evaluated for MRD, the high rate of measured MRD negativity in bone marrow (in 100% of 16 patients with a partial response or better who could be evaluated for MRD) is encouraging. By comparison, a recent study of daratumumab plus pomalidomide–dexamethasone for relapsed or refractory multiple myeloma showed that 35% and 29% of 17 patients with a complete response who could be evaluated for MRD had MRD negativity at a threshold of 10−4 and 10−5 nucleated cells, respectively.40 However, unlike analyses in previous studies that examined MRD status only in patients with a complete response, MRD analyses in our study were scheduled in all the patients, so direct comparisons across studies are difficult. Despite the high rate observed here, the occurrence of MRD negativity did not translate into continuous remission for all patients, possibly owing to the advanced, refractory nature of the myeloma in this patient population. Analyses of mechanisms of relapse in patients with initial MRD-negative responses are ongoing.
The expansion and persistence of bb2121 CAR T cells were also notable. Peak expansion was observed within 11 days at doses of at least 150×106 CAR+ T cells. A modest correlation was observed between the CAR+ CD4:CD8 T-cell ratio in the bb2121 drug product and that observed at peak expansion in vivo. Peak expansion was greater in patients who had a response than in those who did not have a response, a finding similar to those of other studies of CAR T-cell therapy.41,42 Durable persistence was observed in 57% of the patients at 6 months and 20% at 12 months, which is longer than the 3 months of persistence reported for CAR T cells with CD28 costimulatory domains.43
To date, the safety profile of bb2121 has been assessed at doses as high as 800×106 CAR+ T cells. Cytokine release syndrome was mostly of grade 1 or 2; the two grade 3 events (in 6% of the patients) resolved within 24 hours. Cytokine release syndrome was treated with tocilizumab in 21% of the patients and glucocorticoids in 12%. Such treatment did not appear to negatively affect CAR T-cell expansion or treatment response. The incidence of cytokine release syndrome of grade 3 or higher that has been reported with CD19-directed CAR T cells (23 to 49% with tisagenlecleucel and 13% with axicabtagene ciloleucel)33 is higher than that with BCMA-directed CAR T cells in our study. In a phase 1 study of another BCMA-targeting CAR construct with CD28 costimulation (instead of 4–1BB), 38% of the patients had cytokine release syndrome of grade 3 or higher, and the study was amended to treat only patients with myeloma who had less than 30% marrow plasma cells.25 The overall frequency of grade 3 or 4 neurologic toxic effects with bb2121 was also low (3%), with no fatal events; two cases of neurologic toxic effects of any grade (in 6% of the patients) were treated with glucocorticoids. Although comparisons among studies are complicated by differences in patient populations, CAR constructs, administered doses, and grading scales of toxic effects, the results observed with bb2121 indicate a favorable safety profile.
In conclusion, in a heavily pretreated population of patients with multiple myeloma, bb2121 showed promising efficacy at dose levels of 150×106 or more CAR+ T cells. Nonhematologic toxic effects were primarily of grade 2 or lower.
Supplementary Material
Acknowledgments
Supported by Bluebird Bio and Celgene.
We thank all the study participants, especially the patients and their families; and Johna Van Stelten, Ph.D., of Bio Connections for medical writing assistance with an earlier version of the manuscript.
Footnotes
REFERENCES
- 1.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med 2011;364:1046–60. [DOI] [PubMed] [Google Scholar]
- 2.Rajkumar SV. Treatment of multiple myeloma. Nat Rev Clin Oncol 2011;8:479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia 2014;28:1122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldschmidt H, Ashcroft J, Szabo Z, Garderet L. Navigating the treatment landscape in multiple myeloma: which combinations to use and when? Ann Hematol 2019;98:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chim CS, Kumar SK, Orlowski RZ, et al. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia 2018;32:252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar S Treatment of newly diagnosed multiple myeloma in transplant-eligible patients. Curr Hematol Malig Rep 2011;6:104–12. [DOI] [PubMed] [Google Scholar]
- 7.Kumar SK, Lee JH, Lahuerta JJ, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter International Myeloma Working Group study. Leukemia 2012;26:149–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sonneveld P Management of multiple myeloma in the relapsed/refractory patient. Hematology Am Soc Hematol Educ Program 2017;2017:508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nijhof IS, van de Donk NWCJ, Zweegman S, Lokhorst HM. Current and new therapeutic strategies for relapsed and refractory multiple myeloma: an update. Drugs 2018;78:19–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makita S, Yoshimura K, Tobinai K. Clinical development of anti-CD19 chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma. Cancer Sci 2017;108:1109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neelapu SS, Locke FL, Go WY. CAR T-cell therapy in large B-cell lymphoma. N Engl J Med 2018;378:1065. [DOI] [PubMed] [Google Scholar]
- 12.Mikkilineni L, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood 2017;130:2594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015;385:517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 2016;126:2123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park JH, Rivière I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med 2018;378:449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kochenderfer JN, Somerville RPT, Lu T, et al. Long-duration complete remissions of diffuse large B cell lymphoma after anti-CD19 chimeric antigen receptor T cell therapy. Mol Ther 2017;25:2245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018;378:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther 2017;25:285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maude SL, Teachey DT, Rheingold SR, et al. Sustained remissions with CD19-specific chimeric antigen receptor (CAR)-modified T cells in children with relapsed/refractory ALL. J Clin Oncol 2016; 34:Suppl:3011. abstract. [Google Scholar]
- 22.Jain MD, Bachmeier CA, Phuoc VH, Chavez JC. Axicabtagene ciloleucel (KTE-C19), an anti-CD19 CAR T therapy for the treatment of relapsed/refractory aggressive B-cell non-Hodgkin’s lymphoma. Ther Clin Risk Manag 2018;14:1007–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tai YT, Anderson KC. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015;7:1187–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Novak AJ, Darce JR, Arendt BK, et al. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood 2004;103:689–94. [DOI] [PubMed] [Google Scholar]
- 25.Brudno JN, Maric I, Hartman SD, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol 2018;36:2267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali SA, Shi V, Maric I, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016;128:1688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res 2013;19:2048–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedman KM, Garrett TE, Evans JW, et al. Effective targeting of multiple B-cell maturation antigen-expressing hematological malignances by anti-B-cell maturation antigen chimeric antigen receptor T cells. Hum Gene Ther 2018;29:585–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guidance for industry: gene therapy clinical trials — observing subjects for delayed adverse effects Silver Spring, MD: Food and Drug Administration, November 2006. (https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm078719.pdf). [Google Scholar]
- 30.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014;124:188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guidance for industry: clinical trial endpoints for the approval of cancer drugs and biologics Silver Spring, MD: Food and Drug Administration, December 2018. (https://www.fda.gov/downloads/drugsGuidanceComplianceRegulatoyInformation/Guidance/UCM071590.pdf). [Google Scholar]
- 32.Kymriah (tisagenlecleucel). Prescribing information East Hanover, NJ: Novartis Pharmaceuticals, 2018. [Google Scholar]
- 33.Yescarta (axicabtagene ciloleucel). Prescribing information Santa Monica, CA: Kite Pharma, 2017. [Google Scholar]
- 34.Miguel JS, Weisel K, Moreau P, et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol 2013;14;1055–66. [DOI] [PubMed] [Google Scholar]
- 35.Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016;387:1551–60. [DOI] [PubMed] [Google Scholar]
- 36.Chen C, Siegel D, Gutierrez M, et al. Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom macroglobulinemia. Blood 2018;131:855–63. [DOI] [PubMed] [Google Scholar]
- 37.Vogl DT, Dingli D, Cornell RF, et al. Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J Clin Oncol 2018;36:859–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trudel S, Lendvai N, Popat R, et al. Targeting B-cell maturation antigen with GSK2857916 antibody-drug conjugate in relapsed or refractory multiple myeloma (BMA117159): a dose escalation and expansion phase 1 trial. Lancet Oncol 2018; 19:1641–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao WH, Liu J, Wang BY, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol 2018;11:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chari A, Suvannasankha A, Fay JW, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 2017;130:974–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fraietta JA, Lacey SF, Orlando EJ, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018;24:563–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mueller KT, Waldron E, Grupp SA, et al. Clinical pharmacology of tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin Cancer Res 2018;24:6175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maus MV, June CH. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin Cancer Res 2016;22:1875–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.