

Abstract
An extract of the plant Anacolosa clarkii was obtained from the NCI Natural Products Repository, and it showed cytotoxic activity toward several types of pediatric solid tumor cell lines. Bioassay-guided fractionation led to the purification of eight new clerodane diterpenes [anacolosins A–F (1–6) and corymbulosins X and Y (7 and 8)] and two known compounds (9 and 10) that contained an isozuelanin skeleton. The structures of the new natural products were determined using 1D and 2D NMR and HRESIMS data, while the relative and absolute configurations of the compounds were assessed using a combination of 1H NMR coupling constant data, ROESY experiments, ECD (electronic circular dichroism) and VCD (vibrational circular dichroism) spectroscopy, chemical methods (including Mosher and 2-naphthacyl esterification), and chiral HPLC analyses. The purified natural products exhibited a range of cytotoxic activities against cell lines representing four pediatric cancer types (i.e., rhabdomyosarcoma, Ewing sarcoma, medulloblastoma, and hepatoblastoma) with total growth inhibitory (TGI) values in the range 0.2–4.1 μM. The rhabdomyosarcoma and medulloblastoma cell lines showed higher sensitivity to compounds 1–4, which are the first compounds reported to contain an isozuelanin skeleton and feature keto carbonyl groups at the C-6 positions. In contrast, the hepatoblastoma cell line was modestly more sensitive to 7–10, which contained a C-6 hydroxy group moiety.
Graphical Abstract
Despite numerous clinical advances, cancer remains a significant cause of death among children and adolescents across the world.1 In the United States alone, >10,000 cases of cancer are reported in children each year.2 While childhood solid cancers are often grouped based solely on patient age, studies reveal a growing list of genomic features that enable the separation of childhood cancers into distinct groups and subtypes. These results also demonstrate that many of the molecular drivers found in childhood cancers are different than those observed in adult cancers. While high mutational burdens define adult solid cancers, few mutations are found in childhood cancers, highlighting the significant differences between pediatric and adult cancers.2,3 The implication of these findings is that childhood cancers can be targeted pharmacologically based on their unique genomic and molecular features. A key consequence of the improved molecular characterization of pediatric cancers is the identification of molecular targets and cognate inhibitors that can be used to selectively affect the often unique defects that drive childhood solid cancers. Therefore, efforts invested in identifying compounds that selectively target specific childhood cancer types offer new opportunities to develop drugs that are potentially more effective at treating younger cancer patients.
Acting on this premise, our consortium of investigators from the University of Oklahoma, the University of Texas Health Science Center at San Antonio, and the Natural Products Branch of NCI has investigated plant and fungal natural products as a resource for compounds that exhibit selective activity profiles against pediatric cancer cell lines. As part of this drug discovery process, we have identified 10 compounds from the plant Anacolosa clarkii Pierre (Olacaceae) that display cytotoxic activities against pediatric solid tumor cell lines. An investigation of the literature showed that the genus Anacolosa is a relatively unexplored group of plants, with only two published accounts appearing in SciFinder that reported secondary metabolites from this species.4,5 This report provides details of the metabolites obtained from A. clarkii and describes their differential activities against pediatric cancer cell lines representing four distinct pediatric solid tumor types.
RESULTS AND DISCUSSION
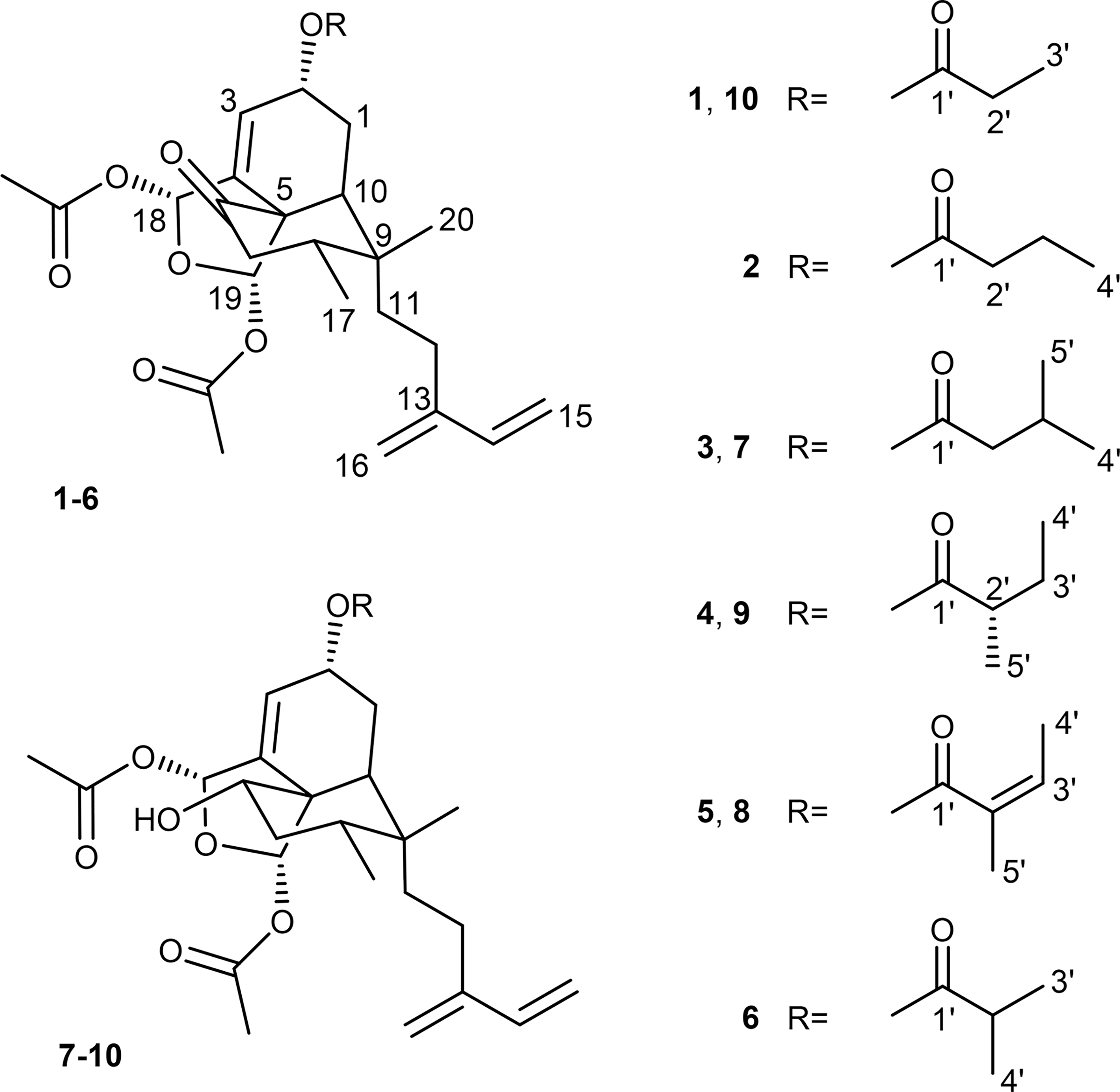
Examination of the LC-PDA-MS data collected for the A. clarkii extract revealed that the sample contained an abundance of metabolites that exhibited λmax values occurring near 220 nm and molecular weights averaging ~500 Da. Bioassay-guided fractionation using a combination of vacuum-liquid-chromatography (HP20ss), preparative HPLC (C18), and semipreparative HPLC (C18 and pentafluorophenyl) led to the purification of metabolites 1–10 (Chart 1).
Chart 1.
Anacolosin A (1) was purified as a colorless oil. Its molecular formula was determined to be C27H36O8 based on HRESIMS data (m/z 511.2304 [M + Na]+, calcd for 511.2302), which indicated 10 degrees of unsaturation. Based on the HMBC correlations between the singlets at δH 1.84 and 2.05 (Table 1) and the carbonyl carbons at δC 168.6 and 170.0 (Table 2), the presence of two acetoxy groups was proposed. The occurrence of a propionate group was confirmed by the 1H−1H COSY correlations between H-2′ (δH 2.36, m) and H-3′ (δH 1.06, t, J = 7.5 Hz) and the HMBC correlations from H-2′/3′ and H-2 (δH 5.31, m) to C-1′ (δC 173.0) (Figure 1). Beyond the aforementioned resonances, an additional 20 carbon spins remained (Table 2), which led to an initial conjecture that this metabolite might be a diterpenoid. The carbon signals at δC 122.4 (C-3), 141.7 (C-4), 144.8 (C-13), 140.1 (C-14), 112.8 (C-15), and 115.9 (C-16), as well as the proton resonances at δH 6.05 (brd, J = 3.8 Hz, H-3), 6.45 (dd, J = 10.8, 17.6 Hz, H-14), 5.23 (d, J = 17.5 Hz, H-15a), 5.06 (brd, J = 10.7 Hz, H-15b), 5.11 (s, H-16a), and 5.05 (s, H-16b), supported the presence of three double bonds in the structure. Taking into consideration the six units of unsaturation that were confirmed in 1 (two acetoxy groups, a propionate group, and three double bonds), as well as 13C NMR-derived evidence for the presence of a keto carbonyl (δC 207.9, C-6), it was concluded that the remaining three units of unsaturation could be accounted for as three rings. Bearing this in mind, an analysis of the 1H–1H COSY and HMBC correlation data (Figure 1) led to the proposal that 1 featured a tricyclic clerodane diterpene core.6–12 More specifically, compound 1 belongs to a subgroup of clerodane diterpenes that possess an isozuelanin skeleton.10 However, in contrast to the reported isozuelanin-containing natural products that bear a methylene, a hydroxy, or an alkoxy group at C-6, metabolite 1 is the first member of this class to feature a C-6 keto carbonyl group.10
Table 1.
1H NMR Data for Compounds 1–8 (δ in ppm, J in Hz)
| no. | 1a | 2b | 3c | 4d | 5d | 6d | 7d | 8d |
|---|---|---|---|---|---|---|---|---|
| 1ax | 1.42 ddd (3.9, 14.8, 15.1) | 1.53 ddd (4.0, 14.8, 14.9) | 1.54 ddd (4.0, 14.2, 14.3) | 1.53 dd (3.9, 13.7, 14.5) | 1.54 ddd (4.0, 14.7, 14.8) | 1.51 ddd (3.9, 14.9, 14.9) | 1.94 m | 1.94 m |
| 1eq | 1.86 d (14.8) | 2.04 brd (14.0) | 2.05 brd (11.7) | 2.06 brd (14.9) | 2.06 brd (14.9) | 2.04 m | 2.03 m | 2.03 m |
| 2 | 5.31 m | 5.37 m | 5.36 brs | 5.36 m | 5.42 brs | 5.35 m | 5.42 m | 5.46 m |
| 3 | 6.05 brd (3.8) | 6.12 brd (4.6) | 6.14 brd (4.6) | 6.13 brd (4.8) | 6.15 brd (4.7) | 6.11 brd (4.8) | 6.02 brd (4.5) | 6.02 brd (4.3) |
| 6 | 3.82 dd (4.3, 12.1) | 3.84 dd (4.3, 12.1) | ||||||
| 7ax | 3.18 dd (14.7, 15.1) | 2.95 dd (14.0, 14.0) | 2.96 dd (14.0, 13.7) | 2.96 dd (14.1, 13.7) | 2.97 dd (13.6, 14.1) | 2.95 dd (13.7, 14.0) | 1.72 ddd (12.9, 12.6, 12.6) | 1.73 ddd (12.8, 12.6, 12.6) |
| 7eq | 2.04 m | 2.19 m | 2.19 m | 2.20 m | 2.20 m | 2.19 m | 1.63 ddd (3.7, 4.0, 13.4) | 1.64 ddd (3.7, 3.9, 13.3) |
| 8 | 2.06 m | 2.15 m | 2.15 m | 2.17 m | 2.16 m | 2.15 m | 1.87 m | 1.86 m |
| 10 | 2.40 m | 2.59 dd (2.5, 13.7) | 2.62 dd (2.3, 13.6) | 2.63 dd (2.4, 13.6) | 2.68 dd (2.4, 13.6) | 2.62 dd (2.5, 13.7) | 2.36 dd (3.6, 13.7) | 2.41 dd (3.5, 13.6) |
| 11a | 1.62 m | 1.78 m | 1.78 m | 1.79 m | 1.79 m | 1.78 m | 1.53 m | 1.54 m |
| 11b | 1.62 m | 1.70 m | 1.71 m | 1.71 m | 1.71 m | 1.70 m | 1.28 m | 1.30 m |
| 12 | 2.10 m | 2.25 m | 2.24 m | 2.25 m | 2.26 m | 2.24 m | 2.12 m | 2.13 m |
| 14 | 6.45 dd (10.8, 17.6) | 6.50 dd (11.0, 17.6) | 6.51 dd (11.0, 17.6) | 6.51 dd (11.1, 17.8) | 6.52 dd (11.1, 17.7) | 6.50 dd (11.1, 17.7) | 6.48 dd (10.9, 17.6) | 6.48 dd (10.9, 17.6) |
| 15a | 5.23 d (17.5) | 5.27 brd (17.4) | 5.26 d (17.6) | 5.24 dd (0.9, 17.5) | 5.25 dd (1.0, 17.7) | 5.24 dd (1.0, 17.6) | 5.23 dd (1.1, 17.6) | 5.22 dd (1.1, 17.6) |
| 15b | 5.06 d (10.7) | 5.08 d (10.0) | 5.08 d (10.2) | 5.08 d (10.3) | 5.08 brd (11.5) | 5.06 brd (10.1) | 5.05 brd (11.0) | 5.05 brd (10.9) |
| 16a | 5.11 s | 5.07 brs | 5.07 brs | 5.08 brs | 5.08 brs | 5.07 brs | 5.02 brs | 5.03 brs |
| 16b | 5.05 s | 5.07 brs | 5.07 brs | 5.08 brs | 5.08 brs | 5.07 brs | 4.94 brs | 4.95 brs |
| 17 | 0.90 d (6.4) | 1.02 d (6.2) | 1.03 d (6.7) | 1.03 d (6.7) | 1.04 d (6.7) | 1.03 d (6.7) | 0.95 d (6.8) | 0.95 d (6.8) |
| 18 | 6.45 brs | 6.59 t (1.6) | 6.59 brs | 6.61 t (1.6) | 6.61 t (1.6) | 6.60 t (1.6) | 6.66 dd (1.6, 1.7) | 6.68 dd (1.7, 1.7) |
| 19 | 6.58 s | 6.54 s | 6.54 s | 6.55 s | 6.55 s | 6.54 s | 6.44 s | 6.44 s |
| 20 | 0.93 s | 1.06 s | 1.06 s | 1.05 s | 1.06 s | 1.05 s | 0.96 s | 0.95 s |
| OAc-18 (Me) | 2.05 s | 2.05 s | 2.04 s | 2.06 s | 2.07 s | 2.06 s | 2.04 s | 2.06 s |
| OAc-19 (Me) | 1.84 s | 1.90 s | 1.90 s | 1.90 s | 1.91 s | 1.90 s | 1.84 s | 1.85 s |
| 2′ | 2.36 m | 2.37 t (7.2) | 2.26 m | 2.46 m | 2.62 m | 2.26 m | ||
| 3′ | 1.06 t (7.5) | 1.70 m | 2.11 m | 1.69 m; 1.58 m | 6.91 qq (7.0, 1.5) | 1.22 d (7.0) | 2.12 m | 6.90 qq (1.5, 7.2) |
| 4′ | 1.01 t (7.4) | 1.03 d (6.7) | 0.99 t (7.5) | 1.85 dq (7.1, 1.2) | 1.19 d (6.7) | 1.03 d (6.7) | 1.84 dq (7.1, 1.2) | |
| 5′ | 1.02 d (6.7) | 1.18 d (6.9) | 1.88 m | 1.02 d (6.7) | 1.88 m |
Obtained on a 400 MHz Varian instrument in a 5 mm NMR tube; solvent was DMSO-d6.
Obtained on a 600 MHz Varian instrument in a 5 mm NMR tube; solvent was MeOH-d4.
Obtained on a 600 MHz Varian instrument in a 3 mm NMR tube; solvent was MeOH-d4.
Obtained on a 500 MHz Varian instrument in a 3 mm NMR tube; solvent was MeOH-d4.
Table 2.
13C NMR Data for Compounds 1–8 (δ in ppm)
| no. | 1a | 2b | 3c | 4d | 5d | 6e | 7d | 8e |
|---|---|---|---|---|---|---|---|---|
| 1 | 26.7 CH2 | 28.1 CH2 | 28.0 CH2 | 28.0 CH2 | 28.2 CH2 | 27.6 CH2 | 27.7 CH2 | 27.7 CH2 |
| 2 | 65.2 CH | 67.3 CH | 67.4 CH | 67.3 CH | 67.3 CH | 67.0 CH | 68.2 CH | 67.9 CH |
| 3 | 122.4 CH | 123.8 CH | 123.8 CH | 123.8 CH | 123.9 CH | 123.7 CH | 122.5 CH | 122.2 CH |
| 4 | 141.7 C | 143.4 C | 143.4 C | 143.5 C | 143.4 C | 143.1 C | 147.6 C | 147.3 C |
| 5 | 65.1 C | 67.0 C | 66.9 C | 67.0 C | 67.1 C | 66.7 C | 55.1 C | 55.4 C |
| 6 | 207.9 C | 209.0 C | 209.0 C | 209.0 C | 209.0 C | 208.9 C | 73.4 CH | 73.0 CH |
| 7 | 43.2 CH2 | 44.4 CH2 | 44.4 CH2 | 44.4 CH2 | 44.4 CH2 | 44.0 CH2 | 37.8 CH2 | 37.5 CH2 |
| 8 | 38.8 CH | 40.9 CH | 40.9 CH | 41.0 CH | 41.0 CH | 40.4 CH | 38.4 CH | 38.2 CH |
| 9 | 37.0 C | 38.8 C | 38.8 C | 38.8 C | 38.8 C | 37.9 C | 38.6 C | 38.4 C |
| 10 | 38.4 CH | 40.4 CH | 40.4 CH | 40.3 CH | 40.5 CH | 40.0 CH | 37.9 CH | 37.8 CH |
| 11 | 27.9 CH2 | 29.6 CH2 | 29.6 CH2 | 29.5 CH2 | 29.5 CH2 | 29.3 CH2 | 29.2 CH2 | 29.0 CH2 |
| 12 | 23.3 CH2 | 24.8 CH2 | 24.8 CH2 | 24.8 CH2 | 24.8 CH2 | 24.4 CH2 | 25.1 CH2 | 24.8 CH2 |
| 13 | 144.8 C | 146.6 C | 146.6 C | 146.5 C | 146.5 C | 146.5 C | 147.0 C | 146.3 C |
| 14 | 140.1 CH | 141.6 CH | 141.6 CH | 141.6 CH | 141.7 CH | 141.4 CH | 141.7 CH | 141.4 CH |
| 15 | 112.8 CH2 | 112.9 CH2 | 112.9 CH2 | 112.8 CH2 | 112.8 CH2 | 112.7 CH2 | 112.8 CH2 | 112.3 CH2 |
| 16 | 115.9 CH2 | 116.1 CH2 | 116.1 CH2 | 116.1 CH2 | 116.1 CH2 | 116.0 CH2 | 115.7 CH2 | 115.5 CH2 |
| 17 | 15.7 CH3 | 16.3 CH3 | 16.2 CH3 | 16.2 CH3 | 16.2 CH3 | 15.9 CH3 | 16.1 CH3 | 15.8 CH3 |
| 18 | 94.0 CH | 96.0 CH | 96.0 CH | 96.0 CH | 95.9 CH | 95.7 CH | 97.1 CH | 96.9 CH |
| 19 | 96.9 CH | 98.8 CH | 98.8 CH | 98.8 CH | 98.7 CH | 98.7 CH | 99.8 CH | 99.3 CH |
| 20 | 24.3 CH3 | 25.2 CH3 | 25.1 CH3 | 25.2 CH3 | 25.3 CH3 | 24.9 CH3 | 25.8 CH3 | 25.7 CH3 |
| OAc-18 (C=O) | 170.0 C | 171.7 C | 171.6 C | 171.7 C | 171.7 C | 171.3 C | 172.0 C | 171.7 C |
| OAc-18 (Me) | 21.0 CH3 | 20.9 CH3 | 20.9 CH3 | 20.9 CH3 | 20.9 CH3 | 20.5 CH3 | 21.1 CH3 | 20.8 CH3 |
| OAc-19 (C=O) | 168.6 C | 170.8 C | 170.7 C | 170.7 C | 170.7 C | 170.3 C | 171.5 C | 171.2 C |
| OAc-19 (Me) | 21.0 CH3 | 21.5 CH3 | 21.5 CH3 | 21.4 CH3 | 21.4 CH3 | 21.1 CH3 | 21.9 CH3 | 21.7 CH3 |
| 1′ | 173.0 C | 174.5 C | 173.9 C | 177.3 C | 168.5 C | 177.3 C | 174.1 C | 167.9 C |
| 2′ | 27.3 CH2 | 37.2 CH2 | 44.5 CH2 | 42.5 CH | 129.9 C | 34.9 CH | 44.6 CH2 | 129.5 C |
| 3′ | 9.2 CH3 | 19.8 CH2 | 27.4 CH | 28.2 CH2 | 138.9 CH | 19.3 CH3 | 27.4 CH | 138.5 CH |
| 4′ | 14.0 CH3 | 22.8 CH3 | 12.2 CH3 | 14.5 CH3 | 18.6 CH3 | 22.8 CH3 | 14.2 CH3 | |
| 5′ | 22.8 CH3 | 17.1 CH3 | 12.1 CH3 | 22.8 CH3 | 11.9 CH3 |
Obtained on a 100 MHz Varian instrument in 5 mm NMR tube; solvent was DMSO-d6.
Obtained on a 100 MHz Varian instrument in a 5 mm NMR tube; solvent was MeOH-d4.
Obtained on a 150 MHz Varian instrument in a 3 mm NMR tube; solvent was MeOH-d4.
Obtained on a 100 MHz Varian instrument in a 3 mm NMR tube; solvent was MeOH-d4
Extracted from HSQC and HMBC spectra which were obtained on a 500 MHz Varian instrument in a 3 mm NMR tube; solvent was MeOH-d4.
Figure 1.
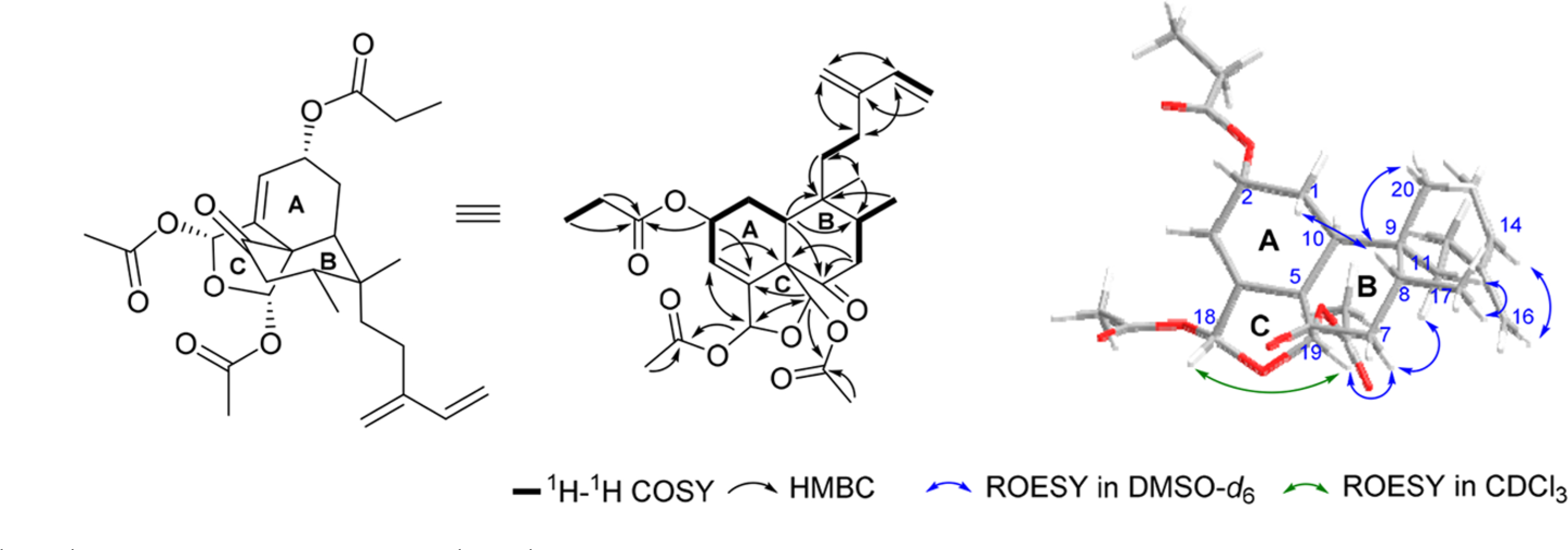
Selected 1H–1H COSY, HMBC, and 1H–1H ROESY correlations used to determine the structure of compound 1.
The relative configuration of 1 was determined based on a combination of 1H–1H ROESY correlations (Figure 1) and coupling constants between key protons (Table 1). We prioritized establishing the relative configuration of ring B (Figure 1) because ascertaining the relative configuration of this portion of the compound was crucial to mapping the spatial relationships of all the protons throughout the molecule. The two large J values measured for H-7ax (14.7 and 15.1 Hz) were interpreted as having arisen from a geminal coupling with H-7eq, as well as a vicinal (trans-diaxial) coupling with H-8. Additionally, a ROESY correlation from H-7ax across ring B to H-11 indicated that both groups were on the same face of the cyclohexanone system. Expanding on these observations, a series of ROESY correlations was detected that extended from H-8 to the H-20 methyl protons (attached to ring B) and H-1ax (part of ring A). Proton H-1ax was determined to be at a critical junction point in the molecule since it presented diagnostic couplings with one geminal (H-1eq, 2J = 14.8 Hz) and two vicinal (H-10, 3Jtrans = 15.1 and H-2, 3Jcis = 3.9 Hz) protons that were key to establishing the relative configuration of ring A. Returning to H-7ax, an additional ROESY correlation was identified with H-19, and this in turn displayed a ROESY correlation with H-18, which helped finalize the relative configuration of the ring C system. The geometry of the conjugated diene was determined based on the ROESY correlations between H-14 and H-16b, as well as between H-12 and H-15a.
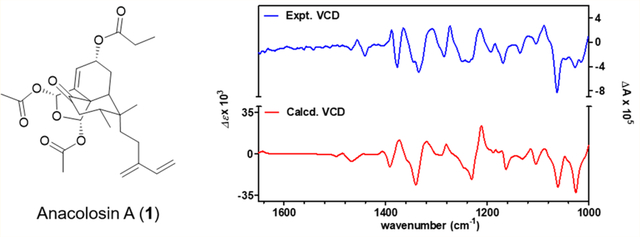
With the relative configuration of 1 established, we proceeded to evaluate the absolute configuration of the molecule, which included comparing the experimental and calculated (theoretical) ECD and VCD spectra for 1 (Figures 2 and 3, respectively). A theoretical ECD spectrum for 1 was generated at the CAM-B3LYP/SVP level, and the results compared to an experimentally derived spectrum, revealing a strong match between both data sets. Similarly, a theoretical VCD spectrum was prepared using optimized geometries of the molecule obtained from a conformational search employing ComputeVOA (BioTool, Inc.) with MMFF and further optimized at the density functional theory (DFT) level (B3LYP/6–31+G(d,p)) with Gaussian software (a computational package for electronic structure modeling).13 The calculated and experimentally derived VCD data revealed a strong match between the two data sets obtained. Therefore, the absolute configuration of the metabolite was established as shown for compound 1.
Figure 2.
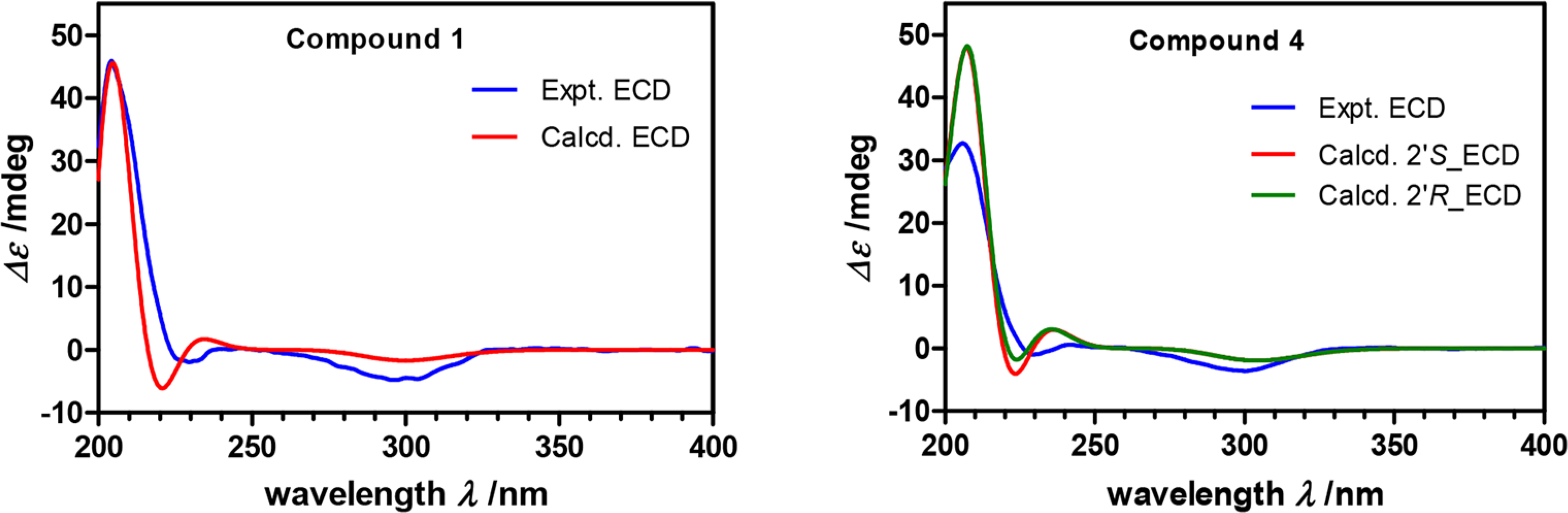
Calculated and experimental ECD curves for compounds 1 and 4.
Figure 3.
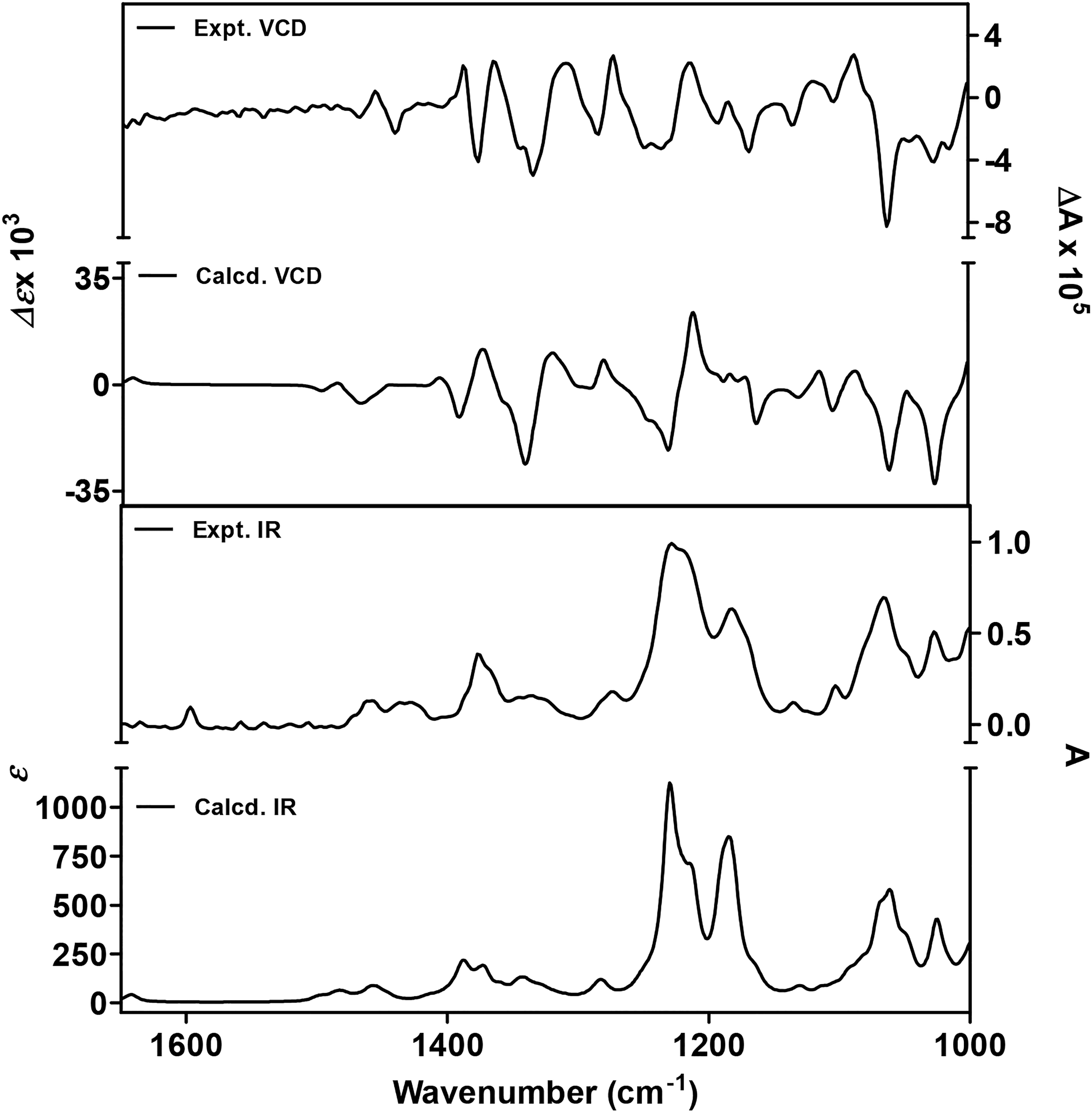
Calculated and experimental VCD/IR spectra for compound 1.
Anacolosin B (2) was purified as a colorless oil. The HRESIMS (m/z 525.2464), together with the 1H and 13C NMR data (Tables 1 and 2), led to its proposed molecular formula, C28H38O8. This corresponded to one additional methylene unit in 2 compared to 1. The 1H and 13C NMR data for 2 (Tables 1 and 2) were similar to those obtained for 1; however, some modest differences were observed corresponding to the chemical shifts attributable to the propionate group. Specifically, both an additional methylene carbon signal and proton resonance were found that were determined to be part of a butyrate group [δC 174.5 (C-1′, C=O), 37.2 (C-2′, CH2), 19.8 (C-3′, CH2), and 14.0 (C-4′, CH3); δH 2.37 (2H, t, J = 7.2, H-2′), 1.70 (2H, m, H-3′), and 1.01 (3H, t, J = 7.4 Hz, H-4′)]. The placement and identity of the butyrate were established using 1H–1H COSY and HMBC correlation data (Figure S1, Supporting Information). The relative and absolute configurations of 2 were also confirmed by 1H–1H ROESY data (Figure S2, Supporting Information), coupling constants, and ECD and VCD analyses (Figures S3 and S4, Supporting Information). Based on these experiments, compound 2 was confirmed to have the same absolute configuration as 1.
Anacolosin C (3) was obtained as a colorless oil, and its molecular formula was established to be C29H40O8 based on the HRESIMS data (m/z 539.2613 [M + Na]+, calcd for 539.2615). Focusing on the small changes in the 1H and 13C NMR resonances for 3 versus 2 (Tables 1 and 2), our attention was guided to the ester group moiety with which a new methyl signal was associated. The 1H–1H COSY and HMBC correlation data (Figure S1, Supporting Information) confirmed the position of the methyl group belonging to a methylbutanoate moiety in 3. The relative configuration of 3 was confirmed by 1H–1H ROESY and coupling constants analyses, whereas its absolute configuration was established based on the significant similarity between the ECD spectra of 1 and 2 to data obtained for 3.
Anacolosin D (4) was determined to have the same molecular formula as metabolite 3, which suggested the new compound was isomeric. Analysis of the 1H and 13C NMR data (Tables 1 and 2) led to the conclusion that the structural changes in metabolite 4 were once again confined to the ester moiety. In this case, a 2-methylbutanoate group was proposed, which was supported by 1H–1H COSY and HMBC correlation data (Figure S1, Supporting Information). Whereas ECD data were appropriate for determining the absolute configuration of the molecule’s isozuelanin core, the configuration of the C-2′ stereocenter remained undefined. To probe this last stereocenter, compound 4 was subjected to hydrolysis to liberate 2-methylbutyric acid, which was converted to its corresponding naphthacyl ester with α-bromo-2-acetonaphthone (11) (Scheme 1).14,15 Similarly, the naphthacyl esters made from authentic samples of S- and R/S-2-methylbutyric acid (12) were also prepared, and the products were analyzed by chiral HPLC. The retention times for S- and R-13 were determined to be 12.8 and 13.6 min, respectively, on a Phenomenex Lux 5 μm Cellulose-3 chiral column. With these data in hand, the product generated from the hydrolysis of compound 4 was analyzed and found to match the S isomer. Thus, the structure of the new metabolite was secured as shown for 4.
Scheme 1.
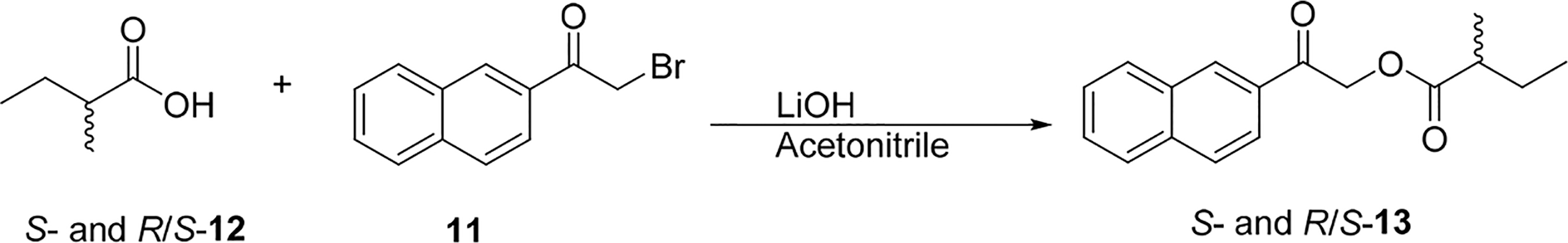
Synthesis of 2-Naphthacyl Esters (S- and R/S-13) of S- and R/S-2-Methylbutyric Acid (S- and R/S-12)
Anacolosin E (5) was obtained as a colorless oil. The sodiated molecular ion observed by HRESIMS at m/z 537.2455 (calcd 537.2459) suggested the metabolite’s molecular formula was C29H38O8. Examination of the 1H and 13C NMR data (Tables 1 and 2) focused our attention on the ester moiety as the site where this compound varied from other metabolites in the series. In this case, the ester group exhibited rather substantial changes including an upfield carbonyl group (δC 168.5, C-1′), two olefinic carbons (δC 129.9, C-2′; δC 138.9, CH-3′), and two methyl carbons (δC 14.5, CH3-4′; δC 12.1, CH3-5′). Analysis of the chemical shift data and HMBC correlations confirmed the identity of the new ester group as a 2-methylbut-2-enoate. 1H–1H ROESY correlation data (δH 6.91, H-3′ and δH 1.88, H-5′) supported a Z configuration for the alkene. A combination of 1H–1H ROESY and ECD helped verify that compound 5 shared the same absolute configuration with other compounds in this series.
Anacolosin F (6) was determined to have the molecular formula C28H38O8 based on HRESIMS, which yielded a [M + Na]+ ion at m/z 525.2456 (calcd 525.2459). An examination of the 1H and 13C NMR data (Tables 1 and 2) for the ester moiety led to the conclusion that 6 was a constitutional isomer of compound 2, with the former compound bearing an isobutyrate group. The limited quantity of 6 obtained from the extract precluded the collection of a ROESY data set; however, we propose that this molecule shared the same absolute configuration as metabolites 1–5 based on (i) its co-occurrence with these metabolites, (ii) an ECD spectrum that was similar to the previously described compounds, and (iii) 1H NMR coupling constants that were a good match to the isozuelanin portions of the other natural products.
Purified corymbulosin X (7) appeared as a colorless oil and was found to possess the molecular formula C29H42O8 as determined by HRESIMS, which indicated that the metabolite contained nine degrees of unsaturation. An examination of the 1H and 13C NMR data for 7 and 3 (Tables 1 and 2, respectively) showed the two compounds to be quite similar, with one major exception: the C-6 keto carbonyl signal in 3 was missing and it was replaced by a new oxygenated methine (δC 73.4). The relative configuration of ring B was established based on an analysis of J-coupling (Table 1) and 1H–1H ROESY correlation data (Figure S2, Supporting Information). The experimental ECD spectrum (Figure S3, Supporting Information) was consistent with data calculated for 7, as well as published data for similar metabolites,8 leading to the proposed absolute configuration as depicted. Mosher ester analysis was carried out on 7 (Figure S7, Supporting Information) confirming its 6S configuration.
Corymbulosin Y (8) appeared as a colorless oil, and its HRESIMS data (m/z 539.2638 [M + Na]+, calcd 539.2615) supported a molecular formula of C29H40O8, which meant the compound contained 10 degrees of unsaturation. The 1H and 13C NMR data indicated that 7 and 8 shared the same clerodane diterpene skeleton, but the two molecules differed in their respective ester groups. The 1H–1H COSY and HMBC data confirmed that 8 and 5 shared the same pendant (Z)-2-methylbut-2-enoate ester moiety. Based on its ECD data (Figure S3, Supporting Information), compound 8 was proposed to have the same absolute configuration as the other metabolites purified from this plant.
Two previously described metabolites, 9 and 10,8 were also purified from the bioactive extract. The identities of both compounds were confirmed based on comparisons of the experimental and reported NMR chemical shift data, as well as analyses of their ECD spectroscopic data. The configuration of the C-2′ stereocenter in compound 9 was determined to be S as previously reported in the literature.8 To the best of our knowledge, a trivial name had not been previously assigned to compound 9, whereas compound 10 had been assigned the name caseamembrin S.8
The antiproliferative and cytotoxic effects of metabolites 1 – 10 were evaluated in cell lines representing four pediatric cancer types: Ewing sarcoma (A-673), rhabdomyosarcoma (SJCRH30), medulloblastoma (D283), and hepatoblastoma (Hep293TT). The microtubule-destabilizing chemotherapeutic vinorelbine, which has demonstrated particular efficacy in sarcomas from among a panel of patients with pediatric solid tumors,16 was used as a positive control. As expected, vinorelbine had potent and selective activity against the Ewing and rhabdomyosarcoma cells, with only cytostatic effects observed in the hepatoblastoma and medulloblastoma cells (Table 3, Figure 4). Compounds 1–10 demonstrated a distinct profile of efficacy with cytotoxicity observed in each of the 4 cell lines with TGI values ranging from 0.2 to 4.1 μM (Table 3, Figure 4); however, certain cell lines exhibited greater sensitivities to one or more of the natural products. The A-673 cells were the least sensitive to each of the 10 compounds, whereas SJCRH30 and D283 cells were more sensitive to each compound throughout the entire concentration response curve (Figure 4). This shift was associated with a modest, but consistent, 1.4–2.5-fold difference in TGI values between these cell lines (Table 3). Among the C-6 keto-carbonyl-group-containing compounds (i.e., compounds 1–6), compounds 5 and 6 were the least potent, with TGI values 2–5-fold higher than 1–4.
Table 3.
TGI Values of Compounds 1–10 in Pediatric Cancer Cell Linesa
| cell line |
||||
|---|---|---|---|---|
| compound | A-673 | SJCRH30 | D283 | Hep293TT |
| 1 | 1.1 ± 0.1 | 0.52 ± 0.03 | 0.7 ± 0.1 | 1.0 ± 0.1 |
| 2 | 1.0 ± 0.1 | 0.50 ± 0.02 | 0.6 ± 0.1 | 0.9 ± 0.2 |
| 3 | 1.1 ± 0.1 | 0.67 ± 0.02 | 0.66 ± 0.04 | 1.0 ± 0.1 |
| 4 | 1.2 ± 0.1 | 0.73 ± 0.02 | 0.66 ± 0.04 | 0.8 ± 0.1 |
| 5 | 3.1 ± 0.1 | 1.9 ± 0.1 | 2.0 ± 0.2 | 1.8 ± 0.2 |
| 6 | 4.1 ± 0.5 | 2.3 ± 0.2 | 2.3 ± 0.1 | 3.2 ± 0.6 |
| 7 | 0.7 ± 0.1 | 0.34 ± 0.01 | 0.36 ± 0.02 | 0.22 ± 0.02 |
| 8 | 1.0 ± 0.2 | 0.44 ± 0.04 | 0.7 ± 0.1 | 0.28 ± 0.02 |
| 9 | 1.7 ± 0.2 | 0.8 ± 0.1 | 1.1 ± 0.2 | 0.6 ± 0.1 |
| 10 | 0.9 ± 0.1 | 0.36 ± 0.03 | 0.5 ± 0.1 | 0.3 ± 0.1 |
| vinorelbine | 0.006 ± 0.002 | 0.0060 ± 0.0004 | ~0.08 | ~0.08 |
Concentration (μM) that caused total growth inhibition (TGI) for each pediatric cancer cell line as determined by the SRB assay. n = 3; mean ± SEM.
Figure 4.
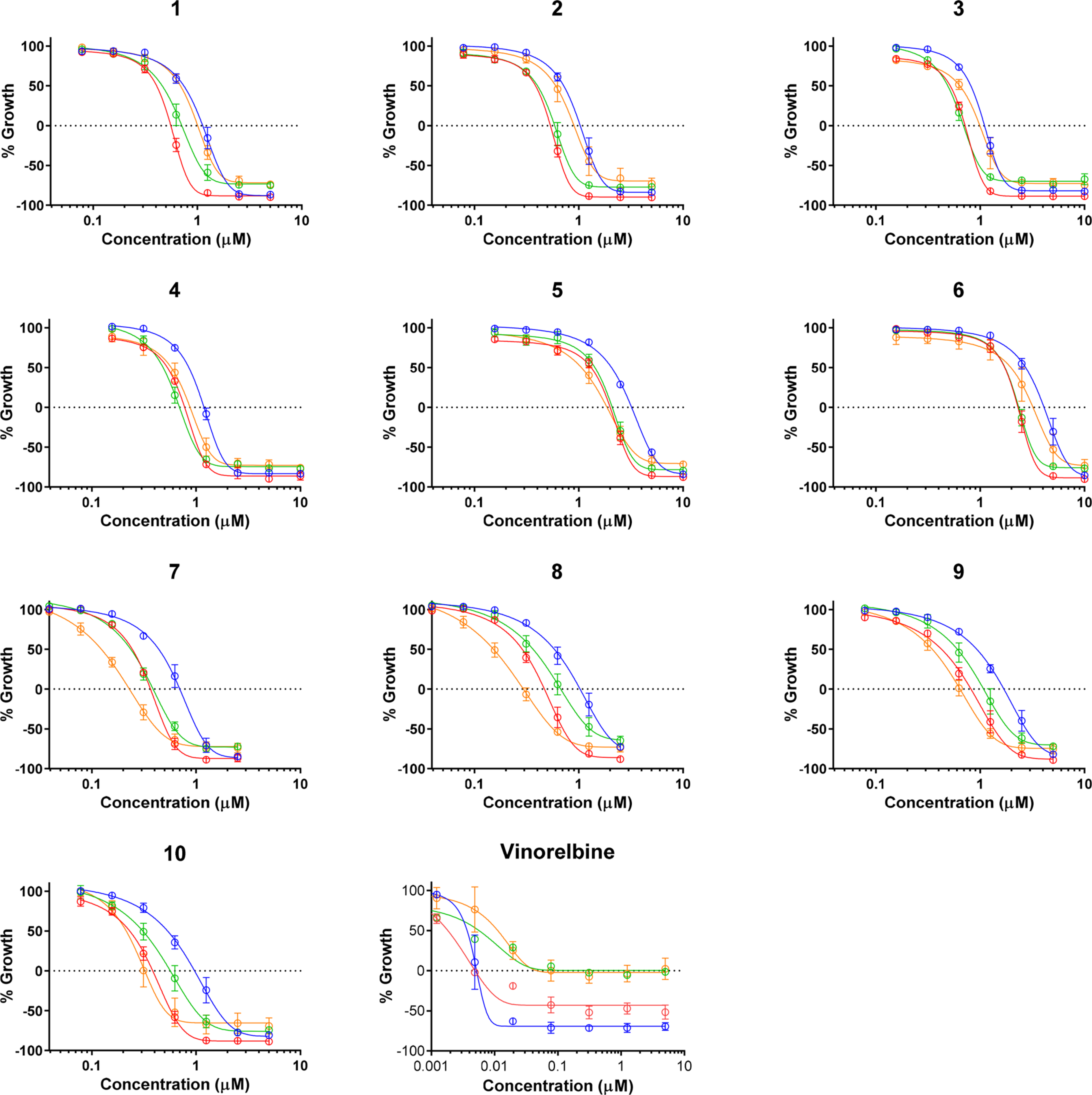
Concentration–response data from the SRB assay for compounds 1–10 and vinorelbine against A-673 (blue), SJCRH30 (red), D283 (green), and Hep293TT (orange) cell lines. n = 3; mean ± SEM.
The Hep293TT hepatoblastoma cells were some of the most resistant to compounds 1–6. However, the cell line selectivity profile was distinct for the C-6 hydroxy-group-containing compounds 7–10, with the Hep293TT cells being the most sensitive (TGI values of 0.2–0.6 μM) followed by the SJCRH30 (TGI values 0.3–0.8 μM), D283 (TGI values 0.4–1.1 μM), and A-673 (TGI values 0.7–1.7 μM) cells lines. The finding that Hep293TT cells were selectively sensitive to each of the C-6 hydroxy compounds (i.e., 7, 8, 9, and 10), but not the analogous C-6 keto carbonyl compounds (i.e., 3, 5, 4, and 1), demonstrated the importance of this moiety in conferring sensitivity for hepatoblastoma cells as compared to cells representing other pediatric cancer types. Importantly, although the windows of selectivity for these compounds is narrow, each of these compounds was highly effective, with maximal efficacy values of essentially 100% in each of the pediatric cancer cell lines. The results were reproducible and significant, which suggests that medicinal chemistry efforts would have the potential to identify analogues with even greater selectivity based on this scaffold.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotation data were obtained on a Rudolph Research AUTOPOL III automatic polarimeter. ECD data were obtained on a JASCO J-715 CD instrument. VCD spectra were recorded on a BioTools, Inc. ChiralIR-2X VCD spectrometer, equipped with a dual PEM accessory. NMR data were collected on Varian 400, 500, and 600 MHz NMR spectrometers. LC-MS data were obtained on a Shimadzu LC-MS 2020 system (ESI quadrupole) coupled to a photodiode array detector, with a Phenomenex Kintex 2.6 μm C18 column (100 Å, 75 × 3.0 mm, 0.4 mL/min). The preparative HPLC system utilized SCL-10A VP pumps and a system controller with a Phenomenex Gemini 5 μm C18 column (110 Å, 250 × 21.2 mm, 10 mL/min), and the analytical and semipreparative HPLC systems utilized Waters 1525 binary pumps with Waters 2998 photodiode array detectors and Phenomenex Gemini 5 μm Gemini C18, Phenomenex Kinetex 5 μm biphenyl, and Phenomenex Kinetex 5 μm pentafluorophenyl (250 × 4.6 mm, 1 mL/min and 250 × 10 mm, 4 mL/min) columns. Accurate mass data were collected on an Agilent 6538 HRESI QTOF MS coupled to an Agilent 1290 HPLC. Chiral HPLC analyses were performed on a system composed of Waters 1525 binary pumps, a Waters 2998 photodiode array detector, and a Phenomenex Lux 5 μm Cellulose-3 column (250 × 4.6 mm, 1 mL/min). The S- and R/S-2-methylbutyric acid (12) and α-bromo-2-acetonaphthone (11) were purchased from Sigma-Aldrich. All solvents were of ACS grade or better.
Plant Material.
Fruit, leaf, and twigs of the plant Anacolosa clarkii were collected in the Bana Forest Preserve in Danang, Vietnam, by Dr. Djaja D. Soejarto (University of Illinois at Chicago) under contract to the Natural Products Branch for the National Cancer Institute, on December 3, 2003. Voucher specimens for this collection [Voucher #0GHA5499] are housed at the Smithsonian Institution, Washington, DC.
Extraction and Isolation.
An organic extract of the plant A. clarkii (N135363) was produced as reported previously17 and provided by NCI Natural Products Branch (Frederick, MD, USA). The crude extract (1.0 g) was fractionated by vacuum-liquid chromatography (HP20ss) eluted with a MeOH–H2O step gradient (30:70, 50:50, 70:30, 90:10, and 100:0) and washed with CH2Cl2–MeOH (50:50). The fourth fraction (MeOH–H2O, 90:10) was found to be bioactive, and it was further fractionated by C18 preparative HPLC (gradient elution using MeOH–H2O, 30:70 to 100% MeOH in 30 min) followed by semipreparative HPLC using a pentafluorophenyl column (isocratic CH3CN−H2O, 55:45 or 60:40), to obtain 1 (20.0 mg), 2 (8.6 mg), 3 (3.7 mg), 4 (1.0 mg), 5 (0.6 mg), 6 (0.3 mg), 7 (0.6 mg), 8 (0.4 mg), 9 (0.7 mg), and 10 (0.5 mg).
Anacolosin A (1): colorless oil; [α]20D +2.6 (c 1.02, MeOH); 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 511.2304 [M + Na]+ (calcd for C27H36O8Na, 511.2302).
Anacolosin B (2): colorless oil; [α]20D +7.4 (c 0.43, MeOH); 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 525.2464 [M + Na]+ (calcd for C28H38O8Na, 525.2459).
Anacolosin C (3): colorless oil; [α]20D +9.7 (c 0.12, MeOH); 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 539.2613 [M + Na]+ (calcd for C29H40O8Na, 539.2615).
Anacolosin D (4): colorless oil; 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 539.2610 [M + Na]+ (calcd for C29H40O8Na, 539.2615).
Anacolosin E (5): colorless oil; 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 537.2455 [M + Na]+ (calcd for C29H38O8Na, 537.2459).
Anacolosin F (6): colorless oil; 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 525.2456 [M + Na]+ (calcd for C28H38O8Na, 525.2459).
Corymbulosin X (7): colorless oil; [α]20D +16 (c 0.05, MeOH); 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 541.2798 [M + Na]+ (calcd for C29H42O8Na, 541.2772).
Corymbulosin Y (8): colorless oil; 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 539.2638 [M + Na]+ (calcd for C29H40O8Na, 539.2615).
Analysis of the Ester Group Chirality for Compound 4.
α-Bromo-2-acetonaphthone (11, 45 mg, 0.18 mmol) was added to an acetonitrile solution of S-2-methylbutyric acid (S-12) [20 μL (18.8 mg, 0.18 mmol)] in 0.8 mL of acetonitrile, followed by the addition of 0.2 mL of an aqueous solution of LiOH (1.0 mg/mL). The solution was stirred at room temperature for 3 h before the solvent was removed. The residue was analyzed by LC-MS, and analytical chiral HPLC was used to confirm the identity of the product [S-2-methylbutyric acid 2-naphthacyl ester (S-13)]. S-13 (27 mg) was purified via semipreparative HPLC, and its structure was confirmed by 1H and 13C NMR (Figures S67 and S68, Supporting Information) and ESIMS m/z 271 [M + H]+. Similarly, the R/S-2-methylbutyric acid (R/S-12) was used as described above to prepare the R/S-2-methylbutyric acid 2-naphthacyl esters (R/S-13) (NMR: Figures S65 and S66, Supporting Information, and ESIMS m/z 271 [M + H]+).
Compound 4 (0.2 mg) was added to 0.2 mL of an aqueous LiOH solution (1 mg/mL) and stirred for 3 h followed by the addition of α-bromo-2-acetonaphthone (5 mg dissolved in 0.8 mL of acetonitrile). The mixture was stirred overnight at room temperature, at which point the presence of 13 was confirmed by analysis of the extracted ion chromatogram (Figure S5, Supporting Information). The target product (13) was purified from the reaction residue by semipreparative HPLC.
The esters from the hydrolysate of 4, S-13, and R/S-13 were analyzed by chiral HPLC (Phenomenex Lux 5 μm Cellulose-3 column, 1 mL/min, 60:40 acetonitrile−water), and the retention times of the compounds were recorded as 12.8 min (ester from compound 4), 12.8 min (S-13), and 12.8 and 13.6 min (R/S-13), which revealed a match between the natural-product-derived material and the S-form of the authentic standard. This conclusion was further confirmed by co-injection of the material prepared from metabolite 4 and S-13, which showed both compounds eluted at the same time and had superimposable PDA chromatograms.
VCD Calculations for Compounds 1 and 2.
Anacolosins A and B (1 and 2) were dissolved separately in CDCl3 (50 mg/mL for 1 and 43 mg/mL for 2) and placed in a 100 μm path-length cell with BaF2 windows. IR and VCD spectra were recorded on a ChiralIR-2X VCD spectrometer (BioTools, Inc.) coupled with a single PEM accessory, with 4 cm−1 resolution, and instrument optimization at 1400 cm−1 (5 h data collection for each sample, as well as a CDCl3 blank). A preliminary conformational search was executed with ComputeVOA (BioTools, Inc.) at the molecular mechanics level (MMFF). The geometry, frequency, IR, and VCD intensity calculations of the low-energy conformers were carried out at the B3LYP/6–31+G(d,p) level with Gaussian 09 (Gaussian Inc., Wallingford, CT, USA).13 The calculated IR and VCD spectra of the stable conformers with populations greater than 1% were added together after Boltzmann statistical weighting using SpecDis 1.71 (scaled by 0.97 and a half-bandwidth of 6 cm−1). The resulting calculated IR and VCD spectra were compared against the experimental VCD curves obtained for 1 and 2 and are shown in Figures 3 and S4 (Supporting Information), respectively.
ECD Calculations for Compounds 1–5, 7, and 8.
A preliminary conformational search was executed with ComputeVOA (BioTools, Inc.) at the molecular mechanics level (MMFF). The conformers obtained were further optimized at the B3LYP/6–31+G(d,p) level with Gaussian 09 (Gaussian Inc.).13 Stable conformers with populations greater than 1% were submitted to ECD calculation by time-dependent DFT at the CAM-B3LYP/SVP level. The ECD spectra were added together after a Boltzmann statistical weighting using SpecDis 1.71 (sigma value of 0.3 eV). After applying a UV-shift correction, the computed ECD spectra were compared with the experimentally derived ECD curves (Figures 2 and S3, Supporting Information).
Pediatric Cancer Cell Lines.
The rhabdomyosarcoma (SJCRH30), Ewing sarcoma (A-673), and medulloblastoma (D283) cell lines were purchased from the ATCC (Manassas, VA, USA). The hepatoblastoma cell line (Hep293TT) was obtained directly from Dr. Gail Tomlinson.18 The SJCRH30, A-673, and Hep293TT cells were grown in RPMI-1640 medium (Corning) with 5% fetal bovine serum (Corning) and 0.5% gentamycin (Gibco), while the D283 cells were grown in IMEM (Gibco) with 5% fetal bovine serum and 0.3% gentamycin. Cells were grown at 37 °C in a humidified incubator with 5% CO2. Cells were used within four months of retrieval from liquid nitrogen. The SJCRH30, D283, and A-673 cells lines were validated by STR profiling (Genetica).
Determination of TGI Values against Pediatric Cancer Cell Lines Using a Sulforhodamine B Cytotoxicity Assay.
Compounds were evaluated for antiproliferative and cytotoxic activities using a sulforhodamine B (SRB) assay as previously described.19,20 Cells were plated at a predetermined density of 4000–6000 cells per well in 96-well tissue culture plates and allowed to adhere overnight. Compounds were dissolved in DMSO and added to triplicate wells with final DMSO volumes of 0.5% v/v with vehicle controls included on each plate. Cells were treated for 48 h, after which the medium was removed, and the adherent cells were fixed with 10% TCA. Due to the low adherence of the D283 cells, 180 μL of 20% TCA was added to the tissue culture medium of each well for these cells. At the time of compound addition, a separate “time 0” plate was fixed to allow calculations of TGI and cytotoxicity. The absorbance values were measured and compared to vehicle and time 0 controls to determine concentrations that caused total growth inhibition for each experiment using the nonlinear regression function in Prism 6 (GraphPad).
Supplementary Material
ACKNOWLEDGMENTS
Support for this project was provided by NIH grant R01GM107490 to R.H.C. and S.L.M. and the Greehey Endowment (S.L.M.). The LC-MS instrument used for this project was provided in part by a Challenge Grant from the Office of the Vice President for Research, University of Oklahoma, Norman Campus, and an award through the Shimadzu Equipment Grant Program (R.H.C.). Computational data for ECD and VCD experiments were obtained through the University of Oklahoma Supercomputing Center for Education & Research (OSCER). This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.8b01015.
Summaries of 2D NMR correlation data, experimental and calculated ECD curve comparisons, comparison of experimental and calculated VCD curves for compound 2, Mosher’s reaction results for compound 7, LC-MS analysis of the reaction-product residue for compound 4, C18 and chiral HPLC analysis of synthetic product for compound 4 and the synthetic standards, 1D and 2D NMR spectra for compounds 1–8, 1D NMR spectra for synthetic standards, and the HRESIMS data (PDF)
REFERENCES
- (1).Pritchard-Jones K; Pieters R; Reaman GH; Hjorth L; Downie P; Calaminus G; Naafs-Wilstra MC; Steliarova-Foucher E Lancet Oncol. 2013, 14, e95–e103. [DOI] [PubMed] [Google Scholar]
- (2).Siegel RL; Miller KD; Jemal A Ca-Cancer J. Clin 2016, 66, 7–30. [DOI] [PubMed] [Google Scholar]
- (3).Gröbner SN; Worst BC; Weischenfeldt J; Buchhalter I; Kleinheinz K; Rudneva VA; Johann PD; Balasubramanian GP; Segura-Wang M; Brabetz S; Bender S; Hutter B; Sturm D; Pfaff E; Hübschmann D; Zipprich G; Heinold M; Eils J; Lawerenz C; Erkek S; Lambo S; Waszak S; Blattmann C; Borkhardt A; Kuhlen M; Eggert A; Fulda S; Gessler M; Wegert J; Kappler R; Baumhoer D; Burdach S; Kirschner-Schwabe R; Kontny U; Kulozik AE; Lohmann D; Hettmer S; Eckert C; Bielack S; Nathrath M; Niemeyer C; Richter GH; Schulte J; Siebert R; Westermann F; Molenaar JJ; Vassal G; Witt H; Burkhardt B; Kratz CP; Witt O; van Tilburg CM; Kramm CM; Fleischhack G; Dirksen U; Rutkowski S; Fruhwald M; von Hoff K; Wolf S; Klingebiel T; Koscielniak E; Landgraf P; Koster J; Resnick AC; Zhang J; Liu Y; Zhou X; Waanders AJ; Zwijnenburg DA; Raman P; Brors B; Weber UD; Northcott PA; Pajtler KW; Kool M; Piro RM; Korbel JO; Schlesner M; Eils R; Jones DTW; Lichter P; Chavez L; Zapatka M; Pfister SM Nature 2018, 555, 321–327. [DOI] [PubMed] [Google Scholar]
- (4).Bourjot M; Leyssen P; Eydoux C; Guillemot JC; Canard B; Rasoanaivo P; Guéritte F; Litaudon M Fitoterapia 2012, 83, 1076–1080. [DOI] [PubMed] [Google Scholar]
- (5).Alimboyoguen AB; Castro-Cruz KAD; Shen C-C; Ragasa CY Res. J. Pharm. Biol. Chem. Sci 2014, 5, 1189–1191. [Google Scholar]
- (6).Kanokmedhakul S; Kanokmedhakul K; Kanarsa T; Buayairaksa M J. Nat. Prod 2005, 68, 183–188. [DOI] [PubMed] [Google Scholar]
- (7).Kanokmedhakul S; Kanokmedhakul K; Buayairaksa M J. Nat. Prod 2007, 70, 1122–1126. [DOI] [PubMed] [Google Scholar]
- (8).Aimaiti S; Suzuki A; Saito Y; Fukuyoshi S; Goto M; Miyake K; Newman DJ; O’Keefe BR; Lee K-H; Nakagawa-Goto K J. Org. Chem 2018, 83, 951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Suzuki A; Saito Y; Fukuyoshi S; Goto M; Miyake K; Newman DJ; O’Keefe BR; Lee K-H; Nakagawa-Goto K J. Nat. Prod. 2017, 80, 1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li R; Morris-Natschke SL; Lee K-H Nat. Prod. Rep 2016, 33, 1166–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Xu J; Zhang Q; Wang M; Ren Q; Sun Y; Jin D-Q; Xie C; Chen H; Ohizumi Y; Guo Y J. Nat. Prod 2014, 77, 2182–2189. [DOI] [PubMed] [Google Scholar]
- (12).Calderón C; Ford CD; Castro V; Merfort I; Murillo R J. Nat. Prod 2014, 77, 455–463. [DOI] [PubMed] [Google Scholar]
- (13).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- (14).Molinski TF; Reynolds KA; Morinaka BI J. Nat. Prod 2012, 75, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bray AM; Maeji NJ; Valerio RM; Campbell RA; Geysen HM J. Org. Chem 1991, 56, 6659–6666. [Google Scholar]
- (16).Minard-Colin V; Ichant J-L; Nguyen L; Paci A; Orbach D; Bergeron C; Defachelles A-S; André N; Corradini N; Schmitt C; Tabone M-D; Blouin P; Sirvent N; Goma G; Geoerger B; Oberlin O Eur. J. Cancer 2012, 48, 2409–2416. [DOI] [PubMed] [Google Scholar]
- (17).McCloud TG Molecules 2010, 15, 4526–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chen TT-L; Rakheja D; Hung JY; Hornsby PJ; Tabaczewski P; Malogolowkin M; Feusner J; Miskevich F; Schultz R; Tomlinson GE Pediatr. Blood Cancer 2009, 53, 1040–1047. [DOI] [PubMed] [Google Scholar]
- (19).Skehan P; Storeng R; Scudiero D; Monks A; McMahon J; Vistica D; Warren JT; Bokesch H; Kenney S; Boyd MR J. Natl. Cancer Inst. 1990, 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
- (20).Vichai V; Kirtikara K Nat. Protoc 2006, 1, 1112–1116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.