

Abstract
Progressive tumor growth is associated with deficits in the immunity generated against tumor antigens. Vaccines targeting tumor neoepitopes have the potential to address qualitative defects; however, additional mechanisms of immune failure may underlie tumor progression. In such cases, patients would benefit from additional immune-oncology agents targeting potential mechanisms of immune failure. This study explores the identification of neoepitopes in the MC38 colon carcinoma model by comparison of tumor to normal DNA and tumor RNA sequencing technology, as well as neoepitope delivery by both peptide- and adenovirus-based vaccination strategies. To improve antitumor efficacies, we combined the vaccine with a group of rationally selected immune-oncology agents. We utilized an IL15 superagonist to enhance the development of antigen-specific immunity initiated by the neoepitope vaccine, PD-L1 blockade to reduce tumor immunosuppression, and a tumor-targeted IL12 molecule to facilitate T-cell function within the tumor microenvironment. Analysis of tumor-infiltrating leukocytes demonstrated this multifaceted treatment regimen was required to promote the influx of CD8+ T cells and enhance the expression of transcripts relating to T-cell activation/effector function. Tumor-targeted IL12 resulted in a marked increase in clonality of T-cell repertoire infiltrating the tumor, which when sculpted with the addition of either a peptide or adenoviral neoepitope vaccine promoted efficient tumor clearance. In addition, the neoepitope vaccine induced the spread of immunity to neoepitopes expressed by the tumor but not contained within the vaccine. These results demonstrate the importance of combining neoepitope-targeting vaccines with a multifaceted treatment regimen to generate effective antitumor immunity.
Introduction
The development of vaccines targeting nonsynonymous mutations uniquely expressed by a tumor has become a viable therapeutic strategy, and the potential of expanding a pool of high-avidity T cells capable of mediating tumor clearance holds the promise of revolutionizing the treatment of cancer. However, the presence of regulatory T cells (1), dysregulation of immune checkpoints (2), and an immune-suppressive tumor microenvironment (3) impact the ability of reactive T cells expanded by neoepitope vaccines to cause the regression of established tumors.
The selection of appropriate neoepitopes to target continues to be a challenge. In this study, we observed that using a combination of in silico analysis, along with both in vitro and in vivo studies aimed at enriching for immunogenic neoepitopes, was not sufficient to identify neoepitopes capable of mediating tumor regression when utilized as a single agent. To improve the clinical efficacy of this neoepitope-targeted vaccine, we combined it with three additional immune-oncology agents. Together, these agents act to (i) initiate the immune response, (ii) potentiate systemic antitumor immunity, (iii) reduce tumor-associated immune-suppression, and (iv) facilitate the expansion and function of T cells within the tumor microenvironment.
Previously, Yadav and colleagues (4) identified three neoepitopes in the MC38 colon cancer model and demonstrated that vaccination was able to induce tumor regression; tumors were induced using a low dose of MC38 tumor cells that results in a slow-growing tumor, which is exceptionally responsive to immune interventions such as PD-L1 blockade (5). In those studies, adjuvant alone induced tumor regression in 40% of animals. In this study, we implanted animals with a higher dose of MC38 tumor cells that results in a faster growing tumor, which is more resistant to immune interventions (6). To ensure our therapeutic regimen has a clear translational path, we employed agents that have undergone or are undergoing clinical evaluation (7–9). In this study, the antitumor immune response was initiated using either a peptide- or adenoviral-based vaccine targeting immunogenic neoepitopes identified in the MC38 colon carcinoma cell line. This immunity was potentiated systemically with the addition of the IL15 superagonist N-803 (previously known as ALT-803), which has been shown to be 4- to 5-fold more potent than IL15 (10, 11), and promotes the expansion, survival, and function of high-avidity memory T cells, without expanding the regulatory T-cell compartment (12–17). PD-L1 blockade was added to our treatment regimen to inhibit tumor immune evasion, help prevent T-cell exhaustion and promote T-cell function within the tumor microenvironment. The immunocytokine NHS-IL12 comprises IL12 fused with a necrosis-targeting antibody, which targets IL12 to the tumor to promote T-cell expansion and function within the tumor microenvironment (18). We observed that each agent acted in concert to promote the generation and maintenance of the tumor-specific immune response capable of mediating efficient tumor regression.
The expression of tumor neoepitopes can vary greatly among cells comprising the tumor mass (19–25). Thus, the generation of a diverse immune repertoire capable of recognizing numerous neoepitopes is well suited to control the growth of numerous tumor variants. In this study, we observed that vaccines targeting neoepitopes induced the in situ spread of immunity to other neoepitopes expressed by the tumor, but not incorporated into the vaccine. Our data suggest that this epitope spreading is an essential aspect of an effective antitumor immune response. This report demonstrates the importance of a multifaceted immunotherapy treatment regimen targeting different potential facets of immune failure in the development and maintenance of a diverse, effective antitumor immune response capable of mediating tumor regression.
Our studies demonstrate that (i) the effectiveness of neoepitope-targeted vaccines can be improved via the addition of rationally selected immune-oncology agents, (ii) adenoviral-based vaccines encoding neoepitopes, when used in combination with additional immune-mediators, can mediate tumor regression, (iii) vaccination of tumor-bearing animals with vaccines targeting either neoepitopes or a tumor-associated antigen can mediate epitope spreading, and (iv) this diversification of immunity correlates with an effective antitumor immune response.
Materials and Methods
Cell culture
MC38 and RMA-S cells were grown in RPMI1640 with l-glutamine (Corning) supplemented with 10% (v/v) FBS (Atlanta Biologicals) and 1% (v/v) antibiotic/antimitotic solution (Corning). All cells were cultured at 37°C, 5% CO2 for less than 1 month. The identity of the MC38 cell line was confirmed utilizing whole-genome DNA sequencing, and comparing identified neoepitopes to those previously published for this cell line (4).
Animals and tumor implantation
Mice were handled in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines, and under the approval of the NIH Intramural Animal Care and Use Committee. Mice were bred and housed at the NIH. Tumors were induced by implanting 3 × 105 tumor cells subcutaneously. All studies utilized female C57BL/6 animals.
Identification of tumor variants and neoepitopes
Single-nucleotide variants (SNV) and insertions/deletions (indel) were identified as previously described (26). Neoepitopes were identified by creating all possible permutations of 9-mer amino acid sequences derived from an identified nonsilent SNV or indel. Neoepitopes were ranked by RNA expression as well as allele frequency of the observed coding variant to offset issues arising from tumor heterogeneity. NetMHC 3.4 (http://www.cbs.dtu.dk/services/NetMHC-3.4/; refs. 27, 28) was used to predict neoepitope binding to a specific MHC H-2 allele. Neoepitopes with predicted binding affinities <500 nmol/L were retained for further analysis. Raw sequencing data from high-throughput sequencing was deposited in BioProject Accession PRJNA551473 (https://www.ncbi.nlm.nih.gov/bioproject/551473).
Peptide synthesis
Peptides were synthesized by Bio-Synthesis or GenScript to >85% purity.
Cell isolation and preparation
Spleens were harvested, dissociated through 70-μm filters, and subjected to ACK lysis to obtain splenocytes for analysis. Tumors were harvested, cut into small pieces, and incubated for 1 hour in a digestion cocktail composed of RPMI supplemented with 5% (v/v) FBS, 2 mg/mL Collagenase Type I (Worthington Biochemical Corporation), and 40 U/mL DNase I (Calbiochem). Following digestion, tumors were ground through 70-μm filters and tumor-infiltrating leukocytes (TIL) were enriched using a 40%/70% Percoll (Sigma) gradient. Peripheral blood mononuclear cells (PBMC) were isolated from whole, anticoagulated mouse blood by layering over lymphocyte separation medium (MP Biomedicals) and collecting lymphocyte layer.
Flow cytometric assays
All antibodies used for flow cytometric analysis are fully described in Supplementary Table S1. Peptide-binding assays were performed using RMA-S cells incubated with individual peptides at 50 μg/mL overnight. Following incubation, cells were stained with MHC antibodies. Data were acquired using a FACS Calibur (BD Biosciences) and reported as an in vitro binding score, which is the percentage of RMA-S cells expressing MHC on their surface. TILs were stained for immune cell subsets and data were collected using an Attune NxT Flow Cytometer (Thermo Fisher Scientific) or BD LSRFortessa (BD Biosciences). Cell populations were identified as follows: CD8+ T cells: live/CD45+/CD3+/CD8+; macrophages: live/CD45+/CD3−/CD11b+/F4/80+; central memory: CD44+/CD62L+; effector: CD44+/CD62L−/CD127−; effector memory: CD44+/CD62L−/CD127+. For intracellular cytokine staining, splenocytes were cultured with indicated peptides for 4 hours, at which time GolgiPlug/GolgiStop (BD Biosciences) were added. Cultures were incubated an additional 20 hours, and fixed, permeabilized, and stained for TNF and IFNγ production. Data were collected using an Attune NxT flow cytometer.
Vaccination and treatment with immunomodulators
Animals were vaccinated with pools of 9-mer or 25-mer neoepitope peptides (100 μg each peptide), emulsified in Montanide ISA 51 VG (Seppic), administered subcutaneously. Adenoviral vectors encoding TWIST1 or neoepitopes were kindly produced and provided by ImmunityBio and NantOmics Corporations. Viral particles (1010) encoding the multiepitope virus were administered subcutaneously. The admixed virus was administered by injecting animals subcutaneously with 1010 viral particles, each virus encoding a single neoepitope. N-803 was kindly provided by Immunity Bio, and 1μg was administered subcutaneously into animals. Anti–PD-L1 (10F.9G2, BioXCell, 200 μg) was administered intraperitoneally. Murine NHS-IL12 was kindly provided by EMD Serono and was administered at a dose of 50 μg. Reagents provided by ImmunityBio, NantWorks, and EMD Serono were kindly provided as part of Collaborative Research and Development Agreements (CRADA) with the NCI.
Assessment of immunity
Splenocytes or PBMCs were harvested and ex vivo antigen-dependent cytokine secretion was assessed using an IFNγ (BD Biosciences) or TNFα (Cellular Technology Ltd.) ELISPOT. Assays were performed according to the manufacturers’ instructions. Target peptides (10 μg/mL final concentration) were incubated with 0.5–1.0 × 106 splenocytes overnight. ELISPOT data are adjusted to the number of spots/million splenocytes after subtracting the number of spots in paired wells containing a control peptide.
IHC
Tumors, fixed in Z-fix (Anatech), were paraffin-embedded and sectioned. Slides were stained for CD8a (5μg/mL; Clone: 4SM16) using the Opal Multiplex Immunohistochemistry Kits (PerkinElmer). Images were acquired using an Axio Scan.Z1 Slide Scanner (Zeiss).
Depletion studies
Depletions were performed as described previously (29) using antibodies listed in Supplementary Table S1. Briefly, depletions were started prior to either the first vaccination (early depletion) or NHS-IL12 administration (late depletion). CD4 (100 μg, i.p.) and CD8 (100 μg, i.p.) antibodies were administered on days 1, 2, and 3, followed by once weekly for the duration of the experiment. Natural killer (NK) antibodies (200 μg, i.p.) were administered on days 1 and 3, followed by every 5 days throughout the duration of the experiment. Depletions were monitored in representative mice using flow cytometric analysis on peripheral blood. Cell populations were defined as follows: CD8+ T cells: CD3+/CD8b+; CD4+ T cells: CD3+/CD4+; NK cells: CD3−/CD49b+/NKp46+.
NanoString and T-cell receptor sequencing
Isolated tumor-infiltrating leukocytes were enriched using a CD45+ or CD4+/CD8+ murine TIL Microbead Kit (Miltenyi Biotech). RNA was purified from CD45+ cells using the RNeasy Mini Kit (Qiagen). Gene expression was assayed using the murine nCounter PanCancer Immune Profiling Panel (NanoString). NanoString data were normalized using the nSolver Analysis Software 4.0. Genes for which expression was altered by at least 2-fold in biological replicates compared with nontreated control animals were considered to be significant. T-cell receptor (TCR) diversity was assessed using genomic DNA purified from tumor-infiltrating CD4+ and CD8+ T cells using the QIAamp DNA Mini Kit (Qiagen). TCRβ chain sequencing was performed by Adaptive Biotechnologies and analyzed using the Immunoseq analyzer. The top 100 TCR sequences were analyzed. TCR sequences are available at https://clients.adaptivebiotech.com/pub/hamilton-2019-CIR DOI: 10.21417/DH2019CIR.
Data and statistical analysis
Flow cytometry data were analyzed using FlowJo (v10, BD Biosciences). All hierarchical clusters were generated using the Partek Genomics Suite. Statistical analyses were performed using GraphPad Prism (v7; GraphPad Software). All data points represent the mean ± SEM and P ≤ 0.05 was considered significant. Significance is indicated within figures as follows: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
Results
Effective targeting of tumor neoepitopes expressed by MC38 tumors in vivo
Whole-exome DNA and RNA sequencing was performed on two MC38 tumors, and expressed nonsynonymous mutations were identified using a tumor-normal DNA analysis from splenocytes harvested from the same animals (Fig. 1A; ref. 30). Although both tumors were induced using MC38 cells harvested from the same culture flasks, we observed a 2-fold difference in number of DNA mutations, and potential neoepitopes identified in each of the tumors analyzed (Table 1). This observation is supportive of previous studies in which the MC38 tumor cell line has been identified as being microsatellite-unstable (31). In total we identified 51 potential neoepitopes, representing 43 unique nonsynonymous mutations, shared among both tumors assayed (Table 1; Supplementary Table S2). There continues to be no defined method of selecting appropriate nonsynonymous mutations to target with a vaccine; in our study, we assumed that an ideal neoepitope would be both highly expressed and have a high affinity for binding MHC. Based upon this metric, we initially ranked potential neoepitopes by dividing the relative expression by predicted MHC binding affinity. In addition, we also utilized a second metric, which ranked neoepitopes solely by their predicted MHC binding affinity. Thirteen peptides, representing the top 10 peptides from both metrics, were synthesized (sequences highlighted in Supplementary Table S2). Seven of these 13 neoepitopes were capable of binding MHC in vitro (Fig. 1B), and the immunogenicity of each peptide was assessed by vaccinating non–tumor-bearing mice with pools of neoepitope peptides emulsified in Montanide ISA 51 VG Adjuvant (Montanide). Using an ex vivo IFNγ ELISPOT assay, 6 of 13 of the neoepitopes were found to induce immune responses in repeated experiments using non–tumor-bearing animals (Fig. 1C, reactive peptides highlighted with shaded bars). Based upon these observations, we chose to utilize a vaccination strategy incorporating a pool of four 9-mer neoepitope peptides (Jak1, Olfr99, Ptgfr, and Trp53) emulsified in Montanide.
Figure 1.
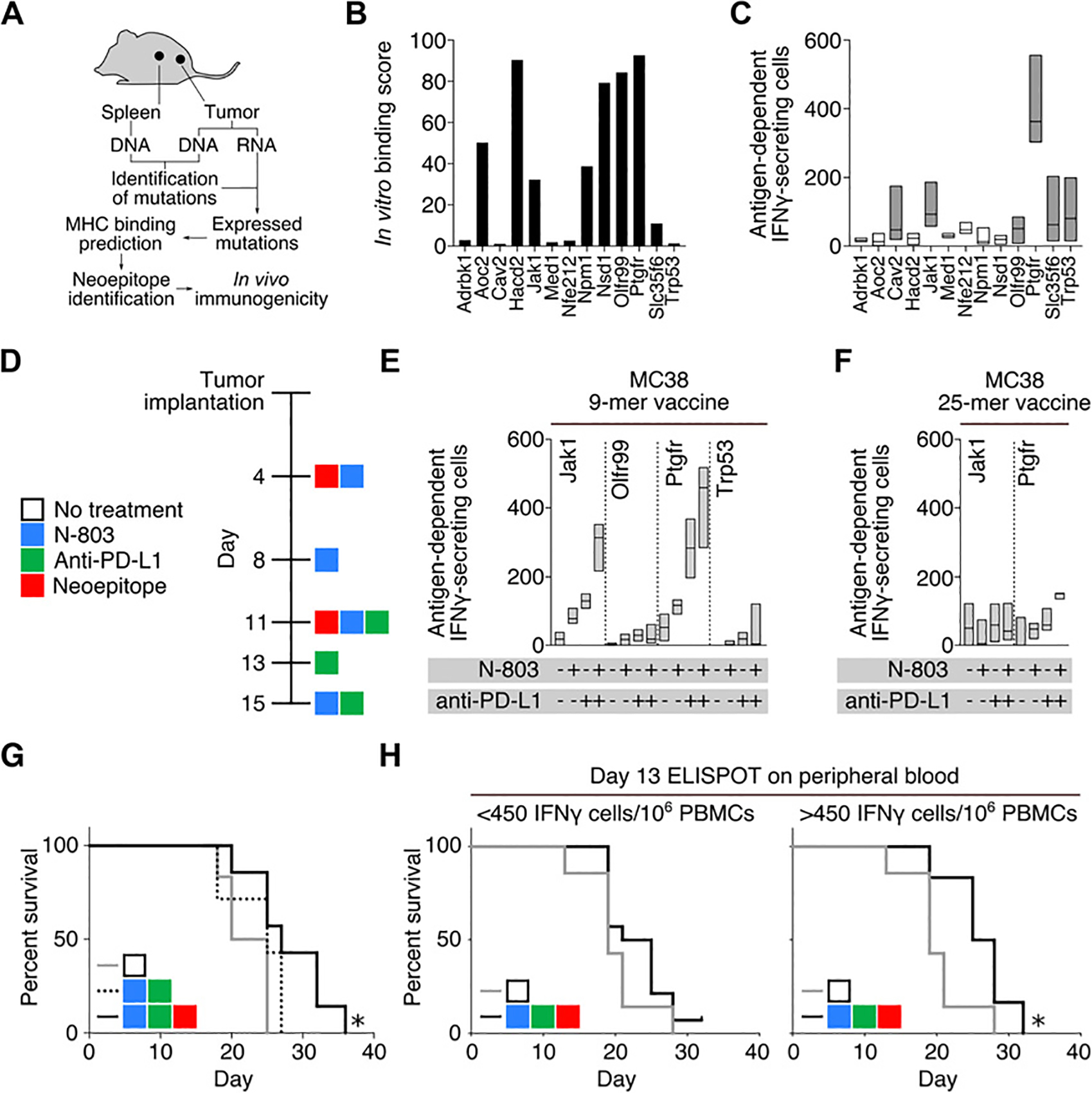
Identification of immunogenic neoepitopes. A, Workflow for neoepitope discovery in subcutaneously implanted MC38 tumors. B, In vitro binding score of MC38 neoepitopes. C, IFNγ ELISPOT analysis of naïve mice vaccinated twice with pools of 9-mer neoepitopes. Splenocytes were harvested 1 week following the second vaccination. Shaded bars represent peptides that induced robust immune responses in repeated experiments (n = 4). D, Treatment schedule. Day 18 IFNγ ELISPOT analysis of mice vaccinated with four 9-mer (n = 3; E) or two 25-mer neoepitopes (n = 3; F), alone or in combination with N-803 and/or anti–PD-L1. G, Survival curves of tumor-bearing mice treated with N-803, anti–PD-L1, and four 9-mer neoepitope peptides (black line), N-803, anti–PD-L1, and PBS (dashed line), or no treatment (gray line) (n = 6–7). H, Survival curves of mice treated with 9-mer neoepitope vaccine, N-803, and anti–PD-L1 stratified by antigen-specific IFNγ-secreting cells per 106 cells in the peripheral blood on day 13, *, P < 0.05, (n = 7–9). Data are representative of 1 to 2 independent experiments.
Table 1.
Characteristics of neoepitopes identified in two MC38 tumors
| Tumor 1 | Tumor 2 | |
|---|---|---|
| Number of nonsynonymous mutations | 16,828 | 8,676 |
| Number of expressed nonsynonymous mutations | 7,098 (42.2%) | 3,548 (40.9%) |
| Number of nonsynonymous mutations predicted to bind MHC | 124 (0.7%) | 66 (0.8%) |
| Number of shared neoepitopes | 51 (0.3%–0.5%) | |
When administered as a single agent, the neoepitope vaccine induced a low level of immunity, but failed to provide any survival benefit as compared with control animals (Supplementary Fig. S1A). In an effort to enhance the effectiveness of the neoepitope vaccine, we combined it with the IL15 superagonist fusion protein N-803 and PD-L1 MAb. Using the treatment regimen outlined in Fig. 1D, N-803 and anti–PD-L1 synergized to increase the immunogenicity of the 9-mer, but not 25-mer neoepitope vaccines in tumor-bearing animals (Fig. 1E and F). This enhancement in immunogenicity seen when combining our 9-mer neoepitope vaccine with N-803 and anti–PD-L1 correlated with only a slight, but significant increase in the survival of treated as compared with control animals (Fig. 1G). This survival benefit was seen only in animals mounting a robust immune response against vaccine components, as assessed by performing an ELISPOT using peripheral blood collected from animals on day 13 of tumor growth (Fig. 1H).
Neoepitope vaccination combined with immunomodulators promotes tumor regression
Using the treatment regimen depicted in Fig. 2A, we assayed the immune response in vaccinated animals on days 11, 18, and 25 of tumor growth, and observed that the magnitude of the immunity generated against vaccine components decreased over time. We observed that the neoepitope vaccine also resulted in the in situ expansion of T cells reactive against neoepitopes not contained in the vaccine. However, the magnitude of these de novo immune responses also diminished over time despite continued vaccination (Fig. 2B). In an effort to promote the maintenance of neoepitope-reactive cells within the tumor microenvironment, we incorporated a single injection of NHS-IL12 to the vaccine regimen on day 18 of tumor growth. Animals treated with the combination of anti–PD-L1, NHS-IL12, N-803, and neoepitope vaccine were able to maintain a robust immune response against neoepitopes Ptgfr and Trp53, both of which are components of the neoepitope vaccine, along with additional neoepitopes expressed by MC38 tumors, but not included in the vaccine (Fig. 2B). Treatment of mice with N-803, anti–PD-L1, and NHS-IL12, but without vaccine, promoted only the expansion of T cells specific for a peptide (P15e) derived from GP70, an endogenous retrovirus protein expressed by MC38 cells, but not to any of the neoepitopes assayed (Fig. 2B).
Figure 2.
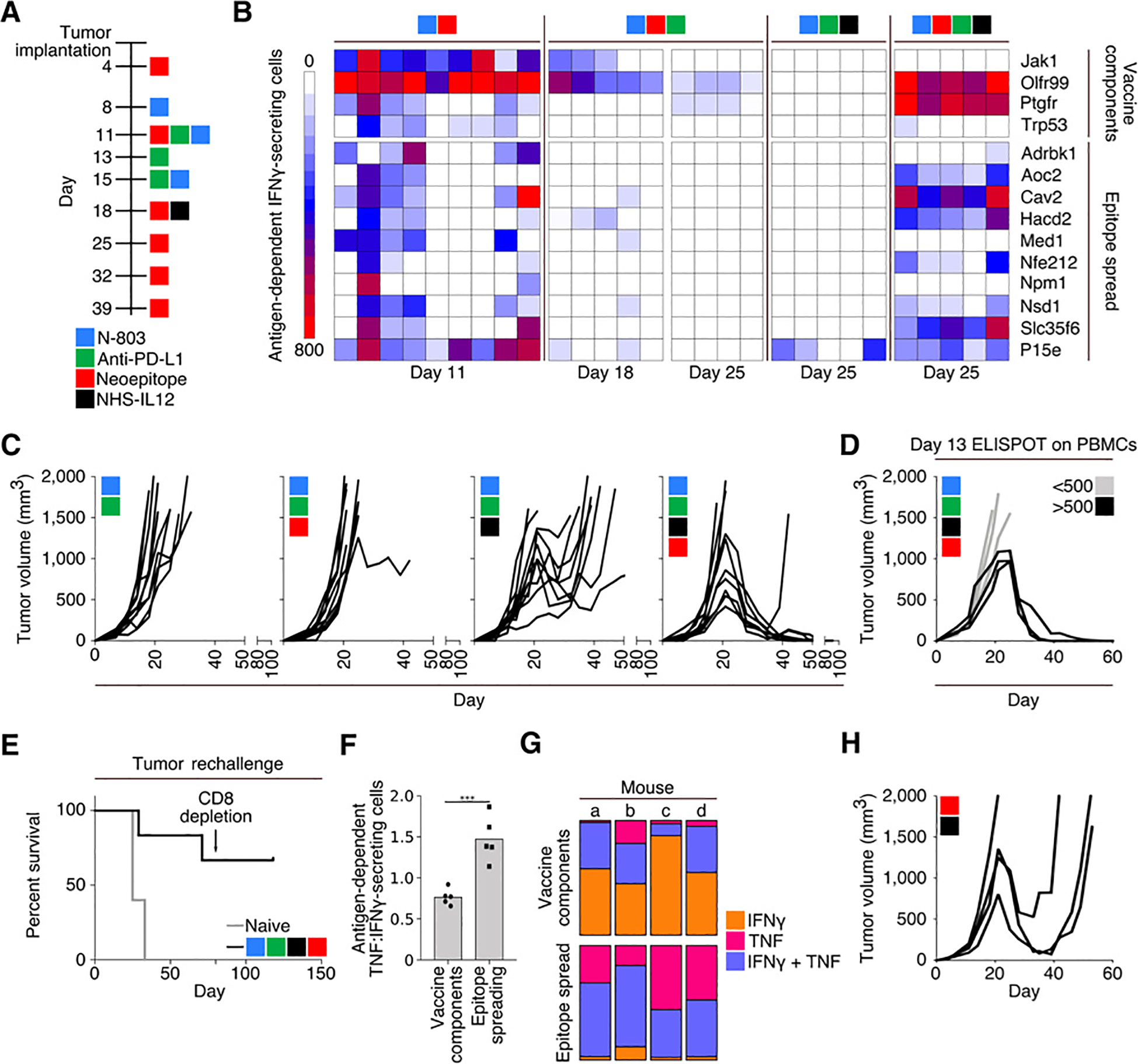
Combination therapy using a 9-mer neoepitope vaccine, N-803, anti–PD-L1, and NHS-IL12. A, Treatment schedule. B, IFNγ ELISPOT analysis on days 11, 18, and 25 of tumor growth against peptides contained within the vaccine (top) or MC38 neoepitopes not contained within the vaccine or P15e (bottom). Each column represents 1 mouse (n = 4–9). C, Tumor growth curves (n = 10). D, Tumor growth in mice treated with 9-mer neoepitope vaccine, N-803, anti–PD-L1, and NHS-IL12 stratified by antigen-specific IFNγ-secreting cells per 106 cells in the peripheral blood on day 13 (n = 6). E, Survival curves after rechallenge of naïve animals or those with a previously regressed MC38 tumor following indicated treatment. Rechallenged animals were implanted with MC38 tumors on day 0 of survival curve and received no subsequent therapies. Arrow indicates depletion of CD8+ cells (n = 6). F and G, Analysis of splenocytes from mice treated with 9-mer neoepitope vaccine, N-803, anti–PD-L1, and NHS-IL12, harvested on day 25 and stimulated overnight with either vaccine components or cascade antigens. F, ELISPOT analysis of the ratio of antigen-dependent TNFα:IFNγ-secreting splenocytes (n = 5). G, Flow cytometric analysis of percent of CD8+ cells that were single IFNγ (orange), single TNFα (pink), or double (purple) producers (n = 4). H, Tumor growth in mice treated with 9-mer neoepitope vaccine and NHS-IL12 according to the schedule in A (n = 4). Data are representative of 1 to 4 independent experiments.
In the presence or absence of vaccination, animals treated with N-803 and anti–PD-L1 showed no evidence of tumor control (Fig. 2C); in addition, animals treated with the combination of N-803, anti–PD-L1, and NHS-IL12, and no vaccine, had transient tumor control (Fig. 2C), with the median overall survival increasing from 21 days in nontreated animals to 39 days in animals treated with the triple combination. The addition of a neoepitope vaccine to this combination, however, resulted in regression of 6 of 10 MC38 tumors (Fig. 2C, right). There was also a correlation as to which animals responded to this treatment via assessing the magnitude of the immune response generated against vaccine components using peripheral blood collected from animals on day 13 of tumor growth, prior to the administration of NHS-IL12 (Fig. 2D). Animals whose tumors resolved following treatment remained tumor-free after the cessation of vaccination on day 39, and 4 of 6 animals subsequently resisted tumor rechallenge with MC38 on day 76. These rechallenged animals likely were able to completely clear any residual MC38 cells, as we did not observe tumor outgrowth, even after depleting animals of CD8+ T cells 81 days after the second tumor implantation (Fig. 2E). We observed a higher proportion of T cells producing TNFα in response to cascade neoepitopes as compared with those contained within the vaccine, as assessed by ELISPOT (Fig. 2F). Similarly, flow cytometric analysis demonstrated a larger predominance of polyfunctional T cells producing antigen-dependent IFNγ and TNFα in T cells reactive against cascade neoepitopes as compared with those contained within the vaccine (Fig. 2G). Animals treated with vaccine and NHS-IL12, using the same treatment timeline outlined in Fig. 2A, were able to mediate some degree of tumor regression in the absence of N-803 and anti–PD-L1 treatment; however, all of the treated animals eventually succumbed to progressive tumor growth (Fig. 2H).
The regression of tumors in the combinatorial treatment group was associated with limited toxicity (Supplementary Table S3). All tissues examined were histologically normal with the exception of focal areas of glandular epithelial necrosis without inflammation in the duodenum, and hypercellularity in the small bowel of a treated animal. There was also a transient, slight increase in serum liver enzyme levels in treated mice that was not associated with any liver pathology (Supplementary Table S3).
Multiepitope vaccines are required to promote tumor regression
To ascertain whether a vaccine consisting of a single neoepitope was capable of mediating tumor regression, tumor-bearing animals were treated with either a single 9-mer or a pool of all four neoepitope peptides in combination with N-803, anti–PD-L1, and NHS-IL12. The pool of neoepitopes was more efficient at inducing the regression of MC38 tumors than any of the single-peptide vaccinations (Fig. 3A). To determine the cell populations mediating effective antitumor immunity, animals were depleted of NK1.1+, CD4+, or CD8+ T cells either beginning on day 1 of tumor growth (early depletion) or day 15 of tumor growth (late depletion). Animals depleted of NK1.1+ cells early during tumor growth were able to resolve MC38 tumors with kinetics similar to those seen in nondepleted animals. Early depletion of CD4+ T cells in treated animals was associated with a more rapid regression of tumors as compared with nondepleted animals. Late depletion of NK1.1+ cells was associated with only 2 of 10 animals controlling tumor growth. Late depletion of CD4+ cells was associated with a rate of tumor resolution similar to that seen in nondepleted treated animals. As expected, both early and late depletion of CD8+ cells were associated with a lack of response to treatment, resulting in progressive tumor growth (Fig. 3B; Supplementary Fig. S1B).
Figure 3.
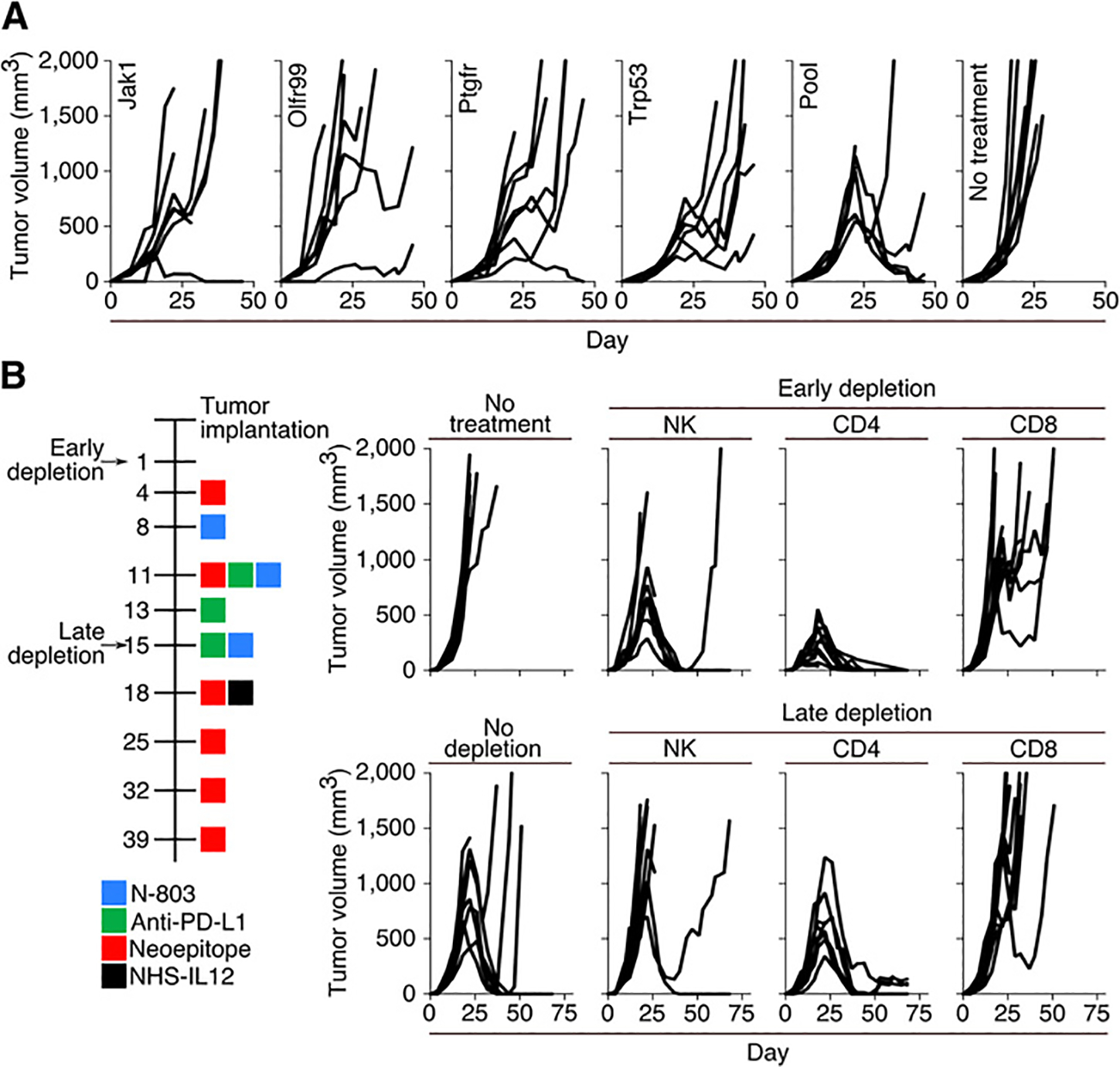
Tumor progression in mice treated with combination therapy using single-peptide vaccines or immune cell depletions. A, Tumor growth in mice treated with N-803, anti–PD-L1, NHS-IL12, and a neoepitope vaccine consisting of a single 9-mer neoepitope or a pool of four 9-mer neoepitopes. Mice were treated according to the schedule in Fig. 2A (n = 6–7). B, Timeline (left) and tumor growth (right) of depletion studies. Tumor-bearing mice were depleted of NK, CD4+ T cells, or CD8+ T cells starting 3 days prior to the first vaccine (early depletion) or NHS-IL12 administration (late depletion) (n = 7–10).
Tumor regression associates with increased CD8+ T-cell infiltration of tumors
Using the same treatment regimen depicted in Fig. 2A, we assessed which components of the immune system were modulated by each of the different agents and their combinations within the tumor microenvironment. The quadruple treatment regimen was shown to maximally enhance the infiltration of CD8+ T cells into the tumor (Fig. 4A and B). The addition of NHS-IL12 to the treatment regimen had a profound impact on both the innate and adaptive immune cells within the tumor microenvironment. It promoted the expansion of M1 macrophages with a coordinate contraction of M2 macrophages (Fig. 4C); also observed was a trend in the reduction of effector and central memory CD8+ T cells, and an expansion of CD8+ T effector memory cells (Fig. 4D). Treatment with vaccine and NHS-IL12 in combination with either N-803 or anti–PD-L1 was able to induce regression in 60% of MC38 tumors, as compared with 80% observed with the quad-therapy (Supplementary Fig. S2A). Protective immunologic memory was generated more efficiently in animals treated with either the quad-therapy or the combination of vaccine, anti–PD-L1, and NHS-IL12; all animals whose tumors regressed following treatment were able to resist a subsequent tumor rechallenge. In comparison, in animals treated with N-803, vaccine, and NHS-IL12, we observed that only 66% of animals with complete tumor regression were able to resist tumor rechallenge (Supplementary Fig. S2B).
Figure 4.
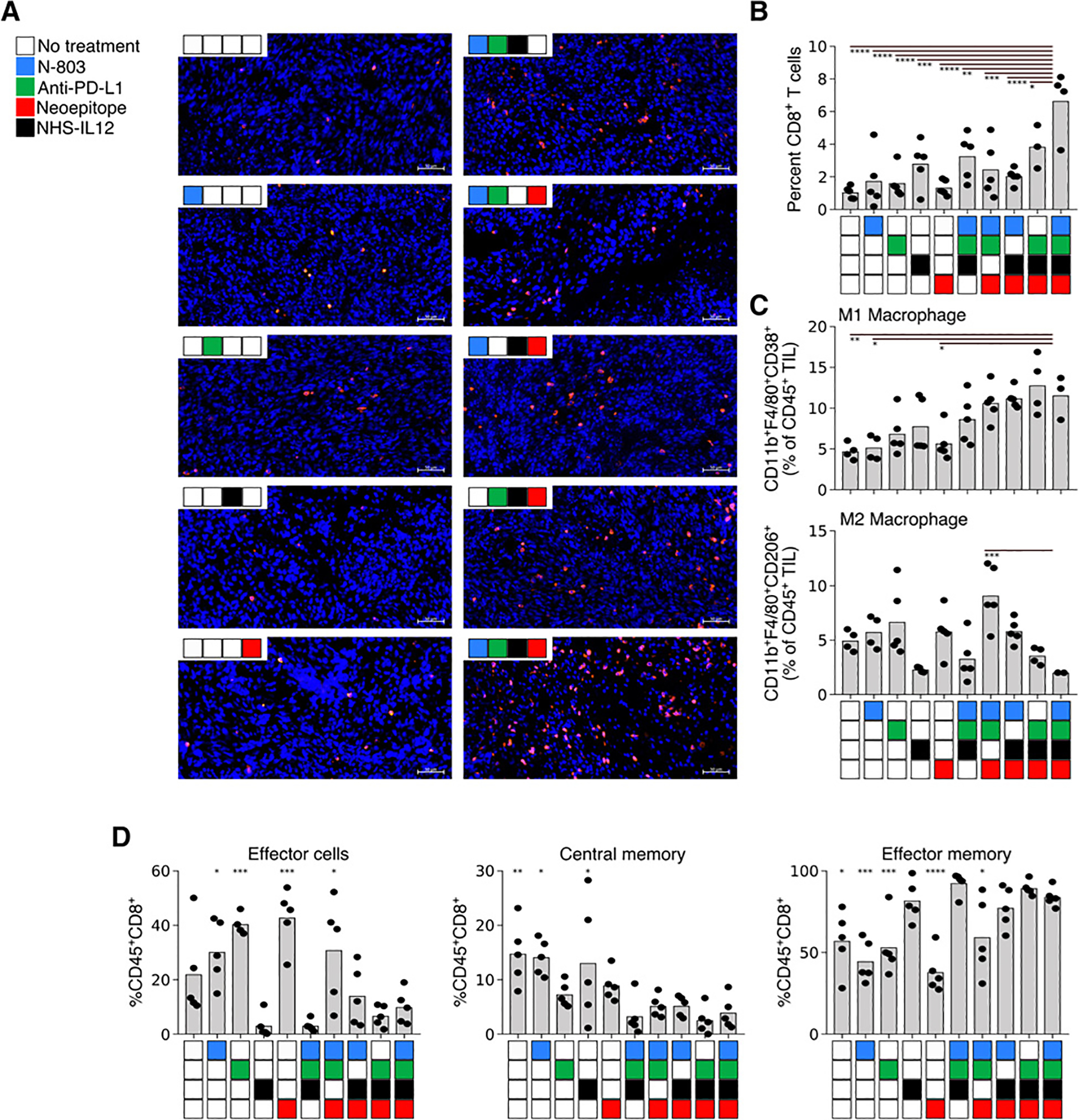
Tumor progression and tumor-infiltrating immune cells in mice treated with single, triple, or quadruple combinations of N-803, anti–PD-L1, NHS-IL12, and 9-mer neoepitope vaccine. Mice were treated as described previously, and tumors were harvested on day 22 posttumor implantation. Tumors were analyzed via immunofluorescent analysis (A and B) or flow cytometry (C and D). A, Representative immunofluorescent images of CD8+ (red) cells in zinc formalin-fixed paraffin-embedded tumor sections. Blue corresponds to DAPI staining. Scale bar, 50 μm (n = 4–5). B, Percent CD8+ T cells (of total DAPI+ cells) in immunofluorescent sections (n = 4–5). C, Intratumoral M1 macrophages (top, CD11b+/F4/80+/CD38+) and M2 macrophages (CD11b+/F4/80+/CD206+) (n = 3–5). D, CD8+ TIL maturation (CD44/CD62L/CD127). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (n = 5). Data are representative of 1 to 2 independent experiments.
To better assess the impact of the treatment regimen on immune cells infiltrating the tumor, we performed an analysis of gene expression in CD45+ cells isolated from day 25 tumors. Treatment of the combination of N-803, anti–PD-L1, and neoepitope vaccine had little observable impact on the expression of immune-related genes as compared with cells isolated from untreated tumors (Fig. 5A). The addition of NHS-IL12 to the N-803 and anti–PD-L1 treatment regimen correlated with an increased expression of a large number of genes primarily related to enhancement of the innate immune system. The addition of a neoepitope vaccine to this treatment, which was required to induce maximum tumor clearance, was associated with a greater than 5-fold increase in transcripts relating to T-cell activation and effector functions (Fig. 5A).
Figure 5.
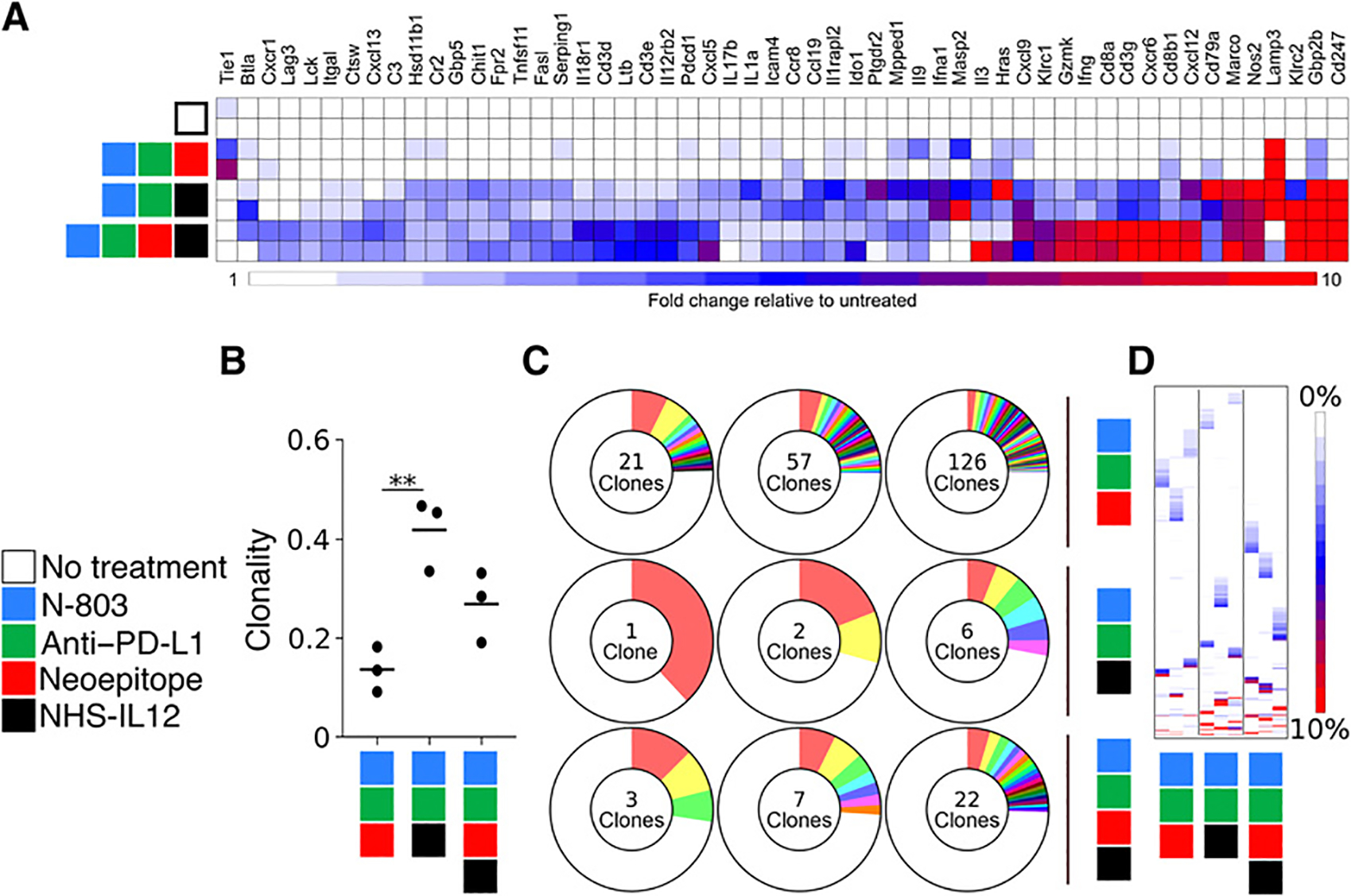
Gene expression and clonality of tumor-infiltrating immune cells after treatment with a 9-mer neoepitope vaccine, N-803, anti–PD-L1, and NHS-IL12. A, Gene expression analysis of tumor-infiltrating leukocytes (n = 2). B, Clonality of TCRβ chains detected in tumor infiltrates **, P < 0.001 (n = 3). C, Number of TCRβ clones that comprise the top 25% of detected sequences (n = 3). D, Frequency of the top 100 TCRβ sequences detected in each sample. Each column represents 1 mouse, and each row represents a unique clone (n = 3). Data are representative of 1 to 2 independent experiments.
To assess the impact of the treatment on the diversity of the immune repertoire of cells infiltrating the tumor, we sequenced the beta chain of the T-cell receptor (TCRβ) of CD4+ and CD8+ T cells isolated from day 25 tumors following treatment with indicated therapies. The use of NHS-IL12, N-803, and anti–PD-L1 in the treatment regimen resulted in a 3-fold increase in clonality of T cells within the tumor, which is modestly decreased 2-fold upon incorporation of the neoepitope vaccine (Fig. 5B). To gain a better sense of the clonality of the T-cell infiltrates within each tumor, we examined the number of clones required to make up the top 25% of the productive clones. In mice treated with the neoepitope vaccine, N-803, and anti–PD-L1, the top 25% of productive rearrangements was composed of 21, 57, and 126 clones. The number decreased to 1, 2, and 6 in animals treated with N-803, anti–PD-L1, and NHS-IL12. With the addition of the neoepitope vaccine, which is required for tumor regression, the numbers of clones were 3, 7, and 22. These results indicate that NHS-IL12 drove the expansion of a limited number of clones, whereas the neoepitope vaccine broadened the repertoire, which was associated with tumor clearance (Fig. 5C). An analysis of the top 100 TCRβ sequences detected in each sample revealed that each animal, regardless of treatment, had a unique T-cell repertoire (Fig. 5D).
Multiepitope adenoviral vectors are required to promote tumor regression
Recombinant adenoviral vectors were produced that encoded either single neoepitopes or four neoepitopes in a single viral vector (Fig. 6A). Following the treatment schedule outlined in Fig. 6B, tumor-bearing animals were treated using an admix of four vectors with each single neoepitope vector administered at spatially separated injection sites, or a single viral vector encoding four neoepitopes (multiepitope). Both the admix and multiepitope vectors resulted in comparable immunity generated against the four neoepitopes comprising the vaccine (Fig. 6C); however, vaccination with the multiepitope vector was more efficient at promoting the in situ spread of immunity to neoepitopes expressed by the tumor, but not incorporated into the vector (Fig. 6D). We hypothesized that one could facilitate epitope spreading induced by the admixed vaccine by mixing the four preparations of viral particles prior to injection. However, as depicted in Supplementary Fig. S3, even when all four admixed neoepitopes vaccines are administered together, and presumably activating T cells within the same draining lymph nodes, the admixed vaccine continues to be inefficient at mediating epitope spreading as compared with the multiepitope vaccine. This increased epitope spreading observed with the administration of the multiepitope vaccine was associated with a more efficient tumor resolution in animals vaccinated with the multiepitope vector, as compared with the admix of single neoepitope vectors (Fig. 6E). This protective antitumor immune response correlated with increased tumor infiltration of CD8+ T cells, along with a higher CD8+ T cell: CD4+ T-cell ratio (Fig. 6F and G). We did not observe any differences in the presence of tumor-infiltrating T-regulatory cells among the two experimental groups (Fig. 6H). Animals whose tumors resolved after treatment remained tumor-free even after the cessation of vaccination on day 39, and all animals (4/4) subsequently resisted tumor rechallenge.
Figure 6.
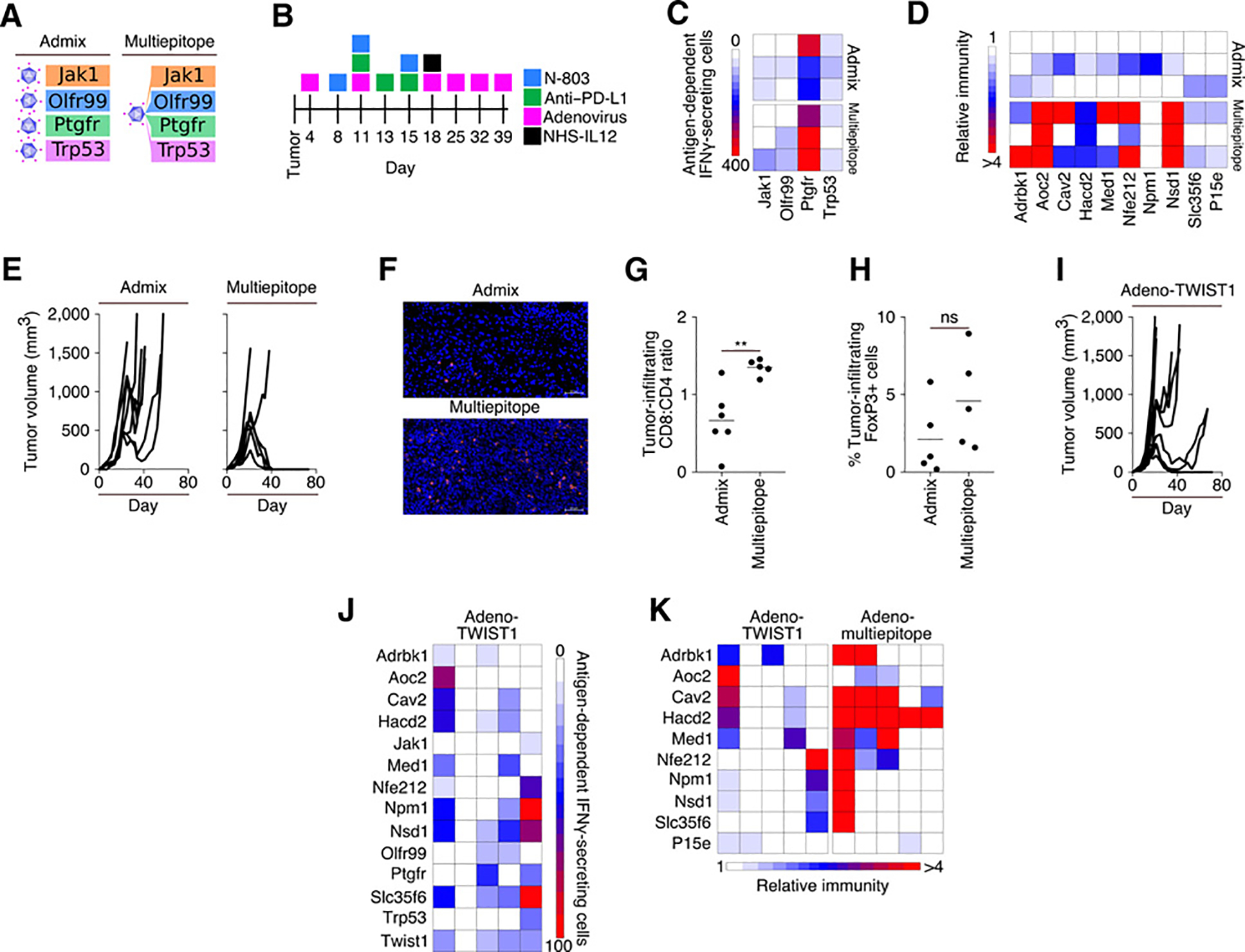
Combination therapy utilizing adenoviral vectors. A, Visual representation of admixed and multiepitope adenoviral vaccines targeting neoepitopes. B, Treatment schedule. C, IFNγ ELISPOT analysis on day 25 of tumor growth against neoepitopes contained within the vaccine. Each row represents 1 mouse (n = 3). D, Immune responses generated in animals vaccinated with multiepitope adenovirus relative to paired neoepitopes from animals treated with admixed adenovirus. Each row represents 1 mouse (n = 3). E, Tumor growth curves (n = 7). F, Representative immunofluorescent images of CD8+ (red) cells in zinc formalin-fixed, paraffin-embedded tumor sections. Blue corresponds to DAPI staining. Scale bar, 50 μm (n = 5–6). G, Ratio of CD8+:CD4+ TIL in immunofluorescent sections (n = 5–6). H, % FoxP3+ cells within immunofluorescent sections (n = 5). I, Tumor growth in mice treated as indicated in B utilizing an adenovirus targeting TWIST1 (n = 10). J, IFNγ ELISPOT analysis on day 25 of tumor growth against neoepitopes identified in the MC38 cell line. Each column represents 1 mouse (n = 5). K, Relative immunity of the immune responses generated in animals against neoepitopes in the adeno multiepitope vaccinated animals as compared with animals vaccinated with adeno-TWIST1 vaccine. Each column represents 1 mouse (n = 5). Data are representative of 1 to 2 independent experiments.
We sought to determine whether the degree of epitope spreading detected on day 25 of tumor growth associated with a change in the rate of tumor growth in animals treated with the combination of neoepitope vaccine, N-803, anti–PD-L1, and NHS-IL12. As shown in Supplementary Fig. S4, we observed a positive association between a decreased rate of tumor growth and the generation of epitope spreading to numerous neoepitopes. There was no correlation of a decreased rate of tumor growth with the magnitude of immunity generated against the vaccine components or the P15e peptide.
Neoepitope, not tumor-associated antigen, vaccines are most efficient at tumor regression induction
Utilizing the same treatment schedule and regimen including N-803, anti–PD-L1, and NHS-IL12 outlined in Fig. 6B, mice were vaccinated with an adenoviral vector encoding the “self” tumor-associated antigen TWIST1, which is expressed by the MC38 cell line (32), and has been used in prior anticancer vaccine studies (33, 34). Tumor regression was observed in only 2 of 10 animals treated with a regimen incorporating the TWIST1-targeted vaccine (Fig. 6I). TWIST1 vaccination was associated with the expansion of T cells reactive against tumor neoepitopes (Fig. 6J); however, the neoepitope vaccine is more efficient at mediating epitope spreading than a vaccine targeting the tumor-associated antigen (Fig. 6K).
Discussion
The barriers to the development of effective antitumor immunity range from a hostile tumor microenvironment to quantitative/qualitative defects in the response to tumor antigens. Neoepitope vaccines, when used as a monotherapy, offer a promising avenue to overcome quantitative defects in the antitumor immune response; however, they may not offer clinical benefit when other mechanisms of immune failure are also at play. Combining neoepitope vaccines with immune-oncology agents targeting other mediators of immune failure should improve their efficacy. In the MC38 colon carcinoma model, we observed that robust immunity generated against neoepitopes was not sufficient, requiring the addition of N-803, NHS-IL12, and anti–PD-L1 to mediate tumor regression. This highlights the benefit of multifaceted immunotherapeutic treatment regimens for the treatment of cancer.
The majority of neoepitopes are passenger mutations for which expression varies within primary and metastatic lesions (19–25). This is especially relevant in cases where tumors like the MC38 cell line are microsatellite unstable (31), as shown by the variable number of mutations detected in the two MC38 tumors analyzed that originated from the same culture flask. Thus, in situ diversification of the immune response following the administration of a neoepitope-targeted vaccine is important. A diversified immune response can better target the breadth of neoepitopes expressed by the tumor and reduce the potential development of resistant, antigen-loss variants. Early assessment of immunity demonstrated an association between immunity generated against vaccine components and increased survival in treated animals, suggesting that epitope spreading occurs early following vaccination and correlates with the magnitude of the immune response generated to vaccine components. At later time points, however, the degree of epitope spreading observed on day 25 of tumor growth negatively correlated with tumor growth in animals treated with a neoepitope target vaccine in combination with N-803, anti–PD-L1, and NHS-IL12, suggesting that immunity generated by epitope spreading is responsible for the control of tumor growth. This slowing of the rate of tumor growth precedes the administration of NHS-IL12 and demonstrates that targeting IL12 to the tumor microenvironment increases the magnitude of preexisting immunity to levels capable of rejecting the tumor.
T cells expanded during this process of epitope spreading were polyfunctional as compared with a primarily IFNγ response in those T cells specific for the vaccine components. Although previous studies suggest that neoepitope vaccines may promote epitope spreading (35, 36), this study provides direct experimental in vivo evidence and biologic consequence for this phenomenon.
In this study we also demonstrated that a vaccine targeting TWIST1, a tumor-associated antigen, could induce an in vivo diversification of the immune response to include tumor neoepitopes. Epitope spreading was observed only in animals that generated a measurable immune response against the vaccine target, TWIST1. However, a vaccine targeting cancer neoepitopes was more efficient at inducing epitope spreading as compared with a vaccine targeting a tumor-associated antigen. Our studies using a peptide-based neoepitope vaccine demonstrated that single neoepitopes are unable to mediate tumor regression when used in combination with N-803, anti–PD-L1, and NHS-IL12. It is thus possible that vaccination with more than one tumor-associated antigen may be able to mediate epitope spreading more efficiently and promote tumor regression at a greater efficiency.
In the absence of vaccination, we observed a significant survival benefit in animals treated with N-803, anti–PD-L1, and NHS-IL12; however, none of the animals cleared their tumors, and we did not observe the development of immunity to any of the neoepitopes studied in this report. We hypothesized that the lack of detectable immunity observed in animals not receiving a vaccine was due to the immunosuppressive tumor microenvironment inhibiting the effective expansion and diversification of the antitumor immune response.
Here, we also report the use of recombinant adenoviral vaccines targeting neoepitopes and demonstrate that such vectors induced immunity against encoded neoepitopes and, when used in combination with N-803, anti–PD-L1, and NHS-IL12, resulted in tumor regression at a rate similar to that observed when using a peptide-based neoepitope vaccine.
The multifaceted therapeutic strategy required to induce tumor clearance had a large impact on the genes expressed by tumor-infiltrating leukocytes. Treating animals with the combination of N-803, anti–PD-L1, and NHS-IL12 promoted an expansion of transcripts associated with activation of the innate immune system; with the addition of neoepitope vaccine, we observed a marked expansion of transcripts associated with activation/effector functions of the adaptive immune system. In addition to these changes in gene expression, treatment with a neoepitope-targeted therapeutic quartet resulted in distinct changes in the T-cell repertoire present within the tumor. NHS-IL12 appears to be the agent primarily responsible for the increased clonality of the immune response observed in treated animals; however, increased clonality was insufficient to promote tumor clearance in the absence of a neoepitope-targeted vaccine.
In an analysis of the abundance of TCR sequences within the tumor, virtually no sequences overlapped among any of the tumors assayed. We propose that neoepitope vaccination promotes tumor clearance by facilitating the expansion of T cells whose repertoire is capable of targeting the diverse, ever-changing panel of neoepitopes expressed by the tumor. Our observations support the premise that T cells generated via epitope spreading are those responsible for mediating tumor regression.
The work presented here demonstrates the utility of combining neoepitope vaccines with other immune-oncology agents in a murine model to enhance immune responses to these neoepitopes and facilitate tumor clearance. In the development of clinically effective therapies for so-called “immunologically cold tumors” that employ neoepitope vaccines, one should consider the results presented in this study: (i) the expansion alone of T cells against neoepitopes may not be sufficient to induce tumor regression; (ii) multifaceted immunotherapeutic regimens are necessary to improve response rates following administration of a neoepitope vaccine; (iii) vaccines targeting multiple neoepitopes, whether via a peptide- or vector-based treatment strategy, are more effective at mediating tumor regression than vaccines composed of a single peptide; and (iv) following administration of a vaccine targeting either a tumor-specific neoepitope or a tumor-associated antigen, we observed the spreading of immunity to neoepitopes expressed by the tumor. These observations may provide rationale in the design of clinical trials that aim to target cancer neoepitopes to improve the likelihood of a promising clinical outcome.
Supplementary Material
Acknowledgments
The authors thank Laura Minang and Kristen McCampbell for their technical assistance and Debra Weingarten for her editorial assistance in the preparation of this manuscript. This study was supported by the Intramural Research Program of the Center for Cancer Research, NCI, NIH, and through Cooperative Research and Development Agreements (CRADAs) between NantBioScience and the NCI, NIH; EMD Serono and the NCI, NIH; and Syndax and the NCI, NIH.
Footnotes
Disclosure of Potential Conflicts of Interest
S.C. Benz is the president, genomics at NantCell and has ownership interest (including stocks and patents) in NantOmics, LLC and NantHealth, Inc. A. Nguyen is the director computational immunology at NantCell and is a bioinformatic scientist at NantOmics and has ownership interest (including stocks and patents) in NantCell and NantOmics. J.Z. Sanborn is an executive VP at NantCell and has ownership interest (including stocks and patents) in NantOmics and NantHealth. P. Ordentlich is the CSO and has ownership interest (including stocks and patents) in Syndax Pharmaceuticals. S. Rabizadeh is the CSO at NantBio & ImmunityBio and NantOmics. P. Soon-Shiong is the chairman and CEO and has ownership interest (including stocks and patents) at NantHealth, NantKwest, ImmunityBio, NantWorks, LLC & Affiliates. K. Niazi is the CTO at NantBio & ImmunityBio and has ownership interest (including stocks and patents) in NantBio & ImmunityBio and Agenus. No potential conflicts of interest were disclosed by the other authors.
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
References
- 1.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 2006;6:295. [DOI] [PubMed] [Google Scholar]
- 2.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013;138:105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014;515:572–6. [DOI] [PubMed] [Google Scholar]
- 5.Juneja VR, McGuire KA,Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med 2017;214: 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 2012;72: 917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aucouturier J, Dupuis L, Deville S, Ascarateil S, Ganne V. Montanide ISA 720 and 51: a new generation of water in oil emulsions as adjuvants for human vaccines. Expert Rev Vaccines 2002;1:111–8. [DOI] [PubMed] [Google Scholar]
- 8.Cox JC, Coulter AR.Adjuvants–a classification and review of their modes of action. Vaccine 1997;15:248–56. [DOI] [PubMed] [Google Scholar]
- 9.Wilson-Welder JH, Torres MP, Kipper MJ, Mallapragada SK, Wannemuehler MJ, Narasimhan B. Vaccine adjuvants: current challenges and future approaches. J Pharm Sci 2009;98:1278–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han KP, Zhu X, Liu B, Jeng E, Kong L, Yovandich JL, et al. IL-15:IL-15 receptor alpha superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 2011;56:804–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu X, Marcus WD, Xu W, Lee HI, Han K, Egan JO, et al. Novel human interleukin-15 agonists. J Immunol 2009;183:3598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim PS, Kwilas AR, Xu W, Alter S, Jeng EK, Wong HC, et al. IL-15 superagonist/IL-15RalphaSushi-Fc fusion complex (IL-15SA/IL-15RalphaSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget 2016;7:16130–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, et al. First-in-human Phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 2018;131: 2515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu W, Jones M, Liu B, Zhu X, Johnson CB, Edwards AC, et al. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: interleukin-15 receptor alphaSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res 2013;73:3075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A, et al. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med 2002;195:1541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schluns KS, Klonowski KD, Lefrancois L. Transregulation of memory CD8 T-cell proliferation by IL-15Ralpha+ bone marrow-derived cells. Blood 2004;103:988–94. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity 1998;8:591–9. [DOI] [PubMed] [Google Scholar]
- 18.Fallon J, Tighe R, Kradjian G, Guzman W, Bernhardt A, Neuteboom B, et al. The immunocytokine NHS-IL12 as a potential cancer therapeutic. Oncotarget 2014;5:1869–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mansfield AS, Ren H, Sutor S, Sarangi V, Nair A, Davila J, et al. Contraction of T cell richness in lung cancer brain metastases. Sci Rep 2018;8:2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010;464:999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A 2008;105:4283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009;461:809–13. [DOI] [PubMed] [Google Scholar]
- 24.Tao Y, Ruan J, Yeh SH, Lu X, Wang Y, Zhai W, et al. Rapid growth of a hepatocellular carcinoma and the driving mutations revealed by cell-population genetic analysis of whole-genome data. Proc Natl Acad Sci U S A 2011;108:12042–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010; 467:1114–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanborn JZ, Chung J, Purdom E, Wang NJ, Kakavand H, Wilmott JS, et al. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc Natl Acad Sci U S A 2015;112:10995–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res 2008;36:W509–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class IMHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics 2008;24:1397–8. [DOI] [PubMed] [Google Scholar]
- 29.Knudson KM, Hicks KC, Luo X, Chen JQ, Schlom J, Gameiro SR. M7824, a novel bifunctional anti-PD-L1/TGFβ Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology 2018;7:e1426519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rabizadeh S, Garner C, Sanborn JZ, Benz SC, Reddy S, Soon-Shiong P. Comprehensive genomic transcriptomic tumor-normal gene panel analysis for enhanced precision in patients with lung cancer. Oncotarget 2018; 9:19223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H, et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat Commun 2018;9:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamilton DH, Litzinger MT, Fernando RI, Huang B, Palena C. Cancer vaccines targeting the epithelial-mesenchymal transition: tissue distribution of brachyury and other drivers of the mesenchymal-like phenotype of carcinomas. Semin Oncol 2012;39:358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ardiani A, Gameiro SR, Palena C, Hamilton DH, Kwilas A, King TH, et al. Vaccine-mediated immunotherapy directed against a transcription factor driving the metastatic process. Cancer Res 2014;74:1945–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwilas AR, Ardiani A, Dirmeier U, Wottawah C, Schlom J, Hodge JW. A poxviral-based cancer vaccine the transcription factor twist inhibits primary tumor growth and metastases in a model of metastatic breast cancer and improves survival in a spontaneous prostate cancer model. Oncotarget 2015;6:28194–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015;348:803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014;14:135–46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.