

Abstract
Therapies targeting VEGF have proven only modestly effective for the treatment of proliferative sickle cell retinopathy (PSR), the leading cause of blindness in patients with sickle cell disease. Here, we shift our attention upstream from the genes that promote retinal neovascularization (NV) to the transcription factors that regulate their expression. We demonstrated increased expression of HIF-1α and HIF-2α in the ischemic inner retina of PSR eyes. Although both HIFs participated in promoting VEGF expression by hypoxic retinal Müller cells, HIF-1 alone was sufficient to promote retinal NV in mice, suggesting that therapies targeting only HIF-2 would not be adequate to prevent PSR. Nonetheless, administration of a HIF-2–specific inhibitor currently in clinical trials (PT2385) inhibited NV in the oxygen-induced retinopathy (OIR) mouse model. To unravel these discordant observations, we examined the expression of HIFs in OIR mice and demonstrated rapid but transient accumulation of HIF-1α but delayed and sustained accumulation of HIF-2α; simultaneous expression of HIF-1α and HIF-2α was not observed. Staggered HIF expression was corroborated in hypoxic adult mouse retinal explants but not in human retinal organoids, suggesting that this phenomenon may be unique to mice. Using pharmacological inhibition or an in vivo nanoparticle-mediated RNAi approach, we demonstrated that inhibiting either HIF was effective for preventing NV in OIR mice. Collectively, these results explain why inhibition of either HIF-1α or HIF-2α is equally effective for preventing retinal NV in mice but suggest that therapies targeting both HIFs will be necessary to prevent NV in patients with PSR.
Keywords: Angiogenesis, Ophthalmology
Keywords: Hypoxia, Mouse models, Retinopathy
Introduction
Several multicenter randomized controlled clinical trials have demonstrated the benefit of monthly (or bimonthly) injections with biological molecules directed against the vasoactive mediator VEGF to treat macular edema in patients with ischemic retinopathies (IRs). However, less than 50% of treated patients demonstrate a major improvement in vision (i.e., a gain of at least 15 letters on the ETDRS vision chart) despite monthly treatment (1). This may be due, in part, to the contribution of nonischemic factors (e.g., hyperglycemia, advanced glycation end products, oxidative stress, inflammation) to the development of macular edema in IRs. Subsequent studies have demonstrated a significant reduction in the progression to neovascularization (NV) in some — but not all — patients with IR receiving anti-VEGF therapy (2–4). Collectively, these clinical studies suggest that other vasoactive mediator(s) may participate in the development of macular edema and pathological angiogenesis in patients with IR.
Emerging preclinical and post hoc clinical data have also raised concerns regarding the consequences of chronic VEGF inhibition (5). VEGF produced by the retinal pigment epithelium is essential in maintaining the health of the underlying choriocapillaris, the vascular bed that supplies the metabolically active retinal photoreceptors (6–9). It is also postulated that VEGF may play a direct role as a neurotrophic factor for the neurosensory retina (10) and that chronic inhibition of VEGF may impair its normal physiological roles in the eye (11, 12). In a post hoc analysis of patients with age-related macular degeneration, treatment with anti-VEGF therapy was associated with the development of retinal atrophy in a subset of patients (13). Recent data raise the additional concern that anti-VEGF therapy may also contribute to the development of sustained elevation of intraocular pressure (and glaucoma) in vulnerable patients (14). Collectively, these observations emphasize the importance of ongoing efforts to identify additional therapeutic targets for the safe and effective treatment of IRs.
In this regard, a family of transcriptional activators, HIFs, has emerged as a master regulator of the expression of hypoxia-regulated angiogenic stimulators (15). HIFs are heterodimeric proteins composed of an exquisitely oxygen-sensitive α subunit and a ubiquitously expressed β subunit (16). HIF-1α was the first HIF α subunit isoform to be identified (17), and its role has been extensively studied in retinal vascular disease (18). Two other isoforms, HIF-2α and HIF-3α, have since been reported; the amino acid sequence of HIF-2α is closely related to that of HIF-1α, and HIF-2α also activates hypoxia-inducible gene transcription, whereas HIF-3α is more distantly related and is expressed as alternative splice isoforms that can either activate or repress HIF target genes (19).
The stability and transcriptional activity of HIF-1α and HIF-2α are regulated by O2-dependent hydroxylation. Under standard tissue culture conditions (95% air/5% CO2 = 20% O2), proline residues 402 and 564 of the human HIF-1α subunit (residues 405 and 531 of the human HIF-2α subunit), are hydroxylated by a family of HIF prolyl hydroxylases (20). Hydroxylated HIF-1α and HIF-2α bind to the von Hippel-Lindau (VHL) tumor suppressor protein, which is the substrate recognition subunit of a protein-ubiquitin ligase that ubiquitinates HIF-1α and HIF-2α and targets them for degradation by the proteasome (21). An additional level of regulation is provided by an asparaginyl hydroxylase, factor inhibiting HIF-1 (FIH-1; refs. 22, 23). FIH-1 hydroxylates asparagine residue 803 of HIF-1α (851 of HIF-2α) and prevents binding of the transcriptional coactivator p300, thereby inhibiting HIF transcriptional activity.
Under hypoxic conditions (<5% O2), as occurs in the ischemic retina, the ability of prolyl hydroxylases and FIH-1 to hydroxylate HIF-1α or HIF-2α is impaired. In the absence of hydroxylation, VHL does not bind to either HIF-1α or HIF-2α to trigger their degradation, whereas p300 binds to both HIFs to enhance their transcriptional activity (24–26). The resulting active HIF-1α and HIF-2α proteins localize to the nucleus and heterodimerize with HIF-1β to form HIF-1 and HIF-2, respectively, which are capable of binding to the hypoxia response elements of specific hypoxia-inducible genes, thereby activating broad changes in gene expression that mediate acclimation of cells, tissues, and the organism to conditions of low oxygen tension (27). In ischemic retinal disease (including sickle cell retinopathy, diabetic eye disease, retinal vein occlusions, and retinopathy of prematurity), many of these gene products (e.g., VEGF) promote pathological angiogenesis (resulting in retinal NV). This has prompted speculation that inhibition of HIF accumulation in ischemic retina could broadly reduce the expression of multiple angiogenic gene products back to physiological (preischemic) levels but would not influence their basal (HIF-independent) expression, providing a potentially safer and more effective treatment of NV.
Although the role of HIF-1 and HIF-2 in IRs has been an area of intense investigation, the relative contribution of each of these transcription factors to disease pathogenesis is not well understood. Here, we focused on the role of hypoxia and the accumulation of HIF-1α and HIF-2α to the development of the classic IR, proliferative sickle cell retinopathy (PSR), the most common cause of severe vision loss in patients with sickle cell disease. Using PSR as a model, we set out to examine the relative contribution of HIF-1 versus HIF-2 to the development of retinal NV to investigate the potential of targeting HIF-1 and/or HIF-2 as a therapeutic approach for the treatment of patients with IR.
Results
Retinal ischemia is not sufficient to predict which patients with sickle cell disease develop retinal NV.
Patients with sickle cell disease who are homozygous for Hb S or who are heterozygous for Hb S and either Hb C or β-thalassemia are at increased risk for occlusions of the small peripheral retinal vessels resulting in localized anterior (peripheral) retinal nonperfusion, often in the setting of normal posterior retinal perfusion. These peripheral vascular occlusions cause tissue ischemia and the release of angiogenic mediators (28, 29) that promote the development of retinal NV, initiating PSR, a classic example of an IR. However, why some patients with sickle cell disease develop NV whereas others do not remains unclear.
To investigate whether the severity of peripheral retinal ischemia alone is predictive of whether NV develops in patients with PSR, we performed a retrospective study of patients with sickle cell disease who underwent ultra-wide-field fluorescein angiography (UWF FA) to characterize the relationship between retinal ischemia and the development of retinal NV. We examined UWF FA images from 55 eyes of 28 patients with sickle cell disease (Tables 1 and 2), including patients with sickle cell SS (32 eyes), SC (15 eyes), and Sβ-thalassemia (8 eyes; Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI139202DS1). Peripheral nonperfusion was determined by measuring the area of nonperfused (peripheral) retina observed in a single image (Supplemental Figure 1B). The retinal ischemic index was calculated by dividing the area of ischemic retina by the total retina area in the image (Supplemental Figure 1C). Two independent graders evaluated images from all 55 eyes, and the intraclass correlation between the 2 graders was 0.96 (Supplemental Figure 2). When the average ischemic index from the 2 graders was plotted, we observed an increase in the ischemic index in patients with sickle cell disease with PSR compared with patients without PSR (Figure 1A). This increase persisted when we examined patients with sickle cell disease with SC or SS (Figure 1B). The ischemic index of patients with Sβ-thalassemia was lower than that for patients with SS or SC, consistent with the observation that no patients with Sβ-thalassemia developed PSR.
Table 1. Characteristics of patients with sickle cell.

Table 2. Characteristics of patients with sickle cell included in retrospective study.
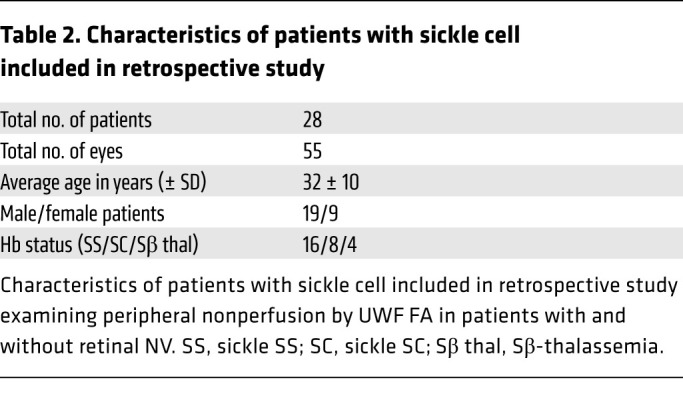
Figure 1. Peripheral ischemia in patients with sickle cell disease is not sufficient to promote the development of NV.

(A) Retinal ischemic index in all patients with sickle cell disease without PSR (no PSR) and with PSR (PSR). Blue and red dotted lines identify mean retinal ischemic index in patients with and without PSR, respectively. (B) Retinal ischemic index in patients with sickle cell disease divided by hemoglobin status (SS, SC, and Sβthal) without (–) and with (+) PSR. Blue arrows identify patients without PSR with a retinal ischemic index greater than the average for patients with PSR. Red arrows identify patients with PSR with retinal ischemic index less than the average for patients without PSR. The ischemic indices of patients without PSR and with PSR were compared using a 2-tailed Student’s t test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Interestingly, there were many patients with a high ischemic index (defined as an ischemic index greater than the mean for PSR eyes) who did not develop PSR (Figure 1, A and B, blue arrows), and patients with a relatively low ischemic index (defined as an ischemic index less than the mean for non-PSR eyes) who did develop PSR (Figure 1, A and B, red arrows). Moreover, despite the fact that less than one-third of eyes (17 of 55) developed PSR, 100% of all eyes from adult patients with sickle cell disease (55 of 55) had evidence of peripheral nonperfusion on UWF FA (Table 3). Collectively, these results demonstrated that the area of retinal ischemia alone was not predictive of the presence of PSR.
Table 3. Retinal ischemic index, PSR, and peripheral ischemia in sickle cell eyes.

Expression of HIF-1α and HIF-2α is increased in the inner retina underlying retinal NV in the eyes of patients with PSR.
The presence of peripheral ischemia in 100% of adult patients with sickle cell disease — whether or not they had PSR — suggests that an explanation for why regions of ischemic retina promote NV in patients with sickle cell disease will require closer examination of nonperfused retina in patients with PSR. To examine in more detail the histopathology of the peripheral ischemic retina of PSR eyes in areas with and without NV, we obtained paraffin-embedded autopsy eyes from 5 nondiabetic, sickle cell (SS) disease patients with untreated PSR (i.e., no prior history of scatter laser photocoagulation or intravitreal anti-VEGF therapy), and no history of another ischemic retinal disease (28). We then examined the peripheral ischemic retina in areas with and without overlying NV compared with posterior (perfused) retina (Figure 2A). We observed abundant superficial, intermediate, and deep (CD34-positive) vessels in posterior (perfused) retina in all 5 patients with sickle cell disease (Figure 2B). In peripheral (ischemic) retina, we observed few, intermittent, and scattered superficial and intermediate vessels in the absence of NV but a complete absence of inner retinal vessels underlying retinal NV (Figure 2B). IgG was used as a negative control (Figure 2C). Expression of 2 HIF-regulated angiogenic mediators, VEGF and ANGPTL4, were undetectable in posterior retina and modestly expressed in the inner nuclear layer (INL) — but not in the outer nuclear layer (ONL) — in ischemic retina in the absence of overlying NV (Figure 2, D and E). However, we observed robust expression of VEGF and ANGPTL4 in the INL and, to a lesser extent the ONL, in areas underlying retinal NV (Figure 2, D and E).
Figure 2. Expression of HIF-1α and HIF-2α and HIF-regulated angiogenic mediators in the ischemic retina of patients with sickle cell disease.

(A) Fluorescein angiogram from a patient with PSR demonstrating areas of normal perfusion (pink arrow) posteriorly (i) and nonperfusion (blue arrow) peripherally (ii and iii); the margin between perfused and nonperfused retina demarcated in red. Areas without (ii) and with (iii) retinal NV (red arrows) are seen adjacent to one another. (B) Representative images from immunohistochemical analysis for the endothelial cell marker CD34 in posterior perfused and peripheral ischemic retina, the latter without and with NV (red arrows). In the posterior retina, vessels are noted in the superficial, intermediate, and deep vascular plexuses (pink arrows). In the ischemic retina (without NV), rare vessels are noted in the superficial and intermediate vascular plexuses (pink arrows), but vessels are absent in the deep vascular plexus (blue arrows). In ischemic retina with overlying retinal NV (red arrows), there was a complete absence of retinal vessels in all vascular plexuses (blue arrows). (C) IgG was used as a negative control. (D–G) Expression of the HIF-regulated angiogenic mediators VEGF (D) and ANGPTL4 (E), HIF-1 (F), and HIF-2α (G) in these same regions of sickle cell eyes. n = 5 eyes. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium. Scale bar: 100 μm.
We next examined the expression of HIF-1α and HIF-2α, which regulate expression of angiogenic genes in ischemic retina (18) in eyes of patients with sickle cell disease. We did not observe expression of either HIF-1α or HIF-2α in the posterior, perfused retina of 5 of 5 eyes examined (Figure 2, F and G). In the periphery, in the absence of overlying retinal NV, we observed modest expression of HIF-1α and rare expression of HIF-2α within the INL. In peripheral ischemic inner retina underlying retinal NV, we observed robust expression of HIF-1α throughout the INL, but sparse expression of HIF-2α within a subset of cells within the INL (Figure 2, F and G). These results demonstrated that expression of HIF-1 and, to a lesser extent HIF-2α, is increased within the INL in ischemic retina in the setting of retinal NV and suggest that both HIF-1 and HIF-2 could contribute to the expression of angiogenic genes (e.g., VEGF and ANGPTL4), which in turn could help promote the development of retinal NV in patients with sickle cell disease.
HIF-1α and HIF-2α are coexpressed in hypoxic Müller cells in human inducible pluripotent stem cell–derived 3D retinal organoids.
To further characterize the expression of HIF-1α and HIF-2α in human retinal cells after hypoxic injury, we generated human induced pluripotent stem cell–derived (hiPSC-derived) 3D retinal organoids. After 40 days in culture, iPSCs developed into 3D retinal cups (Figure 3A). By 120 days of differentiation (D120), the inner and outer retinal layers were clearly defined (Figure 3B) and contained the precursors of the major retinal cell types, including outer retina photoreceptors (expressing recoverin), few newly differentiating bipolar cell precursors (lacking expression of recoverin and Pax6), amacrine cells (expressing high levels of Pax6), as well as Müller cells (expressing CRALBP; Figure 3, C and D and ref. 30). Exposure of D120 retinal organoids to hypoxia (1% O2) for 24 hours resulted in robust expression of HIF-1α throughout the hypoxic retina (Figure 3E), with nuclear localization of HIF-1α concentrated within CRALBP-expressing cells within the INL of the retina (Figure 3F). Similar results were observed for expression of HIF-2α (Figure 3, G and H); however, nuclear localization of HIF-2α was not readily detected. Although retinal organoids (Figure 4A) exposed to prolonged hypoxia lost the distinction between retinal layers, they nonetheless demonstrated sustained expression of HIF-1α and HIF-2α (Figure 4B). Coexpression of HIF-1α and HIF-2α was observed in most retinal cells, including CRALBP-expressing retinal Müller cells (Figure 4C). These results demonstrated that HIF-1α and HIF-2α were coexpressed in hypoxic human retinal organoids, similar to what we observed in human tissue.
Figure 3. Expression of HIF-1α or HIF-2α in hypoxic retinal cells in hiPSC-derived 3D retinal organoids.

(A) D40 and (B) D120 3D retinal organoids derived from hiPSCs containing (C) outer retina photoreceptors (expressing recoverin, REC), mitotic retinal progenitors within the neuroblastic layer (expressing low levels of Pax6), amacrine cells (expressing high levels of Pax6), and (D) Müller glial cells (expressing CRALBP). (E and F) Exposure of D120 3D retinal organoids to hypoxia (24 hours) induced HIF-1α expression throughout the retina (E), including nuclear accumulation (white arrows) in CRALBP-expressing retinal Müller cells (F). (G and H) Expression of HIF-2α and CRALBP in D120 3D retinal organoids after treatment with hypoxia. INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; IS/OS, inner/outer segments. Scale bars: 25 μm.
Figure 4. Simultaneous expression of HIF-1α and HIF-2α in hypoxic retinal cells in hiPSC-derived 3D retinal organoids.

(A) H&E staining of D120 3D retinal organoid prior to treatment with prolonged hypoxia. (B) Expression of HIF-1 and HIF-2α over time in boxed region (from A) of D120 3D retinal organoids exposed to hypoxia for 2 hours to 4 days. (C) HIF-1α, HIF-2α, and CRALBP expression in hypoxic hiPSC-derived retinal organoids treated with hypoxia. Scale bars: 25 μm.
Inhibition of both HIF-1α and HIF-2α is required to block the expression of VEGF mRNA in vitro.
Two key HIF-regulated angiogenic mediators, VEGF and ANGPTL4, have been previously reported to play an important role in promoting retinal NV (31, 32). Accordingly, we observed an increase in VEGF and ANGPTL4 mRNA expression in hiPSC-derived retinal organoids treated with hypoxia over time (Figure 5, A and B). Treatment of retinal organoids with the pharmacological inhibitor digoxin to inhibit both HIF-1 and HIF-2 resulted in complete inhibition of VEGF and ANGPTL4 mRNA (Figure 5, A and B). We and others have previously demonstrated that activated retinal Müller cells participate in the expression of HIF-regulated genes in ischemic retinal disease (29, 33, 34). We therefore isolated Müller cells from human retinal organoids (Figure 5C) and exposed these cells to hypoxia. Accumulation of HIF-1α and HIF-2α in hypoxic hiPSC-derived Müller cells was blocked with administration of digoxin (Figure 5D). This, in turn, resulted in a complete inhibition of VEGF and ANGPTL4 mRNA expression by these cells (Figure 5, E and F), similar to what was observed in the hiPSC-derived retinal organoids.
Figure 5. HIF-dependent expression of ANGPTL4 and VEGF in retinal organoids.

(A and B) D120 hiPSC-derived retinal organoids were treated with hypoxia (1–5 days) in the presence or absence of digoxin (30 μM) and the expression of VEGF and ANGPTL4 mRNA was examined by qPCR. (C) Müller cells isolated from retinal organoids expressed key Müller cell markers (vimentin and CRALBP). (D) Accumulation of HIF-1α and HIF-2α protein in hiPSC-derived Müller cells treated with hypoxia (12 hours) was blocked by digoxin (at indicated doses). (E and F) Expression of VEGF (E) and ANGPTL4 (F) mRNA in hiPSC-derived Müller cells treated with hypoxia (12 hours) inhibited by digoxin (3 μM). One-way ANOVA with Bonferroni’s multiple-comparison test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
We next examined the expression of HIF-1α and HIF-2α in an immortalized human retinal Müller cell line (MIO-M1; ref. 35) in the presence of hypoxia. We observed early accumulation of HIF-1α (within 2 hours, peaking by 24 hours) and delayed accumulation of HIF-2α (first noted at 8 hours) in hypoxic MIO-M1 cells (Figure 6, A and B). We next used RNAi to knock down expression of HIF-1α, HIF-2α, or both (Figure 6C). RNAi targeting HIF-1α blocked ANGPTL4 but did not affect VEGF mRNA expression (Figure 6, D and E). RNAi targeting HIF-2α did not affect ANGPTL4 expression but resulted in a modest decrease in VEGF mRNA expression (Figure 6, D and E). Simultaneous inhibition of HIF-1α and HIF-2α did not affect ANGPTL4 mRNA expression compared with inhibition of HIF-1α alone (Figure 6D), but it abolished VEGF mRNA expression (Figure 6E). Accordingly, inhibition of both HIF-1α and HIF-2α expression more effectively blocked VEGF protein secretion compared with inhibition of either HIF-1α or HIF-2α alone (Figure 6F). Collectively, these results suggested that in hypoxic retinal Müller cells, both HIF-1 and HIF-2α cooperate to promote VEGF expression, but only HIF-1 is responsible for the expression of ANGPTL4.
Figure 6. HIF-1α accumulation is necessary to promote ANGPTL4 and VEGF expression in retinal Müller cells.

(A and B) Accumulation of HIF-1α and HIF-2α in MIO-M1 cells cultured in hypoxia by WB (A) and IF (B). (C) Expression of HIF-1α and HIF-2α by WB in MIO-M1 cells exposed to hypoxia after knockdown of HIF-1α, HIF-2α, or both, by RNAi. (D and E) ANGPTL4 (D) and VEGF (E) mRNA expression in response to RNAi targeting HIF-1α, HIF-2α, or both, in MIO-M1 cells exposed to hypoxia. (F) VEGF protein expression in response to RNAi targeting HIF-1α, HIF-2α, or both, in MIO-M1 cells exposed to hypoxia. Data are shown as mean ± SD. Statistical analyses were performed 2-way ANOVA with Bonferroni’s multiple-comparison test (D–F). h, hours; NS, nonsignificant. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Accumulation of HIF-1α alone is sufficient to promote VEGF and ANGPTL4 mRNA expression in vitro and retinal NV in vivo.
The observation that HIF-1α is necessary to promote the expression of both VEGF and ANGPTL4, 2 key angiogenic mediators in the promotion of retinal NV in ischemic retinal disease (31, 32, 34), prompted us to explore whether accumulation of HIF-1α alone can promote retinal NV. To this end, we took advantage of Ad-CA5, an adenovirus expressing a previously characterized, constitutively active (normoxia-stable) HIF-1α mutant (36). Ad-CA5 enabled us to examine the consequence of HIF-1α accumulation in the absence of hypoxia or other hypoxia-triggered responses. Infection of MIO-M1 cells with Ad-CA5 resulted in an increase in the accumulation of HIF-1α under normoxic (20% O2) conditions in contrast to Ad-LacZ, an adenovirus expressing E. coli β-galactosidase (Figure 7A). Within 24 hours of infection with adenovirus, we observed a nonspecific increase in VEGF and ANGPTL4 mRNA expression (Figure 7, B and C). However, 48 hours after infection, Ad-CA5–infected MIO-M1 cells demonstrated an increase in both VEGF and ANGPTL4 mRNA expression compared with Ad-LacZ–infected MIO-M1 cells (Figure 7, B and C). We next injected Ad-LacZ or Ad-CA5 into the eyes of 10-week-old male C57BL/6 mice and examined the retina for the development of NV. Six days after intravitreal injection, we observed an increase in the accumulation of HIF-1α (Figure 7D) in cross-sections of the inner retina. We further observed an increase in isolectin-B4–labeled and CD31-labeled vessel length in the inner retina of mice injected with Ad-CA5 (Figure 7, E and F). Similar results were obtained in retinal flat mounts from mice injected with Ad-CA5 (Figure 7, G and H). Collectively, these results demonstrated that HIF-1α was sufficient to promote retinal NV and further suggest that inhibition of HIF-1α will be necessary to prevent the development of NV in ischemic retinal disease.
Figure 7. HIF-1α accumulation alone is sufficient to promote retinal NV in vivo.

(A) Accumulation of HIF-1α in MIO-M1 cells cultured in normoxia (20% O2) and infected with adenovirus expressing a constitutively active (normoxia-stable) HIF-1α mutant (Ad-CA5) led to increased expression of (B) VEGF and (C) ANGPTL4 mRNA expression. (D) Accumulation of HIF-1α in mouse retinas 6 days after intravitreal injection with Ad-CA5 or Ad-LacZ. (E and F) Representative isolectin-B4–labeled (red) and CD31-labeled (green) mouse retinas after intraocular injection with Ad-CA5 (E) and quantitation of retinal vessel (based on vessel length, F). (G and H) Representative isolectin-B4–labeled retinal flat mounts after intraocular injection with Ad-CA5 or Ad-LacZ in an adult mouse eye (G) and quantitation of retinal neovascularization (based on relative fluorescence intensity; H). Data are shown as mean ± SD. Statistical analyses were performed by 2-way ANOVA with Bonferroni’s multiple-comparison test (B and C) or 2-tailed unpaired Student’s t test (F and H). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. h, hours. Scale bar: 100 μm.
Inhibition of HIF-2α alone is sufficient to prevent retinal NV in the OIR model.
To further characterize the contribution of HIF-1 versus HIF-2 to the promotion of retinal NV in ischemic retina in vivo, we used the classic OIR model characterized by Smith and colleagues (37). In stage 2 of this model, P12 pups that had been exposed to hyperoxia (75% O2) for 5 consecutive days (P7–P12; resulting in obliteration of the posterior retinal vasculature) were returned to room air (21% O2). The resulting relative ischemia promoted the expression of angiogenic mediators that stimulated the development of retinal NV (peaking at P17; Figure 8A). Daily i.p. injections of the pharmacological HIF inhibitor digoxin (0.5 mg/kg; P12 to P16) inhibited the development of NV (Figure 8A and refs. 31, 33, 38) and the expression of VEGF mRNA (Figure 8B) at P17, demonstrating that OIR animals reproduced the HIF-dependent promotion of retinal NV observed in patients with IR.
Figure 8. Inhibition of HIF-2α modestly affects VEGF expression in vitro but markedly reduces retinal NV in vivo.

(A) Representative retina flat mounts demonstrating the promotion of retinal NV at P17 in the OIR mouse model after daily i.p. injection (P12–P16) with the HIF-1 and HIF-2 inhibitor digoxin (0.5 mg/kg). (B) Expression of VEGF mRNA expression in MIO-M1 cells treated with increasing doses of PT2385 for 24 hours. (C) Binding of endogenous HIF-1β to HIF-1α or HIF-2α in MIO-M1 cells treated with the HIF-2–specific inhibitor PT2385 (at the specified doses) or vehicle (DMSO) and exposed to 20% or 1% O2 for 24 hours was detected by co-IP. (D) Expression of Vegf mRNA expression in retina from OIR mice after daily i.p. injection (P12–P16) with digoxin. (E) Representative retina flat mounts demonstrating the inhibition of retinal NV at P17 in the OIR mouse model after daily oral gavage (P13–P16) with PT2385 (30 mg/Kg) or vehicle. (F) Quantitation of retinal avascular area (left) and retinal NV (right) at P17. (G) Expression of Vegf mRNA expression in retina from OIR mice after daily oral gavage (P12–P16) with PT2385. (H) IF of primary retinal Müller cells isolated from mice demonstrating expression of key Müller cell markers, vimentin (above) and GS (below). (I) Expression of Vegf mRNA expression in primary mouse Müller cells treated with PT2385 (at the specified doses) for 16 hours. Data are shown as mean ± SD. Statistical analyses were performed by 1-way ANOVA with Bonferroni’s multiple-comparison test (B and G), 2-way ANOVA with Bonferroni’s multiple-comparison test (D and I), or 2-tailed unpaired Student’s t test (F). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Scale bars: 500 μm (A and E).
We next took advantage of the recent development of a HIF-2–specific small-molecule inhibitor, PT2385 (39), to examine the contribution of HIF-2 to angiogenic gene expression. PT2385 selectively binds HIF-2α (kD < 50 nM) — but not HIF-1α — to prevent it from binding to HIF-1β and has shown promise in preclinical studies (39); it is currently under investigation for the treatment of patients with renal cell carcinoma (40, 41). This has inspired speculation about the therapeutic potential of PT2385 for the treatment of ocular neovascular disease. We therefore examined whether PT2385 can affect VEGF mRNA expression in MIO-M1 cells. We first confirmed that PT2385 effectively inhibits binding of HIF-2α — but not HIF-1α — to HIF-1β in MIO-M1 cells (Figure 8C). Similar to HIF-2α knockdown by RNAi, we observed only a partial reduction in VEGF mRNA expression in MIO-M1 cells treated with effective doses of PT2385 (Figure 8D).
Collectively, these data predict that inhibition of HIF-2 with PT2385 would not be effective for the treatment of retinal NV in ischemic retinal disease. To interrogate this hypothesis, we examined whether PT2385 would influence the development of retinal NV in the OIR model of ischemic retinal disease. Surprisingly, we observed complete inhibition of retinal NV in OIR mice treated with PT2385 by twice daily oral gavage (P12–P16; Figure 8, E and F), identical to what was observed with daily treatment with the HIF-1/HIF-2 inhibitor digoxin. Indeed, we observed complete inhibition of VEGF mRNA expression at P17 in OIR mice treated with PT2385 (Figure 8G). To examine whether this was due to an increased sensitivity of mouse retinal cells to PT2385, we isolated Müller cells from mice (Figure 8H) and treated them with increasing doses of PT2385. Similar to MIO-M1 cells, PT2385 only partially inhibited VEGF mRNA expression in primary mouse Müller cells (Figure 8I). Contrary to in vitro studies in MIO-M1 cells, hiPSC-derived Müller cells, and primary mouse Müller cells, and inconsistent with immunohistochemical studies in hiPSC-derived retinal organoids and PSR eyes, results from the OIR model suggest that HIF-2 inhibition alone may be sufficient to prevent the development of retinal NV in patients with IR.
HIF-1α and HIF-2α expression is coordinated but segregated in the OIR model.
To understand the discordance of the findings in human tissue, retinal organoids, and cell-based models with that observed in the OIR model, we carefully examined the hypoxic induction of HIF expression in the OIR model. The obliteration of the posterior inner retinal vasculature (Figure 9A) that occurred during the hyperoxic stage (stage 1) of the OIR model resulted in acute and marked hypoxia (as demonstrated by the markedly increased staining with the hypoxia-sensitive nitroimidazole Hypoxyprobe) in the posterior retina upon return to room air (Figure 9B and refs. 33, 34, 42). Revascularization of the retina occurred from the periphery toward the posterior pole over time (43) and resulted in a decrease in the total area of hypoxic retina (Figure 9C). In the posterior nonvascularized retina, there was marked hypoxia in the inner retina (Figure 9C) extending from the internal limiting membrane to the INL. By contrast, in the peripheral vascularized (perfused) anterior retina and in the outer retina, no significant hypoxia was detected. Tissue ischemia promoted retinal NV (observed as early as P14 and peaking at P17; Figure 9D) at the margin between perfused and nonperfused retina, similar to the NV observed in patients with PSR.
Figure 9. Coordinated expression of HIF-1α and HIF-2α promotes VEGF expression in the OIR mouse model.

(A and B) Retina flat mounts demonstrating vaso-obliteration (demarcated by white line in A) and hypoxia (as measured by Hypoxyprobe staining in B) in the posterior retina at the initiation of the ischemic stage (P12) of the OIR model. Cyan dashed line represents the location of cross-sections used for subsequent IF studies. (C) Representative images of cross-section of OIR eye demonstrating decrease in hypoxia (as measured by Hypoxyprobe) in the posterior inner retina from P12.5 to P14 with revascularization in the peripheral retina (red arrowheads) that progressed posteriorly. (D) Representative image of retinal NV at P17. (E and F) The accumulation of HIF-1α (E) and HIF-2α (F) in the hypoxic inner retina during the ischemic stage of the OIR model (P12–P17). (G and H) Nuclear (white arrows) versus cytoplasmic (yellow arrows) accumulation of HIF-1α (G) and HIF-2α (H) in the INL. (I) Increased Vegf mRNA expression in the ischemic inner retina beginning at P13 and peaking between P15 and P16 by RNAscope. n = 4–6 animals. on, optic nerve; GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer. Scale bars: 500 μm (A–D); 100 μm (E–J).
We next examined the accumulation of HIF-1α and HIF-2 in the hypoxic inner retina during the ischemic stage of the OIR model (P12–P17). Expression of HIF-1α was detected in the posterior inner retina as early as P12.5 (Figure 9E), but its accumulation faded rapidly and was only detected in rare cells after P13. Close examination demonstrated nuclear accumulation of HIF-1 initially detected at P12.5 in scattered cells within the middle of the INL, consistent with the localization of retinal Müller cells (44). Expression of HIF-1α peaked by P13, at which time nuclear expression was detected more diffusely in the INL and in the retinal ganglion cell layer (GCL). At P14, 24 hours later, expression of HIF-1α was no longer detected in the INL (Figure 9E).
Unlike HIF-1α, expression of HIF-2α was not detected at P12.5 or P13 of the OIR model, despite marked hypoxia (and robust expression of HIF-1α) in the inner retina. Expression of HIF-2α was first detected in the inner retina at P14 (when HIF-1α was no longer detected) and persisted to the end of the ischemic stage at P17 (Figure 9F). Expression of HIF-2α was simultaneously detected broadly within the INL and the GCL. Nuclear accumulation of HIF-2α in the INL rapidly diminished and was no longer detectable by P16 (Figure 9F). Conversely, nuclear accumulation of HIF-2α in the GCL peaked at P14 but persisted into P17 (Figure 9F). Collectively, these results demonstrated a rapid but transient accumulation of HIF-1α and a delayed but sustained accumulation of HIF-2α in ischemic retinal cells. Close examination of HIF-1α (Figure 9G) and HIF-2α (Figure 9H) expression in the INL of OIR eyes demonstrated predominantly cytoplasmic or perinuclear accumulation of HIF-1α at P12.5, with peak nuclear accumulation at P13. By P14, HIF-1α expression was no longer detected and was replaced by robust nuclear accumulation of HIF-2α, which was detected in only rare cells by P15.
Examination of Vegf mRNA transcripts by in situ hybridization (Figure 9I) in OIR mice demonstrated an increase of Vegf mRNA in the ischemic inner retina beginning at P13 that plateaued at P15–P16. Increased Vegf mRNA expression was observed at P13 within the middle of the INL, corresponding to the accumulation of HIF-1α within this layer and consistent with the localization of retinal Müller cells. By P14, expression of Vegf mRNA expanded to the entire INL and by P15 was detected in the GCL, consistent with the expression pattern of HIF-2α. This correlation between the localization of HIF-1α and HIF-2α accumulation and Vegf mRNA expression was consistent with in vitro studies demonstrating the cooperative upregulation of VEGF by both HIF-1 and HIF-2. However, the mutually exclusive expression of HIF-1α and HIF-2α was inconsistent with our in vitro data and our observations of simultaneous expression of both HIF-1α and HIF-2α in PSR eyes and their coexpression in retinal cells in hypoxic hiPSC-derived retinal organoids.
Coordinated but segregated expression of HIF-1α and HIF-2α is reproduced in adult mouse retinal explants exposed to hypoxia.
We next set out to determine whether the segregated expression of HIFs in the OIR model was specific to mouse retinal cells. To confirm the accumulation of HIFs in retinal Müller cells in OIR mice, we examined the coexpression of HIF-1α or HIF-2α (at P13 and P14, respectively) in cells expressing the Müller cell markers glutamate synthetase (GS) and vimentin. We observed accumulation of both HIF-1α and HIF-2α in inner retinal cells expressing GS (Figure 10, A and B) and vimentin (Figure 10, C and D). We next isolated primary retinal Müller cells from mice and examined the expression of HIF-1α and HIF-2α over time in response to hypoxia. We observed rapid accumulation of HIF-1α and delayed accumulation of HIF-2α (Figure 10E), identical to what was observed in human MIO-M1 cells in vitro. Both HIF-1α and HIF-2α were required to promote Vegf mRNA expression (Figure 10F), also similar to human MIO-M1 cells. Unlike cells within the INL of OIR mice, we observed coexpression of both HIF-1α and HIF-2α in primary mouse Müller cells in culture (Figure 10G).
Figure 10. Expression of HIF-1α and HIF-2α in OIR mice Müller cells and mouse retinal explants treated with hypoxia.

(A–D) IF demonstrating the expression of HIF-1α (A and C) and HIF-2α (B and D) in Müller cells expressing glutamine synthetase (GS; A and B) or vimentin (C and D) in the INL (yellow arrows) at P13 or P14. White arrows point to cells coexpressing GS and HIF-1α or HIF-2α. (E) Expression of HIF-1α and HIF-2α over time by Western blot in primary mouse Müller cells treated with hypoxia. (F) Expression of VEGF mRNA expression in hypoxic primary mouse Müller cells after RNAi knockdown of HIF-1α, HIF-2α, or both. (G) Coexpression of HIF-1α and HIF-2α by IF in primary mouse Müller cells treated with hypoxia. (H and I) Rapid but transient expression of HIF-1α (H) and delayed expression of HIF-2α (I) in adult mouse retinal explants treated with hypoxia for 8 to 24 hours. Data are shown as mean ± SD. Statistical analyses were performed by 2-way ANOVA with Bonferroni’s multiple-comparison test. *P < 0.05; **P < 0.01; ***P < 0.001. n = 4 to 6 animals. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; h, hours. Scale bar: 100 μm.
To explore whether the segregated expression pattern of HIF-1α and HIF-2α in OIR mice may require an intact mouse retina, we examined HIF-1α and HIF-2α expression in adult mouse retinal explants exposed to hypoxia. Similar to retinal cells in OIR mice, there was a strict time-dependent expression pattern for HIF-1α and HIF-2α. Mouse retinal explants exposed to hypoxia for as little as 8 hours demonstrated increased expression of HIF-1α in the inner retina (Figure 10H). HIF-1α expression in the INL peaked at 16 hours but faded rapidly and was largely absent by 24 hours (Figure 10H). HIF-2α expression in the INL of hypoxic mouse retinal explants was observed in rare cells at 16 hours but was markedly increased by 24 hours (Figure 10I); this rapid but transient expression of HIF-1α and delayed expression of HIF-2α closely resembled the expression pattern observed in OIR mice. Collectively, these results suggest that the segregated expression of HIF-1α and HIF-2α in the hypoxic inner retinal cells observed in OIR mice and reproduced in adult mouse retinal explants may be specific to acute ischemic injury in the mouse retina.
Transient pharmacological inhibition of peak expression of either HIF-1 or HIF-2 within the INL is sufficient to inhibit the development of retinal NV in OIR mice.
Although the conflicting observations between the expression pattern of HIFs in human tissue from patients with PSR (and reproduced in human retinal organoids) compared with the OIR model (and reproduced by mouse retinal explants) raises questions as to whether ischemic injury to the human retina is accurately reproduced by this widely used mouse model, the discrete segregated phases of HIF-1α and HIF-2α accumulation in INL cells in OIR mice provided a unique opportunity to further dissect their relative contribution to retinal NV in vivo. We therefore performed transient inhibition of the HIF-1α or HIF-2α accumulation in the INL using i.p. injection with the pharmacological inhibitor digoxin according to the time-dependent hypoxic-induction of HIF-1α and HIF-2α accumulation in the OIR model (Figure 11A). Digoxin is a potent but short-acting inhibitor of both HIF-1 and HIF-2 in vivo, thereby providing transient inhibition of HIFs in OIR mice after i.p. administration. We examined the effect of inhibiting the peak accumulation of HIF-1α within the INL on P13 by treating OIR mice with i.p. digoxin on day P12.5 (designated treatment “D1”; Figure 11, A and B) or the peak accumulation of HIF-2α within the INL on P14 by treating OIR mice with i.p. digoxin on day P13.5 (designated treatment “D2”; Figure 11A), the latter resulting in an inhibition of HIF-2α accumulation for 48 hours (Figure 11C). Inhibition of peak accumulation of either HIF-1α or HIF-2α with a single i.p. treatment with digoxin on P12.5 or P13.5 (D1 or D2, respectively) resulted in a potent inhibition of retinal NV on P17 without influencing the area of avascular retina (Figure 11D). Despite the redundancy in the ability of HIF-1 and HIF-2 to upregulate VEGF expression, these results suggested that the rapid but transient increase in HIF-1α expression (peaking at P13) and the delayed but sustained expression of HIF-2α (peaking at P14) within the INL were both essential for the promotion of retinal NV in OIR mice.
Figure 11. Inhibition of HIF-1α or HIF-2α prevents the development of retinal neovascularization in OIR mice.

(A) Schematic summarizing the time-dependent and cell-specific hypoxic induction of HIF-1α and HIF-2α accumulation in inner retinal cells defines 2 phases of the ischemic stage of the OIR model. In the first phase (P12–P14), the onset of hypoxia (green) corresponded with the rapid accumulation of Hif-1α mRNA (cyan) and protein (blue) expression in retinal glial cells. In the second phase (P14–P17), the rapid accumulation of HIF-2α was observed in the INL (solid purple) and GCL (dashed red lines). This resulted in the accumulation of VEGF expression (orange) and retinal neovascularization (red). (B) Injection (i.p.) with digoxin (2 mg/kg) at P12.5 (designated D1) provided transient (24-hour) pharmacological inhibition of HIF-1α in OIR mice. (C) Retinal neovascularization (outlined) at P17 after inhibition of peak expression of HIF-1α or HIF-2α with a single i.p. injection of digoxin (2 mg/kg) on P12.5 (D1) or P13.5 (designated D2), respectively, compared with vehicle (DMSO) control. (D) Quantitation of avascular retina and retinal neovascularization at P17 after D1 or D2 treatment compared with vehicle control in OIR mice. Data are shown as mean ± SD. Statistical analyses were performed by 1-way ANOVA with Bonferroni’s multiple-comparison test. *P < 0.05; **P < 0.01; ***P < 0.001. P, postnatal; NS, nonsignificant. Scale bar: 500 μm.
In vivo nanoparticle-mediated RNAi specifically targeting either HIF-1α or HIF-2α prevents the development of retinal NV in OIR mice.
To corroborate these results, we next set out to examine the effect of specifically inhibiting either HIF-1α or HIF-2α accumulation on retinal NV in the OIR model. However, given that expression of HIFs by retinal glial cells contributes to the normal vascular development in the postnatal developing mouse retina (45), it is difficult to distinguish between the impact of loss of HIF expression on developmental vasculogenesis versus pathological angiogenesis using KO approaches. To overcome this obstacle, we developed a nanoparticle-based RNAi approach to specifically knock down expression of genes in vivo. To this end, we generated siRNA-encapsulated nanoparticles (Figure 12A) using reducible branched ester amine quadpolymers (rBEAQs; Supplemental Figure 3, A and B and ref. 46). These biodegradable nanoparticles are designed to release siRNA cargo in an environmentally triggered manner upon cleavage of disulfide bonds in the polymer backbone in the reducing cytosolic environment. Nanoparticle hydrodynamic diameter was measured by nanoparticle tracking analysis and zeta potential was measured by electrophoretic mobility (Supplemental Figure 3, C and D). Transmission electron microscopy imaging of rBEAQ-siRNA nanoparticles demonstrated relatively uniformly sized nanoparticles (Supplemental Figure 3E). Using NIH-3T3 cells stably expressing GFP, we established that delivery of a 100 nM GFP siRNA (NP-siGFP) dose with a nanoparticle formulation in which the nucleic acid was formulated to be approximately 4.8% of the total material by mass (20 wt/wt) enabled approximately 80% gene knockdown in vitro as assessed by flow cytometry (Figure 12B) and immunofluorescence (Figure 12C).
Figure 12. In vivo nanoparticle-mediated RNAi targeting either HIF-1α or HIF-2α prevents the development of retinal NV in OIR mice.

(A) rBEAQ polymer for in vivo delivery of RNAi. (B) rBEAQ polymer nanoparticle-mediated knockdown of GFP reporter gene in NIH-3T3 cells stably expressing GFP in vitro. (C) Fluorescence micrograph of NIH-3T3 cells stably expressing GFP treated with rBEAQ nanoparticles encapsulating a nontargeting scrambled control siRNA (NP-scr) or with nanoparticles encapsulating siRNA targeting GFP (NP-siGFP). Images were taken 1 day after transfection and show sequence-specific GFP knockdown. (D and E) Expression of fluorophore in retinal cross-section (D) and flat mount (E) of mice 1 day after intravitreal injection with NP-scr or NP-scr conjugated to Cy5. (F) Schematic demonstrating use of NP-siGFP to knock down expression of GFP in photoreceptors in rho-GFP mice. (G and H) Expression of GFP mRNA (G) and protein (H) in rho-GFP mice 3 days after intravitreal injection with NP-siGFP versus NP-scr. (I) Hif1a and Hif2a mRNA expression 1 and 3 days after a single intravitreal injection with RNAi targeting HIF-1α or HIF-2α (NP-siHIF1 or NP-siHIF2), respectively. (J) Retinal NV (outlined) at P17 after inhibition of HIF-1α or HIF-2α expression with a single intravitreal injection with NP-siHIF1 or NP-siHIF2, respectively (above). Quantitation of avascular retina and retinal NV at P17 (below) after inhibition of HIF-1α or HIF-2α expression with a single intravitreal injection with NP-siHIF1 or NP-siHIF2 compared with NP-scr. Data are shown as mean ± SD. Statistical analyses were performed by 2-way ANOVA with Bonferroni’s multiple-comparison test (I) or 1-way ANOVA with Bonferroni’s multiple-comparison test (J). *P < 0.05; **P < 0.01. n = 4–6 animals. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; IS/OS, inner/outer segments; RPE, retinal pigment epithelium; h, hours; NS, nonsignificant. Scale bar: 100 μm (D, E, and H); 500 μm (J).
Intraocular injection with the nanoparticle encapsulating scrambled RNAi (NP-scr) conjugated to a fluorophore (Cy5) demonstrated effective transfection of retinal cells within all retinal layers (Figure 12D) after a single intravitreal injection (Figure 10E). To examine the ability of NP-RNAi to knock down gene expression in vivo, we injected NP-siGFP into the eyes of mice expressing GFP under the rod-specific rhodopsin promoter (rho-Gfp mice; ref. 47 and Figure 12F). We observed efficient knockdown of GFP mRNA (Figure 12G) and protein (Figure 12H) expression in retinal photoreceptors after a single injection of NP-siGFP compared with NP-scr control.
We next used the nanoparticles to encapsulate Hif-1α or Hif-2α RNAi (NP-siHIF1 and NP-siHIF2, respectively). We titrated the nanoparticles to transiently (24–48 hours) knock down Hif1a mRNA expression but to maintain sustained (>72 hours) knockdown of Hif2a mRNA expression (Figure 12I). Intravitreal injection with NP-scr at P12 reduced the levels of NV compared with untreated mice, consistent with a prior report demonstrating the ability of RNAi to reduce retinal NV independent of its target (48). After a single intravitreal injection at P12, we observed a marked inhibition of retinal NV with either NP-siHIF1 or NP-siHIF2 (without influencing the area of avascular retina) compared with NP-scr (Figure 12J). Consistent with our pharmacological studies, these results suggest that the early accumulation of HIF-1α and the late accumulation of HIF-2α are both essential for the development of retinal NV in OIR mice.
Discussion
Since it was first described in 1954 (49), the OIR model has served as the foundation for our understanding of pathological angiogenesis in the eye (50). Consequently, this powerful model has played a fundamental role in studies examining the pathogenesis of retinal NV in patients with IR. We demonstrated here that the ischemic stage (P12–P17) of the classic OIR model can be subdivided into 2 distinct phases based on the expression of HIF-1α and HIF-2α. The first phase begins upon the initiation of the ischemic stage (P12) and is characterized by marked hypoxia and the rapid accumulation of HIF-1α within the INL and GCL. Nuclear accumulation of HIF-1α was initially detected at P12.5 within the middle of the INL, corresponding to the location of retinal Müller cells. HIF-1α accumulation in the INL peaked at P13 and rapidly disappeared by P14. Interestingly, accumulation of HIF-1α was not observed within the INL at P14 despite persistent hypoxia, suggesting that although acute hypoxia triggered the accumulation of HIF-1α within the INL, hypoxia was not sufficient to sustain its continued nuclear accumulation in these cells in OIR mice. This precise (and transient) upregulation of HIF-1α expression suggests that this transcription factor — and the genes it regulates — may play a specific role in the “acute” stages after ischemic injury to the neurosensory retina in mice.
In the second phase of the ischemic stage, HIF-2α accumulation was noted simultaneously in the INL (diffusely) and the GCL beginning at P14. This suggests a marked difference in the regulation of HIF-1α and HIF-2α in the neurosensory retina after acute ischemic injury. Similar to the expression of HIF-1α, the nuclear accumulation of HIF-2α in the INL was also transient: HIF-2α expression in the INL was markedly diminished after 24 hours (P15). However, HIF-2α expression was maintained in the GCL until the end of the ischemic stage (P17). This further suggests that different cell populations differentially regulate HIF-2α expression in the ischemic inner retina. Collectively, these results suggest that HIF-2α — and HIF-2–regulated gene products — may play important and distinct role(s) in “subacute” hypoxic retinal cells in different layers of the inner retina of OIR mice.
It has previously been reported that HIF-1 and HIF-2 can regulate different genes in the same cell (51) and can regulate genes differentially in different cell types (36). Our results introduce an additional layer of complexity for HIF regulation in ischemic tissue. Despite evidence that HIF-1α and HIF-2α were expressed within the INL in OIR mice, we were unable to detect simultaneous expression of both HIFs in the same cells at the same time in the INL. Remarkably, this coordinated transient expression of HIFs in the INL resulted in the robust and sustained expression of the HIF-regulated angiogenic mediator VEGF throughout the ischemic phase. The expression of Vegf mRNA followed the pattern (both in terms of timing and localization) of HIF-1α and HIF-2α expression within the INL and GCL, supporting a cooperative and complementary role for HIFs in regulating angiogenic gene expression in this mouse model. Our observation that HIF-1α and HIF-2α were not simultaneously expressed within the INL at the same time in the OIR model further suggests that HIF-1 and HIF-2 may have mutually exclusive roles in regulating the expression of specific genes in retinal tissue at different stages after acute ischemic injury of the mouse retina.
Interestingly, staggered expression of HIF-1α and HIF-2α was not observed in tissue from patients with chronic hypoxia. This suggests that extrapolation of studies examining the role of HIFs using the OIR model alone should be approached cautiously. This observation has broad implications because much of our understanding of the regulation of gene expression after hypoxic injury in IRs has come from studies using the OIR model. Arguably, the hyperoxia-induced retinopathy observed in the developing retina of OIR mice may better reproduce early events observed in infants with retinopathy of prematurity; however, the expression pattern of HIFs in hiPSC-derived 3D retinal organoids exposed to hypoxia — which also models acute ischemic injury of the developing human retina — was more consistent with the expression pattern in adults with chronic ischemic retinal disease than in the OIR model. Moreover, the observation that staggered expression of HIF-1α and HIF-2α was reproduced in adult mouse retinal explants exposed to hypoxia raises questions as to whether mouse models of acute ischemic injury, in general, accurately reproduce the chronic ischemia observed in patients with IRs (at least with regard to the contribution of HIF-1 and HIF-2).
In the developing mouse retina, expression of VEGF by astrocytes was previously reported to promote the proliferation and radial migration of endothelial cells along the astrocyte network on the vitreoretinal surface, forming the superficial vascular plexus (P1–P7; ref. 52). However, 2 of 3 subsequent studies using astrocyte-specific Vegf-KO mouse lines did not corroborate a role for astrocytes in the radial growth of the superficial retinal vasculature (53, 54). In a recent study, Rattner and colleagues set out to reconcile these conflicting reports and demonstrated that VEGF secreted by astrocytes did, indeed, promote the radial extension of the superficial capillary plexus (P1–P7; ref. 45). They further showed that expression of HIF-1α in interneurons (horizontal and/or amacrine cells) promotes the development of the intermediate capillary plexus, and expression of HIF-2α in Müller cells promotes the development of the deep capillary plexus (P8–P15). It is important to note that a role for an astrocyte network in driving vasculogenesis was not observed in tissue from developing human fetal retina; instead, CD34+ vascular cords were observed that were assembled in advance of GFAP+/PAX2+ astrocytes (55), demonstrating differences in mouse versus human retinal vascular development.
Because the postnatal retinal vascular development described above (P1–P15) overlaps with both the hyperoxic (P7–P12) and ischemic (P12–P17) stages of the OIR model, it can be challenging to unravel the contribution of HIFs to developmental versus pathological angiogenesis using the OIR model with KO mice. Therefore, we instead used 2 complementary approaches that exploited the segregated expression of HIFs in the OIR model to dissect the contribution of HIF-1 versus HIF-2 to retinal NV. First, we used a pharmacological approach to transiently inhibit peak expression of HIF-1α or HIF-2α in the INL of OIR mice. These results suggested that peak expression of HIF-1α and HIF-2α were both essential for the development of NV in OIR mice. We then engineered a nanoparticle-based knockdown approach to transiently inhibit Hif-1α or Hif-2α gene expression. Using the latter approach, we knocked down expression of either HIF-1α or HIF-2α after stage 1 (hyperoxia stage) of the OIR model and observed that both HIFs were essential for the promotion of retinal NV. Although these methods have their own inherent limitations (e.g., possible off-target effects and absence of cell specificity) the concordance of results from these studies supports a cooperative role for HIFs in the promotion of retinal NV in the OIR model and further demonstrates the potential efficacy of therapies targeting HIFs to treat (or prevent) NV.
While our results from the OIR model might lead to the conclusion that targeting either HIF would be equally effective for the treatment of PSR (and other IRs), this observation may be unique to mouse models in which staggered (nonoverlapping) expression of HIF-1α and HIF-2α does not reproduce a (potential) redundant role for HIFs in patients with IRs. Indeed, we demonstrated that both HIF-1α and HIF-2α were simultaneously expressed in the ischemic inner retina — and, in particular, in areas underlying NV — in PSR eyes, as well as in human retinal organoids treated with hypoxia. These studies suggest that therapies targeting both HIFs may be required for the treatment of retinal NV in patients with PSR and expose an incongruence with one of the most commonly used animal models and the human diseases it is purported to reproduce.
In this regard, organoids have emerged as a powerful tool to study human disease, and retinal organoids may be considered a more suitable model for studying retinal disease. Here, we demonstrated coexpression of HIF-1α and HIF-2α in human retinal organoids exposed to hypoxia, similar to what was observed in tissue from patients with chronic ischemia. However, neurons in retinal organoids (when present) are immature. Moreover, we were unable to readily detect nuclear accumulation of HIF-2α within the retinal organoids, although this may be a limitation of the antibodies used rather than retinal organoid as a model. Regardless, because retinal organoids do not contain blood vessels (or endothelial cells), they do not reproduce physiological retinal conditions in humans nor pathological conditions in ischemic retinal disease. Caution is therefore needed when interpreting data from retinal organoids and translating these findings to human physiological or pathological conditions. Collectively, our observations emphasize the importance of using a combination of approaches (e.g., human tissue, retinal organoids, and in vitro studies) in addition to mouse models in preclinical studies to assess the potential efficacy of therapies targeting HIFs or HIF-regulated genes for the treatment of patients with IRs.
Methods
Cell culture, reagents, and assays.
Details for cell culture, reagents, co-IP assay, Western blot, ELISAs, and reverse transcription and quantitative real-time PCR (RT-qPCR) are provided in Supplemental Methods.
Mice.
Details for in vivo studies are provided in Supplemental Methods.
Retinal organoids.
An hiPSC line derived from CD34+ cord blood was used in this study (A18945, Thermo Fisher Scientific; ref. 56). Undifferentiated hiPSCs and derived retinal organoids were routinely tested for Mycoplasma contamination by PCR. Cell culture, retinal differentiation, and organoid formation were conducted as previously described (30). Retinal organoids at 120 days of differentiation were used for experiments.
siRNA studies.
HIF-1α and HIF-2α siRNA and a nontargeting control were purchased from Ambion. For in vitro knockdown experiments, cells were transfected with Lipofectamine 2000. The efficiency of siRNA was confirmed by immunoblot and/or RT-qPCR assays.
Adenoviral vectors.
Details for adenoviral vectors are provided in Supplemental Methods.
Studies on mouse and human tissue.
Details for antibodies are provided in Supplemental Table 1.
Details for immunofluorescence assays, Hypoxyprobe, H&E staining, in situ hybridization, and immunohistochemistry are provided in Supplemental Methods.
Nanoparticles.
Details for nanoparticles are provided in Supplemental Methods.
Imaging studies.
Inclusion criteria for patients with sickle cell disease included a known diagnosis of sickle cell disease (by hemoglobin electrophoresis) and a clear view to the posterior pole (facilitating imaging studies using UWF FA). Exclusion criteria included diabetes, any other ischemic retinal disease, uveitis, retinal detachment, or NV from another cause.
Autopsy eyes.
Five eyes from 5 patients with sickle cell disease (documented by hemoglobin electrophoresis) with a history of untreated PSR (i.e., no history of scatter laser photocoagulation or prior intravitreal anti-VEGF therapy) and no known history of diabetes or ischemic retinal disease were selected for examination.
Statistics.
In all cases, results are shown as mean ± SD from at least 3 independent experiments. Statistical analysis was performed with Microsoft Excel and Prism 8.0 software (GraphPad). To calculate statistical significance, 2-tailed Student’s t test, 1-way ANOVA, or 2-way ANOVA followed by Bonferroni’s multiple-comparison test were used as indicated in the figure legends. Ischemic indices from patients with sickle cell disease were manually generated by 2 separate graders and verified for consistency by Pearson’s correlation. Analysis of this data was performed in MATLAB and Excel. Comparisons between PSR and non-PSR groups were made using a 2-tailed Student’s t test. P values less than 0.05 were considered significant.
Study approval.
IRB approval from the Johns Hopkins University School of Medicine was obtained for all patient images used in this study and for all autopsy eyes used in this study. All experiments involving animals were performed in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and formally reviewed and approved by the Johns Hopkins University Animal Care and Use Committee Animal Research Reporting.
Author contributions
AS was the primary contributor to research design. JZ, YQ, MM, MFB, MR, AD, MD, YQ, SAD, YR, SYT, SS, and SF were responsible for research execution and were contributors to data acquisition. AS, JZ, MM, MFB, AD, XC, SS, JG, VCS, and SM were the primary contributors to data analysis and interpretation. AS prepared the manuscript with revisions provided by VCS, JG, GLS, and SM.
Supplementary Material
Acknowledgments
This work was supported by the National Eye Institute, NIH grants R01EY029750 to AS, R01EY025705 to SM and AS, R01CA228133 to JG, and EY001765 (the Wilmer Core Grant for Vision Research, Microscopy and Imaging Core Module); the Research to Prevent Blindness Inc. Special Scholar Award to AS, Sybil B. Harrington Stein Innovation Award to GLS, and unrestricted grants to the Wilmer Eye Institute at the Johns Hopkins School of Medicine and the Department of Ophthalmology at the University of Colorado; the CellSight Development Fund to VCS; the Doni Solich Family Chair in Ocular Stem Cell Research to VCS; the National Science Foundation to YR; and the Branna and Irving Sisenwein Professorship in Ophthalmology to AS. The funding organizations had no role in the design or conduct of this research.
Version 1. 06/15/2021
Electronic publication
Funding Statement
No change from below
Footnotes
Conflict of interest: AS and GLS are cofounders of and hold equity in HIF Therapeutics Inc. This arrangement has been reviewed and approved by Johns Hopkins University in accordance with its conflict-of-interest policies.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(12):e139202.https://doi.org/10.1172/JCI139202.
Contributor Information
Jing Zhang, Email: jzhan192@jhu.edu.
Yaowu Qin, Email: qinyaowu715@126.com.
Mireya Martinez, Email: mireyamvelez@gmail.com.
Miguel Flores-Bellver, Email: m.flores-bellver@cuanschutz.edu.
Murilo Rodrigues, Email: murilowrj@yahoo.com.br.
Aumreetam Dinabandhu, Email: adinabandhu@umaryland.edu.
Xuan Cao, Email: xuancao1@gmail.com.
Monika Deshpande, Email: monamd@yahoo.com.
Yu Qin, Email: qinyubb@126.com.
Yuan Rui, Email: yrui2@jhmi.edu.
Shaima Salman, Email: ssalman7@jhmi.edu.
Jin Yuan, Email: yuanjincornea@126.com.
Jordan J. Green, Email: green@jhu.edu.
M. Valeria Canto-Soler, Email: valeria.canto-soler@cuanschutz.edu.
Gregg L. Semenza, Email: gsemenza@jhmi.edu.
Silvia Montaner, Email: smontaner@umaryland.edu.
Akrit Sodhi, Email: asodhi1@jhmi.edu.
References
- 1.Krispel C, et al. Ranibizumab in diabetic macular edema. World J Diabetes. 2013;4(6):310–318. doi: 10.4239/wjd.v4.i6.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bressler SB, et al. Exploratory analysis of the effect of intravitreal ranibizumab or triamcinolone on worsening of diabetic retinopathy in a randomized clinical trial. JAMA Ophthalmol. 2013;131(8):1033–1040. doi: 10.1001/jamaophthalmol.2013.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ip MS, et al. Long-term effects of ranibizumab on diabetic retinopathy severity and progression. Arch Ophthalmol. 2012;130(9):1145–1152. doi: 10.1001/archophthalmol.2012.1043. [DOI] [PubMed] [Google Scholar]
- 4.Writing Committee for the Diabetic Retinopathy Clinical Research Network. et al. Panretinal photocoagulation vs intravitreous ranibizumab for proliferative diabetic retinopathy: a randomized clinical trial. JAMA. 2015;314(20):2137–2146. doi: 10.1001/jama.2015.15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Usui-Ouchi A, Friedlander M. Anti-VEGF therapy: higher potency and long-lasting antagonism are not necessarily better. J Clin Invest. 2019;129(8):3032–3034. doi: 10.1172/JCI129862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaauwgeers HG, et al. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris. Evidence for a trophic paracrine relation. Am J Pathol. 1999;155(2):421–428. doi: 10.1016/S0002-9440(10)65138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marneros AG, et al. Vascular endothelial growth factor expression in the retinal pigment epithelium is essential for choriocapillaris development and visual function. Am J Pathol. 2005;167(5):1451–1459. doi: 10.1016/S0002-9440(10)61231-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saint-Geniez M, et al. VEGF expression and receptor activation in the choroid during development and in the adult. Invest Ophthalmol Vis Sci. 2006;47(7):3135–3142. doi: 10.1167/iovs.05-1229. [DOI] [PubMed] [Google Scholar]
- 9.Saint-Geniez M, et al. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci U S A. 2009;106(44):18751–18756. doi: 10.1073/pnas.0905010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, et al. Vascular endothelial growth factor-A: a multifunctional molecular player in diabetic retinopathy. Int J Biochem Cell Biol. 2009;41(12):2368–2371. doi: 10.1016/j.biocel.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 11.McLeod DS, et al. Relationship between RPE and choriocapillaris in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009;50(10):4982–4991. doi: 10.1167/iovs.09-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurihara T, et al. Targeted deletion of Vegfa in adult mice induces vision loss. J Clin Invest. 2012;122(11):4213–4217. doi: 10.1172/JCI65157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grunwald JE, et al. Risk of geographic atrophy in the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2014;121(1):150–161. doi: 10.1016/j.ophtha.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bakri SJ, et al. Intraocular pressure in eyes receiving monthly ranibizumab in 2 pivotal age-related macular degeneration clinical trials. Ophthalmology. 2014;121(5):1102–1108. doi: 10.1016/j.ophtha.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 15.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007;2007(407):cm8. doi: 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- 17.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270(3):1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 18.Paulus YM, Sodhi A. Anti-angiogenic therapy for retinal disease. Handb Exp Pharmacol. 2017;242:271–307. doi: 10.1007/164_2016_78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan C. Hypoxia-inducible factor 3 biology: complexities and emerging themes. Am J Physiol Cell Physiol. 2016;310(4):C260–C269. doi: 10.1152/ajpcell.00315.2015. [DOI] [PubMed] [Google Scholar]
- 20.Kaelin WG., Jr The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem Biophys Res Commun. 2005;338(1):627–638. doi: 10.1016/j.bbrc.2005.08.165. [DOI] [PubMed] [Google Scholar]
- 21.Samanta D, et al. Systems biology of oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. 2017;9(4):10.1002/wsbm.1382. doi: 10.1002/wsbm.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lando D, et al. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16(12):1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahon PC, et al. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001;15(20):2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arany Z, et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci U S A. 1996;93(23):12969–12973. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ebert BL, Bunn HF. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol Cell Biol. 1998;18(7):4089–4096. doi: 10.1128/MCB.18.7.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kallio PJ, et al. Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998;17(22):6573–6586. doi: 10.1093/emboj/17.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Semenza GL, Prabhakar NR. The role of hypoxia-inducible factors in oxygen sensing by the carotid body. Adv Exp Med Biol. 2012;758:1–5. doi: 10.1007/978-94-007-4584-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jee K, et al. Expression of the angiogenic mediator, angiopoietin-like 4, in the eyes of patients with proliferative sickle retinopathy. PLoS One. 2017;12(8):e0183320. doi: 10.1371/journal.pone.0183320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodrigues M, et al. Expression pattern of HIF-1α and VEGF supports circumferential application of scatter laser for proliferative sickle retinopathy. Invest Ophthalmol Vis Sci. 2016;57(15):6739–6746. doi: 10.1167/iovs.16-19513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong X, et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat Commun. 2014;5:4047. doi: 10.1038/ncomms5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babapoor-Farrokhran S, et al. Angiopoietin-like 4 is a potent angiogenic factor and a novel therapeutic target for patients with proliferative diabetic retinopathy. Proc Natl Acad Sci U S A. 2015;112(23):E3030–E3039. doi: 10.1073/pnas.1423765112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sodhi A, Montaner S. Angiopoietin-like 4 as an emerging therapeutic target for diabetic eye disease. JAMA Ophthalmol. 2015;133(12):1375–1376. doi: 10.1001/jamaophthalmol.2015.3723. [DOI] [PubMed] [Google Scholar]
- 33.Rodrigues M, et al. VEGF secreted by hypoxic Müller cells induces MMP-2 expression and activity in endothelial cells to promote retinal neovascularization in proliferative diabetic retinopathy. Diabetes. 2013;62(11):3863–3873. doi: 10.2337/db13-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xin X, et al. Hypoxic retinal Muller cells promote vascular permeability by HIF-1-dependent up-regulation of angiopoietin-like 4. Proc Natl Acad Sci U S A. 2013;110(36):E3425–E3434. doi: 10.1073/pnas.1217091110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Limb GA, et al. In vitro characterization of a spontaneously immortalized human Müller cell line (MIO-M1) Invest Ophthalmol Vis Sci. 2002;43(3):864–869. [PubMed] [Google Scholar]
- 36.Kelly BD, et al. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93(11):1074–1081. doi: 10.1161/01.RES.0000102937.50486.1B. [DOI] [PubMed] [Google Scholar]
- 37.Smith LE, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35(1):101–111. [PubMed] [Google Scholar]
- 38.Yoshida T, et al. Digoxin inhibits retinal ischemia-induced HIF-1alpha expression and ocular neovascularization. FASEB J. 2010;24(6):1759–1767. doi: 10.1096/fj.09-145664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen W, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 2016;539(7627):112–117. doi: 10.1038/nature19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Courtney KD, et al. Phase I dose-escalation trial of PT2385, a first-in-class hypoxia-inducible factor-2α antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J Clin Oncol. 2018;36(9):867–874. doi: 10.1200/JCO.2017.74.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Courtney KD, et al. HIF-2 complex dissociation, target inhibition, and acquired resistance with PT2385, a first-in-class HIF-2 inhibitor, in patients with clear cell renal cell carcinoma. Clin Cancer Res. 2020;26(4):793–803. doi: 10.1158/1078-0432.CCR-19-1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mowat FM, et al. HIF-1alpha and HIF-2alpha are differentially activated in distinct cell populations in retinal ischaemia. PLoS One. 2010;5(6):e11103. doi: 10.1371/journal.pone.0011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Connor KM, et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat Protoc. 2009;4(11):1565–1573. doi: 10.1038/nprot.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bringmann A, et al. Müller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25(4):397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 45.Rattner A, et al. Roles of HIFs and VEGF in angiogenesis in the retina and brain. J Clin Invest. 2019;130(9):3807–3820. doi: 10.1172/JCI126655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rui Y, et al. Reducible branched ester-amine quadpolymers (rBEAQs) codelivering plasmid DNA and RNA Oligonucleotides enable CRISPR/Cas9 genome editing. ACS Appl Mater Interfaces. 2019;11(11):10472–10480. doi: 10.1021/acsami.8b20206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan F, et al. Knock-in human rhodopsin-GFP fusions as mouse models for human disease and targets for gene therapy. Proc Natl Acad Sci U S A. 2004;101(24):9109–9114. doi: 10.1073/pnas.0403149101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kleinman ME, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452(7187):591–597. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerschman R, et al. Effect of high oxygen concentrations on eyes of newborn mice. Am J Physiol. 1954;179(1):115–118. doi: 10.1152/ajplegacy.1954.179.1.115. [DOI] [PubMed] [Google Scholar]
- 50.Kim CB, et al. Revisiting the mouse model of oxygen-induced retinopathy. Eye Brain. 2016;8:67–79. doi: 10.2147/EB.S94447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wiesener MS, et al. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood. 1998;92(7):2260–2268. [PubMed] [Google Scholar]
- 52.Stone J, et al. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci. 1995;15(7 pt 1):4738–4747. doi: 10.1523/JNEUROSCI.15-07-04738.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scott A, et al. Astrocyte-derived vascular endothelial growth factor stabilizes vessels in the developing retinal vasculature. PLoS One. 2010;5(7):e11863. doi: 10.1371/journal.pone.0011863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weidemann A, et al. Astrocyte hypoxic response is essential for pathological but not developmental angiogenesis of the retina. Glia. 2010;58(10):1177–1185. doi: 10.1002/glia.20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lutty GA, McLeod DS. Development of the hyaloid, choroidal and retinal vasculatures in the fetal human eye. Prog Retin Eye Res. 2018;62:58–76. doi: 10.1016/j.preteyeres.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burridge PW, et al. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS One. 2011;6(4):e18293. doi: 10.1371/journal.pone.0018293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.