

Abstract
Hypoxia is a hallmark of solid tumors that promotes cell growth, survival, and metastasis and confers resistance to chemo and radiotherapies. Hypoxic responses are largely mediated by the transcription factors hypoxia-inducible factor 1α (HIF-1α) and HIF-2α. Our work demonstrates that HIF-2α is essential for colorectal cancer (CRC) progression. However, targeting hypoxic cells is difficult, and tumors rapidly acquire resistance to inhibitors of HIF-2α. To overcome this limitation, we performed a small molecule screen to identify HIF-2α–dependent vulnerabilities. Several known ferroptosis activators and dimethyl fumarate (DMF), a cell-permeable mitochondrial metabolite derivative, led to selective synthetic lethality in HIF-2α–expressing tumor enteroids. Our work demonstrated that HIF-2α integrated 2 independent forms of cell death via regulation of cellular iron and oxidation. First, activation of HIF-2α upregulated lipid and iron regulatory genes in CRC cells and colon tumors in mice and led to a ferroptosis-susceptible cell state. Second, via an iron-dependent, lipid peroxidation–independent pathway, HIF-2α activation potentiated ROS via irreversible cysteine oxidation and enhanced cell death. Inhibition or knockdown of HIF-2α decreased ROS and resistance to oxidative cell death in vitro and in vivo. Our results demonstrated a mechanistic vulnerability in cancer cells that were dependent on HIF-2α that can be leveraged for CRC treatment.
Keywords: Metabolism, Oncology
Keywords: Cancer, Cell stress, Hypoxia

Introduction
Colorectal cancer (CRC) is the third most common cancer and one of the leading causes of cancer-related deaths globally (1, 2). Cancer cells expand rapidly, and all solid tumors experience hypoxia due to inadequate vascularization (3). Hypoxia plays a critical role in cancer progression via increasing angiogenesis, glycolysis, apoptotic resistance, therapy resistance, genomic instability, and tumor invasion/metastasis (4–6). Hypoxic responses are transcriptionally controlled by hypoxia-inducible factor 1α (HIF-1α), HIF-2α, and HIF-3α, which are members of the basic helix–loop-helix-PER-ARNT-SIM (bHLH–PAS) family (7). HIFs regulate multiple pathways involved in cell proliferation, survival, apoptosis, migration, metabolism, and inflammation (8, 9). HIF-1α and HIF-2α exhibit distinct roles in CRCs (10–12).
HIF-1α is positively associated with the malignant progression of various tumor entities (13). However, the role of HIF-1α in CRC is controversial, and disruption or constitutive activation of HIF-1α in intestinal epithelial cells did not alter colon adenoma formation (14). In contrast, HIF-2α is essential for CRC growth and progression in cell culture and in vivo (15–18). Activation of intestinal epithelial HIF-2α induces a potent epithelial proinflammatory response by regulating the expression of inflammatory cytokines and chemokines (19). Our recent work has demonstrated an essential role for epithelial HIF-2α–elicited inflammation and regulation of intratumoral iron homeostasis in CRC (16). Recently, PT2385, a selective, potent, and orally active small molecule was shown to selectively inhibit HIF-2α by blocking dimerization with its partner protein, aryl hydrocarbon receptor nuclear translocator (ARNT) (20). PT2385 efficiently inhibits the expression of target genes in clear cell renal cell carcinoma (ccRCC) cells and tumor xenografts (21, 22) and is in phase 2 clinical trials in patients with advanced ccRCC. However, other strategies are needed to target HIF-2α–expressing cancer cells, as resistance to PT2385 arises rapidly (21, 22). Moreover, there is an urgent need to increase clinical benefits of known anticancer therapies by recognizing tumor cell–specific vulnerabilities. Herein, we performed an unbiased screen in tumor enteroids with HIF-2α overexpression with the aim of identifying hypoxic tumor cell–specific vulnerabilities. Interestingly, HIF-2α expression potently sensitized tumor enteroids to ferroptotic activators, such as erastin, RSL3, and sorafenib, and to dimethyl fumarate (DMF). Ferroptosis is a nonapoptotic, iron-dependent form of oxidative cell death (23) characterized by the loss of lipid peroxide repair by glutathione peroxidase 4 (GPX4), an increase in free iron, and the oxidation of polyunsaturated fatty acid–containing (PUFA-containing) phospholipids (24). Activating ferroptosis has emerged as a potent mechanism for targeting cancer cells in vivo (25–27).
DMF is a cell-permeable dimethyl ester of fumaric acid and an FDA-approved drug for the treatment of relapsing forms of multiple sclerosis and psoriasis due to its immunomodulatory properties (28, 29). Numerous studies have shown that DMF has antioxidant and cytoprotective effects in nonmalignant models (28, 30, 31) via its activation of the nuclear factor erythroid 2–related (NF-E2 related) factor 2 (NRF2) pathway (28, 30–33). In parallel, several in vitro studies have suggested that DMF is cytotoxic to a variety of cancer cell lines (34–37). However, the underlying mechanisms have remained obscure.
In the present study, we report that HIF-2α activation is essential in the cellular vulnerability to oxidative cell death. Hypoxia or hypoxic mimetics via HIF-2α stabilization synergize with prooxidants to potentiate an increase in ROS and lipid ROS and a decrease in glutathione (GSH) production, inducing cell death. Mechanistically, we reveal that activation of HIF-2α increases cellular iron to enhance ferroptosis and/or irreversible cysteine oxidation leading to cell death, which can be protected by hydrogen sulfide (H2S) or its precursor, 3-mercaptopyruvate (3-MP). These findings highlight HIF-2α–dependent tumor lethality via iron-dependent lipid and protein oxidation that has implications for the development of novel therapeutics for the improved treatment of CRC.
Results
Drug screen identifies synthetic vulnerability to HIF-2α in tumor enteroids.
Adenomatous polyposis coli (APC) is a tumor-suppressor protein mutated in more than 80% of patients with sporadic CRC (38). The Cdx2-ERT2Cre; Apcfl/fl mouse model enables tamoxifen-inducible deletion of both Apc alleles in intestinal epithelial tissues. These mice were crossed with HIF2αLSL/LSL mice, which harbor oxygen-stable HIF-2α alleles flanked by the loxP-Stop-loxP cassette (Figure 1A and ref. 39). In Cdx2-ERT2Cre; Apcfl/flHIF2αLSL/LSL mice, tamoxifen treatment results in a robust induction of HIF-2α and disrupts Apc specifically in colon epithelial cells. Enteroids were isolated from these 2 mouse models and cultured with a panel of chemotherapeutics; growth was monitored for 5 days (Figure 1A). Colon tumor enteroids overexpressing HIF-2α were resistant to carboplatin, cisplatin, cyclophosphamide, and oxaliplatin compared with enteroids from Cdx2-ERT2Cre; Apcfl/fl mice (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI143691DS1). These data are consistent with the well-known role of HIF signaling in chemo resistance (5). Tumor enteroids from Cdx2-ERT2Cre; Apcfl/fl mice were highly sensitive to drugs such as doxorubicin, mitoxantrone, irinotecan, and eribulin unlike tumor enteroids from Cdx2-ERT2Cre; Apcfl/flHIF2αLSL/LSL mice (Figure 1B). RSL3, sorafenib, erastin, and DMF ranked as the most effective small molecules that significantly reduced the growth of tumor enteroids from Cdx2-ERT2Cre; Apcfl/flHIF2αLSL/LSL in comparison to CDX2-CreERT2Apcfl/fl mice (Figure 1C). Erastin and RSL3 are classic ferroptosis activators (23) that inhibit xCT (encoded by the Slc7a11 gene and is a component of system xC–) and GPX4, respectively. DMF is not a known regulator of ferroptosis and is a cell-permeable mitochondrial derivative that is cytotoxic in several cancer cell lines (Figure 1D). Together, these results suggest that HIF-2α–expressing tumors can be selectively targeted by oxidative stress activators.
Figure 1. Screening of compounds that exhibit reduction in growth of HIF-2α–overexpressing tumor enteroids.

(A) Schematic of enteroids isolated from a sporadic CRC mouse model (Cdx2-ERT2Cre; Apcfl/fl) and CRC HIF-2α–overexpressing mouse model (Cdx2-ERT2Cre; Apcfl/flHIF2αLSL/LSL). (B and C) The log2 fold change in AUC from cell viability dose-response curves for each compound in the library, signifying the most sensitive (sensitivity rank 1) to least sensitive (sensitivity rank 35) compounds (n = 10). (D) Schematic of known oxidative cell death pathways of the 4 most significant compounds from the screen, indicating ferroptosis activators, such as erastin, RSL3, and sorafenib, that inhibit Slc7a11 or GPX4 and induce lipid peroxides. The process of lipid ROS (Li-ROS) accumulation can be inhibited by ferroptosis inhibitors, such as ferrostatins and liproxstatins, which directly eliminate lipid peroxide formation. Small molecules such as DMF are known to mediate oxidative stress and cell death by depletion of GSH.
HIF activation synergizes with ferroptotic activators in CRC cells.
Erastin and RSL3 are classical inducers of ferroptosis that were originally identified in a screen for small molecules that are selectively lethal to cancer cells (23). Recently, ccRCC-derived cell lines were reported to require HIF-2α activation to exhibit vulnerability to ferroptosis (40). We demonstrate that the hypoxic mimetic FG4592 or hypoxia significantly potentiated cell death following treatment of ferroptosis inducers erastin (Supplemental Figure 2) and RSL3 (Supplemental Figure 3) in a HIF-2α–dependent manner (Supplemental Figure 4, A–C). It is important to note that FG4592 treatment alone had no effect on growth in cell lines (Supplemental Figure 4D), whereas hypoxia decreased growth in CRC cells (Supplemental Figure 4E). Expression of 2 lipid genes important in ferroptosis sensitization in renal cancers, hypoxia inducible lipid droplet associated protein (HILPDA) and perilipin 2 (PLIN2), and aberrant generation of lipid ROS were increased following FG4592 treatment (Supplemental Figure 5, A and B), and this increase was HIF-2α dependent (Supplemental Figure 5C), implicating the role of HIF-2α in driving ferroptosis through the accumulation of oxidized lipids. To examine the role of HIF-2α in regulating ferroptosis in vivo, Villin-CreERT2-HIF2αLSL/LSL were crossed with Slc7a11fl/fl mice. Tamoxifen treatment enabled the intestine-specific deletion of Slc7a11 (Supplemental Figure 6A) and overexpression of HIF-2α (Supplemental Figure 6B). The colonic tissue from these mice was analyzed for histological changes 14 days following the last tamoxifen dose (Figure 2A). The deletion of Slc7a11 or overexpression of HIF-2α alone were indistinguishable from what occurred in control littermates (Figure 2B). However, disruption of Slc7a11 in combination with HIF-2α overexpression led to colonic epithelial degeneration and vacuolization (Figure 2B). Lipid peroxide–induced oxidative stress was measured by 4-hydroxy 2-nonenal (4-HNE) staining (Figure 2C). The Villin-CreERT2Slc7a11fl/fl; HIF2αLSL/LSL mice showed a robust increase in histological score (Figure 2D) and 4-HNE intensity (Figure 2E), clearly indicating increased oxidative stress and epithelial cell loss in these mice in comparison with their littermate controls. Furthermore, HILPDA and PLIN2 mRNA levels were significantly increased in Villin-CreERT2-HIF2αLSL/LSL mice compared with littermate controls (Figure 2F). Since iron accumulation is involved in lipid-induced oxidative stress and cell death, we also measured iron levels in liver and intestinal tissue of these mice. We observed higher levels of both liver and intestinal iron in Villin-CreERT2-HIF2αLSL/LSL mice compared with littermate controls (Figure 2G). The liver iron increase was due to increased iron absorption from the small intestine. These data confirm the role of HIF-2α in ferroptosis sensitization in vivo and suggest that drugs that induce ferroptosis could be highly efficacious in killing hypoxic cells.
Figure 2. HIF-2α activation potentiates ferroptosis in vivo.

(A) Schematic of temporal activation of intestinal HIF-2α and deletion of Slc7a11 in the colon following tamoxifen treatment (100 mg/kg). (B) Representative H&E staining and (C) immunohistochemistry analysis showing 4-HNE of colons from a Slc7a11fl/fl, Vil-ERT2Cre; Slc7a11fl/fl, Vil-ERT2Cre; HIF2αLSL/LSL, and Vil-ERT2Cre; Slc7a11fl/fl; HIF2αLSL/LSL mice. Quantitation of histology score (D and E) 4-HNE (n = 3 in each group). One-way ANOVA followed by Tukey’s multiple comparisons test was used for comparison between groups. **P < 0.01; ***P < 0.001; ****P < 0.0001. (F) Quantitative real-time PCR (qRT-PCR) analysis for HILPDA and PLIN2 in HIF-2α+/+ (n = 6) and Vil-ERT2HIF2αLSL/LSL mice (n = 6). (G) Iron levels measured in liver (n = 4) and intestinal tissue (n = 6) in HIF-2α+/+ and Vil-ERT2HIF2αLSL/LSL mice. Data are represented as mean ± SD from 3 independent experiments. P values were determined using unpaired t test. *P < 0.05; **P < 0.01; ***P < 0.001.
HIF activation promotes DMF-induced cell death in CRC cells.
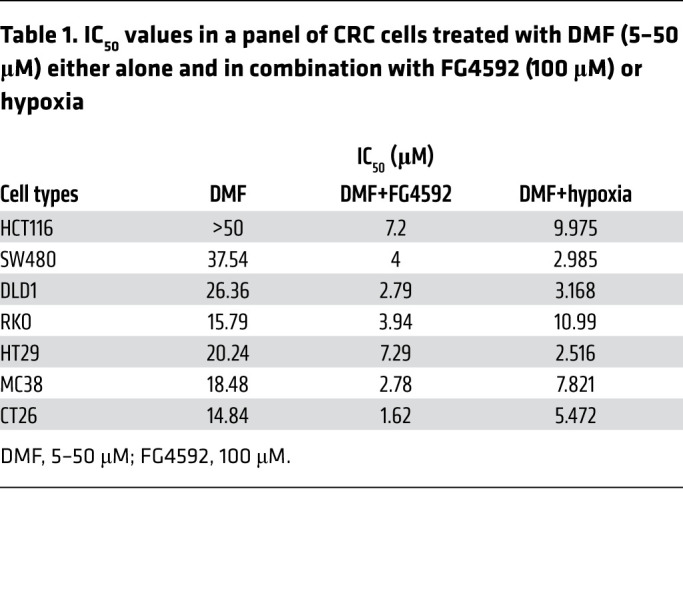
Current ferroptosis inducers have poor in vivo bioavailability in contrast with DMF, an FDA-approved oral drug for the treatment of multiple sclerosis that has fewer side effects than several other drugs (28, 29, 41). Our screen identified DMF as a small molecule effective in reducing the growth of hypoxic tumor enteroids (Figure 1C). A panel of CRC cell lines was treated with DMF either alone or in combination with hypoxia or hypoxia mimetic, FG4592. Our data showed a dose-dependent inhibition of CRC cell growth by DMF (Supplemental Figure 7A). FG4592 potentiated DMF-induced CRC cell death as assessed by MTT and long-term clonogenic survival assays (Figure 3, A–C). Additionally, the cells treated with DMF and cultured in hypoxia were less viable (Figure 3, D and E) and showed decreased cell growth (Supplemental Figure 7B) in comparison with those cultured in normoxia. HIF-2α levels were monitored following treatment and showed increases in FG4592 or DMF and FG4592 (Supplemental Figure 7C). The effect of DMF and FG4592 or hypoxia combination was highly potent in inhibiting CRC cell growth, as confirmed by IC50 (Table 1). Consistent with the erastin and RSL3 data, HIF-2α was essential for promoting DMF-mediated inhibition of CRC cell growth (Figure 3F).
Figure 3. Hypoxia mimetic contributes to DMF-induced growth inhibition in CRC cells.

(A) Cell-growth assay following FG4592 (100 μM) and DMF cotreatment. Error bars represent mean ± SD. Statistical significance was calculated using 1-way ANOVA followed by Tukey’s multiple comparisons test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (B) Representative images and (C) quantitation of colony-forming assays in HCT116 and SW480 cells treated with DMF (25 μM), or FG4592 (100 μM) or cotreated with DMF and FG4592. (D) Representative images and (E) quantitation of colony-forming assays in HCT116 and SW480 cells treated with DMF (25 μM) and cultured under normoxic and hypoxic conditions. Quantitative data are represented as mean ± SD from 3 independent experiments. Statistical significance was calculated using 1-way ANOVA with Tukey’s multiple comparisons. **P < 0.01; ***P < 0.001; ****P < 0.0001. (F) Growth assay of HIF-1α and HIF-2α knockdown HCT116 and SW480 cells treated with DMF alone or in combination with FG4592 (100 μM). Quantitative data are represented as mean ± SD from 3 independent experiments. Statistical significance was calculated using 2-way ANOVA with Tukey’s multiple comparisons. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001
Table 1. IC50 values in a panel of CRC cells treated with DMF (5–50 μM) either alone and in combination with FG4592 (100 μM) or hypoxia.
DMF induces cell death independently of ferroptosis.
Since DMF was effective in decreasing hypoxic cell growth along with other ferroptotic activators (Figure 1C), we assessed whether DMF mediates ferroptotic cell death. For this, HCT116 and SW480 cells were treated with DMF with or without the ferroptosis inhibitors ferrostatin-1 (Fer-1) or liproxstatin-1 (Lip-1) (23, 29). Fer-1 and Lip-1 did not rescue cell death and viability following DMF treatment, whereas RSL3-mediated cell death was rescued by Fer-1 and Lip-1 (Figure 4A and Supplemental Figure 8A). Moreover, DMF only marginally increased lipid ROS in HCT116 cells, and Fer-1 did not prevent this induction, whereas in SW480, DMF did not increase lipid ROS (Figure 4B). In contrast, lipid ROS induction by RSL3 was significantly attenuated by Fer-1 (Figure 4B). DMF can react directly with the antioxidant GSH, leading to decreased NADPH levels and enhanced ROS (42). Consistent with these data, DMF significantly decreased cellular GSH levels so that they were comparable to the levels of erastin and RSL3 (Supplemental Figure 8B). To assess whether the HIF-dependent potentiation of cell death is due to synergizing with compounds that reduce GSH pools, we used buthionine sulfoximine (BSO), which inhibits de novo GSH biosynthesis. DMF, either alone or in combination with BSO, decreased GSH levels in HCT116 and SW480 cells (Supplemental Figure 8, B and C). Treatment with FG4592 alone or in combination with BSO also decreased cellular GSH levels (Supplemental Figure 8, B and C). However, cell viability was not decreased with the cotreatment of BSO and FG4592 (Supplemental Figure 8D), suggesting GSH depletion is not the major mechanism of cell death after DMF and FG4592 cotreatment.
Figure 4. DMF is not a ferroptotic inducer in CRC cells.

(A) HCT116 and SW480 cells were treated with RSL3 (1 and 5 μM) or DMF (25 and 100 μM) alone or in combination with FG4592 (100 μM) with or without ferroptotic inhibitors Fer-1 (0.5 μM) or Lip-1 (1 μM) for 24 hours, and cell death was assayed. Data are represented as mean ± SEM from 3 independent experiments. Statistical significance was calculated using 2-way ANOVA with Tukey’s multiple comparison test. *P < 0.05; **P < 0.01; ***P < 0.001. (B) HCT116 and SW480 cells were treated with RSL3 (2 μM), DMF (50 μM), or in combination with FG4592 (100 μM) with or without Fer-1 (1 μM) for 12 hours. Lipid ROS was determined in these cells through staining with ferroptosis-dependent C11-BODIPY581/591. Data are plotted as the mean ± SD. P values were determined using 1-way ANOVA with Tukey’s multiple comparison. *P < 0.05; **P < 0.01; ****P < 0.0001. (C) Schematic of glycolysis pathway in cells showing DMF, CGP 3466B as inhibitors of GAPDH, and 2-DG as inhibitor of hexokinase. (D) Heatmap showing the relative abundance of glycolytic intermediates in HCT116 and SW480 cells treated with FG4592 (100 μM) or DMF (50 μM) either alone or in combination. (E) HCT116 and SW480 cells were treated with glycolysis inhibitors with or without FG4592, and cell death was assessed using LDH assay.
Fumarate is a TCA cycle metabolite and a potent electrophile (43). DMF inhibits GAPDH via covalent modification of a reactive cysteine and consequent downregulation of aerobic glycolysis (ref. 44 and Figure 4C). We explored the impact of DMF on the metabolome using an liquid chromatography–tandem mass spectrometry–based (LC-MS/MS–based) profiling approach (45, 46). The metabolomics data show an abundance of glycolytic intermediates upstream and downstream of GAPDH in HCT116 and SW480 cells treated with DMF (Figure 4D; full metabolomics data in Supplemental Table 1). Further, and consistent with the well-characterized role of HIF-2α in glycolysis, several glycolytic intermediates were increased with FG4592 treatment. However, cell viability was not decreased with the cotreatment of FG4592 and glycolytic inhibitors (Figure 4E). These results indicate that alternative mechanisms are involved in DMF-mediated cell death of hypoxic cancer cells.
ROS accumulation and iron-toxicity are essential in DMF-induced HIF-2α–mediated cell death.
Our work thus far ruled out ferroptosis, GSH depletion, and inhibition of glycolysis as being relevant to DMF-sensitized cell death. Since DMF is an electrophile, we examined the effect of other electrophiles, dimethyl itaconate (DMI) and 4-octyl itaconate (4-OI) (47), on cell growth. Both DMI and 4-OI decreased cellular growth, which was potentiated by cotreatment with FG4592 (Supplemental Figure 9, A and B).
Supplementation of growth media with the cysteine precursor N-acetyl cysteine (NAC) rescued HCT116 and SW480 cell death and viability of cells treated with DMF with or without FG4592 (Figure 5A and Supplemental Figure 9C). Similar results were observed with DMF treatment and cells maintained in hypoxia (Figure 5B). Interestingly, FG4592 or hypoxia treatment alone increased ROS, as assessed by the cell-permeant 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), which is used as an indicator for ROS (Figure 5, C and D). Cotreatment with DMF potentiated this increase in ROS generation, which was rescued with NAC (Figure 5, C and D). To confirm that the sensitivity toward oxidative cell death is mediated via HIF-2α, shRNA-mediated HIF-1α and HIF-2α knockdown cells were utilized. ROS levels were drastically reduced in HIF-2α knockdown cells (Figure 5E), suggesting the role of HIF-2 in ROS production after DMF treatment. To further confirm the role of HIF-2α, we employed a HIF-2α–specific inhibitor, PT2385 (21, 22), which decreased ROS in FG4592 and DMF-treated CRC cells (Supplemental Figure 9D).
Figure 5. ROS generation and iron accumulation are involved in DMF and FG4592–mediated cell death in CRC cells.

Cell death assay in HCT116 and SW480 cells treated with DMF (25 and 75 μM) (A) cotreated with DMF and FG4592 (100 μM) (B) cultured under hypoxia with or without NAC (5 mM). Data are represented as mean ± SD from 3 independent experiments. Statistical significance was calculated using unpaired t test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. ROS measurements in HCT116 and SW480 cells (C) treated with FG4592 (100 μM), DMF (50 μM), or DMF and FG4592 with or without NAC. (D) Cells treated with DMF and cultured in normoxia and hypoxia with or without NAC. Data are plotted as the mean ± SEM from 3 independent experiments. Statistical significance was calculated using 1-way ANOVA with Tukey’s multiple comparison test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (E) ROS measurements in shRNA-mediated HIF-1α, HIF-2α knockdown, and non–target scrambled HCT116 and SW480 cells treated with DMF (50 μM) either alone or in combination with FG4592 (100 μM). Statistical significance was calculated using 2-way ANOVA with Tukey’s multiple comparison. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (F) Heatmap showing the relative abundance of mitochondrial metabolites in FG4592-treated (100 μM) and DMF-treated (50 μM) HCT116 and SW480 cells. (G) Cell death and (H) ROS measurements using FG4592 (100 μM) and DMF (75 μM) either alone or under cotreated conditions in the presence of normal iron (control) and low iron. Statistical significance was calculated using unpaired t test. **P < 0.01; ***P < 0.001; ****P < 0.0001.
Since HIF activation can lead to changes in mitochondrial metabolism (48) and ROS production, we analyzed whether DMF or FG4592 treatment induced changes in mitochondrial metabolite pools. However, significant changes in the levels of mitochondrial metabolites were not seen, suggesting that HIF-2α influences cellular ROS via other mechanisms (Figure 5F; full metabolomics data in Supplemental Table 1). RNA-Seq data from a HIF-2α overexpression mouse model (49) analyzed for oxidant-generating enzymes revealed upregulation of lysyl oxidase (LOX) and cyclooxygenase (COX) (encoded by Ptgs gene) (Supplemental Figure 10A). However, the viability of HCT116 and SW480 cells treated with DMF and FG4592 was only slightly improved using LOX and COX inhibitors (Supplemental Figure 10B).
We have previously demonstrated that HIF-2α is critical for cellular iron uptake, which leads to ROS generation via the Fenton reaction (50). Consistent with our previous work, FG4592 and hypoxia increased cellular iron in CRC cells (Supplemental Figure 10, C and D). The increase in iron levels after hypoxia or FG4592 treatment was dependent on HIF-2α (Supplemental Figure 10D). Interestingly, the cell death mediated by DMF and FG4592 was rescued in low-iron versus control media (Figure 5G). Moreover, ROS production induced by DMF, FG4592, or DMF and FG4592 cotreatment was attenuated in low-iron medium compared with control (Figure 5H). Together, these data suggest a mechanism linking iron toxicity via HIF-2α and vulnerability to oxidative stress.
Protective persulfidation via H2S can rescue DMF and HIF-2α–induced cell death.
Since metal catalyst oxidation is a central mechanism for DNA damage (51), we checked the expression of γH2AX, a sensitive marker of DNA damage and repair (52). Western blot analysis revealed a robust increase in the expression of γH2AX in HCT116 and SW480 cells treated with DMF; however, the increase was not significant when cells were cotreated with DMF and FG4592 (Supplemental Figure 11A). We next assessed whether irreversible protein oxidation is involved in DMF and FG4592 induced cell death. H2S can protect against overoxidation of cysteine thiols via persulfidation, a posttranslational modification (53) (Figure 6A). We therefore assessed whether Na2S or 3-MP, a substrate for mercaptopyruvate sulfurtransferase (54), by increasing intracellular H2S, protect cells from DMF and FG4592–induced cell death. Both 3-MP and Na2S rescued cell viability following cotreatment with DMF and FG4592 (Supplemental Figure 11, B and C). Interestingly, H2S did not rescue cells from erastin- or RSL-3–induced cell death (Supplemental Figure 11, D and E). To rule out that the protective role of H2S was not due to DMF depletion resulting from the nucleophilic addition of the sulfide anion on DMF, Na2S and 3-MP were added to fresh medium 16 hours after initiation of cell death by DMF and FG4592 or DMF and hypoxia (Figure 6B). Na2S and 3-MP protected against cell death even under these conditions (Figure 6, C and D, and Supplemental Figure 11F). Further, intracellular ROS levels induced by DMF or DMF and FG4592 were decreased upon supplementation with 3-MP and Na2S (Figure 6E). These data are consistent with the model that the potentiation of HIF-induced cell death by DMF involves oxidative protein damage, which is protected by H2S.
Figure 6. H2S can prevent irreversible protein oxidation and rescue cell death mediated by DMF and FG4592.

(A) Schematic showing H2S effects on DMF-mediated cell death. The addition of sulphur group to proteins prevents the cell death mediated by DMF. (B) Schematic showing treatment regime of HCT116 and SW480 cells with DMF, FG4592, 3MP, and Na2S. (C and D) Cell death in HCT116 and SW480 cells treated with DMF (25 and 100 μM) (C) or cotreated with DMF and FG4592 (100 μM) (D) cultured under normoxic and hypoxic conditions with or without Na2S (300 μM). Data are represented as mean ± SD from 3 independent experiments. Statistical significance was calculated using unpaired t test. *P < 0.05; **P < 0.01; ***P < 0.001. (E) ROS measurements in HCT116 and SW480 cells treated with DMF (50 μM) or DMF in combination with FG4592 (100 μM) either alone or in addition with 3MP (5 mM) and Na2S (300 μM) for 16 hours. Data are represented as mean ± SD from 3 independent experiments. Statistical significance was calculated using 2-way ANOVA with Tukey’s multiple comparison. *P < 0.05; **P < 0.01; ***P < 0.001.
FG4592 potentiates DMF-mediated CRC cell death in vivo.
We next assessed the in vivo efficacy of DMF and FG4592 cotreatment in established CRC tumors. For this, HCT116, SW480, and DLD1 cells were implanted subcutaneously into the flanks of immunocompromised mice and allowed to establish for 10 days prior to DMF and FG4592 treatment (Figure 7A). CRC cell xenografts exhibited profound growth inhibition upon DMF and FG4592 treatment; both tumor volume (Figure 7B) and tumor weight (Figure 7C) were significantly reduced in all 3 xenografts treated with DMF and FG4592 compared with those that received vehicle or treatment with the individual drugs. Tumor cell proliferation as assessed by the BrdU incorporation assay was also decreased in HCT116, SW480, and DLD1 xenografts with DMF and FG4592 cotreatment (Figure 7D). However, tumor fumarate levels were not significantly different between the DMF only and the DMF and FG4592 cotreatment groups in all 3 CRC-xenograft mice (Supplemental Figure 12A). The tumors in response to different treatments did not show any major histological changes (Supplemental Figure 12B). However, the percentage of TUNEL-positive apoptotic cells was increased in all 3 xenografts cotreated with DMF and FG4592 (Figure 7E). DMF+FG4592 was not cytotoxic to normal tissues in mice, as confirmed by histological analysis of colon tissue and normal serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels (Supplemental Figure 12, C and D). Consistent with the known roles of hypoxia in erythropoiesis (55, 56), erythropoietin, hematocrits, hemoglobin, and red blood cells were increased with FG4592 treatment alone or in combination with DMF (Supplemental Figure 12E). Together, these data demonstrate that hypoxic tumor cells are highly vulnerable to DMF treatment, highlighting a potential therapeutic window.
Figure 7. DMF and FG4592 potentiate CRC cell death in vivo.

(A) Schematic of xenografts in vivo study. HCT116, SW480, and DLD1 cells were injected subcutaneously into both flanks of C57BL/6 mice (n = 10 for each group). After visible formation of tumor at day 10, the mice were subjected to DMF diet (300 mg/kg of chow) and FG4592 (10 mg/kg of mouse weight). (B) Tumor volume, (C) tumor weight, (D) tumor proliferation, and (E) tumor apoptosis in HCT116, SW480, and DLD1 xenograft mice. Data are represented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (differences between untreated mice and between treated groups). One-way ANOVA with Tukey’s multiple comparison test was used for calculating statistical significance.
DMF-mediated CRC cell death in vivo is HIF-2α dependent.
To confirm the role of HIF-2α in DMF-mediated CRC cell death in vivo, we utilized HIF-2α knockdown HCT116 cells. Stable non–target scrambled and HIF-2α knockdown HCT116 cells were injected subcutaneously into both flanks of immunocompromised mice and allowed to grow for 10 days prior to DMF and FG4592 treatment (Figure 8A). HIF-2α knockdown cells were resistant to DMF and FG4592 treatment. Cells expressing scrambled shRNA showed a significant reduction in tumor volume and weight in mice treated with DMF+FG4592, whereas the HIF-2α knockdown cells were completely resistant (Figure 8, B and C). Similarly, tumor proliferation and apoptosis were not altered in HIF-2α knockdown cells following DMF+FG4592 treatment (Figure 8, D and E). These data demonstrate that HIF-2α activation increases vulnerability to oxidative cell death in vivo.
Figure 8. DMF-mediated CRC cell death in vivo is HIF-2α dependent.

(A) Schematic of HIF-2α knockdown xenograft in vivo study. shRNA-mediated HIF-2α knockdown and non–target scrambled HCT116 cells were injected subcutaneously into both flanks of C57BL/6 mice (n = 8 for each group). After visible formation of tumor at day 10, the mice were subjected to the DMF diet (300 mg/kg of chow) and FG4592 (10 mg/kg of mouse body weight). (B) Tumor volume, (C) tumor weight, (D) tumor proliferation, and (E) tumor apoptosis in HIF-2α knockdown and non–target scrambled HCT116 xenograft mice. Data are represented as mean ± SEM. ***P < 0.001; ****P < 0.0001 (differences between scrambled and HIF-2α knockdown cells for DMF+FG treatment). Unpaired t test was used for calculating statistical significance. (F) Schematic outlining the role of HIF-2α mediating vulnerability to oxidative cell death. HIF-2α mediated iron toxicity and accumulation of lipid ROS, which synergized with ferroptotic activators to enhance CRC cell death. HIF-2α also increased cellular iron and synergize with cellular oxidants such as DMF to enhance irreversible cysteine oxidation and cell death.
Discussion
Our work demonstrates that HIF-2α, via an increase in cellular iron, is an important source of ROS, which coupled to posttranslational protein modification by DMF or lipid-ROS induction by ferroptosis-inducing agents, leads to cell death (Figure 8F). HIF-2α is the major transcriptional regulator of cellular iron levels (50). Our work now connects the HIF-2α–dependent increase in cellular iron with ROS, which enhances the vulnerability of the proteome to oxidative damage and, in the presence of the electrophile DMF, stimulates cell death. A central role of basal HIF-2α is in increasing cellular ROS, which is mimicked by FG4592 in a HIF-2α–dependent manner, and we now demonstrate that this is essential in oxidative cell death pathways.
Apart from leading to oxidative stress, ROS can also transduce signals by reversibly modifying the redox state of cysteine residues in proteins. Cysteine thiols in proteins can be oxidized to sulfenic acid (RSOH), sulfinic acid (RSO2H), and sulfonic acid (RSO3H), which are associated with pathophysiological processes (57). H2S-mediated persulfidation protects proteins from irreversible oxidation (58), as the cysteine persulfide or its oxidation products (e.g., Cys-S-S-sulfonate), can be reduced to a cysteine thiolate by thioredoxin (59, 60). Fumarate itself can covalently modify cysteines, and numerous protein targets of this posttranslational modification have been identified (43). Although considerable heterogeneity in the proteins targeted by fumarate is seen in different cell lines, proteomic analysis reveals that tumor suppressors and signaling proteins are a highly enriched class of modified proteins. Our data demonstrate that protein oxidation might be a major mechanism of cell death following DMF because DMF and FG4592/hypoxia cotreatment is abrogated by protective persulfidation by H2S. While a single bolus administration of H2S was used in the present study, it is important to note that CRCs are exposed to high H2S levels derived from microbial metabolism. High concentrations of H2S inhibit the electron transport chain, and repeated long-term exposure is growth restricting (61). The present work suggests that H2S might confer protection against oxidative stress to tumors, and further work is in needed to define its role in CRCs in vivo.
In vivo disruption of Slc7a11 with a simultaneous increase in HIF-2α led to increased histological intestinal damage and the hallmarks of ferroptosis in normal intestine. Previous work has demonstrated that the susceptibility to ferroptosis following HIF-2α activation is not only iron dependent, but also requires lipid reprogramming via HIF-2α (40). However, pharmacological activation of HIF-2α with DMF did not lead to heightened intestinal injury in normal tissue, illustrating a considerable therapeutic window. Moreover, it was elegantly shown by Taniguchi et al. that activation of HIF-2α was protective in radiation-induced damage (62) and HIF-2α–dependent iron oxidation and cell death were not observed (62). Elevated ROS is a hallmark of most cancers, but need to be precisely balanced by antioxidant genes for tumor progression. There are numerous studies that have highlighted the inhibitory role of HIF-1α toward ROS production. HIF-1α regulates redox homeostasis by regulating the levels of NADPH and GSH. HIF-1α is also known to promote mitochondrial-selective autophagy and thus lower the mitochondrial mass, which suppresses the oxidation of both glucose and fatty acids and decreases mitochondrial ROS production under hypoxic conditions (63). In contrast, in our study we have identified strong association of HIF-2α with ROS production. HIF-2α activation tips the balance toward ROS-induced cell death and thus provides specificity to cancer cells compared with normal tissues.
We show that HIF-2α potentiates cell death via ferroptosis activators and DMF, and both pathways require iron. Interestingly, these oxidative pathways have distinct and nonoverlapping mechanisms of cell death. HIF-2α potentiation of ferroptosis can be completely rescued with lipid ROS scavengers Lip-1 and Fer-1, but these compounds have no effects on DMF- or DMF+FG4592-mediated cell death. Similarly, reversing protein oxidation via H2S can rescue DMF-mediated cell death, but does not protect against ferroptosis induced by erastin or RSL3. It is unclear how the HIF-2α–induced increase in iron-dependent ROS generation regulates distinct cell death pathways; this hence becomes an active area for future studies.
HIF-2α inhibitors that are in clinical trials have shown promise, although acquired resistance to them is a major limitation for long-term efficacy (21, 22). Our work suggests caution if the PT2385 inhibitor is used in an adjuvant setting with known chemotherapeutics that rely on ROS generation. While HIF-2α inhibitors can be used to target cancers that rely on HIF-2α for progression, we demonstrate mechanisms by which HIF-2α activation can be therapeutically leveraged as a synthetic lethality. FG4592 is currently in clinical trials for treating anemia in chronic kidney disease (ClinicalTrials.gov NCT01750190). DMF is clinically used for relapsing multiple sclerosis and psoriasis. Our in vivo study on a mouse xenograft–CRC model shows the potential utility of this drug combination against CRC. However, it is important to note that DMF by itself does not reduce tumor growth in vivo, which could be related to the degree of hypoxia and HIF-2α activation in tumor tissue. Therefore, HIF-2α stabilizers such as FG4592 that induce robust HIF-2α induction are needed to increase the antitumoral efficacy of DMF. Erastin and RSL3 have been used as ferroptosis inducers in mostly in vitro experiments because of the limited in vivo bioavailability and reduced metabolic stability. However, studies have used imidazole ketone erastin (IKE), which is a derivative of erastin with nanomolar potency, increased solubility, and metabolic stability. IKE has been shown to inhibit tumor growth in a diffuse large B cell lymphoma mouse model (64). In conclusion, our study has unmasked the role of HIF-2α in driving the synthetic lethality of hypoxic CRC cells to oxidative stress-inducing compounds such as DMF and exposed the potential for exploiting this intrinsic vulnerability for chemotherapeutic development.
Methods
Animal experiments.
For all experiments, male and female mice, 6 to 8 weeks of age, were used. All mice are on a C57BL/6 background maintained in standard cages in a light- and temperature-controlled room and were allowed a standard chow diet and water ad libitum. Villin-CreERT2 HIF2αLSL/LSL and Apcfl/fl mice have been previously described (16, 39). These mice were crossed with the colon-specific Cre to generate the CDX2-CreERT2-Apcfl/fl; HIF2αLSL/LSL mice. Correctly targeted embryonic stem (ES) cells in which exon 3 of Slc7a11 was flanked by Lox-P (Slc7a11fl/fl) sites were generated by the International Mouse Phenotyping Consortium. Slc7a11fl/fl mice were also crossed to Villin-CreERT2 mice and further crossed to HIF2αLSL/LSL mice to generate Villin-CreERT2-Slc7a11fl/fl;HIF2αLSL/LSL mice. For all experiment, littermate controls were used and the recombinase was activated by i.p. injection with tamoxifen in corn oil (100 mg/kg) for 3 consecutive days; mice were euthanized 1 week or 2 week following the last tamoxifen treatments.
Cell lines and reagents.
HCT116, SW480, DLD1, RKO, HT29, MC38, and CT26 cells were obtained from ATCC and grown in DMEM with l-glutamine, d-glucose, and sodium pyruvate (Gibco, Thermo Fisher Scientific) supplemented with 10% heat-inactivated FBS and 1% antibiotic-antimycotic mix (Invitrogen). All cells were maintained in a humidified environment at 37°C and 5% CO2 in a tissue-culture incubator. For hypoxic treatment, cells were maintained in a humidified environment at 37°C and 1% O2 and 5% CO2 tissue-culture incubator. DMF, BSO, BAPN, and nimesulide were purchased from Sigma-Aldrich. Erastin, RSL3, FG4592, Fer-1, Lip-1, and NAC were purchased from Cayman Chemical. 3-MP was purchased from Santa Cruz Biotechnology Inc.
C11-BODIPY lipid ROS measurement.
HCT116 or SW480 cells (1 × 106) were seeded in 12-well plates and allowed to adhere overnight at 37°C. The day before the experiment, cells were treated with DMSO (vehicle), erastin (5 μM), RSL3 (2 μM) or DMF (50 μM), or Fer-1 (1 μM) with or without FG4592 (100 μM) and incubated for 12 hours at 37°C. Cells were harvested using PBS-EDTA (5 mM), buffer, washed once with HBSS, suspended in HBSS containing 5 μM C11-BODIPY (Thermo Fisher), and incubated at 37°C for 30 minutes. Cells were pelleted, washed, and resuspended in HBSS. Fluorescence intensity was measured on the FITC channel using the Beckman Coulter MoFlo Astrios. A minimum of 20,000 cells were analyzed per condition. Data were analyzed using FlowJo software (Tree Star). Values are expressed as MFI.
ROS detection assay.
The cell-permeable free radical sensor carboxy-H2DCFDA (Invitrogen) was used to measure intracellular ROS levels. For HIF-2α knockdown (shHIF-2α) and nontarget (sh scrambled) HCT116 and SW480 cells, CellROX Deep Red Reagent (Thermo Fisher) was used due to the limitation of plasmid (pGipZ) backbone, which has internal green fluorescence. Cells treated with DMF (50 μM), FG4592 (100 μM), or subjected to hypoxia (1% O2) with or without NAC (5 mM) were harvested by ice-cold PBS-EDTA (5 mM) buffer and incubated with 10 μM carboxy-H2DCFDA or 2.5 μM CellROX dye in PBS at 37°C for 30 to 45 minutes. The cells were washed, resuspended in PBS, and analyzed using the Beckman Coulter MoFlo Astrios flow cytometer. Data were analyzed using FlowJo software. Values are expressed as the percentage of cells positive for DCF fluorescence or MFI in the case of CellROX dye.
MTT cell viability assay.
From 2000 to 3000 cells were seeded in a 96-well plate and allowed to adhere overnight at 37°C. The next day, cells were treated with FG4592 (100 μM) or for 16 hours in cotreatment conditions. The cells were then treated with different agents, as indicated in Figure 3, A and F, and Supplemental Figures 2B, 3B, 8, A and D, 9, A–C, 10B, and 11, B–F. In hypoxia experiments, treated cells were cultured in a hypoxic incubator for 3 days. Twenty-four hours following treatment, a day 0 reading was taken. Following the day 0 read, the corresponding treatment and readings were taken every 24 hours for a 72-hour assay. Cells were incubated for 45 minutes with Thiazolyl Blue Tetrazolium Bromide (MilliporeSigma), then solubilized with dimethyl sulfoxide. Absorbance was taken at 570 nm. All reads were taken in technical triplicate.
Clonogenic assay.
Cells were plated in a 6-well plate in biological triplicate at 300 to 600 cells per well in 2 mL of media. After 48 hours, cells were treated with different reagents, as mentioned in the legends for Figure 3, B and D, and Supplemental Figures 2C and 3C. In the case of hypoxia experiments, cells were treated with DMF and subjected to hypoxia for 48 hours, after which they were maintained in ambient oxygen environments. Assays were concluded after 10 to 15 days by fixing in cold 10% buffered formalin for 10 minutes and staining with 0.5% crystal violet, 20% methanol solution for 30 minutes. Colonies were manually counted via a study-blinded observer.
LDH cell-death assay.
Cell death was measured using the LDH Cytotoxicity Assay Kit from Takara Bio. From 5000 to 10,000 cells were seeded in a 96-well plate and allowed to adhere overnight at 37°C. The next day, cells were treated with FG4592 (100 μM) for 16 hours under cotreatment conditions. The cells were then treated with different agents, as indicated in Figure 4A, Figure 5, A, B, G, and H, Figure 6, C and D, and Supplemental Figures 2E, 3E, and 11F. In the case of hypoxia experiments, cells were treated with different reagents and cultured under hypoxic conditions (1% O2). Supernatants were harvested after 48 hours of treatment, and a media-only control and Triton X-100 (Sigma-Aldrich) were also included. The supernatant was incubated with LDH detection reagent for 30 minutes per the manufacturer’s instructions. Absorbance was taken at 490 nm. All reads were taken in technical triplicate. Cytoxicity/cell death was calculated using the formula mentioned per the manufacturer’s protocol.
Metabolomics.
Cells were plated at 500,000 to 1 million cells per well in 6-well plates. After 24 hours, cells were treated with DMF (50 μM) and FG4592 (100 μM) for another 24 hours. After treatment, cells were washed once with ice-cold 1× PBS, followed by incubation with ice-cold 80% methanol on dry ice for 10 minutes. Cells were then scraped, and the polar metabolite supernatant was collected in a 1.5 mL tube on dry ice. Samples were clarified via centrifugation at 15,000g for 10 minutes at 4°C. The supernatant was then transferred to a fresh tube and stored at –80°C until analysis. Protein concentration was determined by processing a parallel well/dish for each sample and used to normalize metabolite fractions across samples. Based on protein concentrations, cell solutions were transferred to a fresh micro-centrifuge tube and lyophilized using a SpeedVac concentrator. Dried metabolite pellets were resuspended in a 45 μL 50:50 methanol/water mixture for LC-MS/MS analysis. Data were collected using previously published parameters (45, 65). QqQ data were preprocessed with Agilent MassHunter Workstation Quantitative Analysis Software (B0700). Additional analyses were postprocessed for further quality control in the programming language R. Finally, each metabolite abundance level in each sample was divided by the median of all abundance levels across all samples for proper comparisons, statistical analyses, and visualization among metabolites. Statistical significance test was determined by a 2-tailed t test with a significance threshold level of 0.05.
GSH assay.
HCT116 and SW480 cells plated in a 96-well plate were treated with different agents or vehicle for 24 hours. The GSH concentrations were determined using the GSH-Glo Glutathione Assay Kit (Promega) per the manufacturer’s instructions. The luminescence-based assay is based on the conversion of a luciferin derivative into luciferin in the presence of GSH, catalyzed by GSH S-transferase. Luciferase expression was then measured on a Synergy Mx Microplate Reader (BioTek). The signal generated in a coupled reaction with firefly luciferase is proportional to the amount of GSH present in the sample. The concentration was determined through a standard curve using GSH standard solution provided with the kit.
Western blotting.
HCT116 and SW480 cells were seeded at 1 m/mL density in a 6-well plate in triplicate for each condition and allowed to adhere overnight. Cells were lysed with RIPA assay buffer with added protease (1:100 dilution; MilliporeSigma) and phosphatase (1:100 dilution; Thermo Fisher Scientific) inhibitors. After cell lysis, solubilized proteins were resolved on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membrane, blocked with 5% milk in TBST, and immunoblotted with the indicated primary antibodies made at 1:1000 dilution in blocking solution for HIF-1α (catalog ab179483, Abcam), HIF-2α (catalog Bethyl-A700-003, Bethyl Lab), γH2AX (catalog ab2893, Abcam), and β-actin (catalog 66009-1-Ig, Proteintech).
Histology and 4-HNE staining.
Colonic tissues or tumor tissues were rolled and fixed with PBS-buffered formalin for 24 hours, followed by embedding in paraffin. Sections of 5 μM were stained for H&E and mounted with Permount Mounting Medium (Thermo Fisher Scientific). For 4 hydroxy-2-noneal/4-HNE staining, paraffin-embedded tissue sections were subjected to antigen retrieval, followed by blocking with 5% goat serum in PBS, and probed with primary antibody against 4-HNE (1:200 dilutions, BS6313R, Bioss). Sections were then washed 3 times with PBST and were incubated with HRP-conjugated anti-rabbit IgG (1:500 dilution, catalog 7074S, Cell Signaling Technology) for 1 hour. Sections were then washed 3 times with PBST and incubated with DAB substrate solution to sufficiently cover them. After the sample color turned brown, the reaction was stopped by distilled water, and dehydration steps were carried. The slides were mounted using Permount Mounting Medium.
Tumor enteroid screening.
Mouse colonic crypts were isolated using a previously described method (66). Colon was isolated from CDX2-ERT2Cre; Apcfl/fl HIF2αLSL/LSL and CDX2-ERT2Cre; Apcfl/fl mice and cut into 1 cm pieces. Tissue was incubated in 10 mM DTT for 15 minutes at room temperature. Tissues were rinsed with DPBS supplemented with gentamicin and primocin. Tissue was incubated with slow rotation at 4°C for 75 minutes in 8 mM EDTA. EDTA was removed, and tissue was washed 3 times with cold DPBS to release crypts. Isolated crypts were spun down and collected in cold LWRN medium. Crypts were then plated in Matrigel (Corning) in 96-well culture plates in LWRN media and imaged using ImageXpress Micro (Molecular Devices). Enteroids were treated with the indicated drugs/inhibitors (10 μM), and growth was monitored after 5 days.
RNA isolation and RNA-Seq and qPCR analysis.
HCT116 and SW480 cells were seeded at 1 m/mL density in a 6-well plate in triplicate for each condition and allowed to adhere overnight. Cells were then treated with FG4592 (100 μM) for 16 hours. RNA was isolated from cultured tissue or using the TRIzol chloroform extraction method. RNA was reverse transcribed using MMLV reverse transcriptase (Thermo Fisher). Quantitative PCR (qPCR) analysis was done using indicated primers (hHILPDA FP: AAGCATGTGTTGAACCTCTACC, RP: TGTGTTGGCTAGTTGGCTTCT; hPLIN2 FP: ATGGCATCCGTTGCAGTTGAT, RP: GGACATGAGGTCATACGTGGAG; mHILPDA FP: TTTCCTTCTGAGGATCTAGC, RP: GACTCCATCACTCTAACAAAG; mPLIN2 FP: GACAGGATGGAGGAAAGACTGC, RP: GGTAGTCGTCACCACATCCTTC) and Radiant Green qPCR Master Mix (Alkali Scientific Inc.). RNA-Seq analysis was performed using data from HIF-2α–overexpressing colons (NCBI’s Gene Expression Omnibus database (GEO) GSE173363).
Xenograft studies.
Immunocompromised, 6- to 8- or 8- to 10-week-old mice of both sexes were maintained in the facilities of the Unit for Laboratory Animal Medicine (ULAM) at the University of Michigan under specific pathogen–free conditions. For subcutaneous xenograft studies, HCT116, SW480, and DLD1 or sh scrambled and sh HIF-2α HCT116 cells were trypsinized and 2 million cells were implanted into the lower flanks. All treatments began on day 10 after tumors became visible. DMF (300 mg) was mixed in animal chow (1 kg), and this diet was fed to mice. FG4592 was injected i.p. for 7 days continuously at a concentration of 10 mg/kg of mouse body weight. Subcutaneous tumor size was measured with digital calipers at the indicated time points. Tumor volume (V) was calculated as V = 1/2(length × width2). At the endpoint, mice were sacrificed and tumors were excised. The final tumor volume and weight were measured, and tissue was used for proliferation and apoptosis assay.
Statistics.
Data are represented as mean ± SEM unless otherwise indicated. Data are from 3 independent experiments measured in triplicate unless otherwise stated in the figure legend. For statistical analyses, unpaired t tests were conducted to assess the differences between 2 groups. One-way or 2-way ANOVA was used for multiple treatment conditions followed by Tukey’s post hoc test. A P value of less than 0.05 was considered to be statistically significant. All statistical tests were carried out using Prism 8 software (GraphPad).
Study approval.
All animal studies were carried out in accordance with Institute of Laboratory Animal Resources guidelines and approved by the University Committee on the Use and Care of Animals at the University of Michigan (IACUC protocol number: PRO00008292).
Author contributions
YMS and RS conceived the project. RS, SRM, NKD, SAK, PS, SS, AA, and RK performed experiments and analyzed data. KPO, RB, and CAL provided critical reagents for the study and experimental direction. YMS and RS wrote the manuscript with critical input from all authors.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (R01CA148828, R01CA245546, and R01DK095201 to YMS; R37CA237421 and R01CA248160 to CAL; R01CA244931 and R01CA215607 to CAL and KPO); the University of Michigan Comprehensive Cancer Center (UMCCC) Core Grant (P30CA046592 to CAL and YMS; R35GM130183 to RB); the University of Michigan GI SPORE Molecular Pathology and Biosample Core (P50CA130810); the Center for Gastrointestinal Research (DK034933); and the Department of Defense (CA171086 to YMS) and SAK was supported by an NIH F31 Predoctoral Fellowship. PS was supported by an NIH/NCI T32 Training Grant in Cancer Biology (5T32CA009676-27). Metabolomics studies performed at the University of Michigan were supported by NIH grant DK097153, the Charles Woodson Research Fund, and the UM Pediatric Brain Tumor Initiative. The authors thank Megan Radyk (Rogel Cancer Center) and Wesley Huang (Cellular and Molecular Biology, University of Michigan-Medical School) for their help in troubleshooting, suggestions, and advice in 4-HNE staining.
Version 1. 04/29/2021
In-Press Preview
Version 2. 06/15/2021
Electronic publication
Footnotes
Conflict of interest: RB is a paid member of the scientific advisory board of Apneo Therapeutics and owns equity in the company. CAL is an inventor on patents pertaining to Kras-regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting GOT1 as a therapeutic approach (Methods for diagnosing and treating oncogenic Kras-associated cancer, US patent no. 2015126580-A1; Targeting the glutamine to pyruvate pathway for treatment of oncogenic Kras-associated cancer, US patent no. 20190136238; international patent no.: WO2013177426-A2).
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(12):e143691.https://doi.org/10.1172/JCI143691.
Contributor Information
Rashi Singhal, Email: srashi@umich.edu.
Sreedhar R. Mitta, Email: mittasreedhar@gmail.com.
Nupur K. Das, Email: nkdas@med.umich.edu.
Samuel A. Kerk, Email: skerk@umich.edu.
Peter Sajjakulnukit, Email: pnsajja@umich.edu.
Sumeet Solanki, Email: sumeets@med.umich.edu.
Anthony Andren, Email: acandren@umich.edu.
Roshan Kumar, Email: roshank@umich.edu.
Kenneth P. Olive, Email: kenolive@columbia.edu.
Ruma Banerjee, Email: rbanerje@umich.edu.
Costas A. Lyssiotis, Email: clyssiot@med.umich.edu.
Yatrik M. Shah, Email: shahy@umich.edu.
References
- 1.Bhandari A, et al. Colorectal cancer is a leading cause of cancer incidence and mortality among adults younger than 50 years in the USA: a SEER-based analysis with comparison to other young-onset cancers. J Investig Med. 2017;65(2):311–315. doi: 10.1136/jim-2016-000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rawla P, et al. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019;14(2):89–103. doi: 10.5114/pg.2018.81072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu S, et al. Hypoxia promotes colorectal cancer cell migration and invasion in a SIRT1-dependent manner. Cancer Cell Int. 2019;19:116. doi: 10.1186/s12935-019-0819-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krock BL, et al. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. 2011;2(12):1117–1133. doi: 10.1177/1947601911423654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33(4):207–214. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. 2000;35(2):71–103. doi: 10.1080/10409230091169186. [DOI] [PubMed] [Google Scholar]
- 7.Wang GL, et al. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20(1):51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imamura T, et al. HIF-1alpha and HIF-2alpha have divergent roles in colon cancer. Int J Cancer. 2009;124(4):763–771. doi: 10.1002/ijc.24032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keith B, et al. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramakrishnan SK, Shah YM. Role of intestinal HIF-2α in health and disease. Annu Rev Physiol. 2016;78:301–325. doi: 10.1146/annurev-physiol-021115-105202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshimura H, et al. Prognostic impact of hypoxia-inducible factors 1alpha and 2alpha in colorectal cancer patients: correlation with tumor angiogenesis and cyclooxygenase-2 expression. Clin Cancer Res. 2004;10(24):8554–8560. doi: 10.1158/1078-0432.CCR-0946-03. [DOI] [PubMed] [Google Scholar]
- 14.Xue X, et al. Activation of HIF-1α does not increase intestinal tumorigenesis. Am J Physiol Gastrointest Liver Physiol. 2014;307(2):G187–G195. doi: 10.1152/ajpgi.00112.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma X, et al. Hypoxia-inducible factor 2α (HIF-2α) promotes colon cancer growth by potentiating Yes-associated protein 1 (YAP1) activity. J Biol Chem. 2017;292(41):17046–17056. doi: 10.1074/jbc.M117.805655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue X, et al. Iron uptake via DMT1 integrates cell cycle with JAK-STAT3 signaling to promote colorectal tumorigenesis. Cell Metab. 2016;24(3):447–461. doi: 10.1016/j.cmet.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue X, et al. Hypoxia-inducible factor-2α activation promotes colorectal cancer progression by dysregulating iron homeostasis. Cancer Res. 2012;72(9):2285–2293. doi: 10.1158/0008-5472.CAN-11-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shay JE, et al. Inhibition of hypoxia-inducible factors limits tumor progression in a mouse model of colorectal cancer. Carcinogenesis. 2014;35(5):1067–1077. doi: 10.1093/carcin/bgu004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Triner D, Shah YM. Hypoxia-inducible factors: a central link between inflammation and cancer. J Clin Invest. 2016;126(10):3689–3698. doi: 10.1172/JCI84430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scheuermann TH, et al. Artificial ligand binding within the HIF2alpha PAS-B domain of the HIF2 transcription factor. Proc Natl Acad Sci U S A. 2009;106(2):450–455. doi: 10.1073/pnas.0808092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen W, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 2016;539(7627):112–117. doi: 10.1038/nature19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho H, et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature. 2016;539(7627):107–111. doi: 10.1038/nature19795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dixon SJ, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stockwell BR, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lang X, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019;9(12):1673–1685. doi: 10.1158/2159-8290.CD-19-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–274. doi: 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Badgley MA, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368(6486):85–89. doi: 10.1126/science.aaw9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Z, et al. Dimethyl fumarate for multiple sclerosis. Cochrane Database Syst Rev. 2015;(4):CD011076. doi: 10.1002/14651858.CD011076.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stangel M, Linker RA. Dimethyl fumarate (BG-12) for the treatment of multiple sclerosis. Expert Rev Clin Pharmacol. 2013;6(4):355–362. doi: 10.1586/17512433.2013.811826. [DOI] [PubMed] [Google Scholar]
- 30.Ashrafian H, et al. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 2012;15(3):361–371. doi: 10.1016/j.cmet.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pitarokoili K, et al. Dimethyl fumarate ameliorates lewis rat experimental autoimmune neuritis and mediates axonal protection. PLoS One. 2015;10(11):e0143416. doi: 10.1371/journal.pone.0143416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papadopoulou A, et al. Dimethyl fumarate for multiple sclerosis. Expert Opin Investig Drugs. 2010;19(12):1603–1612. doi: 10.1517/13543784.2010.534778. [DOI] [PubMed] [Google Scholar]
- 33.Scannevin RH, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341(1):274–284. doi: 10.1124/jpet.111.190132. [DOI] [PubMed] [Google Scholar]
- 34.Xie X, et al. Dimethyl fumarate induces necroptosis in colon cancer cells through GSH depletion/ROS increase/MAPKs activation pathway. Br J Pharmacol. 2015;172(15):3929–3943. doi: 10.1111/bph.13184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loewe R, et al. Dimethylfumarate impairs melanoma growth and metastasis. Cancer Res. 2006;66(24):11888–11896. doi: 10.1158/0008-5472.CAN-06-2397. [DOI] [PubMed] [Google Scholar]
- 36.Yamazoe Y, et al. Dimethylfumarate inhibits tumor cell invasion and metastasis by suppressing the expression and activities of matrix metalloproteinases in melanoma cells. Cell Biol Int. 2009;33(10):1087–1094. doi: 10.1016/j.cellbi.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 37.Yang M, et al. The emerging role of fumarate as an oncometabolite. Front Oncol. 2012;2:85. doi: 10.3389/fonc.2012.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng Y, et al. Mutant KRAS promotes hyperplasia and alters differentiation in the colon epithelium but does not expand the presumptive stem cell pool. Gastroenterology. 2011;141(3):1003–1013. doi: 10.1053/j.gastro.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solanki S, et al. Temporal induction of intestinal epithelial hypoxia-inducible factor-2α is sufficient to drive colitis. Am J Physiol Gastrointest Liver Physiol. 2019;317(2):G98–G107. doi: 10.1152/ajpgi.00081.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou Y, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617. doi: 10.1038/s41467-019-09277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kappos L, et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;372(9648):1463–1472. doi: 10.1016/S0140-6736(08)61619-0. [DOI] [PubMed] [Google Scholar]
- 42.Sullivan LB, et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51(2):236–248. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perez M, et al. Heterogeneous adaptation of cysteine reactivity to a covalent oncometabolite. J Biol Chem. 2020;295(39):13410–13418. doi: 10.1074/jbc.AC120.014993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kornberg MD, et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science. 2018;360(6387):449–453. doi: 10.1126/science.aan4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee HJ, et al. A large-scale analysis of targeted metabolomics data from heterogeneous biological samples provides insights into metabolite dynamics. Metabolomics. 2019;15(7):103. doi: 10.1007/s11306-019-1564-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halbrook CJ, et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019;29(6):1390–1399. doi: 10.1016/j.cmet.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swain A, et al. Comparative evaluation of itaconate and its derivatives reveals divergent inflammasome and type I interferon regulation in macrophages. Nat Metab. 2020;2(7):594–602. doi: 10.1038/s42255-020-0210-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Semenza GL. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta. 2011;1813(7):1263–1268. doi: 10.1016/j.bbamcr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Triner D, et al. Epithelial hypoxia-inducible factor 2α facilitates the progression of colon tumors through recruiting neutrophils. Mol Cell Biol. 2017;37(5):e00481-16. doi: 10.1128/MCB.00481-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah YM, et al. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 2009;9(2):152–164. doi: 10.1016/j.cmet.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cadet J, Davies KJA. Oxidative DNA damage & repair: an introduction. Free Radic Biol Med. 2017;107:2–12. doi: 10.1016/j.freeradbiomed.2017.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siddiqui MS, et al. Persistent γH2AX: a promising molecular marker of DNA damage and aging. Mutat Res Rev Mutat Res. 2015;766:1–19. doi: 10.1016/j.mrrev.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 53.Filipovic MR, et al. Chemical biology of H2S signaling through persulfidation. Chem Rev. 2018;118(3):1253–1337. doi: 10.1021/acs.chemrev.7b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yadav PK, et al. Thioredoxin regulates human mercaptopyruvate sulfurtransferase at physiologically-relevant concentrations. J Biol Chem. 2020;295(19):6299–6311. doi: 10.1074/jbc.RA120.012616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang GL, Semenza GL. Molecular basis of hypoxia-induced erythropoietin expression. Curr Opin Hematol. 1996;3(2):156–162. doi: 10.1097/00062752-199603020-00009. [DOI] [PubMed] [Google Scholar]
- 56.Wang GL, Semenza GL. General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A. 1993;90(9):4304–4308. doi: 10.1073/pnas.90.9.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Reest J, et al. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat Commun. 2018;9(1):1581. doi: 10.1038/s41467-018-04003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zivanovic J, et al. Selective persulfide detection reveals evolutionarily conserved antiaging effects of S-sulfhydration. Cell Metab. 2019;30(6):1152–1170. doi: 10.1016/j.cmet.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wedmann R, et al. Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem Sci. 2016;7(5):3414–3426. doi: 10.1039/C5SC04818D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Izquierdo-Alvarez A, et al. Differential redox proteomics allows identification of proteins reversibly oxidized at cysteine residues in endothelial cells in response to acute hypoxia. J Proteomics. 2012;75(17):5449–5462. doi: 10.1016/j.jprot.2012.06.035. [DOI] [PubMed] [Google Scholar]
- 61.Libiad M, et al. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J Biol Chem. 2019;294(32):12077–12090. doi: 10.1074/jbc.RA119.009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taniguchi CM, et al. PHD inhibition mitigates and protects against radiation-induced gastrointestinal toxicity via HIF2. Sci Transl Med. 2014;6(236):236ra64. doi: 10.1126/scitranslmed.3008523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Samanta D, Semenza GL. Maintenance of redox homeostasis by hypoxia-inducible factors. Redox Biol. 2017;13:331–335. doi: 10.1016/j.redox.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol. 2019;26(5):623–633. doi: 10.1016/j.chembiol.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yuan M, et al. Ex vivo and in vivo stable isotope labelling of central carbon metabolism and related pathways with analysis by LC-MS/MS. Nat Protoc. 2019;14(2):313–330. doi: 10.1038/s41596-018-0102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dame MK, et al. Human colonic crypts in culture: segregation of immunochemical markers in normal versus adenoma-derived. Lab Invest. 2014;94(2):222–234. doi: 10.1038/labinvest.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.