

Abstract
Chronic cellular stress associated with neurodegenerative disease can result in the persistence of stress granule (SG) structures, membraneless organelles that form in response to cellular stress. In Huntington’s disease (HD), chronic expression of mutant huntingtin generates various forms of cellular stress, including activation of the unfolded protein response and oxidative stress. However, it has yet to be determined whether SGs are a feature of HD neuropathology. We examined the miRNA composition of extracellular vesicles (EVs) present in the cerebrospinal fluid (CSF) of patients with HD and show that a subset of their target mRNAs were differentially expressed in the prefrontal cortex. Of these targets, SG components were enriched, including the SG-nucleating Ras GTPase-activating protein-binding protein 1 (G3BP1). We investigated localization and levels of G3BP1 and found a significant increase in the density of G3BP1-positive granules in the cortex and hippocampus of R6/2 transgenic mice and in the superior frontal cortex of the brains of patients with HD. Intriguingly, we also observed that the SG-associated TAR DNA-binding protein 43 (TDP43), a nuclear RNA/DNA binding protein, was mislocalized to the cytoplasm of G3BP1 granule–positive HD cortical neurons. These findings suggest that G3BP1 SG dynamics may play a role in the pathophysiology of HD.
Keywords: Neuroscience
Keywords: Neurodegeneration
Introduction
Huntington’s disease (HD) is a progressive, inherited neurodegenerative disease caused by an expanded CAG repeat in exon 1 of the huntingtin (HTT) gene (1), encoding an expanded polyglutamine repeat tract within the HTT protein. Although HD is characterized by pathology that most prominently affects neurons of the striatum, human autopsy studies show that other brain areas are also impacted.
The expression of mutant HTT gives rise to cellular stress responses, including oxidative and endoplasmic reticulum (ER) stress, potentially as a neuroprotective strategy against cellular toxicity (2–5), which converge on key translation factors and interfere with general protein synthesis to optimize cell survival and stress recovery (6). One of these translation factors, eIF2α, is inactivated in HD due to ER stress. This alteration is proposed to play an important role in striatal cell death (7). Overall, these studies suggest a direct connection between the regulated expression of translation factors, but any evidence of how these are regulated is not understood.
HD appears to involve progressive spreading of neuropathology (8), and recent findings suggest that the spreading effect that is characteristic of many neurodegenerative disorders could be mediated by extracellular vesicles (EVs) (9, 10). EVs that contain RNA, protein, and lipids can transfer their contents to other cells and mediate intra- and intercellular signaling (11, 12) and could facilitate the spread of pathology between brain regions (9). One type of EVs are exosomes, 40 to 150 nm vesicles of multivesicular body (MVB) origin secreted by several cell types, including neural cells, that can be isolated from biofluids, including the cerebrospinal fluid (CSF) (13). Due to the lack of consensus on specific exosome markers and the recommendations put forth by the International Society of Extracellular Vesicles (14), we will use the generic term EV for cell-secreted nanovesicles.
The composition of EVs is highly regulated. Comparisons between in cellulo RNA content and EV RNAs demonstrate selective enrichment of specific EV RNA (15), suggesting regulation of RNA loading into EVs (16, 17). MicroRNAs, small RNA molecules (~22 nucleotides) that regulate posttranscriptional gene expression by acting as guide molecules to promote the degradation or translational repression of their target mRNAs, are enriched in EVs. The human genome encodes approximately 2000 miRNAs and it is predicted that they collectively regulate one third of the genes in the genome (18). Given that environmental stressors, including heat shock and hypoxia, can modify the miRNA composition of EVs (19), it is plausible that disease-mediated cellular stress, such as that caused in neurodegenerative disease, can modify the miRNA composition of CSF EVs.
Cell-to-cell shuttling of miRNAs via EVs is a critical mediator of transcriptional regulation in recipient cells (9, 20). There is evidence that selected groups of miRNAs are altered in HD cells and tissues (21–24), which suggests that miRNA dysregulation might be involved in gene expression changes detected in the HD brain, and in turn affect cellular function. A recent study identified an EV miRNA secreted from the choroid plexus, a tissue located in the ventricles, which produces the majority of the CSF, that regulates adult neurogenesis at the subependymal zone by repressing translation of neural fate determinants (25). Thus, EV miRNAs secreted into the CSF of patients with HD could be taken up by brain tissues situated near the ventricular zone and regulate the translation of selected proteins, thereby playing a role in the propagation of pathology in the brains of patients with HD.
To investigate the composition of EV miRNAs secreted into the CSF, we isolated EVs from HD and control CSF to assess changes in packaged miRNAs. Among the genes targeted by dysregulated miRNAs are stress granule (SG) components, including the Ras GTPase-activating protein-binding protein 1 (G3BP1), a critical effector of SG assembly (26) and central node of the core SG network (27) in eIF2α-mediated mechanisms of SG formation. Cells respond to stress signals through formation of SGs, cytoplasmic assemblies of protein, and RNAs that form in response to stressors such as hypoxia, heat-shock, and sodium arsenite, which all inhibit translation initiation (28). We examined whether this enrichment of SG targeting miRNAs in CSF could be reflected by SG pathology in HD tissues. We evaluated R6/2 mouse and human HD postmortem brain tissue for altered G3BP1 granule dynamics and found that there is a marked increase in G3BP1 granules in the superior frontal cortex of both R6/2 and human brains. This increase was particularly noted in pyramidal neurons of the superior frontal cortex, and a similar pattern of immunoreactivity was detected in the human parietal cortex and hippocampus. Intriguingly, we also observed TAR DNA-binding protein 43 (TDP43) mislocalization from having exclusively nuclear staining in control tissue to nuclear and cytoplasmic staining in HD brain, similar to that observed in ALS/FTD, where persistent SG formation is also observed (29).
Results
Characterization of HD and control CSF extracellular vesicles.
Because EV-packaged miRNAs distributed via the CSF can alter gene expression (9) and have potential to serve as biomarkers (30), we investigated whether differential miRNA packaging into CSF EVs occurs to alter the miRNA profile of patients with HD compared with unaffected individuals. EVs were isolated by membrane affinity column centrifugation from 10 HD patient and 10 control CSF samples, and RNA contents extracted for miRNA sequencing. CSF samples, provided through HDClarity, were acquired by lumbar puncture (see Methods). Patient demographic characteristics are summarized in Table 1. Given that the profile of CSF miRNAs varies between vesicular and nonvesicular fractions, and that the distribution of their mode of transport can differ between normal and pathological conditions (31, 32), we employed a membrane affinity-based approach to isolate miRNAs from CSF EVs, which ensures that nonvesicular CSF miRNAs are washed out prior to final elution. To characterize the size distribution of the EV fractions collected, we performed fluorescent nanoparticle tracking analysis (F-NTA) on a subset of 5 HD and control CSF samples. This NTA method prevents the inclusion of protein aggregates, membrane fragments, and background particles present in heterogeneous biofluid samples. Each sample was measured in triplicate, and videos of data collection (Supplemental Videos 1–20; supplemental material available online with this article; https://doi.org/10.1172/JCI140723DS1) were analyzed to give the mean, mode, and estimated concentration for each particle size (Supplemental Figure 1). Because EV subtypes are generally characterized by size, and the CSF is likely to contain a heterogeneous mixture of EVs, we analyzed EV particle concentration by EV size increments of 25 nm. We did not detect a significant difference in concentration of any EV size subtype between HD and control CSF samples (Figure 1A). Similarly, we did not detect a significant difference in mode diameter size, or overall particle concentration (Figure 1, B and C).
Table 1. Clinical features of patient CSF samples used for EV miRNA extraction and next generation sequencing studies.
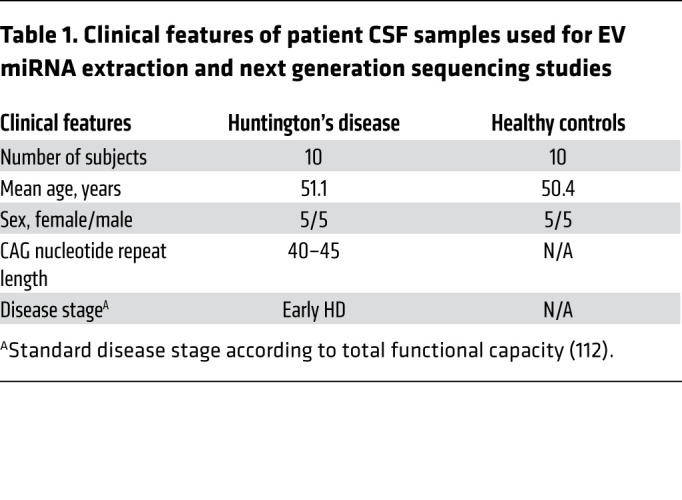
Figure 1. Characterization of CSF EV concentration and size using NTA.

(A) A subset of CSF samples (n = 5 HD, 5 control) was used to determine the size distribution of EVs using F-NTA. CSF EV particle concentration was characterized by EV size subgroups in increments of 25 nm (2-way ANOVA, Bonferroni’s multiple comparisons test, P > 0.05), as well as by (B) mode size (Student’s t test, unpaired, 2-tailed, P > 0.05), and (C) concentration (Student’s t test, unpaired, 2-tailed, P > 0.05). Data are representative of 1 independent experiment performed in triplicate with 5 HD and 5 control samples. Error bars depict mean ± SEM.
Cerebrospinal fluid extracellular vesicles from patients with HD contain miRNAs that target SG-associated genes.
During the miRNA Library Kit construction process, each individual miRNA molecule was tagged with a Unique Molecular Index (UMI). Following sequencing and trimming, reads were analyzed for the presence of UMIs and an average of approximately 10 million reads was generated per mapped sample. Differential expression analysis of the HD versus control samples was carried out using DESeq2. We did not detect any statistically significant differences after correcting for multiple comparisons using the Benjamini-Hochberg method. A subsequent sample size analysis (33) was carried out using a coefficient of variation estimated from this DESeq2 differential expression analysis, which suggested that 143 samples per group would be needed to observe significant differences of about a 2-fold change in expression. This calculation suggested a limitation in the ability to identify statistically significant expression changes. However, we sought to investigate the potential functional relevance of the CSF EV miRNAs detected in our studies by overlapping the predicted gene targets of the miRNAs detected in CSF EVs with a publicly available data set of genes that are differentially expressed (DEGs) in the prefrontal cortex of patients with HD (34), a brain region proximal to the CSF which has the potential to serve as the recipient of CSF EV cargo. Our reasoning here is that miRNAs in the CSF may be coming from different cell types and thus some miRNAs could be very highly expressed in locally released EVs to impart biology in close-proximity target cells, all the while being undetectable from bulk CSF isolation.
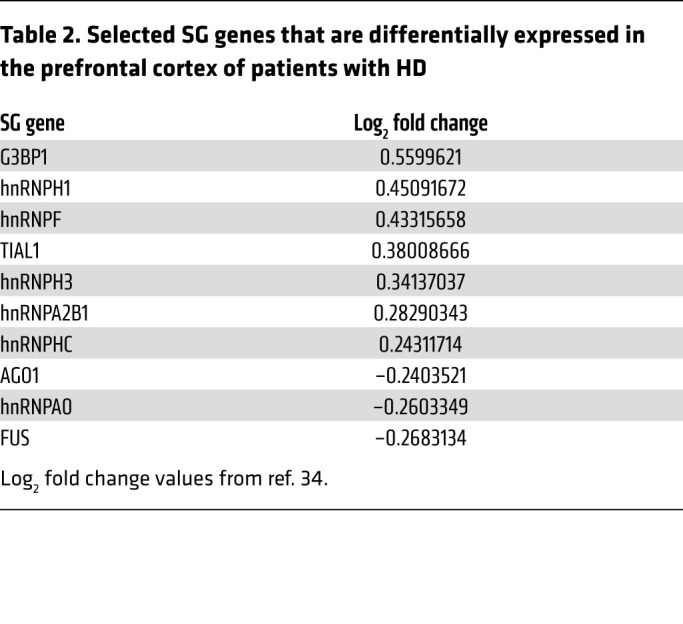
Previous studies have integrated miRNA and mRNA expression profiles to better understand miRNA–mRNA interactions in specific biological contexts (35, 36). Similarly, using the results from the DESeq2 differential expression analysis of CSF EV miRNAs, we generated a list of 22 differential miRNAs (Supplemental Table 1) with a P value less than 0.05 before correcting for multiple comparisons, and another list of 59 miRNAs (Supplemental Table 2) with log2FC values larger than a magnitude of 2 (81 miRNAs total) (Figure 2A). We next performed a target analysis on the 81 miRNAs and generated a list of predicted mRNA targets. The list of mRNA targets was then overlapped with a data set of DEGs from the prefrontal cortex of patients with HD (34), and the number of targets ranged from 72 to 2509 DEGs for each miRNA. Since the expression level of a miRNA is negatively correlated to the expression level of its target gene, we filtered out miRNAs whose fold changes were not the inverse of their target genes 50% or more of the time. This resulted in a list of 41 miRNAs that target DEGs in the HD prefrontal cortex, and whose differential expression in HD CSF EVs is negatively correlated to at least 50% of their prefrontal cortex DEG targets (Supplemental Table 3, GO enrichment analysis of predicted gene targets in Supplemental Table 4). A high number of the miRNAs targeted the SG gene G3BP1, which is a key SG nucleator, acting as the molecular switch that triggers phase separation during SG formation (27). Therefore, we used this filtering approach to evaluate whether SG-related genes are targets of these miRNAs detected in CSF EVs. In the context of SG component regulation, miRNAs may either directly repress the translation of SG mRNAs, or indirectly induce the translation of SG mRNAs by silencing upstream negative regulators of SG components. We asked whether SG genes are significantly enriched in the list of gene targets belonging to these 41 miRNAs, and found that 120 of 4689 CSF EV miRNA gene targets belong to the list of 464 mammalian SG genes (18), which is significantly higher than expected by chance (P = 0.002) (Figure 2B). G3BP1 was one of the SG genes that fit these criteria and is upregulated in the HD prefrontal cortex, together with other SG genes including TIAL1, FUS, and various hnRNP RNA binding proteins (Table 2). Of note, one of the 41 miRNAs was miR-1322, identified as a potential prodromal biomarker for HD in CSF samples containing both vesicular and extravesicular miRNAs (37). The majority of miR-1322 binding sites are located in their targets’ coding domain sequences (CDS), many of which code for polyglutamine repeats, and include HTT (38). Overall, these results suggest that miRNAs packaged in vesicles may serve as regulators of stress response genes. As an initial step to validate whether they do indeed target G3BP1, we investigated whether miRNA overexpression is sufficient for the inhibition of G3BP1-positive granules in vitro.
Figure 2. EV miRNAs in the CSF of patients with HD target SG-related mRNAs that are differentially expressed in the prefrontal cortex of patients with HD.

(A) Workflow used to identify CSF EV miRNAs with likelihood of functional relevance based on overlap with RNA sequencing data from the prefrontal cortex of patients with HD (34). (B) Overlap of 4689 CSF EV mRNA targets and 464 mammalian SG genes (74) is significantly higher than what is expected based on a genome larger than 21,000 genes (P < 0.002 using Fisher’s exact test).
Table 2. Selected SG genes that are differentially expressed in the prefrontal cortex of patients with HD.
G3BP1 protein levels and G3BP1-positive granule density are reduced in 293T cells with the overexpression of miRNAs that are predicted to target G3BP1.
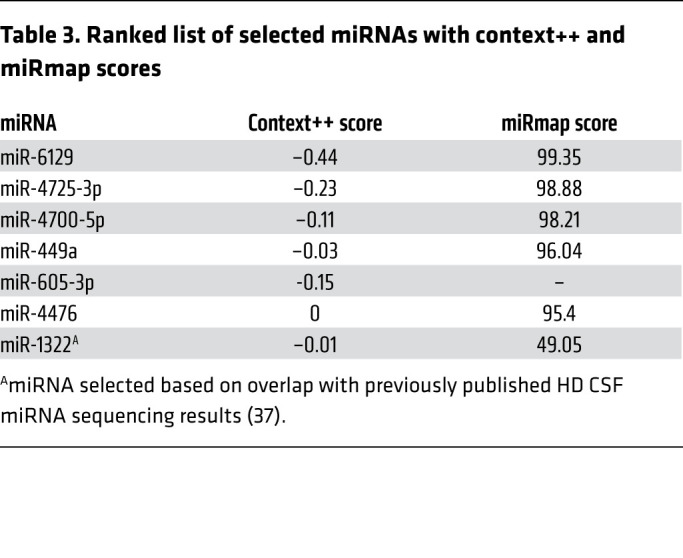
To determine whether the miRNAs predicted to target G3BP1 could in fact regulate mRNA and protein expression of G3BP1, we tested sets of miRNA mimics in human immortalized 293T cells. The 293T cells have a high transfection efficiency and are commonly employed to investigate SG dynamics in vitro (39). The filtered CSF EV miRNAs obtained from patient CSF were ranked based on predicted strength of miRNA repression on G3BP1, or G3BP1 seed strength, using TargetScan and miRmap (40, 41). We created a composite list of ranked miRNAs with highest seed strength toward G3BP1 (Figure 3 and Table 3) and selected the following miRNAs to carry out transfection studies in vitro: 4 miRNAs that were upregulated (Set 1: miR-6129, miR-4725-3p, miR-4700-5p, miR-449a) and 3 that were downregulated (Set 2: miR-605-3p, miR-4476, and miR-1322) in HD CSF EVs. There is evidence that combinatorial miRNA overexpression can achieve greater specificity and minimize off-target effects (42, 43). Therefore, to test the effect of these miRNAs on G3BP1 protein expression, we overexpressed locked nucleic acid (LNA) miRNA mimics in the 293T cells using a combinatorial approach. 293T cells were transfected with either a negative control miRNA, Set 1 miRNAs, or Set 2 miRNAs, and protein expression was evaluated by Western analysis. Protein expression of G3BP1 was significantly reduced in Set 1–treated cells (P = 0.0190, Supplemental Figure 2, A and B; see complete unedited blots in the supplemental material), and trended lower in cells treated with Set 2 but was not statistically significant, potentially because these 3 mRNAs had a lower combined G3BP1 seed strength compared with the miRNA cocktail consisting of 4 miRNAs. Although protein loading levels were normalized to total protein load, as an additional control we measured hnRNPA2/B1 protein expression, which was not predicted to be a cotarget of the miRNA cocktail based on seed strength scores. Indeed, hnRNPA2/B1 protein expression did not change with miRNA treatment (Supplemental Figure 2, A and C–E). To investigate the temporal dynamics mediated by Set 1 miRNAs, we used 293T cells that express G3BP1 as a fusion protein with GFP (293T-G3BP1-GFP; ref. 44) and monitored overall GFP signal using the Incucyte S3 imaging system over a period of 36 hours in the presence of the set of microRNA mimics with the strongest effect (Set 1) or nontargeting miRNA negative control. Cells transfected with the Set 1 miRNA mimics had significantly lower G3BP1-GFP levels at the 11, 12, 15, 17, and 25 hour time points when compared with cells transfected with the nontargeting miRNA negative control (P < 0.0001; Supplemental Figure 2, F and G). Of note, there were small frame shifts in the imaging plane that resulted in a small population of cells not being imaged repeatedly, which could explain the difference in statistical significance between time points. We also performed an ordinary 2-way ANOVA with Bonferroni’s multiple comparison, which showed significant differences at all time points after 6 hours (P < 0.0001).
Figure 3. Selection of G3BP1-targeting miRNAs for overexpression studies in HEK293T cells.

Workflow of miRNA ranking, and selection, based on predicted strength of miRNA repression on G3BP1, or G3BP1 seed strength.
Table 3. Ranked list of selected miRNAs with context++ and miRmap scores.
We hypothesized that treatment with the Set 1 miRNA cocktail would result in lower SG seed formation (45) and thus decrease SG density upon cellular stress induction. SGs can be induced in mammalian cells as a result of sodium arsenite treatment, a form of oxidative stress (46, 47). To test whether the miRNA cocktail treatments have an effect on SG formation in vitro, we repeated the transfection experiments in 293T cells using both sets of miRNA mimics and subjected them to sodium arsenite treatment to induce SG formation, detected by immunofluorescence with an antibody against G3BP1. To quantitate SGs in 3D space, we employed a masking method using the Imaris software that allowed for the detection of punctate structures over diffuse background signal. Specifically, the Imaris surface rendering tool was used to create a mask of G3BP1 SGs in vitro (Figure 4A). Stressed cells that were treated with the 4-miRNA cocktail in Set 1 had a significantly lower SG density (P = 0.0026) compared with cells transfected with a negative control miRNA. Similarly, stressed cells treated with the 3-miRNA cocktail in Set 2 also had a significantly lower SG density compared with the negative control treated cells (P = 0.0294) (Figure 5, A and B, and Supplemental Figure 3). These findings suggest that miRNAs identified in CSF EVs can modulate G3BP1 expression and associated G3BP1 granule biology, and that SG formation in HD may be dysregulated.
Figure 4. Quantitation of G3BP1 SGs using the Imaris 3D masking method.

(A) SGs were induced in vitro with SA treatment, and detected by immunofluorescence with an antibody against G3BP1 (green). The Imaris surface rendering tool was used to detect G3BP1 SG puncta (gray). Scale bar: 10 μm. (B) Using in vitro–induced SGs, parameters were set for the detection of G3BP1 granules in brain tissue. Z stacks were obtained at a thickness of 0.5 μm per slice, and analyzed using the surface rendering tool for quantitation of G3BP1 puncta. The number of puncta was normalized to the number of nuclei (DAPI) per frame. (C) The same pipeline was applied to detect G3BP1 granules in brain tissue, allowing for the quantitation of G3BP1 puncta and not diffuse G3BP1 background signal. Original magnification ×100.
Figure 5. G3BP1-mediated stress granule induction is regulated by miRNAs in 293T cells treated with sodium arsenite.

(A) G3BP1 immunofluorescence (green) of unstressed cells and cells stressed with sodium arsenite (SA). Cells were transfected with either a negative control miRNA (Neg. control), a 3-miRNA cocktail (Set 1), or a 4-miRNA cocktail (Set 2) (n = 3 per condition). Scale bar: 40 μm. (B) Quantitation of SG density (normalized to DAPI-stained nuclei) in both stressed and unstressed conditions suggests that treatment with SA resulted in a significant SG density increase within each condition (2-way ANOVA with Sidak’s multiple comparison test, *P < 0.0001; **P = 0.0283; ***P = 0.0025). Furthermore, SG density is significantly decreased in Set 1 and Set 2 treated cells compared with cells treated with the negative control miRNA (2-way ANOVA with Sidak’s multiple comparison test, #P = 0.0026; ##P = 0.0294). Data are representative of at least 3 independent experiments and analysis was done using 3 frames per condition, per replicate (n = 3 for each set). Error bars depict mean ± SEM.
Increased G3BP1 SG density and immunoreactivity in the R6/2 mouse cortex and hippocampus.
Based on our finding that G3BP1 is a target of CSF EVs from patients with HD, the notion that HD induces a form of chronic stress, and that SGs have been detected in other neurodegenerative diseases using established SG markers (29, 48, 49), we investigated whether G3BP1-associated granules are present as a consequence of chronic mutant HTT expression in vivo. We evaluated whether G3BP1 granule formation could be detected in brain tissue from 12-week-old R6/2 mice, which express a transgene encoding human amino terminal exon1 and have a rapidly progressing phenotype (50). First, to quantitate granules in 3D space, we employed a masking method using the Imaris surface rendering tool as above using SA-induced SGs in 293T cells (Figure 4A) and developed the parameters to detect G3BP1 granules in brain tissue (Figure 4, B and C), allowing for the quantitation of G3BP1 puncta and not diffuse G3BP1 background. Using this method on 12-week-old R6/2 mouse brain sections, localization and intensity of the G3BP1 protein and granule density were evaluated by immunofluorescence and confocal microscopy. A statistically significant increase in G3BP1 immunoreactivity and granule density was detected in the cortex (P = 0.021 and 0.001, respectively; Figure 6, A–C), and of granule density in region CA1 of the hippocampus (P = 0.028; Figure 6, D–F). We did not detect any significant differences in G3BP1 staining or density in the striatum at the 12- or 8-week time points (Supplemental Figures 4 and 5).
Figure 6. Increased G3BP1 granule density in the 12-week R6/2 cortex and hippocampus.

(A and D) G3BP1 immunoreactivity (green) is higher in the R6/2 cortex and hippocampus CA1 region compared with the nontransgenic controls (boxed in red). Images shown are the same. (B and E) High magnification images of cortical and hippocampal CA1 regions suggest that G3BP1 immunoreactivity varies between neural cell subtypes. (C) G3BP1 granule density and G3BP1 immunoreactivity are significantly higher in the R6/2 cortex (Student’s t test, unpaired, 2-tailed, *P = 0.0014; **P = 0.0210), calculated using Imaris image analysis software surface tool and CellProfiler, respectively, and normalized to the number of nuclei per frame (DAPI in blue). (F) The same analysis was used to analyze the CA1 region of the hippocampus (boxed in red in D), which led to the detection of significantly higher granule density in the R6/2 (Student’s t test, unpaired, 2-tailed, ***P = 0.0285), but not G3BP1 immunoreactivity. Immunofluorescence was repeated at least 3 times and quantitation was done for representative samples from each group using 4 frames per mouse brain (n = 3 R6/2; 4 NT). Error bars depict mean ± SEM. Scale bars: 500 μm (A); 10 μm (B, D); 200 μm (C).
Because HTT aggregation has been implicated in the fibrillation of the SG marker TIA1 in R6/2 hippocampus (51, 52), we assessed the potential colocalization of G3BP1-positive granules with HTT at 12 weeks. We first tested the EM48 antibody, which recognizes HTT inclusions (53), but did not detect G3BP1 colocalization with EM48-positive nuclear inclusions (Supplemental Figure 6A). However, we detected modest degrees of fluorophore colocalization between G3BP1 and HTT with 3B5H10 (which recognizes monomeric and small oligomeric polyQ species of mutant HTT, ref. 54), 5490 (which recognizes wild type and mutant HTT, ref. 55), and the polyQ antibody 1C2 (which preferentially binds expanded polyQs, refs. 56, 57) (Supplemental Figure 6, A–D). To confirm that the observed increase in G3BP1 immunoreactivity in R6/2 cortex and hippocampus is not due to a nonspecific increase in antibody binding due to the tissue preparation and fixation methods, we investigated the immunoreactivity of the RNA binding protein hnRNPA2/B1 (58) based on our observations that its immunoreactivity is decreased in the R6/2 hippocampus (Supplemental Figure 7) and find that hnRNPA2/B1 immunoreactivity is not higher in the R6/2 brain. Lastly, because it has been suggested that G3BP2, a G3BP1 homolog expressed in mouse brain, also contributes to the formation of SGs (59, 60), we evaluated whether G3BP2 is involved in SG pathology in the R6/2 brain. Using the same analyses, we did not detect G3BP2-positive granular structures in R6/2 or nontransgenic brains (Supplemental Figure 8), suggesting that G3BP1 and G3BP2 might not be functionally redundant in in vivo SG formation. Overall, these results point toward stress-induced G3BP1-SG formation in R6/2 HD mouse cortex and hippocampus and a potential association with HTT.
Increased G3BP1-specific SG density in the superior frontal cortex of patients with HD.
We next investigated whether a G3BP1 phenotype is detected in human HD patient brain tissue. While HD neurodegeneration is most overt in the striatum, neuronal loss has also been detected in other areas, including pyramidal projection neurons of the cortex (61). Topologically selective cortical changes are thought to explain some of the clinical heterogeneity among patients (62, 63), with the superior frontal and parietal cortices exhibiting the highest overall cortical loss (62), suggesting that these regions might be especially vulnerable to mutant HTT-mediated cellular stress. We therefore investigated G3BP1 pathology in the superior frontal and parietal cortices, as well as hippocampus, based on pyramidal neurons being one of the principal cell types of this region and the fact that memory dysfunction is a clinical feature of HD (64).
First, we examined the superior frontal and parietal cortices, and the hippocampus of 2 HD (pathological grade 2, which designates initial gross striatal atrophy, ref. 45) and 2 control postmortem brains (Table 4). We observed high G3BP1 immunoreactivity in the HD patient brains (Supplemental Figures 9–12), particularly in the superior frontal cortex, where overall neuronal loss is also highest (62). We repeated the G3BP1 granule analysis performed in the R6/2 mice in 6 HD (pathological grade 3) and 6 control superior frontal cortex postmortem samples (Table 4). We found a statistically significant G3BP1 granule density increase in the superior frontal cortex of HD brains compared with controls (P = 0.008), suggesting that this brain region is particularly reactive to cellular stress in HD (Figure 7, A and B, and Supplemental Figures 13–15). Because SGs are compositionally diverse and their components depend on the type of stress the cell is exposed to (45), we investigated the colocalization of G3BP1-positive granules with other SG markers.
Table 4. Patient brain tissue samples.
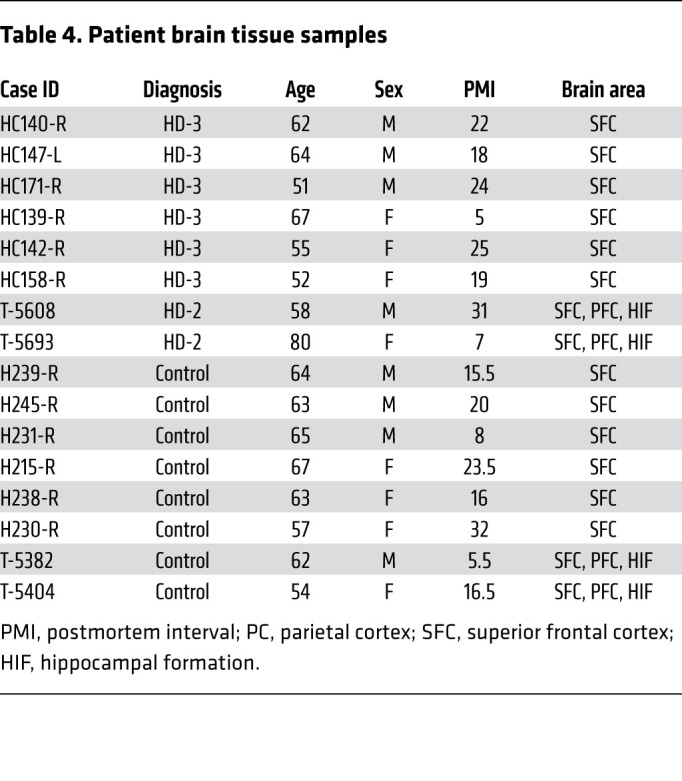
Figure 7. Increased G3BP1 granule density in the superior frontal cortex of HD human brain.

G3BP1 (in green) granules were immunostained, their density calculated using Imaris image analysis software surface tool, and granules normalized to the number of nuclei per frame (DAPI in blue). (A and B) G3BP1 granule density is significantly higher in the superior frontal cortex of HD brains (grade 3) compared with controls (Student’s t test, unpaired, 2-tailed, *P = 0.0085). Data are representative of 6 HD and 6 control human brain samples, and quantitation was done using 3 frames per sample (n = 6, n = 3). (C) Costaining of SG markers G3BP1 and eIF3eta (red) in the superior frontal cortex demonstrates colocalization in an HD case compared with control. Data are representative of 2 HD and 2 control samples (n = 2). Error bars depict mean ± SEM. Scale bars: 20 μm (A); 10 μm (C).
We first tested TIA1 antibodies. However, were not able to detect TIA1 staining in vivo as has been described for ALS tissue (65, 66). We found that some G3BP1 granules colocalize with the translation initiation factor and SG marker eIF3eta (Figure 7C and Supplemental Figure 16), which colocalizes with poly(GR) dipeptide repeat protein in the brains of c9FTD/ALS patients (49) and supports the identification of the G3BP1 granules as SGs. This colocalization is not present in all cases, which could indicate that each of these markers represents a different SG subtype under mutant HTT-mediated cellular stress, and that in vivo SGs may assemble via more than just one canonical mechanism. TDP43 is a predominantly nuclear SG-associated RBP that is mislocalized to the cytoplasm and recruited to cytoplasmic SGs upon stress induction (67), and its cytoplasmic mislocalization has been implicated as a key pathogenic feature in neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (68). To determine whether the G3BP1 granule phenotype shows colocalization with cytoplasmic TDP43, we costained human superior frontal cortex samples with G3BP1 and TDP43 (Figure 8A). Intriguingly, while TDP43 staining remained largely nuclear in cells from control brain tissue, we observed mislocalized TDP43 in HD tissues that are immunoreactive for G3BP1, suggesting that TDP43 pathology characteristic of ALS/FTD also occurs in HD, particularly under conditions of stress leading to formation of G3BP1 SGs in neurons. Colocalization of G3BP1 and TDP43 was not detected.
Figure 8. SG-associated protein TDP43 is mislocalized to the cytoplasm in the superior frontal cortex of HD human brain, and the P body marker DCP1a does not colocalize with G3BP1.

(A) The nuclear RBP TDP43 (red) was detected in the cytoplasm of cells that are immunoreactive for G3BP1 (green), in the superior frontal cortex of patients with HD. Comparatively, the localization of TDP43 in cells from control patient brains was largely nuclear. (B) DCP1a (red), a P body marker, was not found to colocalize with G3BP1 (green) puncta. (C) Imaris SPOT analysis of G3BP1 and DCP1a puncta was done using the Imaris software. Neurons were identified by MAP2-positive staining (gray mask), and the location of G3BP1 (in green) and DCP1a (in red) puncta was determined within the neuronal cytoplasm. Data are representative of 3 HD and 3 control human brain samples (n = 3). Scale bars: 10 μm (A); 3 μm (B, C).
Processing bodies (P bodies) are membraneless organelles that form through liquid–liquid phase separation. Unlike SGs, they are enriched with factors related to mRNA degradation and decay (69). Previous work has shown that the assembly of SGs and P bodies is regulated independently by different pathways, and that they can physically interact under certain stresses (69, 70). To our knowledge, the cellular localization of G3BP1 SGs relative to P bodies has not been investigated in the human HD brain. We costained for G3BP1 and the P body–specific protein decapping enzyme subunit 1a (DCP1a) in the human superior frontal cortex (Figure 8B) and found that DCP1a and G3BP1 granules do not colocalize with each other. However, they were observed in close proximity to each other within MAP2-positive neurons (Figure 8C), suggestive of potential interactions within the cytoplasm.
Mutant HTT and G3BP1 each interact with the SG-associated cell cycle associated protein 1 (CAPRIN1) in vitro under thapsigargin-mediated ER stress (71). We costained for G3BP1 and CAPRIN1 in the human HD cortex, and found that while CAPRIN1 granules were present in G3BP1-positive cells, CAPRIN1 did not colocalize with G3BP1 in 3D space (Supplemental Figure 17). This discordance can be explained by the fact that the earlier study was done in vitro and used an exogenous ER stressor, which may not adequately represent the mechanism involved in the formation of G3BP1 granules in the brains of patients with HD.
Cells highly immunoreactive to G3BP1 display pyramidal neuron features.
Pyramidal cell loss is detected in various cortical regions in human HD, including the superior frontal cortex (62), the region where we observed a significant increase in G3BP1 density. It has been proposed that pyramidal neurons residing in the deep layers of the cortex, which project directly to the striatum, are selectively vulnerable to mutant HTT-mediated toxicity (72). While G3BP1 appears to be widely expressed throughout the HD brain, a subgroup of cells demonstrates higher G3BP1 immunoreactivity. These cells have pyramid-shaped cell bodies, are immunoreactive for the pyramidal neuron marker Ca2+/calmodulin-dependent protein kinase 2 (CaMK2) and are primarily located in the cortical ribbon of the cortex and areas CA1/CA2/CA3 of the hippocampus (Figure 9). These findings suggest that pyramidal neurons might be especially vulnerable to cellular stress in HD.
Figure 9. Highly immunoreactive G3BP1-positive cells have pyramid-shaped cell bodies and express CaMK2.

Costaining of G3BP1 (green) and the pyramidal neuron marker CaMK2 (red), as well as cell morphology, suggest that cells with high-density G3BP1-positive granules are likely to be pyramidal neurons. This pattern of reactivity was observed for HD and control cases. Nuclei were stained with DAPI (blue). Data are representative of 2 HD and 2 control samples. Scale bar: 20 μm.
Taken together, these findings show that CSF EV miRNAs from patients with HD are enriched for targeting the SG nucleator G3BP1, and that a subset of these miRNAs modulate G3BP1 protein expression and G3BP1 granule density in vitro. Additionally, our results show aberrant G3BP1-specific pathology in HD mice and human brain tissue, and cytoplasmic mislocalization of TDP43 in G3BP1-positive cells. Overall, our findings suggest that SG dynamics might play a role in the pathophysiology of HD.
Discussion
SGs are assemblies of protein and RNAs that form in the cytoplasm in response to stressors such as hypoxia, heat-shock, and sodium arsenite, which all inhibit translation initiation (73). Their elimination, in turn, depends on ATP-driven disaggregases, such as Hsp40 and Hsp70, and granulophagy through the ATP-dependent VCP-autophagy pathway (74, 75). Pathologic SG accumulation and persistence has been implicated in several neurodegenerative diseases (29, 68). However, although in vitro experiments using fragments of the HTT protein suggest that expanded HTT interacts with SG-associated proteins and is redistributed to SGs under ER-stress conditions (71), it has not been demonstrated for HD in human brain. In addition, bioinformatic studies determined that the expression of 395 of 464 putative SG related components are altered in HD (76); of these 395 components, 195 are induced and 200 are repressed in HD patient brain. These findings support the need for further investigations into potentially pathologic SG dynamics in HD.
Perturbations of SG dynamics have been implicated in neurodegenerative diseases such as ALS, FTD, and Alzheimer’s disease (29), although the consequence of these changes is not yet clear. Our study provides evidence for the presence of a G3BP1 phenotype in the brains of the R6/2 mouse model and human HD, and the identification of miRNAs that may modulate G3BP1 granule density via targeting expression of G3BP1. Our findings also show that EV miRNAs altered in the CSF of patients with HD are predicted to target mRNAs that are differentially expressed in the HD prefrontal cortex, with SG genes being significantly overrepresented.
The majority of our knowledge of SG biology is currently based on in vitro experiments using a stress time course that is likely shorter than what an organism experiences in the context of a chronic neurodegenerative disease. Under acute stress conditions, SGs appear to provide a prosurvival benefit and are highly dynamic and show punctate structures (73, 77). Current hypotheses regarding mechanism include the regulation of translation of a specific subgroup of mRNAs or activation of prosurvival signaling pathways (78). Efforts have been made to better understand SG biology in the context of chronic stress in vitro by using prolonged nutrient starvation as a stressor, suggesting that chronic stress SGs differ substantially from acute stress SGs by way of their contents, decreased exchange with cytoplasmic mRNP pools, and promotion of cell death (79). Interestingly, SG depletion in the context of chronic starvation resulted in increased cell survival, corroborating other findings in fly and mouse models of neurodegeneration (80–82), and providing a rationale for further investigating therapies to target SG pathology in neurodegenerative disease.
SG assembly is promoted by RNA binding proteins that oligomerize in response to cellular stress. One of these key proteins is G3BP1 (28), which is dephosphorylated and oligomerizes as a response to cellular stress, resulting in the nucleation of SGs (26). Recently, G3BP1 has been identified as a central node of the protein-RNA interaction network that triggers RNA-dependent liquid–liquid phase separation during SG formation under eIF2α-mediated cellular stress (27). Previous reports demonstrate that mutant HTT-mediated ER stress results in PERK activation, leading to the phosphorylation of eIF2α and inhibition of general protein synthesis (7), all of which are involved in SG formation in vitro (45). This study found both total and phosphorylated eIF2α levels to be higher in the HD mouse cortex compared with striatum, perhaps explaining why we detected a substantial increase of G3BP1 granule density in the cortex but not striatum. That is, if the available eIF2α concentration is not large enough, the threshold for SG liquid–liquid phase separation might not be reached. A limitation of this study is that we exclusively investigated G3BP1-mediated SG pathology, and the question remains whether the density of other SG subtypes is affected when G3BP1-positive granules are repressed with miRNAs. Similar to other studies (65, 66), we were unable to detect TIA1 staining in human tissue, but we did observe eIF3eta costaining of G3BP1 granules as shown previously for in vivo SG formation (49, 83). Therefore, we propose that the G3BP1 granules identified in vivo may represent SGs distinct from canonical SGs that form in vitro. For example, there is increasing evidence that G3BP1 has multiple functions aside from regulating translation, including interacting with lysosomes and mitochondria (84, 85), pathways which are implicated in HD pathogenesis (86, 87). Lastly, the miRNA cocktails used to target G3BP1 were designed based on whether the miRNAs were upregulated or downregulated in HD CSF EVs. However, the strength of target repression could potentially be further optimized by grouping miRNAs in such a way that increases the combined G3BP1 seed-strength. Employing this type of in vitro system will narrow down miRNAs that are highly effective at modulating SG densities and can be used to inform future studies that focus on the impact of SGs on disease progression.
The protein profile of SGs is stress- and cell type–specific (39, 88), suggesting that SG characterization experiments done in vitro may not adequately represent the state of SGs in the human brain. TDP43 has been associated with ALS/FTD, where it is mislocalized from the nucleus to the cytoplasm and forms cytoplasmic aggregates that, in some cases, colocalize with SG markers (67, 88, 89). In the human HD cortex, TDP43 inclusions have been detected in cytoplasmic inclusions and dystrophic neurites (90), but it remained a question whether these colocalized with SG markers. Our results suggest that, while TDP43 is mislocalized to the cytoplasm in the human HD cortex, it does not colocalize with G3BP1 SGs. The cytoplasmic mislocalization of TDP43 likely has important functional implications with dysregulation of its critical nuclear roles, including mRNA maturation, repression of cryptic exon expression, splicing, and DNA double stranded break (DSB) response (89, 91). Indeed, missplicing and cryptic exon usage of Stathmin 2 through TDP-43 mislocalization has emerged as a potential biomarker for expanded repeat C9orf72-associated ALS (92). An area of future investigation will include staging of TDP-43 pathology in brain areas impacted in HD, similar to that summarized for ALS, FTD, and Alzheimer’s disease, which show TDP-43 deposits in different brain areas across the different disorders (summarized in ref. 93). It is important to note that we did not investigate modified forms of TDP43, which may affect its localization and functional properties (94). We also show that G3BP1 SGs did not colocalize with the P body marker DCP1a in the HD cortex, but these 2 structures were found to be adjacent to each other, consistent with previous reports suggesting that although SGs and P bodies are separate entities, they participate in protein and RNA exchange (70).
One potential mechanism through which SGs might be detrimental in diseases where proteostasis is impaired is if SG components, of which a majority are RNA binding proteins, have the potential to cross-seed with protein aggregates via their low complexity domains (51, 95), exacerbating proteotoxicity. Conversely, some studies suggest that SGs might serve a more protective role in disease (48, 96). Testing of small molecule compounds that modulate SG accumulation (44), miRNAs such as described here, and other approaches to target SG formation will be informative in future studies to evaluate the consequence of altered SG dynamics in HD.
Our finding that pyramidal neurons demonstrate high G3BP1 granule immunoreactivity, and previous findings of pyramidal neuron loss in the cortex of patients with HD (62), poses the question of whether pyramidal neurons are especially vulnerable to mutant HTT-mediated cellular stress. If SGs are serving a protective role at any point during HD progression, this finding of high G3BP1 granule formation might also explain why neuron loss in the cortex is not as profound as that which is detected in the striatum. Memory dysfunction is an important feature in the early clinical presentation of HD (97). A meta-analysis of multiple studies identified episodic memory impairments in HD (63), a type of memory that relies heavily on the hippocampus and prefrontal cortex (98). Interestingly, the hippocampus and cortex are areas of the brain with highest G3BP1 expression (99). In the hippocampus, high G3BP1 expression is detected in the cell bodies of the dentate granule cell layer and CA pyramidal regions of hippocampal formation. Furthermore, G3bp1-KO mice demonstrate behavioral defects linked to the CNS as well as altered Ca2+ homeostasis in hippocampal neurons, and behavioral studies suggest that G3BP1 plays a role in synaptic transmission and plasticity in the hippocampus (99). It is tempting to speculate that SG pathology in pyramidal neurons of the cortex and hippocampus could be associated with the memory impairments presented by patients with HD.
Our SG time point study observations in 8- and 12-week-old R6/2 mice suggest that G3BP1 immunoreactivity progressively increases, becoming statistically significant sometime between the 8- and 12-week time points. This has important implications for postmortem brain studies, as many of the available control samples often originate from older individuals. Therefore, both postmortem interval and age matching are important parameters to consider when selecting postmortem cases to study SG pathology in disease. Based on our findings and the potential for HTT aggregates to participate in cross-seeding with SG components, we speculate that the SG pathology in HD may result from an accumulation of G3BP1 SGs that perhaps initially served a protective function, but develop into hyper-stable structures due to chronic mutant HTT-mediated stress and compromised autophagy (74, 100). Specifically, HTT is essential for normal selective autophagy in mice, and loss of WT HTT function may play a role in the dysregulation of SG clearance by granulophagy — a type of selective autophagy (70).
Last, because the EV miRNAs with strongest seed-strength for G3BP1 are upregulated in the CSF of patients with HD, we speculated that G3BP1 upregulation in the prefrontal cortex would be even greater in the absence of these miRNAs. This is corroborated by the fact that of the 75 miRNAs differentially expressed in the HD prefrontal cortex (101), 35 are predicted to target G3BP1, and 22 of those are upregulated in HD (P < 5.993 × 10–10) (Supplemental Table 5). In contrast, there is no overlap between the prefrontal cortex and CSF EV differentially expressed miRNAs, suggesting that both intrinsic and extrinsic miRNA-mediated repression of G3BP1 is at play. Future studies can investigate both the therapeutic potential of targeting SG pathology and to determine whether increased SG density in HD affects the ability of cells to deal with additional environmentally induced stress, as this may elucidate mechanisms though which SG pathology contributes to neurodegeneration.
Methods
Methods for the following categories can be found in the supplemental material: EV RNA isolation, library preparation, and next generation sequencing; EV characterization by F-NTA; 293T cell transfection and SG induction experiments; 293T-G3BP1-GFP microRNA mimic Incucyte S3 imaging; R6/2 mice and tissue processing; free-floating immunofluorescence staining; paraffin-embedded immunofluorescence staining; and microscopy.
Study participants and CSF sample collection.
CSF samples were obtained from HDClarity (ClinicalTrials.gov identifier: NCT02855476). CSF was collected after an overnight fast by lumbar puncture and cleared by centrifugation before storage in polypropylene cryotubes at –80°C.
Analysis of miRNA sequencing data.
Reads containing ambiguous bases or with a mean quality score lower than 30 were removed from analysis using Prinseq (102), and UMIs were identified and extracted from the remaining reads using UMI-Tools (103). Reads were aligned to all human mature miRNAs or pre-miRNAs from miRBase v22 (104) using bowtie2 (105) with very sensitive parameters and a seed length of 8 bp with no mismatches allowed in the seed (106). Primary mappings greater than or equal to 16 bp in length were retained for further analysis. Mapped reads were deduplicated with UMI-Tools (103) using the unique grouping method, considering each unique UMI an individual miRNA. Read counts for each miRNA were determined using samtools (107). High-throughput sequencing data will be deposited in GEO.
Enrichment analysis.
The PANTHER Classification System (108) was used to generate GO terms that are significantly overrepresented in the miRNA targets list versus the background of all genes for all 3 GO aspects. For the enrichment analysis of SG genes in the miRNA targets list versus the 464 SG genes (76), a Fisher’s exact test was used to determine whether the number of overlapping genes was significantly higher than what was expected by chance based on a human genome of more than 21,000 genes.
miRNA target prediction and data set overlap.
An initial list of miRNAs was generated by pooling together miRNAs that were detected in the miRNA sequencing studies with P values less than 0.05 before correcting for multiple comparisons, and miRNAs with log2 fold changes greater than or equal to 2 or less than or equal to –2, which were filtered based on interquartile range of the counts. IQR was calculated from the counts in the HD and control groups, and miRNAs were excluded if any count value was greater than quantile3 + 3*IQR for either group. A list of predicted gene targets for the miRNAs that passed the filtering process was generated using TargetScan (40). The list of predicted gene targets for each miRNA was checked for overlap with a list of DEGs detected in the prefrontal cortex of patients with HD (34), and the number of overlapping targets that had an opposite fold change sign to the miRNA fold change were counted.
Seed strength ranking and LNA miRNA mimics.
Seed strength values for all filtered miRNAs were acquired with TargetScan and miRmap, 2 different open source software programs that combine multiple predictor features to predict the strength of miRNA repression on targeted mRNAs, in this case G3BP1 mRNA (40, 41). miRNAs were ranked from strongest to weakest seed strength values using each predictive model, a composite list was made, and 7 miRNAs with the strongest seed strength values were selected for overexpression experiments. miRCURY LNA miRNA Mimics (Qiagen) were used, as listed in Supplemental Table 6.
Postmortem human brain tissue.
Brain tissue samples from human superior frontal cortex, parietal cortex, and hippocampal formation were obtained from the New York Brain Bank at Columbia University (109). Additional superior frontal cortex samples used for SG density statistical analysis were obtained from the Neurological Foundation of New Zealand Human Brain Bank. Paraffin-embedded samples from healthy controls (n = 8) and patients with Huntington’s disease (pathological grade 2 and 3) (n = 8) were used for immunofluorescence experiments (Supplemental Table 7). Patient demographic and clinical information is described in Table 4.
SG analysis.
SGs were quantified using the Imaris Surface tool (Imaris Single Full software, BITPLANE). Using in vitro–induced SGs, parameters were set for the detection of SGs in brain tissue. Z stacks were obtained at a thickness of 0.5 μm per slice, and analyzed using the surface rendering tool for quantitation of G3BP1 SG puncta. The number of puncta was normalized to the number of nuclei (DAPI) per frame. The same pipeline was applied to detect SGs in brain tissue, allowing for the quantitation of G3BP1 SG puncta and not diffuse G3BP1 background signal. G3BP1 and hnRNPA2/B1 immunoreactivity was quantified using an in-house script (CellProfiler Cell Image Analysis software). Nuclei (DAPI stained) were counted using the Imaris Spots tool and used for normalization of SG and G3BP1 immunoreactivity quantitation. A previously described SG quantitation approach (39) was also used for validation.
Statistics.
All mouse and human tissue immunofluorescence and SG quantitation data were analyzed in GraphPad Prism software using a Student’s 2-tailed t test, assuming equal variance. NTA data were analyzed in GraphPad Prism software using a 2-way ANOVA with Bonferroni’s multiple comparison test or a Student’s 2-tailed t test, assuming equal variance. The overlap of CSF EV miRNA targets with mammalian SG genes was analyzed using a Fisher’s exact test, based on a genome larger than 21,000 genes. Differential expression for HD versus control miRNA sequencing samples was analyzed using the DESeq2 package (110) in R (111) with a significance cutoff of P less than 0.05. Sample size calculation for miRNA sequencing experiments was done using an established statistical model to calculate sample size estimates for RNA sequencing data (33), where given any 4 of Type I error, Type II error/power, sequencing depth, coefficient of variation, and samples per group, the fifth can be calculated. All 293T cell immunofluorescence, Western blot, and SG quantitation data were analyzed in GraphPad Prism software using a 1-way ANOVA with Dunnett’s multiple comparison test, repeated measures 2-way ANOVA with Bonferroni’s multiple comparison test, or 2-way ANOVA with Sidak’s multiple comparison test for SG density comparisons among stressed and unstressed conditions. All data are mean ± SEM with a P less than 0.05 considered statistically significant.
Study approval.
Mouse studies were carried out following the Guide for the Care and Use of Laboratory Animals of the NIH and an approved animal research protocol by the Institutional Animal Care and Use Committee (IACUC) at the University of California, Irvine.
Author contributions
IIS designed research studies, conducted experiments, acquired data, analyzed data, prepared figures, and wrote the manuscript. TBN designed research studies, conducted experiments, and acquired data, and edited the manuscript. WEE and RGL analyzed data and edited the manuscript. AQV and GWY provided the script for data analysis. RM analyzed data. CDF assisted with the design of the research studies and interpretation of experiments. SM generated 293T cells that express G3BP1 as a fusion protein with GFP. LMB processed and arranged shipment of CSF samples. ALL harvested mouse tissue. IO harvested and processed mouse tissue for free-floating immunohistochemistry. RLMF and MAC collected, processed, pathologically analyzed, and arranged ethics approval and consent with the families for the human brain tissue. JCR helped design the research studies and edited the manuscript. EJW provided CSF samples and edited the manuscript. RCS helped design the research studies and wrote the manuscript. LMT designed research studies, established collaborations, and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank the New York Brain Bank, the Neurological Foundation of New Zealand Human Brain Bank, and the UC Irvine Alzheimer’s Disease Research Center for banked tissue samples; Leonard Petrucelli and Yongjie Zhang for providing the eIF3eta antibody; Mark Miller for providing the G3BP1-GFP line; and Michael Neel for sharing the paraffin-embedded immunofluorescence staining protocol. Samples and data used in this work were generously provided by the participants in the HDClarity and HD-CSF studies and made available by CHDI Foundation, Inc. This work was supported in part by a grant from the Medical Research Council, CHDI Foundation (to EJW).Additional information regarding Enroll-HD and HD Clarity can be found in the supplemental material. This work was supported by NIH grants 5P01NS092525 and R35 NS116872 (to LMT), 1T32NS082174 (to IIS and TBN), 5R01MH109588-05 (to RCS), HG004659 (to GWY), and 5R01NS089076, as well as the Chan Zuckerberg Initiative. IIS was supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through grant TL1 TR001415. RCS is a Pew Biomedical Scholar and LMT is a Donald Bren Professor. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. AQV was supported by a National Science Foundation Graduate Research Fellowship.
Version 1. 05/04/2021
In-Press Preview
Version 2. 06/15/2021
Electronic publication
Funding Statement
LMT
LMT
IIS and TBN
LMT
IIS
RCS
GWY
Footnotes
Conflict of interest: EJW reports a grant and personal fees from F. Hoffmann-La Roche Ltd and personal fees from Triplet Therapeutics, PTC Therapeutics, Shire Therapeutics, Wave Life Sciences, Mitoconix, Takeda, Loqus23. All honoraria for the consultancies were paid through the offices of UCL Consultants Ltd., a wholly owned subsidiary of University College London. GWY is a cofounder, member of the Board of Directors, member of the Scientific Advisory Board, equity holder, and paid consultant for Locana and Eclipse BioInnovations.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(12):e140723.https://doi.org/10.1172/JCI140723.
Contributor Information
Isabella I. Sanchez, Email: iisanche@uci.edu.
Thai B. Nguyen, Email: thaibn@uci.edu.
Whitney E. England, Email: wengland@uci.edu.
Ryan G. Lim, Email: limrg@uci.edu.
Anthony Q. Vu, Email: aqvu@ucsd.edu.
Ricardo Miramontes, Email: miramonr@uci.edu.
Lauren M. Byrne, Email: lauren.byrne.14@ucl.ac.uk.
Sebastian Markmiller, Email: smarkmiller@ucsd.edu.
Alice L. Lau, Email: allau@uci.edu.
Iliana Orellana, Email: orellani@uci.edu.
Maurice A. Curtis, Email: m.curtis@auckland.ac.nz.
Richard Lewis Maxwell Faull, Email: rlm.faull@auckland.ac.nz.
Gene W. Yeo, Email: geneyeo@ucsd.edu.
Christie D. Fowler, Email: cdfowler@uci.edu.
Jack C. Reidling, Email: jreidlin@uci.edu.
Edward J. Wild, Email: e.wild@ucl.ac.uk.
Robert C. Spitale, Email: rspitale@uci.edu.
Leslie M. Thompson, Email: lmthomps@uci.edu.
References
- 1.The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 2.Browne SE, et al. Oxidative stress in Huntington’s disease. Brain Pathol. 1999;9(1):147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duennwald ML. Cellular stress responses in protein misfolding diseases. Future Sci OA. 2015;1(2):FSO42. doi: 10.4155/fso.15.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fulda S, et al. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010:214074. doi: 10.1155/2010/214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shacham T, et al. Protein misfolding and ER stress in Huntington’s disease. Front Mol Biosci. 2019;6:20. doi: 10.3389/fmolb.2019.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu B, Qian SB. Translational reprogramming in cellular stress response. Wiley Interdiscip Rev RNA. 2014;5(3):301–315. doi: 10.1002/wrna.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leitman J, et al. ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS One. 2014;9(3):e90803. doi: 10.1371/journal.pone.0090803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rub U, et al. Huntington’s disease (HD): the neuropathology of a multisystem neurodegenerative disorder of the human brain. Brain Pathol. 2016;26(6):726–740. doi: 10.1111/bpa.12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott H. Extracellular microRNAs as messengers in the central and peripheral nervous system. Neuronal Signal. 2017;1(4):NS20170112. doi: 10.1042/NS20170112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bang C, Thum T. Exosomes: new players in cell-cell communication. Int J Biochem Cell Biol. 2012;44(11):2060–2064. doi: 10.1016/j.biocel.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Mathieu M, et al. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. 2019;21(1):9–17. doi: 10.1038/s41556-018-0250-9. [DOI] [PubMed] [Google Scholar]
- 13.Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478):eaau6977. doi: 10.1126/science.aau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thery C, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nolte-’t Hoen ENM, et al. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res. 2012;40(18):9272–9285. doi: 10.1093/nar/gks658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shurtleff MJ, et al. Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife. 2016;5:e19276. doi: 10.7554/eLife.19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villarroya-Beltri C, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980. doi: 10.1038/ncomms3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev. 2015;87:3–14. doi: 10.1016/j.addr.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beninson LA, Fleshner M. Exosomes: an emerging factor in stress-induced immunomodulation. Semin Immunol. 2014;26(5):394–401. doi: 10.1016/j.smim.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, et al. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinformatics. 2015;13(1):17–24. doi: 10.1016/j.gpb.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoss AG, et al. MicroRNAs located in the Hox gene clusters are implicated in huntington’s disease pathogenesis. PLoS Genet. 2014;10(2):e1004188. doi: 10.1371/journal.pgen.1004188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson R, et al. A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol Dis. 2008;29(3):438–445. doi: 10.1016/j.nbd.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Lee ST, et al. Altered microRNA regulation in Huntington’s disease models. Exp Neurol. 2011;227(1):172–179. doi: 10.1016/j.expneurol.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 24.Kocerha J, et al. microRNA-128a dysregulation in transgenic Huntington’s disease monkeys. Mol Brain. 2014;7:46. doi: 10.1186/1756-6606-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lepko T, et al. Choroid plexus-derived miR-204 regulates the number of quiescent neural stem cells in the adult brain. EMBO J. 2019;38(17):e100481. doi: 10.15252/embj.2018100481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tourriere H, et al. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J Cell Biol. 2003;160(6):823–831. doi: 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Yang P, et al. G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell. 2020;181(2):325–345. doi: 10.1016/j.cell.2020.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- 29.Wolozin B, Ivanov P. Stress granules and neurodegeneration. Nat Rev Neurosci. 2019;20(11):649–666. doi: 10.1038/s41583-019-0222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanninen KM, et al. Exosomes as new diagnostic tools in CNS diseases. Biochim Biophys Acta. 2016;1862(3):403–410. doi: 10.1016/j.bbadis.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 31.Kopkova A, et al. Cerebrospinal fluid microRNAs as diagnostic biomarkers in brain tumors. Clin Chem Lab Med. 2018;56(6):869–879. doi: 10.1515/cclm-2017-0958. [DOI] [PubMed] [Google Scholar]
- 32.Yagi Y, et al. Next-generation sequencing-based small RNA profiling of cerebrospinal fluid exosomes. Neurosci Lett. 2017;636:48–57. doi: 10.1016/j.neulet.2016.10.042. [DOI] [PubMed] [Google Scholar]
- 33.Hart SN, et al. Calculating sample size estimates for RNA sequencing data. J Comput Biol. 2013;20(12):970–978. doi: 10.1089/cmb.2012.0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Labadorf A, et al. RNA sequence analysis of human huntington disease brain reveals an extensive increase in inflammatory and developmental gene expression. PLoS One. 2015;10(12):e0143563. doi: 10.1371/journal.pone.0143563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nuzziello N, et al. Integrated analysis of microRNA and mRNA expression profiles: an attempt to disentangle the complex interaction network in attention deficit hyperactivity disorder. Brain Sci. 2019;9(10):288. doi: 10.3390/brainsci9100288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Iterson M, et al. Integrated analysis of microRNA and mRNA expression: adding biological significance to microRNA target predictions. Nucleic Acids Res. 2013;41(15):e146. doi: 10.1093/nar/gkt525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reed ER, et al. MicroRNAs in CSF as prodromal biomarkers for Huntington disease in the PREDICT-HD study. Neurology. 2018;90(4):e264–e272. doi: 10.1212/WNL.0000000000004844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niyazova R, et al. miR-1322 binding sites in paralogous and orthologous genes. Biomed Res Int. 2015;2015:962637. doi: 10.1155/2015/962637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markmiller S, et al. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell. 2018;172(3):590–604. doi: 10.1016/j.cell.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agarwal V, et al. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vejnar CE, Zdobnov EM. MiRmap: comprehensive prediction of microRNA target repression strength. Nucleic Acids Res. 2012;40(22):11673–11683. doi: 10.1093/nar/gks901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bracken CP, et al. A network-biology perspective of microRNA function and dysfunction in cancer. Nat Rev Genet. 2016;17(12):719–732. doi: 10.1038/nrg.2016.134. [DOI] [PubMed] [Google Scholar]
- 43.Cursons J, et al. Combinatorial targeting by microRNAs co-ordinates post-transcriptional control of EMT. Cell Syst. 2018;7(1):77–91. doi: 10.1016/j.cels.2018.05.019. [DOI] [PubMed] [Google Scholar]
- 44.Fang MY, et al. Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron. 2019;103(5):802–819. doi: 10.1016/j.neuron.2019.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Panas MD, et al. Mechanistic insights into mammalian stress granule dynamics. J Cell Biol. 2016;215(3):313–323. doi: 10.1083/jcb.201609081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jain S, et al. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell. 2016;164(3):487–498. doi: 10.1016/j.cell.2015.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kedersha NL, et al. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;147(7):1431–1442. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanderweyde T, et al. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci. 2012;32(24):8270–8283. doi: 10.1523/JNEUROSCI.1592-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang YJ, et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med. 2018;24(8):1136–1142. doi: 10.1038/s41591-018-0071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mangiarini L, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87(3):493–506. doi: 10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 51.Furukawa Y, et al. Cross-seeding fibrillation of Q/N-rich proteins offers new pathomechanism of polyglutamine diseases. J Neurosci. 2009;29(16):5153–5162. doi: 10.1523/JNEUROSCI.0783-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilks N, et al. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell. 2004;15(12):5383–5398. doi: 10.1091/mbc.e04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gutekunst CA, et al. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci. 1999;19(7):2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller J, et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol. 2011;7(12):925–934. doi: 10.1038/nchembio.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lunkes A, et al. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10(2):259–269. doi: 10.1016/S1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- 56.Klein FA, et al. Linear and extended: a common polyglutamine conformation recognized by the three antibodies MW1, 1C2 and 3B5H10. Hum Mol Genet. 2013;22(20):4215–4223. doi: 10.1093/hmg/ddt273. [DOI] [PubMed] [Google Scholar]
- 57.Trottier Y, et al. Cellular localization of the Huntington’s disease protein and discrimination of the normal and mutated form. Nat Genet. 1995;10(1):104–110. doi: 10.1038/ng0595-104. [DOI] [PubMed] [Google Scholar]
- 58.Geuens T, et al. The hnRNP family: insights into their role in health and disease. Hum Genet. 2016;135(8):851–867. doi: 10.1007/s00439-016-1683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cirillo L, et al. UBAP2L forms distinct cores that act in nucleating stress granules upstream of G3BP1. Curr Biol. 2020;30(4):698–707. doi: 10.1016/j.cub.2019.12.020. [DOI] [PubMed] [Google Scholar]
- 60.Matsuki H, et al. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells. 2013;18(2):135–146. doi: 10.1111/gtc.12023. [DOI] [PubMed] [Google Scholar]
- 61.Cudkowicz M, Kowall NW. Degeneration of pyramidal projection neurons in Huntington’s disease cortex. Ann Neurol. 1990;27(2):200–204. doi: 10.1002/ana.410270217. [DOI] [PubMed] [Google Scholar]
- 62.Nana AL, et al. Widespread heterogeneous neuronal loss across the cerebral cortex in Huntington’s disease. J Huntingtons Dis. 2014;3(1):45–64. doi: 10.3233/JHD-140092. [DOI] [PubMed] [Google Scholar]
- 63.Rosas HD, et al. Cerebral cortex and the clinical expression of Huntington’s disease: complexity and heterogeneity. Brain. 2008;131(Pt 4):1057–1068. doi: 10.1093/brain/awn025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Montoya A, et al. Episodic memory impairment in Huntington’s disease: a meta-analysis. Neuropsychologia. 2006;44(10):1984–1994. doi: 10.1016/j.neuropsychologia.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 65.Hirsch-Reinshagen V, et al. Clinical and neuropathological features of ALS/FTD with TIA1 mutations. Acta Neuropathol Commun. 2017;5(1):96. doi: 10.1186/s40478-017-0493-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mackenzie IR, et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron. 2017;95(4):808–816. doi: 10.1016/j.neuron.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Colombrita C, et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111(4):1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- 68.Chew J, et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener. 2019;14(1):9. doi: 10.1186/s13024-019-0310-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Riggs CL, et al. Mammalian stress granules and P bodies at a glance. J Cell Sci. 2020;133(16):jcs242487. doi: 10.1242/jcs.242487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stoecklin G, Kedersha N. Relationship of GW/P-bodies with stress granules. Adv Exp Med Biol. 2013;768:197–211. doi: 10.1007/978-1-4614-5107-5_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ratovitski T, et al. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle. 2012;11(10):2006–2021. doi: 10.4161/cc.20423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sieradzan KA, Mann DM. The selective vulnerability of nerve cells in Huntington’s disease. Neuropathol Appl Neurobiol. 2001;27(1):1–21. doi: 10.1046/j.0305-1846.2001.00299.x. [DOI] [PubMed] [Google Scholar]
- 73.Arimoto K, et al. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol. 2008;10(11):1324–1332. doi: 10.1038/ncb1791. [DOI] [PubMed] [Google Scholar]
- 74.Buchan JR, et al. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 2013;153(7):1461–1474. doi: 10.1016/j.cell.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kedersha N, et al. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol. 2005;169(6):871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nunes C, et al. MSGP: the first database of the protein components of the mammalian stress granules. Database (Oxford) 2019;2019:baz031. doi: 10.1093/database/baz031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Arimoto-Matsuzaki K, et al. TIA1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat Commun. 2016;7:10252. doi: 10.1038/ncomms10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reineke LC, Neilson JR. Differences between acute and chronic stress granules, and how these differences may impact function in human disease. Biochem Pharmacol. 2019;162:123–131. doi: 10.1016/j.bcp.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reineke LC, et al. Chronic starvation induces noncanonical pro-death stress granules. J Cell Sci. 2018;131(19):jcs220244. doi: 10.1242/jcs.220244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim HJ, et al. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46(2):152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Radford H, et al. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015;130(5):633–642. doi: 10.1007/s00401-015-1487-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang K, et al. Stress granule assembly disrupts nucleocytoplasmic transport. Cell. 2018;173(4):958–971. doi: 10.1016/j.cell.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang X, et al. In vivo stress granule misprocessing evidenced in a FUS knock-in ALS mouse model. Brain. 2020;143(5):1350–1367. doi: 10.1093/brain/awaa076. [DOI] [PubMed] [Google Scholar]
- 84.Fung G, et al. Production of a dominant-negative fragment due to G3BP1 cleavage contributes to the disruption of mitochondria-associated protective stress granules during CVB3 infection. PLoS One. 2013;8(11):e79546. doi: 10.1371/journal.pone.0079546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meyer C, et al. The G3BP1-family-USP10 deubiquitinase complex rescues ubiquitinated 40S subunits of ribosomes stalled in translation from lysosomal degradation. Mol Cell. 2020;77(6):1193–1205. doi: 10.1016/j.molcel.2019.12.024. [DOI] [PubMed] [Google Scholar]
- 86.Carmo C, et al. Mitochondrial dysfunction in Huntington’s disease. Adv Exp Med Biol. 2018;1049:59–83. doi: 10.1007/978-3-319-71779-1_3. [DOI] [PubMed] [Google Scholar]
- 87.Martin DD, et al. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015;38(1):26–35. doi: 10.1016/j.tins.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 88.Aulas A, et al. Stress-specific differences in assembly and composition of stress granules and related foci. J Cell Sci. 2017;130(5):927–937. doi: 10.1242/jcs.199240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Suk TR, Rousseaux MWC. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener. 2020;15(1):45. doi: 10.1186/s13024-020-00397-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schwab C, et al. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J Neuropathol Exp Neurol. 2008;67(12):1159–1165. doi: 10.1097/NEN.0b013e31818e8951. [DOI] [PubMed] [Google Scholar]
- 91.Ling JP, et al. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349(6248):650–655. doi: 10.1126/science.aab0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Prudencio M, et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J Clin Invest. 2020;130(11):6080–6092. doi: 10.1172/JCI139741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tan RH, et al. TDP-43 proteinopathies: pathological identification of brain regions differentiating clinical phenotypes. Brain. 2015;138(Pt 10):3110–3122. doi: 10.1093/brain/awv220. [DOI] [PubMed] [Google Scholar]
- 94.Buratti E. TDP-43 post-translational modifications in health and disease. Expert Opin Ther Targets. 2018;22(3):279–293. doi: 10.1080/14728222.2018.1439923. [DOI] [PubMed] [Google Scholar]
- 95.Dobra I, et al. Relation between stress granules and cytoplasmic protein aggregates linked to neurodegenerative diseases. Curr Neurol Neurosci Rep. 2018;18(12):107. doi: 10.1007/s11910-018-0914-7. [DOI] [PubMed] [Google Scholar]
- 96.Goncalves AC, et al. Drug-induced stress granule formation protects sensory hair cells in mouse cochlear explants during ototoxicity. Sci Rep. 2019;9(1):12501. doi: 10.1038/s41598-019-48393-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Josiassen RC, et al. Development of neuropsychological deficits in Huntington’s disease. Arch Neurol. 1983;40(13):791–796. doi: 10.1001/archneur.1983.04050120041005. [DOI] [PubMed] [Google Scholar]
- 98.Eichenbaum H. Prefrontal-hippocampal interactions in episodic memory. Nat Rev Neurosci. 2017;18(9):547–558. doi: 10.1038/nrn.2017.74. [DOI] [PubMed] [Google Scholar]
- 99.Martin S, et al. Deficiency of G3BP1, the stress granules assembly factor, results in abnormal synaptic plasticity and calcium homeostasis in neurons. J Neurochem. 2013;125(2):175–184. doi: 10.1111/jnc.12189. [DOI] [PubMed] [Google Scholar]
- 100.Cortes CJ, La Spada AR. The many faces of autophagy dysfunction in Huntington’s disease: from mechanism to therapy. Drug Discov Today. 2014;19(7):963–971. doi: 10.1016/j.drudis.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hoss AG, et al. miR-10b-5p expression in Huntington’s disease brain relates to age of onset and the extent of striatal involvement. BMC Med Genomics. 2015;8:10. doi: 10.1186/s12920-015-0083-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27(6):863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Smith T, et al. UMI-tools: modeling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 2017;27(3):491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kozomara A, et al. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(d1):D155–D162. doi: 10.1093/nar/gky1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tam S, et al. Optimization of miRNA-seq data preprocessing. Brief Bioinform. 2015;16(6):950–963. doi: 10.1093/bib/bbv019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mi H, Thomas P. PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol Biol. 2009;563:123–140. doi: 10.1007/978-1-60761-175-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vonsattel JP, et al. Twenty-first century brain banking. Processing brains for research: the Columbia University methods. Acta Neuropathol. 2008;115(5):509–532. doi: 10.1007/s00401-007-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Love MI, et al. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. R Core Team. The R Project for Statistical Computing. https://www.r-project.org/ Accessed April 29, 2021.
- 112.Shoulson I. Huntington disease: functional capacities in patients treated with neuroleptic andantidepressant drugs. Neurology. 1981;31(10):1333–1335. doi: 10.1212/WNL.31.10.1333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.