

Abstract
Objectives
Bruton's tyrosine kinase (BTK) plays a non‐redundant signaling role downstream of the B‐cell receptor (BCR) in B cells and the receptors for the Fc region of immunoglobulins (FcR) in myeloid cells. Here, we characterise BIIB091, a novel, potent, selective and reversible small‐molecule inhibitor of BTK.
Methods
BIIB091 was evaluated in vitro and in vivo in preclinical models and in phase 1 clinical trial.
Results
In vitro, BIIB091 potently inhibited BTK‐dependent proximal signaling and distal functional responses in both B cells and myeloid cells with IC50s ranging from 3 to 106 nm, including antigen presentation to T cells, a key mechanism of action thought to be underlying the efficacy of B cell‐targeted therapeutics in multiple sclerosis. BIIB091 effectively sequestered tyrosine 551 in the kinase pocket by forming long‐lived complexes with BTK with t 1/2 of more than 40 min, thereby preventing its phosphorylation by upstream kinases. As a key differentiating feature of BIIB091, this property explains the very potent whole blood IC50s of 87 and 106 nm observed with stimulated B cells and myeloid cells, respectively. In vivo, BIIB091 blocked B‐cell activation, antibody production and germinal center differentiation. In phase 1 healthy volunteer trial, BIIB091 inhibited naïve and unswitched memory B‐cell activation, with an in vivo IC50 of 55 nm and without significant impact on lymphoid or myeloid cell survival after 14 days of dosing.
Conclusion
Pharmacodynamic results obtained in preclinical and early clinical settings support the advancement of BIIB091 in phase 2 clinical trials.
Keywords: B cells, B‐cell receptor, BTK inhibitor, Fc receptor, multiple sclerosis, myeloid cells
In this study, we characterise BIIB091, a novel, potent, selective and reversible small‐molecule inhibitor of Bruton's Tyrosine Kinase (BTK). In vitro, BIIB091‐inhibited BTK‐dependent proximal signaling and distal functional responses in both B cells and myeloid cells, including antigen presentation to T cells, a key mechanism of action thought to be underlying the efficacy of B cell‐targeted therapeutics in MS. In vivo, BIIB091 blocked B‐cell activation, antibody production and germinal center differentiation, however in phase 1 healthy volunteer study, BIIB091 inhibited B‐cell receptor‐mediated activation of naïve and unswitched memory B cells.
Introduction
As a member of the TEC (tyrosine kinase expressed in hepatocellular carcinoma) family of protein tyrosine kinases, BTK is a key signaling node immediately downstream of the B‐cell receptor (BCR) and the receptors for the Fc region (FcR) of immunoglobulins (Igs) which mediate activation and a variety of effector functions in B lymphocytes and myeloid cells. In B cells specifically, BTK is required for B‐cell proliferation and differentiation into memory cells and antibody‐producing cells. 1 An important function associated with BTK‐dependent signaling in B cells is also its proposed requirement for BCR‐mediated antigen presentation to T cells, which is thought to be one of the key mechanisms underlying the recently reported efficacy of B‐cell depleting agents and small molecule inhibitors of BTK in multiple sclerosis (MS). 2 , 3 , 4 , 5 In contrast to B‐cell depleting agents which rely on a cytolytic mechanism of action, BTK is not required for mature B‐cell survival 6 and BTK inhibition is thus not anticipated to directly alter B‐cell numbers. In myeloid cells (including, but not limited to monocytes, macrophages, basophils, dendritic cells, mast cells and granulocytes), BTK is also a key signaling node downstream of the activating Fc receptors which include the FcRs for IgG (FcγR), IgE (FcεR) and IgA (FcαR). In this context, BTK inhibition blocks FcR‐dependent pro‐inflammatory activities including cytokine secretion, reactive oxygen species (ROS) generation and degranulation triggered by binding of immune complexes to FcRs. 7
BTK catalytic activity is low in unstimulated cells, which results in a low, but detectable basal constitutive autophosphorylation of the kinase at tyrosine 223 (Y223) in resting cells. 1 Signal transduction through the BCR and FcRs is dependent on cytoplasmic immunoreceptor tyrosine‐based activation motifs (ITAM) in the cytoplasmic tails of these receptors. Following engagement of those receptors, rapid and transient phosphorylation of tyrosine residues within their ITAMs creates temporary binding sites for Src‐family kinases (SFKs) which in turn activate BTK by trans‐phosphorylation of tyrosine 551 (Y551) in the BTK activation loop. 8 BTK activity is increased by 10‐ to 20‐fold as a result of this post‐translational modification. Upon activation, BTK phosphorylates PLCγ2, 9 which triggers many downstream signaling events among which are calcium‐dependent and nuclear factor‐kappa B (NF‐κB) pathways ultimately leading to the functional activation of effector functions in B cells and myeloid cells.
Here, we characterised a novel reversible inhibitor of BTK, BIIB091, in preclinical and phase 1 clinical trial settings. By using a number of in vitro, in vivo and ex vivo proximal and distal pharmacodynamic readouts, we established that BIIB091 potently inhibits early signaling events as well as downstream cellular effector functions in B cells and myeloid cells in preclinical models and healthy volunteers.
Results
BIIB091 is a highly selective and potent inhibitor of BTK kinase activity and downstream proximal signaling
BIIB091 (PATENT: Hopkins BT, Ma B, Prince R, Marx I, Lyssikatos JP, Zheng F, Peterson M, Patience DB Benzoazepine analogs as inhibiting agents for Bruton's tyrosine kinase and their preparation. PCT Int Appl 2018, WO 2018191577 A1 20181018) (Figure 1a) was discovered during medicinal chemistry efforts targeting BTK, 10 a member of the TEC family of kinases. We reported previously an earlier generation BTK inhibitor (BIIB068) 11 ; however, BIIB091 showed a differentiated profile characterised by an improved potency while maintaining a favorable selectivity profile. 10 Like BIIB068, BIIB091 was designed as a reversible small‐molecule inhibitor of BTK and showed potent inhibitory activity against purified BTK protein in an enzymatic assay with an average IC50 of 450 pm. BIIB091 demonstrated > 500‐fold selectivity for BTK relative to all other kinases from a panel of > 400 individual kinases evaluated in the DiscoverX KINOMEscan. 10
Figure 1.
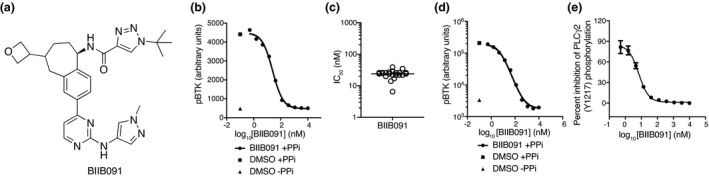
BIIB091 potently inhibits BTK enzymatic activity and downstream proximal signaling events. (a) Chemical structure of BIIB091. (b–d) Detection of phosphorylated BTK and phosphorylated BTK‐associated proteins in whole blood from individual healthy human donors (b, c) or pooled blood from 15 DBA1 mice (d) treated in vitro with dimethyl sulfoxide (DMSO) or titrating concentrations of BIIB091 and lysed in the presence or absence of phosphatase inhibitors (PPi). Representative curve from one individual (b) . Calculated IC50 values across 19 healthy donors (individual data points) are shown along with the mean (24 nm) (c). (e) Phosphorylation levels of PLCγ2 (Y1217) measured in Ramos B cells pre‐treated with titrating concentrations of BIIB091 and stimulated with F(ab′)2 anti‐human IgM (IC50 = 6.9 nm) (Data shown are from one experiment).
To further evaluate the potency of BIIB091 on BTK kinase activity within the cellular context, we measured BIIB091 inhibition of basal BTK tyrosine autophosphorylation in human and mouse whole blood. BIIB091 or vehicle was added to healthy human or mouse whole blood samples and total BTK protein from each sample was captured on a plate with a BTK‐specific monoclonal antibody, and the phosphorylation level of BTK was quantified with an anti‐phosphotyrosine antibody. BIIB091 inhibited the basal autophosphorylation of BTK in a dose‐dependent manner, with an average IC50 of 24 ± 7 nm across 19 human healthy donor whole blood samples (Figure 1b and c) and an IC50 of 4.8 nm in mouse whole blood pooled from 15 mice (Figure 1d).
Among the most proximal pharmacodynamic readout downstream of BTK activation, the activation of PLCγ2 is a critical step in BCR‐coupled signaling 12 and BTK‐dependent phosphorylation of Y753, Y759, Y1197 and Y1217 on PLCγ2 contributes to BCR‐induced activation of PLCγ2. 9 , 13 , 14 BIIB091 showed dose‐dependent inhibition of BCR‐mediated PLCγ2 phosphorylation in Ramos B cells stimulated by cross‐linking the IgM B‐cell receptor (Figure 1e), with an IC50 of 6.9 nm.
BIIB091 selectively blocks human and mouse B‐cell activation in vitro with an inhibitory potency similar to the potency observed in proximal pharmacodynamic readouts
BTK activity is required for BCR‐mediated activation of B cells. 15 Among many gene and protein expression changes induced by BCR‐mediated activation, CD69 upregulation in B cells was demonstrated to be BTK‐dependent using both genetic and pharmacological modulation. 1 , 4 , 16 , 17 To assess the inhibitory potential of BIIB091 on this distal pharmacodynamic readout and surrogate marker for B‐cell activation, we mimicked antigen recognition by the BCR by artificially cross‐linking the BCR (either IgD or IgM) expressed by primary B cells using agonistic antibodies directed against the corresponding receptors. BIIB091 inhibited CD69 induction in anti‐BCR‐stimulated B cells in a dose‐dependent manner, with an average IC50 of 5.4 ± 0.5 nm in PBMCs from two independent healthy donors (Figure 2a and b) and with an average IC50 of 16 ± 11 nm in mouse splenocytes (Figure 2c and d).
Figure 2.
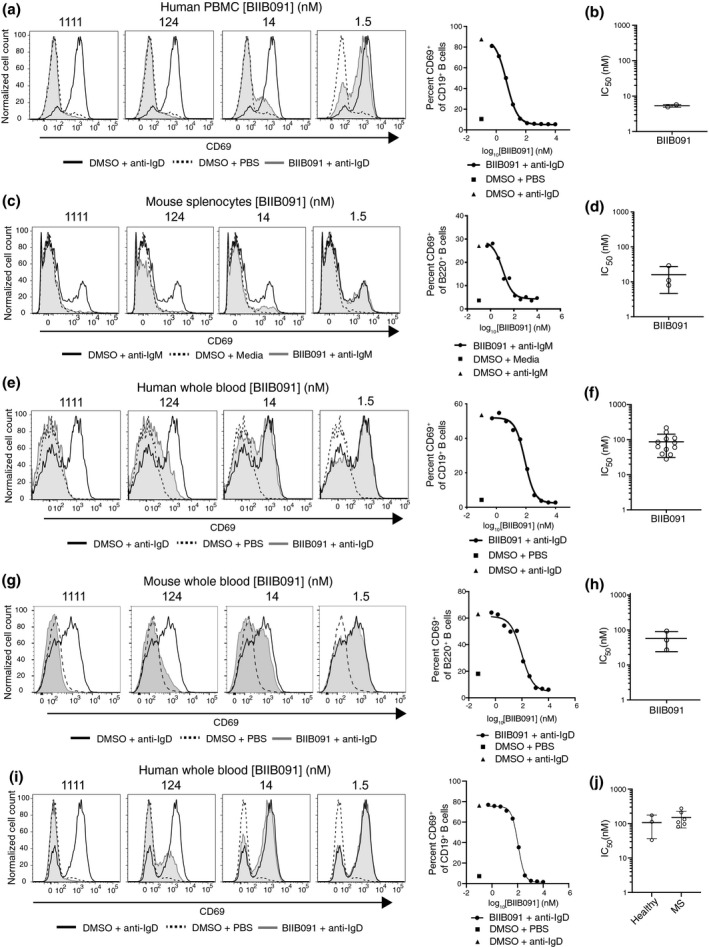
BIIB091 inhibits human and mouse B‐cell activation in vitro. (a–d) Histogram overlays of CD69 expression on CD19+ B cells from human PBMC (a) or on B220+ mouse splenic B cells (c) treated with DMSO or titrating concentrations of BIIB091 and stimulated with polyclonal anti‐human IgD (a) or anti‐mouse‐IgM cross‐linked with a secondary antibody (c). Phosphate‐buffered saline (PBS) was used as control. IC50 values for two healthy donors (b) or from three independent mouse experiments (d) are shown along with mean and SD (5.4 ± 0.5 nm and 16 ± 11 nm, respectively). (e–h) Histogram overlays of CD69 expression on B cells from human (e) or mouse (g) whole blood treated with DMSO or titrating concentrations of BIIB091 and stimulated with PBS or anti‐human IgD (e) or anti‐mouse IgD (g). IC50 values for 12 healthy human donors (f) and from three mouse whole blood independent experiments (h) are shown along with mean and SD (87 ± 56 nm and 57 ± 33 nm, respectively). (i, j) Histogram overlays of CD69 expression on CD19+ B cells from whole blood of patients with MS or healthy volunteers treated with DMSO or titrating concentrations of BIIB091 and stimulated with anti‐human IgD (i). Individual IC50 values for three healthy donors and six patients with MS (individual data points) are shown along with mean and SD (106 ± 70 nm and 151 ± 76 nm, respectively) (j). IC50 differences between the two groups were not statistically significant (unpaired t‐test with Welch's correction).
To assess the potency of BIIB091 at blocking B‐cell activation in whole blood, a more physiologically relevant matrix, the same assay was performed in human and mouse whole blood. In this matrix, anti‐IgD instead of anti‐IgM antibodies were used to activate B cells to avoid titration of anti‐IgM antibodies by soluble immunoglobulins naturally present in mouse and human whole blood. Preincubation of human and mouse whole blood with titrating concentrations of BIIB091 inhibited anti‐IgD‐induced CD69 upregulation in human and mouse B cells in a dose‐dependent manner, with an average IC50 of 87 ± 56 nm (Figure 2e and f) and 57 ± 33 nm (Figure 2g and h), respectively. CD62L downregulation, another marker of B‐cell activation, was also potently inhibited by BIIB091 in mouse whole blood with an average IC50 of 40 ± 24 nm (data not shown). BIIB091 also inhibited B‐cell activation in whole blood from patients with MS with an average IC50 of 151 ± 76 nm, as compared to 106 ± 70 nm in healthy donors tested within the same experiment (Figure 2i and j), altogether establishing the inhibitory potency of BIIB091 in mouse and human B cells from both healthy volunteers and patients with MS. To further define the time window during which BIIB091 had to be present relative to the B‐cell activation event, we added BIIB091 to whole blood from a healthy volunteer either before or at different time points after addition of the stimulating antibody against IgD and measured CD69 expression. As shown in Supplementary figure 1, the inhibitory effect of BIIB091 was rapidly lost (as early as 30 min post‐stimulation) when added after the BCR stimulation event, indicating that BIIB091 had to be present at the time of the BCR‐activation event or within a short time window following BCR engagement to effectively block the B‐cell activation programme.
While biochemical assays indicated that BIIB091 was exquisitely selective for BTK, 10 we next assessed the cellular selectivity of BIIB091's effects by using activation readouts that were known to be BTK‐independent. In a first counter‐screen assay, we focussed on the effect of BIIB091 on T‐cell activation. T cells do not express BTK but do express other TEC family kinase members such as IL‐2 inducible T‐cell kinase (ITK), which functions downstream of the T‐cell antigen receptor (TCR) in an analogous manner to BTK function downstream of the BCR and activating FcRs. Human PBMCs were stimulated with anti‐CD3 and anti‐CD28 overnight and then analysed for induction of CD69 expression on CD4+ or CD8+ T cells (Supplementary figure 2a and b). BIIB091 did not inhibit CD4+ or CD8+ T‐cell activation even at concentrations up to 10 μm. In a second counter‐screen assay, we explored T‐dependent B‐cell co‐stimulation and BCR‐independent B‐cell activation. This type of B‐cell activation can be modelled in vitro by activating T cells in PBMCs with anti‐CD3/anti‐CD28 antibodies. Under these conditions, T cells express soluble factors and surface receptors which co‐stimulate B cells present in the PBMC cultures in an antigen‐independent and BTK‐independent manner. BIIB091 did not inhibit this T cell‐dependent, antigen‐independent B‐cell activation in vitro (Supplementary figure 2c).
Finally, to evaluate the relative potency of BIIB091 on proximal and distal pharmacodynamic readouts, we directly compared whole blood IC50s for the proximal autophosphorylation of BTK with IC50s for the distal upregulation of CD69 B‐cell activation marker. As shown in Figure 3a, the vast majority of a large set of BTK inhibitors generated during our lead optimisation efforts showed weaker potency on the distal readout than on the proximal readout with an average shift between the two readouts of 12‐fold and some compounds being up to 232‐fold less potent on the distal than the proximal readout. Because these assays were performed in the same whole blood matrix, these results directly reflected the differential ability of the compounds to block proximal and distal BTK‐dependent pharmacological readouts. In contrast to our prior lead compound BIIB068 that showed a 26‐fold shift between the two readouts, BIIB091 displayed a more limited 5‐fold shift. While the more potent compounds tended to show shifts of lesser magnitude between proximal and distal readouts (Figure 3a), some very potent compounds were also exhibiting significant shifts between the two readouts. To further investigate the mechanistic basis of this disconnect, we measured the binding kinetics for a set of inhibitors to determine whether the residency time of BTK inhibitors in the kinase pocket and more specifically the half‐life of the BTK:BTK inhibitor complex explained the differential potency observed against these two readouts. While the half‐life of the BTK:BTK inhibitor complexes was only weakly correlated to pBTK IC50s (Figure 3b, R 2 = 0.29, slope = −0.49), there was in contrast a strong correlation between the residency time of BTK inhibitors and the potency against the distal pharmacodynamic readout (Figure 3c, R 2 = 0.75, slope = −1.02). Accordingly, while early generation inhibitors showing poor potency against distal pharmacodynamic readouts had generally a half‐life of less than 10 min, inhibitors such as BIIB091 showing potent inhibition of CD69 readout had longer half‐lives (e.g. of 46 min for BIIB091). Altogether, these results established that BIIB091 was a potent inhibitor of BTK‐dependent proximal and distal pharmacodynamic responses with minimal potency shift attributable to a longer residency time within the kinase pocket.
Figure 3.
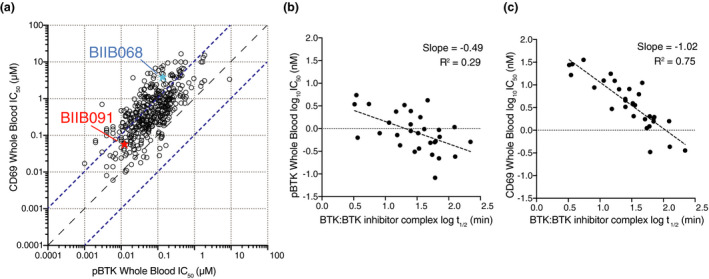
The residency time of BTK inhibitors in the kinase pocket dictates potency against distal B‐cell pharmacodynamic responses. (a) Dot plot showing IC50 values for constitutive BTK phosphorylation in unstimulated cells (pBTK, x‐axis) and CD69 expression in anti‐IgD‐stimulated B cells (CD69, y‐axis) in whole blood (WB) assays. Data from n = 420 Biogen‐reversible BTK inhibitors are shown with BIIB091 and BIIB068 (early generation BTK inhibitor 11 ) highlighted in red and blue, respectively. Dashed lines indicate the equal potency line and the 10‐fold deviation from the equipotency line. (b, c) Dot plots showing plasma protein binding‐corrected IC50 values for constitutive BTK phosphorylation in unstimulated cells (b) and CD69 expression in anti‐IgD‐stimulated B cells (c) against the half‐life of the BTK:BTK inhibitor complex measured by surface plasmon resonance. Data from n = 30 Biogen‐reversible BTK inhibitors are shown.
BIIB091 blocks human B‐cell proliferation and differentiation programmes in vitro
To further explore BIIB091 potency on human B‐cell functional responses, we leveraged a previously reported protocol 18 to promote B‐cell proliferation and model in vitro germinal cent er (GC) B‐cell reactions using BCR stimulation, CD40L cross‐linking and IL‐4 or IL‐21. IL‐4 and IL‐21 are two of the asynchronously secreted cytokines produced by T follicular helper cells during the GC reaction. 19 As an alternative mode of B‐cell activation, we used TLR9 stimulation with TLR9 ligand ODN2006 (CpG‐B).
As previously reported, BCR stimulation alone did not induce B‐cell proliferation whereas TLR9 stimulation or BCR stimulation in combination with CD40 engagement and contextual cytokines IL‐4 or IL‐21 robustly induced B‐cell proliferation as indicated by dilution of the protein‐reactive CFSE dye (Figure 4a). The addition of BIIB091 to these B‐cell cultures at a saturating concentration of 100 nm at the time of activation potently blocked B‐cell proliferative responses following stimulation with each of these stimuli (Figure 4a and b). In agreement with prior results obtained with CD69 (Supplementary figure 1), BIIB091 had to be present at the time of BCR engagement as delayed addition of BIIB091 one day post‐stimulation resulted in no or very limited inhibitory effect (Supplementary figure 3a). We extended these cellular analyses with a more unbiased exploration of the effect of BIIB091 on the entire transcriptional programme induced upon B‐cell activation. Fluorescence‐activated cell sorting (FACS) was used to isolate live B cells from these in vitro cultures 64 h post‐stimulation with the previously described stimuli. BIIB091 inhibited a significant portion of the transcriptional programmes induced by each of these activation triggers (Figure 4c and d, Supplementary figure 3b and c and Supplementary table 2). The magnitude of BIIB091 inhibitory effect on the induced transcriptome varied with each stimulation trigger from almost complete inhibition to partial inhibition. For instance, while BIIB091 inhibited 98% and 79% of the upregulated activation signatures associated with anti‐IgM and CpG‐B stimulation, respectively, BIIB091 only inhibited 23% and 6% of the upregulated activation signatures associated with anti‐IgM, CD40L, IL‐21 stimulation and anti‐IgM, CD40L, IL‐4 stimulation, respectively (Supplementary table 2). Pathway analysis of genes modulated by BIIB091 revealed a clear enrichment in DNA replication and cell cycle‐related genes (Supplementary table 3), in agreement with proliferation results (Figure 4a and b). Altogether, these results indicated that BIIB091 potently blocked human B‐cell proliferative responses and their underlying molecular networks downstream of a variety of activating triggers.
Figure 4.
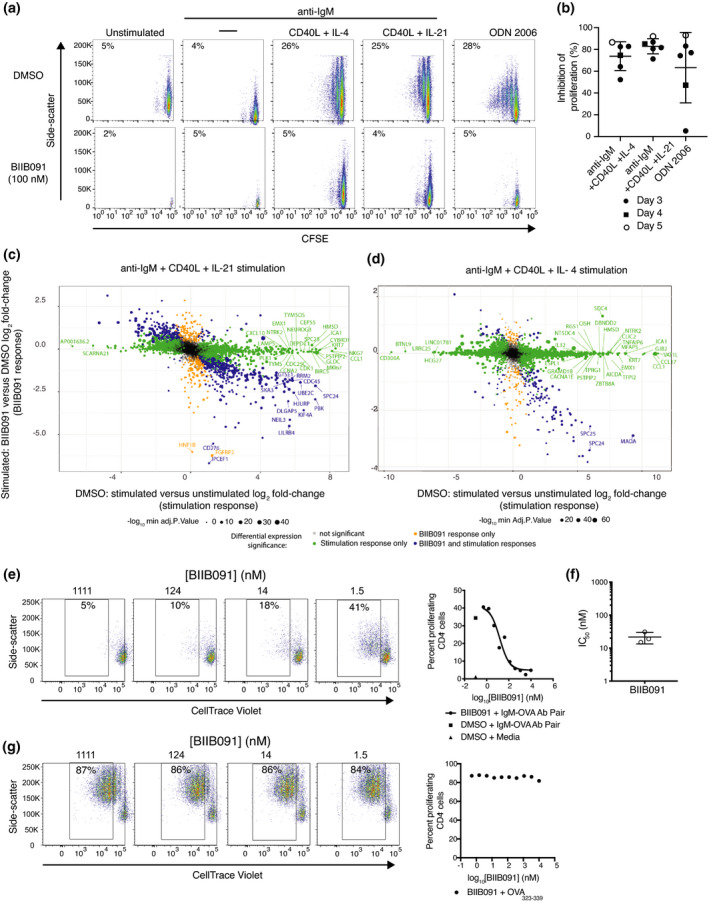
BIIB091 blocks B‐cell proliferation and antigen‐presenting functions in vitro and the associated B‐cell activation transcriptional programme. (a, b) Pseudocolor plots displaying side scatter against the dilution of CFSE used as a measure of proliferation of human B cells left either unstimulated or stimulated for 5 days with anti‐IgM alone, anti‐IgM with CD40L and IL‐4, anti‐IgM with CD40L and IL‐21, or CpG ODN 2006 in the presence or absence of 100 nm BIIB091 (a). Percent inhibition of B‐cell proliferation (relative to proliferation level observed in DMSO control condition) obtained from six independent experiments (individual data points) and measured either three (filled circle), four (square) or five (open circle) days post‐stimulation (b). (c) Log2 fold‐change × fold‐change plot of differentially expressed genes (FDR < 0.05; fold change > |1.2|) observed when comparing anti‐IgM, CD40L and IL‐21‐stimulated human B cells to unstimulated B cells on the x‐axis and anti‐IgM, CD40L and IL‐21‐stimulated B cells treated with 100 nm BIIB091 to stimulated B cells treated with DMSO on the y‐axis. Data from five independent experiments. (d) Log2 fold‐change × fold‐change plot of differentially expressed genes (FDR < 0.05; fold change > |1.2|) observed when comparing anti‐IgM, CD40L and IL‐4‐stimulated human B cells to unstimulated B cells on the x‐axis and anti‐IgM, CD40L and IL‐4 stimulated B cells treated with 100 nm BIIB091 to stimulated B cells treated with DMSO on the y‐axis. Data from five independent experiments. (e–g) Pseudocolor plots displaying side scatter against the dilution of CellTrace Violet used as a measure of proliferation of mouse OTII CD4+ T cell (expressing transgenic T‐cell receptor specific for the ovalbumin (OVA) peptide 323–339) upon B‐cell receptor targeting of OVA protein antigen to B cells using antibodies directed against the B‐cell receptor (e) or upon addition of OVA peptide 323–339 (g) to co‐cultures of splenic B cells and OTII T cells in the presence or absence of titrating concentrations of BIIB091 (left). Dot plot shows the per cent of proliferating OVA323–339‐specific CD4+ T versus the concentration of BIIB091 (right). IC50 values obtained with B‐cell receptor targeting of OVA protein antigen to B cells in three independent experiments are shown along with the mean and SD (22 ± 8 nm) (f).
BIIB091 effectively blocks BCR‐mediated antigen presentation to T cells
Another important functional role of B cells is their ability to efficiently capture, endocytose and present to T cells antigens for which their BCR is specific for. The potency of BIIB091 in blocking this cellular function was evaluated using a model protein antigen, ovalbumin (OVA), that was artificially targeted to the BCR of polyclonal mouse B cells using a pair of antibodies (biotinylated anti‐IgM and OVA‐conjugated anti‐biotin antibodies). To assess the ability of OVA‐targeted B cells to successfully process and present this antigen to T cells, B cells were then cultured with transgenic CD4+ T cells expressing a T‐cell receptor (OTII) specific for the OVA peptide 323–339. T‐cell proliferation was monitored as a measure of successful presentation of the OVA antigen by B cells, that is internalization of OVA protein, intracellular processing into peptides and B‐cell surface expression of the cognate OTII peptide in the context of major histocompatibility complex molecules. BIIB091 inhibited B cell‐mediated antigen presentation to OVA323–339‐specific T cells as shown by the dose‐dependent reduction in the dilution of the Cell Trace Violet tracer used to track proliferation (Figure 4e), with an average IC50 of 22 ± 8 nm (Figure 4f). In contrast, BIIB091 had no effect on the proliferation of the same transgenic T cells in response to the exogenous addition of the OVA323–339 peptide to the cultures (Figure 4g).
BIIB091 blocks myeloid cell functions in vitro
BTK is a crucial signaling node downstream of activating Fc receptors in myeloid cells. 7 In neutrophils, binding of IgG‐containing immune complexes via their Fc region initiates BTK‐dependent signaling leading to cellular activation. One hallmark of immune complex‐induced neutrophil activation is the generation of reactive oxygen species (ROS), a process which is strictly dependent on FcγRIIA signaling. To determine whether BIIB091 could inhibit FcγR‐mediated ROS production, human neutrophils were activated with immune complexes in the absence or presence of BIIB091. Pretreatment with BIIB091 blocked ROS production in a dose‐dependent manner (Figure 5a), with an average IC50 of 4.5 ± 4.9 nm across four healthy donors (Figure 5b).
Figure 5.
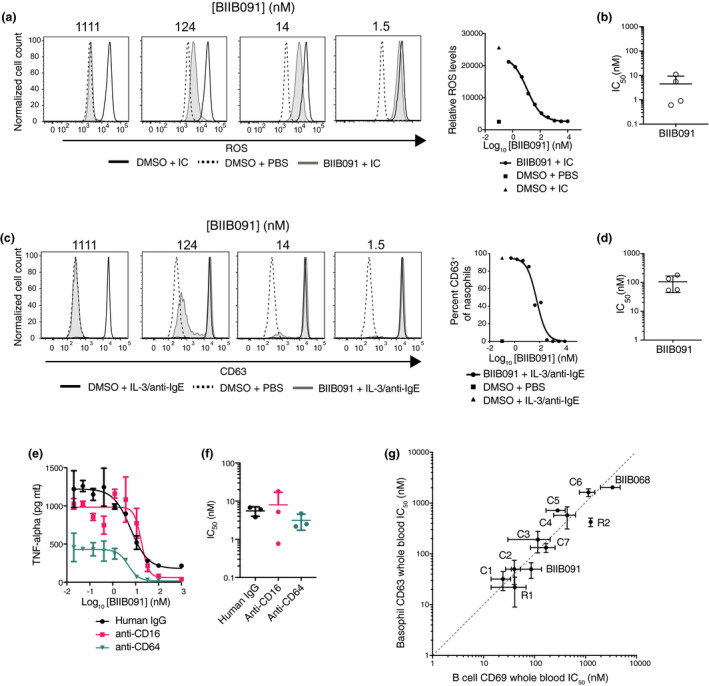
BIIB091 potently inhibits myeloid cell effector functions with IC50s in the same range as in B cells. (a, b) Histogram overlays showing levels of reactive oxygen species (ROS) indicator dihydrorhodamine 123 in purified human neutrophils stimulated with immune complexes (IC) (anti‐ribonucleoprotein (RNP) antibodies complexed with purified RNP) and treated with DMSO or titrating concentrations of BIIB091 (a). IC50 values for four healthy donors (individual data points) are shown along with mean and SD (4.5 ± 4.9 nm) (b). (c, d) Histogram overlays showing CD63 expression levels as a measure of degranulation in CD123+ HLA‐DR− basophils from human whole blood treated with DMSO or titrating concentrations of BIIB091 and stimulated with recombinant human IL‐3 and anti‐human IgE antibodies (c). IC50 values for four healthy donors (individual data points) are shown along with mean and SD (106 ± 62 nm). (e, f) TNFα protein levels measured in supernatant from human primary monocyte cultures treated with DMSO or titrating concentrations of BIIB091 and stimulated with plate‐bound human IgG (circle), anti‐human CD16 (square) or anti‐human CD64 (inverted triangle) antibodies (e). For each stimulating agent, IC50 values for three healthy donors are shown along with mean and SD (5.6 ± 1.5 nm, 8.0 ± 9.0 nm and 3.1 ± 1.4 nm, respectively) (f). (g) Dot plot showing IC50 values (mean and SD) for CD69 expression in anti‐IgD‐stimulated B cells (CD69, x‐axis) and basophil degranulation (CD63, y‐axis) in whole blood assays. Data from n = 11 BTK inhibitors including Biogen BIIB091 and BIIB068 (early generation BTK inhibitor 11 ), 2 reversible competitor inhibitors (R) and 7 covalent competitor inhibitors (C). The dashed line indicates the equipotency line.
BTK is also a crucial signaling node downstream of the FcεRI‐activating receptor for the Fc fragment of IgE. Basophils and mast cells express this receptor and can be activated by cross‐linking of FcεRI‐bound IgE by the specific antigen recognised by the IgE, or experimentally using polyclonal goat anti‐human IgE. The activation of downstream BTK‐dependent signaling pathways leads to rapid degranulation and release of inflammatory mediators. CD63 is a well‐established surrogate measure of basophil and mast cell degranulation and its induction upon FcεR cross‐linking is critically dependent on the kinase activity of BTK. 20 , 21 BIIB091 was evaluated for its ability to inhibit anti‐IgE/FcεRI‐dependent functions using a basophil activation assay in whole blood. Pretreatment of whole blood with BIIB091 completely blocked anti‐IgE‐mediated basophil degranulation in a dose‐dependent manner (Figure 5c) with an average IC50 of 106 ± 62 nm across four healthy donors (Figure 5d).
Finally, human monocytes express at high levels the high‐affinity FcγRI (CD64) receptor for the Fc fragment of IgG while a subset of peripheral non‐classical monocytes also expresses the low‐affinity FcγRIII (CD16) receptor. To assess the effect of BIIB091 on Fc receptor‐dependent effector functions in monocytes, we stimulated human monocytes with plate‐coated FcγR agonists in the presence of titrating concentrations of BIIB091. BIIB091 showed dose‐dependent inhibition of TNFα production in human monocytes (Figure 5e) with average IC50 values of 5.6 ± 1.5 nm, 8.0 ± 9.0 nm and 3.1 ± 1.4 nm following stimulation with human IgG, anti‐human CD16 and anti‐human CD64 antibodies, respectively (Figure 5f).
Importantly, some BTK inhibitors have been reported to have lower potency on Fc receptor‐dependent myeloid cell functions as compared to BCR‐dependent cellular functions in B cells, a property which was attributed to the binding mode of the compounds to BTK and more specifically their ability to sequester Y551 within the kinase pocket away from the activity of upstream SRC kinases. 22 Comparing a large number of competitor and Biogen inhibitors including BIIB091, we could not replicate these findings and instead established that IC50s for inhibition of B‐cell and myeloid cell functions in the same whole blood matrix were within the same range for all inhibitors we tested (Figure 5g), thus indicating that BTK inhibitors were for the most part equipotent in these two cell types irrespective of their binding mode to BTK.
BIIB091 blocks mouse B‐cell activation, antibody secretion and germinal center reactions in vivo
We next explored BIIB091 pharmacodynamic properties in vivo. In a first experimental approach, animals were dosed orally with vehicle or BIIB091 and at 1 h post‐dose, intravenously injected with anti‐IgD antibodies. At 5 h post‐dose, B cells were FACS‐isolated from the spleens for gene expression analysis. Based on PK/PD modelling of the predicted inhibition of B‐cell activation over time post‐dosing, 3 and 30 mg kg–1 dose levels were chosen as they provided an average CD69 inhibition of 70% and 95%, respectively, over the study interval of 4 h (Supplementary figure 4). By comparing gene expression in B cells from IgD‐treated and naïve animals (vehicle groups), we identified 746 differentially expressed genes (DEGs) comprising an IgD‐induced B‐cell activation signature, with 393 upregulated and 353 downregulated DEGs (Figure 6a, Supplementary table 4) (FDR < 0.01, log2 fold change > |1.5|). Pretreatment with BIIB091 dose‐dependently inhibited gene expression changes associated with this in vivo B‐cell activation event, as evidenced by the differential expression (FDR < 0.05) of 499 and 686 out of the 746 IgD signature genes by BIIB091 treatment at 3 and 30 mg kg−1, respectively (Figure 6a). Summarization of the overall changes in expression of the IgD signature genes using z‐scores for each individual gene of the upregulated and downregulated signatures confirmed the dose‐dependent inhibition of the transcriptomic changes associated with B‐cell activation by BIIB091 treatment (Figure 6b). These transcriptomic changes strongly correlated with CD69 levels measured at protein level on the surface of the same B cells during FACS isolation for gene expression analysis (Figure 6b) and thus validated CD69 as a good surrogate marker for the overall transcriptional changes associated with B‐cell activation in vivo. When contrasting gene expression profiles of in vivo IgD‐activated B cells isolated from mice pre‐treated with 30 mg kg−1 BIIB091 versus those isolated from vehicle‐treated mice, Ingenuity Pathway Analysis (IPA) revealed large changes in key cellular pathways including purine nucleotide biosynthesis, cholesterol biosynthesis, BCR signaling, NFAT signaling, cell cycle regulation and tRNA charging (Figure 6c, Supplementary table 5), altogether establishing the inhibitory impact of BIIB091 on cellular and molecular events associated with B‐cell activation.
Figure 6.
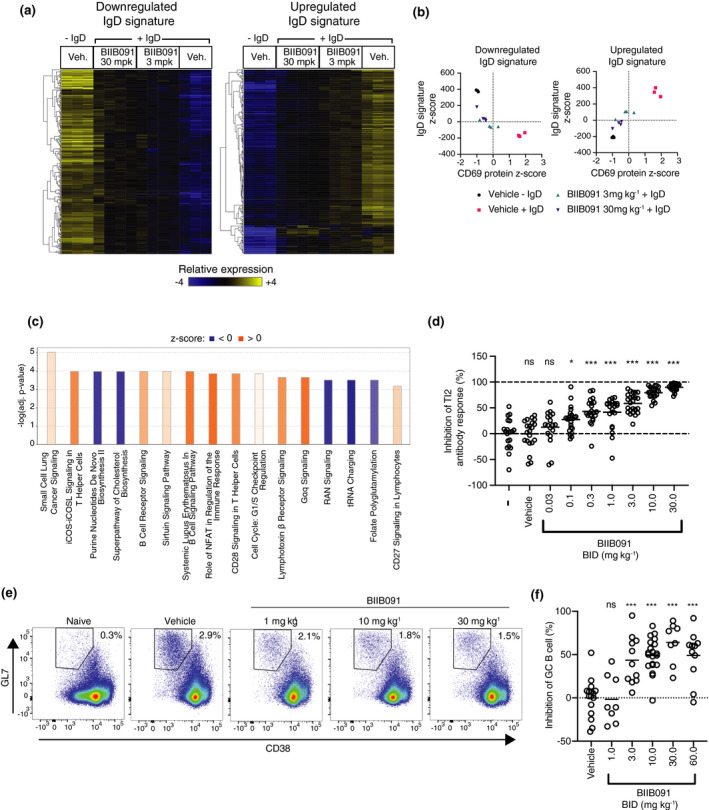
BIIB091 blocks B‐cell activation and differentiation into antibody‐secreting and germinal center B cells in vivo. (a) Hierarchically clustered heatmap of splenic B‐cell gene expression profiles in response to in vivo anti‐IgD stimulation of B cells in mice dosed with either vehicle (Veh.) or BIIB091 at indicated doses. IgD signatures were defined as the genes differentially expressed with FDR < 0.01 and absolute log2 fold change > 1.5 in vehicle‐treated IgD‐stimulated versus vehicle‐treated unstimulated B cells. (b) Scatter plots of IgD signature cumulative z‐scores for the upregulated (right) and downregulated (left) IgD signatures versus CD69 protein z‐scores measured in anti‐IgD‐stimulated (+IgD) or unstimulated (−IgD) B cells isolated from individual mice treated with vehicle or BIIB091 at indicated doses. n = 3 or 4/group. (c) Top differentially regulated canonical pathways identified by IPA in anti‐IgD‐treated B cells isolated from mice dosed with BIIB091 at 30 mpk versus vehicle. Positive (orange) and negative (blue) z‐scores indicate the predicted degree of activation or inhibition for that pathway, respectively. (d) Inhibition of NP‐specific IgM relative titres in mice injected intraperitoneally with 30 μg of NP‐Ficoll and dosed for 10 days with vehicle or BIIB091 at indicated doses. Control groups received either vehicle or no treatment (–). Significance established by 1‐way ANOVA (comparison with vehicle group) followed by Dunnett's multiple comparison test. n = 12/treatment group. (e, f) Representative pseudocolor dot plots (e) and quantification (f) of BIIB091 inhibition of splenic germinal center B‐cell formation identified as CD3−CD19+B220+CD38lowGL7+CD95+ in mice 8 days after immunisation with sheep red blood cells. Number of animals per group from n = 8 to n = 20. Significance established by 1‐way ANOVA (comparison with vehicle group) followed by Dunnett's multiple comparison test. mpk, mg kg−1; Veh, vehicle; ns, not significant; *P < 0.05; ***P < 0.001.
To further assess BIIB091 inhibitory potency on B‐cell functions in vivo, we tested BIIB091 in two well‐established models of BTK‐dependent humoral immunity. In the first model, a thymus‐independent type 2 (TI‐2) antibody response was induced using the hapten antigen NP (4‐hydroxy‐3‐nitrophenyl acetyl) conjugated to Ficoll. Multivalent and highly repetitive epitopes in TI‐2 antigens were demonstrated to induce strictly BTK‐dependent antibody responses. 23 Mice were immunised with NP‐Ficoll and dosed orally BID with BIIB091 or vehicle for 10 consecutive days. A dose‐dependent inhibition of serum NP‐specific IgM antibody titres was observed in mice treated with BIIB091 (Figure 6d). The calculated ED50 of BIIB091 for IgM antibody titres was 1.95 mg kg−1. In the same animals, measurement of pBTK levels in whole blood collected 1 and 4 h post‐last dose using the same assay as described in Figure 1 established a potent and dose‐dependent inhibition of this proximal pharmacodynamic readout in vivo with an IC50 of 3.1 nm (Supplementary figure 5a and b) which was in agreement with the in vitro IC50 of 4.8 nm (Figure 1d).
A thymus‐dependent (TD) immunisation model using the particulate antigen sheep red blood cells (SRBCs) was then used to evaluate the effect of BIIB091 on TD responses. TD responses require thymus‐derived T cells for full activation and subsequent differentiation of B cells into germinal center (GC) B cells and antibody‐producing plasma cells. 24 The SRBC immunisation response model was demonstrated to be BTK‐dependent. 4 , 25 Mice were immunised with SRBCs and dosed orally BID with BIIB091 or vehicle for 8 consecutive days. The frequency of total splenic B cells was unchanged following 8 days of BIIB091 administration at all doses tested (Supplementary figure 6), indicating that inhibition of BTK signaling did not impact B‐cell survival over the duration of this model. In contrast, a dose‐dependent inhibition in the frequency of splenic GC B cells identified as CD19+B220+CD38lowGL7+CD95+ cells was observed in BIIB091‐treated mice with maximal inhibition levels between 49% and 64% observed at doses of 10 mpk and above (Figure 6e and f). Therefore, in contrast to the complete inhibition of T‐independent immunisation responses, BIIB091 only partially inhibited T‐dependent particulate immunisation responses, an observation which was in line with previously published work showing that BTK was strictly necessary for T‐independent B‐cell responses, but only partially required for T‐dependent B‐cell responses. 26 , 27 Altogether, these results demonstrated that BIIB091 inhibited B‐cell activation, antibody secretion and B‐cell differentiation into germinal center B cells in vivo.
BIIB091 blocks B‐cell activation in human healthy volunteers in phase 1 clinical trial
Single and multiple ascending oral doses (SAD/MAD) of BIIB091 were tested in phase 1 healthy participant study (NCT03943056) evaluating safety, tolerability, pharmacokinetics and pharmacodynamic effects. The study details and results were disclosed previously 28 and detailed clinical results along with PK from SAD/MAD will be reported elsewhere (unpublished data). To assess the potential effects of BIIB091 on the survival of circulating immune cells, the abundance of leukocyte subsets was quantified by flow cytometry in whole blood of healthy individuals from the MAD part of the trial on days 1, 7, 14 and on a follow‐up visit 8–10 days post‐last dose (days 22–24). While changes in absolute cell numbers of a few selected cell types were statistically significant by nominal P‐value of 0.05 at some isolated time points (Figure 7a and Supplementary table 6), there was no evidence of consistent trends across doses and time points. More specifically, there was no indication of any decrease in the number of B cells or any other circulating leukocyte subset during duration of BIIB091 administration compared with placebo (Figure 7a and Supplementary table 6).
Figure 7.
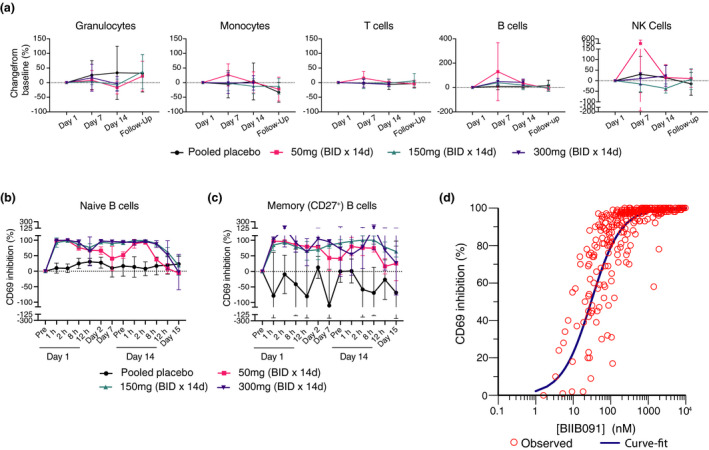
BIIB091 demonstrates complete inhibition of B‐cell activation over the dosing interval in a human phase 1 trial. (a) Changes in leukocyte absolute cell numbers (mean percent change from baseline ± SD) following twice‐daily placebo or BIIB091 dose administration for 14 days in healthy individuals. Day 1, 7 and 14 assessments were done in whole blood samples collected prior to dosing. Follow‐up samples were collected 8–10 days post‐last dose on day 14. Number of tested patient samples for specific cohorts and specific time points varied from n = 3 to n = 6. (b, c) Percent inhibition (SD) of CD69 expression (normalised to day 1 pre‐dose levels) in CD19+ CD27− (naïve) (b) and CD19+ CD27+ (memory) B cells (c) from whole blood of healthy individuals dosed BID with BIIB091 (n = 6/cohort) or placebo (n = 2/cohort) and stimulated ex vivo with anti‐human IgD. Measurements were performed on whole blood samples collected prior to morning dose on day 1 (pre), day 2, day 7 and day 14 (pre). Day 1 (12‐h) assessment was performed on samples collected prior to the evening dose. CD69 expression measurement for the remaining indicated times and day 15 were performed on whole blood samples collected following last morning dose (24 h post‐last administrated dose on day 14 for day 15 time point). Number of tested patient samples varied from n = 2 to n = 6 across different time points and cohorts. (d) Percent inhibition of CD69 expression in total CD19+ B cells from BIIB091‐dosed healthy individuals plotted versus measured plasma BIIB091 concentration across all dose levels and time points (including both SAD and MAD parts of the trial, n = 318 individual data points). BID, twice daily; d, day; h, hour(s).
Next, we evaluated the effect of BIIB091 on B‐cell activation by measuring the upregulation of the activation marker CD69 upon ex vivo stimulation of B cells with anti‐human IgD spiked in whole blood samples collected from the MAD part of the trial. CD69 was chosen as the surrogate biomarker of B‐cell activation based on preclinical results establishing CD69 upregulation on BCR‐stimulated B cells as strictly BTK‐dependent (Figure 2) and correlating with the broader transcriptional programme induced upon BCR‐mediated B‐cell activation (Figure 6). The inhibition of CD69 upregulation in naïve B cells was complete at early time points post‐dosing at the lowest dose tested of 50 mg (at 1 and 2 h post‐dosing), but inhibition levels were not sustained during the dosing interval, as evidenced by pre‐dose average (± SD) CD69 inhibition levels at days 2, 7 and 14 of 66% (± 12), 41% (± 39) and 52% (± 11), respectively (Figure 7b). In contrast, high levels of inhibition were maintained throughout the BID dosing interval at 150 and 300 mg dose levels, as evidenced by ≥ 90% pre‐dose average CD69 inhibition at days 2, 7 and 14, respectively (Figure 7b). Importantly, the inhibitory effect of BIIB091 on B‐cell activation was quickly reversible upon interruption of drug administration with average (± SD) CD69 inhibition levels at 24 h post‐last dose on day 15 ranging from −5.8% (± 2.8) at 50 mg to 19% (± 31) and −2.8% (± 57) at 150 and 300 mg dose levels, respectively. Because of the important role played by memory B cells in MS, we also explored the ability of BIIB091 to inhibit memory B‐cell activation. Only a fraction of memory B cells (49% on average) responded to anti‐IgD stimulation (as opposed to naïve B cells which showed a larger proportion of responding cells with an average frequency of 85% CD69+ cells in this B‐cell subset) (Supplementary figure 7b). This responding memory B‐cell subset corresponded to the fraction of memory B cells which still expressed IgM and IgD and had not switched to other BCR isotypes, a population reported in the literature to represent 10–15% of the total B‐cell pool. 29 While results were intrinsically noisier because of the low frequency of this B‐cell subset, very similar results were obtained as in naïve B cells, establishing the inhibitory potential of BIIB091 in both naïve and unswitched memory B‐cell subsets (Figure 7c and Supplementary figure 7). To characterise the relationship between CD69 inhibition and plasma concentration of BIIB091, a simple E max model was applied. Data from both the SAD and MAD parts of the trial were included in the model and CD69 expression in the total CD19+ B‐cell population was used to model the PK/PD relationship. The estimated human in vivo EC50 value derived from this model was 55 nm with the 95th% confidence interval (CI) [44, 67] (Figure 7c), which was in good agreement with the in vitro whole blood IC50 of 87 nm (± 56) (Figure 2e and f).
Discussion
B cells are a clinically validated target in oncology indications, rheumatoid arthritis, lupus and most recently in MS as evidenced by the recent Phase 3 clinical trials reporting successful results with B‐cell depleting agents in relapsing and primary progressive MS patients. 30 , 31 BTK inhibition was also recently clinically validated with the positive phase 2 trials of Evobrutinib 32 and Tolebrutinib in MS (https://www.sanofi.com/‐/media/Project/One‐Sanofi‐Web/Websites/Global/Sanofi‐COM/Home/common/docs/investors/2020_04_23_BTKi_slides_IR_Call_Final.pdf?la=en&hash=54487F01C86F987F2907BE2B131F73C9). 5 In the context of relapsing MS pathogenesis, the main pathogenic role of B cells is thought to be via BCR‐mediated antigen presentation and stimulation of pro‐inflammatory T‐cell responses in CNS‐draining lymph nodes, a mechanism of action supported by preclinical modelling. 2
From the overall preclinical data presented here, BIIB091 emerges as a novel, potent reversible inhibitor of BTK while maintaining an excellent kinase selectivity characterised by a biochemical potency of 450 pm and a > 500‐fold selectivity relative to more than 400 kinases tested in our kinase‐binding assay. Cellular counter screens confirmed that BIIB091 inhibits specifically BCR and FcR signaling in B cells and myeloid cells, but not ITAM‐containing receptors expressed in other cell types such as T‐cell antigen receptor (TCR) signaling in T cells. In vitro studies testing BIIB091 in assays using purified B cells, myeloid cells and the more physiologically relevant whole blood matrix demonstrated inhibition of proximal signaling such as phosphorylation of BTK itself and PLCγ2, but also and more importantly potent inhibition of distal functional responses. The latter included B‐cell activation, proliferation and its underlying molecular networks as well as FcR‐dependent TNFα secretion by monocytes, reactive oxygen species generation by neutrophils and basophil degranulation (Supplementary table 7). Finally, and more specifically relevant in the context of MS, BIIB091 inhibited BCR‐mediated antigen presentation to T cells, one of the key mechanisms thought to be underlying the efficacy of clinically approved B‐cell depleting agents in this indication (Supplementary table 7).
The potency of BIIB091 in blocking proximal BTK autophosphorylation in unstimulated whole blood with an IC50 of 24 nm is in good agreement with the potency measured upon stimulation of ITAM‐containing immunoreceptors in whole blood assays using more distal readouts such as BCR‐induced B‐cell activation (IC50 = 87 nm) and FcεR‐induced basophil degranulation (IC50 = 106 nm). The fact that the inhibitory potency of BIIB091 is similar in those assays in unstimulated conditions and upon BCR or FcR stimulation indicates that BIIB091 potently inhibits BTK catalytic activity in both resting cells (in which BTK only shows low baseline enzymatic activity) and activated cells (in which BTK activity is increased by 10‐ to 20‐fold 8 ), the latter being most relevant to disease conditions. In addition to reflecting differences between proximal and distal readouts, this residual shift in potency observed for BIIB091 in unstimulated and stimulated cells could be explained by the fact that BTK becomes trans‐phosphorylated by SRC kinases (LYN) on the regulatory tyrosine residue Y551 upon cell stimulation and the fact that this transactivation event is associated with conformational changes altogether leading to the observed increased catalytic activity of the enzyme. 8 Because BIIB091 and most other reversible ‘H3 binders’ compounds do not bind effectively to the Y551‐phosphorylated form of the kinase, 33 the possible mechanistic hypothesis to account for this residual minimal shift as opposed to the larger shifts in potency observed for earlier generation molecules such as BIIB068 is the slower off‐rate (k off) of BIIB091 in the kinase pocket. The longer resulting residency time of BIIB091 in the kinase pocket likely ensures that the competing phosphorylation event by LYN kinase on Y551 which only happens in conditions of BCR or FcR stimulation is largely prevented by the efficient and long‐lasting BIIB091‐mediated sequestration of Y551 within the kinase pocket. This property contrasts and differentiates BIIB091 from prior generation reversible BTK inhibitors with shorter residency times in the kinase pocket and therefore less efficient and more short‐lived sequestration of Y551 away from the activity of upstream SRC kinases.
Based on these considerations, and despite its practical use as a pharmacodynamic biomarker of target engagement in prior clinical studies, the potency of BTK inhibitors should not rely or at least not exclusively rely on the measurement of BTK autophosphorylation on Y223 in unstimulated cells. Further supporting this argument and highlighting the limited functional significance of this pharmacodynamic readout, transgenic expression of a tyrosine (Y) to phenylalanine (F) Y223F mutant of BTK in the B‐cell lineage corrects all B functional defects observed in BTK‐deficient mice. 34 The limitations of pBTK as a pharmacodynamic readout for BTK inhibitors are well illustrated by comparing BIIB068 to BIIB091 inhibitors. While showing potency on pBTK in unstimulated whole blood in the nm range (with IC50s of 165 and 19 nm, respectively), these two inhibitors showed indeed widely distinct potency in stimulated cells in whole blood matrix with a 40‐fold shift in IC50s for B‐cell activation (3.5 µm versus 87 nm for BIIB068 and BIIB091, respectively) and a 19‐fold shift in IC50s for basophil degranulation (2.0 µm versus 106 nm for BIIB068 and BIIB091, respectively). The nanomolar range potency of BIIB091 in both unstimulated and stimulated cells is thus a key differentiating property of this inhibitor compared with prior generation BTK inhibitors. This property is directly relevant to autoimmune disease settings such as MS in which the role of BTK has been tightly associated with the activation of BCR and FcR pathways. 1 In summary, these results indicate that not all reversible BTK inhibitors are equal and that while most show a potency disconnect between proximal and distal readouts, one needs to carefully evaluate the magnitude of shift between proximal and distal readouts and advance preferentially in clinical settings those demonstrating good potency against stimulated cells.
Prior reports suggested that not all inhibitors equally inhibited BCR signaling in B cells and FcR signalling in myeloid cells. 22 Compounds with such disconnect included ibrutinib and evobrutinib with IC50 shifts for inhibition of B‐cell and basophil activation in whole blood assays of 23‐fold and 19‐fold, respectively. 22 , 35 This property was attributed to the differential ability of the BTK inhibitors to sequester effectively Y551, a property which was found to be more critical for inhibition of myeloid cells than B cells. While the molecular mechanism associated with this differential requirement was not elucidated, it was postulated to represent a different signalling requirement in B cells and myeloid cells. We could not replicate these findings here for a number of reversible and covalent competitor BTK inhibitors tested head‐to‐head in whole blood B‐cell activation and basophil degranulation assays. In contrast to this prior study which used anti‐IgM antibodies, we used anti‐IgD antibodies to stimulate B cells in whole blood and compared IC50s obtained in B cells in these stimulation conditions with IC50s obtained in basophils stimulated with anti‐IgE antibodies. One possible hypothesis to explain these divergent results is the titration of the anti‐IgM antibodies used to stimulate B cells in whole blood by the endogenous IgM immunoglobulins (present at 1–4 mg mL−1 concentration in human serum) whereas IgD only exists as a plasma membrane‐associated protein on the surface of B cells and not as a soluble immunoglobulin. Because of this titration phenomenon, anti‐IgM antibodies only lead to a sub‐par and minimal stimulation of B cells when used in whole blood with very minimal change in CD69 expression in whole blood matrix (data not shown). This technical limitation likely led to the overestimation of compound B‐cell potency in these prior studies and the previously reported disconnects for some BTK inhibitors between IC50s in the two cell types. 22 Beyond highlighting the importance of tailoring the B‐cell stimulating agent to the assay matrix and the fact that the use of anti‐IgM antibodies should be precluded when using whole blood assays, these results also reconcile these previously paradoxical conclusions and establish that the molecular requirements for BTK signalling and inhibition are not distinct in B cells and myeloid cells.
In vivo studies with BIIB091 demonstrated target engagement by showing inhibition of basal constitutive BTK tyrosine autophosphorylation in vivo in mouse whole blood in a concentration‐ and time‐dependent manner. The in vivo IC50 of 3.1 nm was in good agreement with the in vitro IC50 of 5 nm for this same proximal readout in mouse whole blood. BIIB091 in vivo inhibitory potency was also established on two types of B‐cell functional responses: antibody production in response to immunization with a thymus‐independent type 2 (TI‐2) antigen; and B‐cell differentiation into GC B cells upon stimulation with a particulate T‐dependent antigen. In agreement with these in vivo preclinical B‐cell functional data, phase 1 results established very potent inhibition of B‐cell activation as measured by CD69 activation marker throughout the entire dosing interval at the two highest BID doses tested of 150 and 300 mg. While prior clinical studies with other BTK inhibitors established the effect of BTK inhibition on B‐cell activation, 36 , 37 , 38 , 39 it is to our knowledge the first demonstration in clinical trial settings that BTK inhibitors block the activation of both naïve and memory B cells, the latter having been postulated as a critical effector B‐cell subset in the context of MS pathogenesis. 40 Because of the nature of the stimulation agent we used (anti‐IgD antibodies), only unswitched IgD+ memory B cells could be assessed in our study, which represents one limitation of this finding. The significant variability that we observed in our measurements in this cell compartment was likely caused by the low abundance of unswitched IgD+ memory B cells. Future analyses should extend these findings to class‐switched memory B cells. Finally, the in vivo IC50 established in human healthy volunteers of 55 nm in phase 1 trial setting is in good agreement with the in vitro preclinical IC50 determined in human whole blood, establishing a good human in vitro to in vivo correlation and supporting the use of this biomarker in future clinical trials.
Like many other B‐cell functions, we established pre‐clinically that BCR‐mediated antigen presentation is potently inhibited by BTK inhibitor BIIB091. Importantly, BIIB091 effect on human B‐cell activation was quickly reversible upon interruption of drug administration and BIIB091 also did not impact the survival of B cells or other immune subsets during the 14‐day dosing period of phase 1 clinical trial. While the impact of BIIB091 on B‐cell survival over longer periods of time will need to be further evaluated in phase 2 trials, inhibiting BTK is predicted to recapitulate the key mechanism underlying the efficacy of B‐cell depleting agents while ensuring reversibility and mitigating safety risks associated with cytolytic B‐cell targeting. In addition to B cells which are now a clinically validated cell type in MS, there is a large body of support for the pathological role of myeloid cells in MS and in particular peripheral CCR2+ monocytes. 41 , 42 , 43 CD16+ monocytes were reported in active MS lesions while detected at lower frequencies in the CSF from patients with MS compared with controls, a result suggesting their preferential recruitment to actively demyelinating lesions. 44 , 45 Genetic ablation of all activating FcγRs or FcγRIII specifically has convincingly established the pathogenic role of FcRs in myeloid cells in MS preclinical models. The role of FcRs in disease pathogenesis is not completely understood, but includes immune complex (IC)‐mediated endocytosis and antigen presentation to T cells as well as the regulation of myeloid cell activation and functions. 46 , 47 , 48 , 49 , 50 By reversibly inhibiting BTK in myeloid cells in addition to its effects in B cells, BIIB091 is thus expected to provide additional clinical benefits over B‐cell only therapeutic agents without the potential safety liability associated with long‐term B‐cell depleting agents or poorly selective covalent BTK inhibitors. Altogether, preclinical and early clinical characterisation of the mechanism and pharmacodynamics of BIIB091 in vitro and in vivo provides support for this therapeutic hypothesis and its use as a differentiated therapy for patients with MS and other B‐cell and/or myeloid cell‐driven autoimmune conditions.
Methods
Clinical trial and human samples
The BIIB091 phase 1 study (NCT03943056) was performed in accordance with Title 21, US CFR Parts 50, 54, 56 and 312 Subpart D; the ICH guideline on GCP (E6); the European Union Clinical Trial Directive 2001/20/EC; and the ethical principles outlined in the Declaration of Helsinki. The IRB approving the study was Midlands IRB (Overland Park, KS, USA). This was a 2‐part, randomised, blinded, placebo‐controlled, dose‐escalation study with single ascending doses and multiple ascending doses of orally administered BIIB091 in healthy adult participants. 28 Detailed clinical results along with SAD/MAD PK data will be reported elsewhere (unpublished data). The objectives of the study were to assess safety, tolerability, pharmacokinetics and pharmacodynamics of orally administered BIIB091. Participants in each cohort were randomly assigned to either BIIB091 or placebo in a 3:1 ratio. Participants in the single ascending dose portion of the study received a single study treatment dose of 50, 150, 300, 600 or 1200 mg. Participants in the multiple ascending dose portion of the trial received study treatment twice daily at a dose level of 50, 150 or 300 mg for up to 14 days. Standard safety assessments were collected daily during the dosing period and at scheduled follow‐up visits, in accordance with the study protocol. Samples for pharmacokinetic (PK) and pharmacodynamic (PD) assessments were collected and assessed as described below.
Whole blood and buffy coat from healthy donors for in vitro preclinical pharmacodynamic readouts were either obtained from the Biogen Blood Donor Program or purchased from Research Blood Components (Watertown, MA) and were processed on the day they were drawn. Whole blood from patients with MS or healthy donors for in vitro preclinical pharmacodynamic readouts were obtained from Sanguine Biosciences (Sherman Oaks, CA) and were processed on the next day following the blood draw. Patients with MS did not have any history of B‐cell depleting therapies or were on Fingolimod for 3 months prior to blood draw.
Mice
This study was performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Research animals at Biogen were housed under specific pathogen‐free conditions in an AAALAC accredited facility with a 12‐h–12‐h light–dark cycle and environmental conditions controlled at 21°C and 40–60% humidity. Animals were handled according to an approved institutional animal care and use committee (IACUC) protocol (IACUC #749). C57BL/6 and B6. Cg‐Tg(TcraTcrb)425Cbn/J (OTII) mice were purchased from Jackson Labs (Bar Harbor, ME). BIIB091 was administered p.o. at the indicated dosage (mg kg−1 body weight) in a volume of 10 mg kg−1 body weight.
For in vivo B‐cell activation studies, mice were dosed i.v. with 200 μL of goat anti‐mouse IgD (MD Biosciences, Oakdale, MN) at 1 h post‐administration of BIIB091. At 5 h post‐BIIB091 (4 h post‐anti‐IgD) administration, mice were euthanised by CO2 inhalation and spleens were dissected and kept on ice until processing.
T cell‐dependent immune responses were elicited by immunisation with sheep red blood (SRB) cells (Cedarlane Labs, Burlington, ON). Beginning on day 1, mice were dosed BID orally with BIIB091 or vehicle at 10 mL kg−1 until day 9. An SRB cell suspension was washed a total of three times with PBS and then resuspended in sterile PBS at a concentration of 1 × 109 cells mL−1 and on day 2, each mouse was injected intraperitoneally with 200 μL of SRB suspension (2 × 109 cells). On day 9, 3 h after final BIIB091 administration, study animals were anaesthetised with isoflurane and euthanised by cardiac puncture until exsanguination. Spleens were collected after euthanasia and stored on ice until processing.
T cell‐independent immune responses were elicited by immunisation with 4‐Hydroxy‐3‐nitrophenylacetic Ficoll (NP‐Ficoll). On day 1, a 0.3 mg mL−1 NP‐Ficoll was mixed at a 1:1 ratio with Imject Alum (Thermo Fisher, Waltham, MA) after preparation, 1 h after the first administration of BIIB091. BIIB091 was administered via oral gavage twice a day on days 1–9 and once on day 10. On day 10, animals were anaesthetised using isoflurane at specified time points relative to the final BIIB091 administration (i.e. pre‐dose, 1 h after final dose or 4 h after final dose; n = 4 animals per treatment group per time point) and blood was collected by cardiac puncture in sodium heparin tubes under deep anaesthesia and until exsanguination.
Flow cytometry and FACS isolation
Human peripheral blood mononuclear cells (PBMCs), isolated B cells and murine splenocytes were first incubated with Fc block (BD Biosciences, San Jose, CA) then stained with fluorochrome‐labelled antibodies in Cell Staining Buffer (Biolegend, San Diego, CA) for 30 min on ice. Whole blood (human and mouse) was directly stained with fluorochrome‐labelled antibodies on ice for 30 min followed by RBC lysis and fixation with Lyse/Fix Buffer (BD Biosciences). Stained cells were analysed on either BD LSRII or LSRFortessa X‐20 flow cytometer (BD Biosciences) along with appropriate compensation controls. Analysis of the data was performed with FlowJo V10 (BD Biosciences). The antibodies used in this study and catalog information are listed in Supplementary table 1.
For FACS isolation of mouse B cells, 2 × 107 splenocytes from each mouse were resuspended in 150 μL of FACS Buffer containing 5 μg mL−1 of anti‐CD16/CD32 and incubated on ice for 10 min. One hundred and fifty microlitre of 2× staining cocktail (anti‐CD3 PE‐Cy7, anti‐CD69‐PE, anti‐CD86 AlexaFluor 647, anti‐B220 Alexa Fluor 488 and anti‐CD62L BV605) was then added to each sample, mixed by gentle vortexing and incubated on ice for 30 min in the dark. Three millilitre of cold FACS Buffer was added, and samples were centrifuged at 300 g for 5 min at 4°C. Supernatant was aspirated, and cells were then resuspended in 500 μL FACS buffer and transferred into 40 μm filter‐top 5‐mL tubes. Prior to sorting, DAPI was added (10 ng mL−1 final) as a viability marker. B cells were cell‐sorted on a BD FACS Aria Fusion™ (BD Biosciences) by selecting for DAPI‐ CD3− B220+ cells, and 1.5 × 105 B220+ CD3− cells were directly sorted into 1.7‐mL tubes containing 500 μL RLT Lysis buffer provided in the RNeasy®Micro RNA Isolation Kit (Qiagen, Hilden, Germany), snap‐frozen on dry ice and then stored at −80°C until RNA isolation.
Live Human B cells were similarly sorted by staining‐activated B cells with DAPI, CD45‐APC, CD20‐BV785 and CD69‐PE. Live CD45+ CD20+ B cells were sorted on a BD FACS Aria Fusion™ into 1.7‐mL tubes containing 500 μL RLT Lysis buffer, snap‐frozen on dry ice and then stored at −80°C until RNA isolation for gene expression profiling.
Surface plasmon resonance spectroscopy
Surface plasmon resonance (SPR) experiments were performed on a Biacore S200 instrument (Cytiva, formerly GE Healthcare, Marlborough, MA) at 25°C using a running buffer composed of 20 mm HEPES pH 7.5, 150 mm NaCl, 0.2 mm TCEP, 0.005% Tween20 and 2% DMSO. Biotin CAPture chip was pre‐conditioned, and oligo streptavidin was captured according to chip manufacturer instructions (Cytiva, formerly GE Healthcare). Full‐length, avi‐tagged active BTK was captured onto the streptavidin at a density around ~1000 RU. Using single‐cycle kinetics compound was injected over BTK surface at various concentrations (top concentration 500 nm, threefold dilution series, five injections), at a flow rate of 70 μL min−1. The associations were set to 1 min followed by up to 60‐min dissociation. The raw data were processed using Biacore evaluation software and the data fitted kinetically to a 1:1 binding model which included a mass transfer limitation. Half‐life was calculated from the off‐rate using the correlation t 1/2 = ln2/K d.
Basal BTK‐associated tyrosine phosphorylation in mouse and human whole blood
Human or mouse whole blood was treated with titrating concentrations of BIIB091 or dimethyl sulfoxide (DMSO) (Sigma‐Aldrich, St. Louis, MO) for 30 min and subsequently lysed at room temperature (RT) for 10 min using lysis buffer (150 mm NaCl, 20 mm Tris pH 7.5, 1 mm EDTA, 1 mm EGTA, 1% Triton‐X‐100) containing protease and phosphatase inhibitors (MSD, Rockville, MD). Negative control (DMSO‐treated) samples were lysed in buffer containing protease inhibitors but lacking phosphatase inhibitors. Lysates were diluted (1:2) in Blocking Buffer (3% Blocker A; MSD) and transferred to plates (goat anti‐rabbit, MSD) which had been coated overnight (4°C) with 0.5 μg mL−1 of rabbit anti‐BTK (D3H5; Cell Signaling Technologies, Danvers, MA) and subsequently blocked with blocking buffer for 2–3 h at RT. Following overnight (4°C) sample incubation, the plate was incubated for 2 h (RT) with biotinylated anti‐phosphotyrosine (diluted 1:400, P‐Tyr‐100; Cell Signaling Technologies) and 1 h (RT) with streptavidin Sulfo‐Tag (diluted 1:500; MSD). Phosphorylated BTK was detected as ECL signal on a QuickPlex Imager (MSD) after the addition of 1× MSD Read Buffer.
BCR‐induced PLCγ2 phosphorylation
Ramos cells (ATCC, Manassas, VA) were treated with titrating concentrations of BIIB091 for 30 min at 37°C in duplicates and subsequently stimulated for 5 min with anti‐human IgM (1 µg mL−1; Jackson ImmunoResearch, West Grove, PA). After centrifugation and removal of the supernatant, the cell pellets were lysed with gentle shaking in cold lysis buffer (Promega, Madison, WI) containing protease (Complete Mini; Roche, Basel, Switzerland) and phosphatase (PhosSTOP™, Roche); and phosphatase inhibition cocktail 2 and 3 (Sigma‐Aldrich) inhibitors. Cell lysates (diluted) were added and incubated for approximately 12 h at 4°C in MSD plate coated with anti‐PLCγ2 antibody (diluted 1:100, B‐10; Santa Cruz Biotechnology, Dallas, TX). Phosphorylated PLCγ2 was detected with rabbit polyclonal anti‐phospho tyrosine 1217 of PLCγ2 (diluted 1:1000; Cell Signaling Technologies) and Sulfo‐Tag goat anti‐rabbit antibody (diluted 1:500; MSD). Levels of phosphorylated PLCγ2 were detected as ECL signal on a Sector S600 (MSD) after the addition of 1× MSD Read Buffer.
Anti‐IgD‐induced B‐cell activation
For human or mouse whole blood, samples were treated with titrating concentrations of BIIB091 or DMSO for 30 min. Mouse whole blood was stimulated with polyclonal goat anti‐mouse IgD anti‐serum (diluted 1:10; eBioscience, San Diego, CA) for 5 h at 37°C. Human whole blood was stimulated with anti‐Human IgD‐dextran (clone 1A62, 0.1 μg mL−1; Fina Biosolutions, Rockville, MD) or F(ab′)2 anti‐human IgD (20 μg mL−1; Agilent, Santa Clara, CA) at 37°C overnight. Density gradient (Ficoll‐Paque, GE Healthcare, Chicago, IL) isolated PBMCs from healthy donors were incubated with titrating concentrations of BIIB091 or DMSO for 30 min followed by overnight stimulation at 37°C with polyclonal rabbit F(ab′)2 anti‐human IgD (5 μg mL−1, Agilent, Santa Clara, CA). The frequency of CD45R+ mouse B cells and CD19+ human B cells expressing B‐cell activation marker CD69 was determined by flow cytometry.
BCR‐mediated antigen presentation to T cells and anti‐IgM‐induced B‐cell activation in mouse splenocytes
Magnetic bead separation was used to isolate B cells (Mouse B Cell Isolation Kit; Miltenyi Biotec, Bergisch Gladbach, Germany) and CD4+ T cells (CD4+ T Cell Isolation kit; Miltenyi Biotec) from mechanically dissociated spleens from OTII mice. CD4+ T cells were labelled with CellTrace™ Violet Proliferation Dye (Thermo Fisher) and pre‐treated with titrating concentrations of BIIB091. Isolated B cells were pre‐treated with titrating concentrations of BIIB091 for 30 min at 37°C. To target OVA to BCR, BIIB091‐treated B cells were incubated with biotin‐conjugated goat F(ab′)2 anti‐mouse IgM (10 μg mL−1; Jackson ImmunoResearch) on ice for 15 min, followed by washing and subsequent incubation with OVA‐conjugated anti‐biotin antibody (OVA Antigen Delivery Reagent; Miltenyi Biotec) for 10 min on ice. Titrating doses of BIIB091 were present during these two incubation steps. OVA‐targeted B cells were then cultured with CellTrace™ Violet‐labelled CD4 T cells in the presence of titrating concentrations of BIIB091 for 72 h at 37°C. Following the 72‐h incubation at 37°C, the B:T Co‐culture was labelled with Zombie UV™ live/dead dye (Biolegend) and stained for flow cytometry. The level of CD4+ T‐cell proliferation and the frequency of CD69+ B220+ B cells was quantified with FlowJo.
FcεR‐mediated basophil degranulation in human whole blood
Human whole blood was pre‐incubated with titrating concentrations of BIIB091 for 30 min at 37°C. Blood was then stimulated with recombinant human interleukin (IL)‐3 (2.22 ng mL−1; R&D Systems, Minneapolis, MN) for 10 min followed by stimulation with polyclonal goat anti‐human IgE (0.23 µg mL−1; Bethyl Laboratories, Montgomery, TX) at 37°C for 20 min. The percentage of CD63+ basophils (HLA‐DR−, CD19−, CD123bright) was determined by flow cytometry.
FcγRIIA‐induced human neutrophil reactive oxygen species generation
Human neutrophils were enriched with Mono‐Poly density gradient (MP Biomedicals, Irvine, CA) and further purified using EasySep® Human Neutrophil Enrichment Kit (STEMCELL Technologies, Cambridge, MA). Neutrophils were labelled with the ROS indicator dihydrorhodamine 123 (DHR123; Thermo Fisher) at a final concentration of 5 μg mL−1 for 5 min at 37°C. Labelled neutrophils were then incubated with titrating concentrations of BIIB091 for 30 min at 37°C and subsequently stimulated with immune complex (IC) for 1–2 h at 37°C. The IC was prepared by incubating equal volumes of human anti‐RNP (RayBiotech Inc., Peachtree Corners, GA), isolated from SLE patient serum and purified RNP antigen (Arotec Diagnostics, Lower Hutt, New Zealand) at 37°C for 30 min. The final dilution/concentrations of the IC components were 1:50 for the anti‐RNP and 16.22 μg mL−1 for the RNP antigen. Following stimulation with IC, neutrophils were stained with live/dead dye TO‐PRO®‐3 (Thermo Fisher) diluted to a final concentration of 10 nm and acquired on a BD LSRFortessa X‐20 flow cytometer. The DHR123 fluorescence, detected in the FITC channel and indicative of ROS generation, was displayed as histogram overlays for the singlet live neutrophil cell population of each sample.
FcγR‐mediated TNFα production by human monocytes
Untouched total monocytes were purified from PBMCs using Pan Monocyte Isolation Kit (Miltenyi Biotec). Monocytes were stimulated overnight with plate‐bound IgG from human serum (Sigma‐Aldrich), anti‐hCD16 antibody (3G8; Biolegend) and anti‐hCD64 antibody (10.1; Biolegend), in the presence of titrating concentrations of BIIB091 in X‐VIVO 10™ medium (Lonza Bioscience, Basel, Switzerland). Supernatants were collected and TNFα levels were quantified by Human TNF‐α Tissue Culture Kit (MSD) according to the manufacturer's instructions.
Human B‐cell isolation and activation
Untouched B cells were purified from healthy donor PBMCs using EasySep® Human B Cell Enrichment Kit (STEMCELL Technologies) and labelled with CellTrace™ CFSE (Life Technologies, Carlsbad, CA). Labelled B cells were then stimulated with TLR9 ligand CpG ODN 2006 (Invivogen, San Diego, CA) or F(ab′)2 goat anti‐human IgM (1 µg mL−1; Jackson ImmunoResearch) or combinations of anti‐IgM)/MEGACD40L (1 μg mL−1; Enzo Life Sciences, Farmingdale, NY) with either recombinant human IL‐4 (20 ng mL−1; R&D Systems) or recombinant human IL‐21 (20 ng mL−1; Miltenyi Biotec) in the presence or absence of 100 nm of BIIB091 in serum‐free X‐VIVO 10™ medium for 3–5 days. At the end of culture period, cells were analysed by flow cytometry and live B cells were FACS‐sorted for gene expression profiling.
Mouse and human gene expression profiling
Total RNA was purified from FACS‐isolated mouse and human B cells using the RNeasy‐Micro RNA Isolation Kit (Qiagen) per manufacturer protocol. Quantity and quality of total RNA samples were measured using a Bioanalyzer Pico RNA chip (Agilent, Santa Clara, CA), and only samples with RNA Integrity Scores (RIN ≥ 8) were included. cDNA for RNA‐Seq libraries was prepared using 10 ng of RNA per sample using the SMART‐Seq v4 Ultra Low RNA Input Library prep protocol (Clontech‐Takara, Kusatsu, Shiga, Japan) according to the manufacturer's instructions, which included 11 cycles of PCR. cDNA was measured on a Life Technologies QuBit 3.0 Fluorometer with the Qubit dsDNS HS Assay kit (Invitrogen Q32854), 150 pg of the resulting cDNA was then tagged and fragmented with the Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA) according to the manufacturer's instructions. Quality of amplified cDNA and fragmented libraries was assessed on a PerkinElmer GX (Waltham, MA) using the DNA extended range lab chip (Perkin Elmer, Waltham, MA) and DNA HiSens Reagent Kit (Perkin Elmer, Waltham, MA) following the manufacturer's instruction. Tagged libraries were pooled (5 nm total DNA) and sequenced on an Illumina HiSeq2500 sequencer (Illumina), with an average depth of 10 million fragments (paired‐end reads). Quality control was performed using Illumina's BaseSpace run summary tool. RNAseq reads were aligned to the murine or human genome using STAR (2.7.0a). 51 Final gene count table was obtained using featureCounts (1.6.4), and TPM table was calculated by RSEM (1.3.1). 52 Differential expression analysis was performed by DESeq2 (1.22.2). 53 A false discovery rate cut‐off of 0.05 was used to select the final list of differentially expressed genes.
Anti‐CD3/anti‐CD28‐mediated T‐cell activation and antigen‐independent B‐cell activation by anti‐CD3/anti‐CD28‐activated T cells
PBMCs were pre‐treated with titrating concentrations of BIIB091 for 30 min and then stimulated with soluble anti‐human CD28 (CD28.2, 5 μg mL−1; BD, Franklin Lakes, NJ) and plate‐bound anti‐human CD3 (HIT3a, 0.5 µg mL−1; BD) overnight at 37°C. At the end of culture, CD69 levels on CD4+, CD8+ T cells and CD19+ B cells were analysed by flow cytometry.
NP‐Ficoll T cell‐independent immune responses
To determine the titres of NP‐specific IgM antibodies in the serum, 96‐well high bind plates were coated with NP‐BSA (20 μg mL−1 in PBS; Biosearch Technologies, Novato, CA) and incubated overnight at 4°C. Using an ELx405 Select plate washer (Bio‐Tek, Winooski, VT), plates were washed three times and were blocked with Blocking Buffer (3% Omniblok in water; American Bioanalytical, Natick, MA) per well for 2–3 h at room temperature. Diluted serum samples were added to NP‐BSA‐coated plates and incubated at 37°C for 1 h with shaking. Plates were subsequently incubated with biotinylated anti‐IgM antibody (0.5 ng mL−1; BD Biosciences) for 1 h, HRP‐conjugated Streptavidin (BD Pharmingen, San Diego, CA) diluted 1:5000 for 1 h and ABTS One Component HRP Microwell Substrate (Surmodics, Eden Prairie, MN) until colour developed, with washing steps between the incubations. 1% SDS Solution was added to stop colour development. Using a SpectraMax Plus plate reader (Molecular Devices, San Jose, CA), sample absorbance values were immediately read at 405 nm.
Phase 1 clinical trial measurements [NCT03943056]
Enumeration of lymphocyte absolute cell counts
Heparinised venous whole blood from patients enrolled in the clinical study was aliquoted (100 μL) into BD Trucount™ tubes (BD Biosciences) and stained for 20 min, at room temperature and protected from light, with fluorochrome‐conjugated lineage‐specific antibodies (Aliquot 1: CD3‐FITC, CD8‐PE, CD45‐PerCP and CD4‐APC; Aliquot 2: CD3‐FITC, CD16‐PE, CD56‐PE, CD45‐PerCP and CD19‐APC). Red blood cells were lysed with 1 mL of 1× FACSLyse (BD Biosciences) solution for 10 min, incubated at room temperature and in the dark. Upon lysis (no washing), tubes were immediately acquired on a properly calibrated BD flow cytometer. Absolute numbers (cells μL−1) of positive cells in the sample were determined by comparing cellular events with the known number of fluorescent beads in the BD Trucount™ tubes.
Ex vivo anti‐IgD‐induced human B‐cell activation
Heparinised venous whole blood from patients enrolled in the clinical study was aliquoted (100 μL) into appropriately labelled 15‐mL conical tubes. One set of aliquots received 0.1 μg mL−1 of anti‐human IgD (clone IA6‐2) conjugated to dextran. All samples were incubated at 37°C/5% CO2 for 18–24 h. Upon incubation, samples were transferred to 12 × 75 mm FACS tubes and stained for 30 min at room temperature with a cocktail of antibodies containing anti‐human CD19‐PE and anti‐human CD69 APC. Red blood cells were lysed by incubating samples for 10 min at room temperature with 2 mL of 1× FACSLyse solution. Upon lysis, remaining cells were washed with 2 mL of PBS supplemented with 1% Bovine Serum Albumin followed by fixation with 500 μL of 1% Paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) solution. Samples were acquired on a properly calibrated BD flow cytometer and analysed for CD69 expression on CD19+, CD19+ CD27− or CD19+ CD27+‐gated B cells. Quantum™ APC MESF beads (Bangs Laboratories, Fishers, IN) were utilised to standardise the fluorescence intensity of CD69. The extent of inhibition was calculated based on pre‐dose samples.
Statistical analyses
Statistical analysis was performed using Prism software (GraphPad, San Diego, CA). IC50 values were derived using a variable slope (3‐ or 4‐parameter) non‐linear regression model. To determine significance values when comparing in vivo mouse drug responses, 1‐way ANOVA followed by Dunnett's multiple comparisons test post hoc was performed. A value of P < 0.05 was considered statistically significant. All data are presented as the mean with standard deviation.
Descriptive statistics were used to summarise data in phase 1 study [NCT03943056]. A mixed model for repeated measure (MMRM) was used to analyse the longitudinal data for each of the lymphocyte subset parameters. The MMRM included variables for treatment, visit and the treatment by visit interaction adjusting for the baseline value. An unstructured covariance matrix was used to model the within‐subject variability. No adjustment for multiple comparisons was made. In this analysis, placebo patients were pooled across cohorts.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Eris Bame: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Resources; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Hao Tang: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Resources; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Jeremy Carlos Burns: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Resources; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Million Arefayene: Conceptualization; Formal analysis; Visualization; Writing‐original draft; Writing‐review & editing. Klaus Michelsen: Data curation; Formal analysis; Investigation; Software; Writing‐review & editing. Bin Ma: Conceptualization; Resources; Writing‐review & editing. Isaac Marx: Resources; Writing‐review & editing. Robin Prince: Resources; Writing‐review & editing. Allie M Roach: Conceptualization; Data curation; Investigation; Methodology; Resources; Validation; Visualization; Writing‐review & editing. Urjana Poreci: Conceptualization; Data curation; Investigation; Methodology; Resources; Visualization; Writing‐review & editing. Douglas Donaldson: Investigation; Resources; Writing‐review & editing. Patrick Cullen: Investigation; Methodology; Resources; Writing‐review & editing. Fergal Casey: Formal analysis; Software; Supervision; Writing‐review & editing. Jing Zhu: Formal analysis; Software; Writing‐review & editing. Thomas Carlile: Formal analysis; Investigation; Methodology; Software; Writing‐review & editing. Dipen Sangurdekar: Formal analysis; Software; Writing‐review & editing. Baohong Zhang: Formal analysis; Software; Writing‐review & editing. Patrick Trapa: Methodology; Supervision; Writing‐review & editing. Joseph Santoro: Investigation; Methodology; Visualization; Writing‐review & editing. Param Murugan: Investigation; Methodology; Visualization; Writing‐review & editing. Alex Pellerin: Investigation; Methodology; Resources; Validation; Visualization; Writing‐review & editing. Stephen Rubino: Conceptualization; Data curation; Investigation; Methodology; Writing‐review & editing. Davide Gianni: Conceptualization; Data curation; Investigation; Methodology; Writing‐review & editing. Bekim Bajrami: Conceptualization; Data curation; Investigation; Methodology; Resources; Validation; Visualization; Writing‐review & editing. Xiaomei Peng: Conceptualization; Writing‐review & editing. Alex Coppell: Project administration; Resources; Writing‐review & editing. Katherine Riester: Formal analysis; Project administration; Software; Visualization; Writing‐review & editing. Shibeshih Belachew: Conceptualization; Supervision; Writing‐review & editing. Devangi Mehta: Conceptualization; Project administration; Supervision; Writing‐review & editing. Mike Palte: Conceptualization; Project administration; Supervision; Visualization; Writing‐review & editing. Brian Hopkins: Conceptualization; Project administration; Supervision; Writing‐review & editing. Matthew Scaramozza: Conceptualization; Project administration; Supervision; Writing‐review & editing. Nathalie Franchimont: Conceptualization; Supervision; Writing‐review & editing. Michael Christian Mingueneau: Conceptualization; Data curation; Formal analysis; Methodology; Project administration; Supervision; Writing‐original draft; Writing‐review & editing.
Supporting information
Supplementary table 1
Supplementary table 2
Supplementary table 3
Supplementary table 4
Supplementary table 5
Supplementary table 6
Supplementary table 7
Acknowledgments
We thank Fengmei Zheng for providing formulation of BIIB091 for in vivo dosing, Attila Fabian for flow cytometry and FACS sorting support, Alison Graham for her help with data analysis, Rebecca Basile, David Koske and Sam Foley for assistance with dosing and necropsy, and Rui Li and Amit Bar‐Or for discussion. This study was funded by Biogen (Cambridge, MA, USA). All Biogen authors are current or prior employees of Biogen, and current employees are shareholders of Biogen.
References
- 1. Corneth OB, Klein Wolterink RG, Hendriks RW. BTK signaling in B cell differentiation and autoimmunity. Curr Top Microbiol Immunol 2016; 393: 67–105. [DOI] [PubMed] [Google Scholar]
- 2. Molnarfi N, Schulze‐Topphoff U, Weber MS et al. MHC class II‐dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin‐specific antibodies. J Exp Med 2013; 210: 2921–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharma S, Orlowski G, Song W. Btk regulates B cell receptor‐mediated antigen processing and presentation by controlling actin cytoskeleton dynamics in B cells. J Immunol 2009; 182: 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Benson MJ, Rodriguez V, von Schack D et al. Modeling the clinical phenotype of BTK inhibition in the mature murine immune system. J Immunol 2014; 193: 185–197. [DOI] [PubMed] [Google Scholar]
- 5. Dolgin E. BTK blockers make headway in multiple sclerosis. Nat Biotechnol 2021; 39: 3–5. [DOI] [PubMed] [Google Scholar]
- 6. Nyhoff LE, Clark ES, Barron BL, Bonami RH, Khan WN, Kendall PL. Bruton's tyrosine kinase is not essential for B cell survival beyond early developmental stages. J Immunol 2018; 200: 2352–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koprulu AD, Ellmeier W. The role of Tec family kinases in mononuclear phagocytes. Crit Rev Immunol 2009; 29: 317–333. [DOI] [PubMed] [Google Scholar]
- 8. Lin L, Czerwinski R, Kelleher K et al. Activation loop phosphorylation modulates Bruton's tyrosine kinase (Btk) kinase domain activity. Biochemistry 2009; 48: 2021–2032. [DOI] [PubMed] [Google Scholar]
- 9. Kim YJ, Sekiya F, Poulin B, Bae YS, Rhee SG. Mechanism of B‐cell receptor‐induced phosphorylation and activation of phospholipase C‐γ2. Mol Cell Biol 2004; 24: 9986–9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hopkins BT. Discovery of BIIB091, a reversible selective potent BTK inhibitor for the treatment of multiple sclerosis. Abstracts of Papers, 260th ACS National Meeting & Exposition, San Francisco, CA, United States, August 23–27, 2020.
- 11. Ma B, Bohnert T, Otipoby KL et al. Discovery of BIIB068: a selective, potent, reversible Bruton's tyrosine kinase inhibitor as an orally efficacious agent for autoimmune diseases. J Med Chem 2020; 63: 12526–12541. [DOI] [PubMed] [Google Scholar]
- 12. Hashimoto A, Takeda K, Inaba M et al. Cutting edge: essential role of phospholipase C‐γ 2 in B cell development and function. J Immunol 2000; 165: 1738–1742. [DOI] [PubMed] [Google Scholar]
- 13. Kurosaki T, Hikida M. Tyrosine kinases and their substrates in B lymphocytes. Immunol Rev 2009; 228: 132–148. [DOI] [PubMed] [Google Scholar]
- 14. Watanabe D, Hashimoto S, Ishiai M et al. Four tyrosine residues in phospholipase C‐γ 2, identified as Btk‐dependent phosphorylation sites, are required for B cell antigen receptor‐coupled calcium signaling. J Biol Chem 2001; 276: 38595–38601. [DOI] [PubMed] [Google Scholar]
- 15. Satterthwaite AB, Li Z, Witte ON. Btk function in B cell development and response. Semin Immunol 1998; 10: 309–316. [DOI] [PubMed] [Google Scholar]
- 16. Rankin AL, Seth N, Keegan S et al. Selective inhibition of BTK prevents murine lupus and antibody‐mediated glomerulonephritis. J Immunol 2013; 191: 4540–4550. [DOI] [PubMed] [Google Scholar]
- 17. Xu D, Kim Y, Postelnek J et al. RN486, a selective Bruton's tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. J Pharmacol Exp Ther 2012; 341: 90–103. [DOI] [PubMed] [Google Scholar]
- 18. Li R, Rezk A, Miyazaki Y et al. Proinflammatory GM‐CSF‐producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med 2015; 7: 310ra166. [DOI] [PubMed] [Google Scholar]
- 19. Weinstein JS, Herman EI, Lainez B et al. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol 2016; 17: 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Knol EF, Mul FP, Jansen H, Calafat J, Roos D. Monitoring human basophil activation via CD63 monoclonal antibody 435. J Allergy Clin Immunol 1991; 88: 328–338. [DOI] [PubMed] [Google Scholar]
- 21. Schafer T, Starkl P, Allard C, Wolf RM, Schweighoffer T. A granular variant of CD63 is a regulator of repeated human mast cell degranulation. Allergy 2010; 65: 1242–1255. [DOI] [PubMed] [Google Scholar]
- 22. Bender AT, Gardberg A, Pereira A et al. Ability of Bruton's tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of Fc receptor but not B‐cell receptor signaling. Mol Pharmacol 2017; 91: 208–219. [DOI] [PubMed] [Google Scholar]
- 23. Mond JJ, Scher I, Mosier DE, Baese M, Paul WE. T‐independent responses in B cell‐defective CBA/N mice to Brucella abortus and to trinitrophenyl (TNP) conjugates of Brucella abortus . Eur J Immunol 1978; 8: 459–463. [DOI] [PubMed] [Google Scholar]
- 24. Vinuesa CG, Chang PP. Innate B cell helpers reveal novel types of antibody responses. Nat Immunol 2013; 14: 119–126. [DOI] [PubMed] [Google Scholar]
- 25. Scher I, Berning AK, Asofsky R. X‐linked B lymphocyte defect in CBA/N mice. IV. Cellular and environmental influences on the thymus dependent IgG anti‐sheep red blood cell response. J Immunol 1979; 123: 477–486. [PubMed] [Google Scholar]
- 26. Ellmeier W, Jung S, Sunshine MJ et al. Severe B cell deficiency in mice lacking the Tec kinase family members Tec and Btk. J Exp Med 2000; 192: 1611–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boswell HS, Nerenberg MI, Scher I, Singer A. Role of accessory cells in B cell activation. III. Cellular analysis of primary immune response deficits in CBA/N mice: presence of an accessory cell‐B cell interaction defect. J Exp Med 1980; 152: 1194–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scaramozza M, Arefayene M, Bame E, Peng X et al. A phase 1 study of BIIB091, a Bruton’s tyrosine kinase (BTK) inhibitor, in healthy adult participants: preliminary results 8th Joint ACTRIMS‐ECTRIMS meeting 2020.
- 29. Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for B cell subpopulations from infancy to adulthood. Clin Exp Immunol 2010; 162: 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hauser SL, Bar‐Or A, Comi G et al. Ocrelizumab versus interferon beta‐1a in relapsing multiple sclerosis. N Engl J Med 2017; 376: 221–234. [DOI] [PubMed] [Google Scholar]
- 31. Montalban X, Hauser SL, Kappos L et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017; 376: 209–220. [DOI] [PubMed] [Google Scholar]
- 32. Montalban X, Arnold DL, Weber MS et al. Placebo‐controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med 2019; 380: 2406–2417. [DOI] [PubMed] [Google Scholar]
- 33. Di Paolo JA, Huang T, Balazs M et al. Specific Btk inhibition suppresses B cell‐ and myeloid cell‐mediated arthritis. Nat Chem Biol 2011; 7: 41–50. [DOI] [PubMed] [Google Scholar]
- 34. Middendorp S, Dingjan GM, Maas A, Dahlenborg K, Hendriks RW. Function of Bruton's tyrosine kinase during B cell development is partially independent of its catalytic activity. J Immunol 2003; 171: 5988–5996. [DOI] [PubMed] [Google Scholar]
- 35. Haselmayer P, Camps M, Liu‐Bujalski L et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J Immunol 2019; 202: 2888–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barf T, Covey T, Izumi R et al. Acalabrutinib (ACP‐196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J Pharmacol Exp Ther 2017; 363: 240–252. [DOI] [PubMed] [Google Scholar]
- 37. Litzenburger T, Steffgen J, Benediktus E et al. Safety, pharmacokinetics and pharmacodynamics of BI 705564, a highly selective, covalent inhibitor of Bruton's tyrosine kinase, in phase I clinical trials in healthy volunteers. Br J Clin Pharmacol 2021; 87: 1824–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee SK, Xing J, Catlett IM et al. Safety, pharmacokinetics, and pharmacodynamics of BMS‐986142, a novel reversible BTK inhibitor, in healthy participants. Eur J Clin Pharmacol 2017; 73: 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Herman AE, Chinn LW, Kotwal SG et al. Safety, pharmacokinetics, and pharmacodynamics in healthy volunteers treated with GDC‐0853, a selective reversible Bruton's tyrosine kinase inhibitor. Clin Pharmacol Ther 2018; 103: 1020–1028. [DOI] [PubMed] [Google Scholar]
- 40. Baker D, Marta M, Pryce G, Giovannoni G, Schmierer K. Memory B cells are major targets for effective immunotherapy in relapsing multiple sclerosis. EBioMedicine 2017; 16: 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Croxford A, Lanzinger M, Hartmann F et al. The cytokine GM‐CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity 2015; 43: 502–514. [DOI] [PubMed] [Google Scholar]
- 42. Mildner A, Mack M, Schmidt H et al. CCR2+Ly‐6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain 2009; 132: 2487–2500. [DOI] [PubMed] [Google Scholar]
- 43. Yamasaki R, Lu H, Butovsky O et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 2014; 211: 1533–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Han S, Lin YC, Wu T et al. Comprehensive immunophenotyping of cerebrospinal fluid cells in patients with neuroimmunological diseases. J Immunol 2014; 192: 2551–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Waschbisch A, Schroder S, Schraudner D et al. Pivotal role for CD16+ monocytes in immune surveillance of the central nervous system. J Immunol 2016; 196: 1558–1567. [DOI] [PubMed] [Google Scholar]
- 46. Abdul‐Majid K‐B, Stefferl A, Bourquin C et al. Fc receptors are critical for autoimmune inflammatory damage to the central nervous system in experimental autoimmune encephalomyelitis. Scand J Immunol 2002; 55: 70–81. [DOI] [PubMed] [Google Scholar]
- 47. Iruretagoyena MI, Riedel CA, Leiva ED, Gutierrez MA, Jacobelli SH, Kalergis AM. Activating and inhibitory Fcγ receptors can differentially modulate T cell‐mediated autoimmunity. Eur J Immunol 2008; 38: 2241–2250. [DOI] [PubMed] [Google Scholar]
- 48. Kinzel S, Lehmann‐Horn K, Torke S et al. Myelin‐reactive antibodies initiate T cell‐mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol 2016; 132: 43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lock C, Hermans G, Pedotti R et al. Gene‐microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 2002; 8: 500–508. [DOI] [PubMed] [Google Scholar]
- 50. Urich E, Gutcher I, Prinz M, Becher B. Autoantibody‐mediated demyelination depends on complement activation but not activatory Fc‐receptors. Proc Natl Acad Sci USA 2006; 103: 18697–18702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dobin A, Davis CA, Schlesinger F et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 2011; 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1
Supplementary table 2
Supplementary table 3
Supplementary table 4
Supplementary table 5
Supplementary table 6
Supplementary table 7