

Abstract
BACKGROUND
Somatic variants in KCNJ5 are the most common cause of primary aldosteronism (PA). There are few patients with PA in whom the disease is caused by germline variants in the KCNJ5 potassium channel gene (familial hyperaldosteronism type III—FH-III).
METHODS
A 5-year-old patient who developed hypertension due to bilateral adrenocortical hyperplasia (BAH) causing PA had negative peripheral DNA testing for any known genetic causes of PA. He was treated medically with adequate control of his PA but by the third decade of his life, due to worsening renal function, he underwent bilateral adrenalectomy.
RESULTS
Focused exome sequencing in multiple nodules of his BAH uncovered a “hot-spot” pathogenic KCNJ5 variant, while repeated Sanger sequencing showed no detectable DNA defects in peripheral blood and other tissues. However, whole exome, “deep” sequencing revealed that 0.23% of copies of germline DNA did in fact carry the same KCNJ5 variant that was present in the adrenocortical nodules, suggesting low level germline mosaicism for this PA-causing KCNJ5 defect.
CONCLUSIONS
Thus, this patient represents a unique case of BAH due to a mosaic KCNJ5 defect. Undoubtedly, his milder PA compared with other known cases of FH-III, was due to his mosaicism. This case has a number of implications for the prognosis, treatment, and counseling of the many patients with PA due to BAH that are seen in hypertension clinics.
Keywords: blood pressure, familial hyperaldosteronism, genetics, hyperaldosteronism, hypertension, KCNJ5, mosaicism, primary aldosteronism
Primary aldosteronism (PA) is recognized as the most common hormonal cause of secondary hypertension.1 Dr Jerome Conn first demonstrated that this condition was linked to adrenal adenomas in 1960s, after adrenalectomy of a unilateral adrenal adenoma resulted in the cure of resistant hypertension in a young female patient. In addition to aldosterone producing adenomas (APAs), PA is also commonly caused by bilateral adrenocortical hyperplasia (BAH), and rarely by unilateral hyperplasia, aldosterone-producing adrenocortical carcinomas, or familial hyperaldosteronism (FH). With the advent of genetic sequencing, more definitive characterization of the underlying genetic defects causing APA was made possible in the last decade.2
Past work has established PA to be either caused by somatic disease-causing variants in adrenal tissue or more rarely, germline defects leading to FH.3,4 FH type I (FH-I), otherwise known as glucocorticoid-remediable aldosteronism (GRA), is due to a cross-over of the promoter of the 11-beta-hydroxylase gene and the coding region of the aldosterone synthase gene causing aldosterone production stimulation by adrenocorticotropic hormone (ACTH). FH type II (FH-II) historically does not have specific genetic defects to define it, though recent work has linked some cases to disease-causing variants in chloride voltage-gated channel 2 (CLCN2), which encode chloride channels in the adrenal glomerulosa.5 The cause of the disease in a subset of families with FH-II has been mapped to chromosome 7p22.6 FH type III (FH-III) is attributed to germline variants in potassium voltage-gated channel subfamily J member 5 (KCNJ5), which codes for an inwardly rectifying potassium channel.4,7 FH type IV (FH-IV) is due to disease-causing variants in calcium voltage-gated channel subunit alpha1 H (CACNA1H), which encodes a T-type calcium channel.3
Choi et al.8 first identified a relatively frequent genetic etiology for APA development: Twenty-two patients with PA due to an unilateral adrenal mass identified on computed tomography (CT) scan underwent genetic analysis. “High probability somatic mutations” were identified via genotyping of tumors on Illumina 1M-Duo chips and were confirmed with Sanger sequencing. After filtering for variants that altered protein function, the gene KCNJ5 was noted to be mutated in multiple patients. Compared against blood DNA (considered the equivalent of germline tissue), these variants were found to be exclusive to the adrenal tumors. Additionally, functional characterization of the disease-causing variants in KCNJ5 demonstrated that the loss of channel selectivity conferred susceptibility to membrane depolarization, leading to calcium influx and consequent stimulation of aldosterone production. Moreover, prolonged calcium stimulation also was found to foster zona glomerulosa growth, initially thought to explain the manifestation of adrenal adenomas.8,9 In the same study, a germline disease-causing variant in KCNJ5 was also shown to cause an inherited form of PA8 associated with bilateral enlargement of the adrenal glands, which histologically was consistent with BAH: a father and his two daughters all developed severe hypertension and PA associated with BAH between ages 4 and 7. The hypertension was resistant to medical therapy and bilateral adrenalectomy was pursued in all three cases. Subsequent work established that somatic disease-causing variants in other genes, such as ATP1A1 and ATP2B3, can lead to APA as well.10 Disease-causing variants in CACNA1D, leading to increased calcium influx, were similarly linked to APA and when in the germline, to BAH. Indeed, these findings confirmed that dysregulated calcium influx is the common last step in APA formation among the different APA- and/or BAH-associated disease-causing variants.2,11
Mosaicism can occur when genetic defects arise postzygotically leading to genetically distinct cell lineages in an individual developing from a single zygote. The clinical presentation in mosaicism may vary according to the type of disease-causing variants, percent of mutated cells, and tissue distribution of the genetic defect. There are three main types of mosaicism: somatic mosaicism which occurs in nongerm cells and cannot be transmitted to the next generation, gonadal or germline mosaicism, which occurs only in germ cells or their precursors and may be inherited by the next generation, and mixed gonadal and somatic mosaicism.12–14 Therefore, identification of mosaicism is of great importance to establish disease diagnosis, assess recurrence risk, and provide genetic counseling.15,16
Here, we show for the first time, a case of BAH due to a mosaic KCNJ5 defect in a patient who was initially thought to have FH-II. Our results showed that, indeed, there was very low-level mosaicism in the germline DNA, whereas all adrenocortical cells tested from 11 different nodules harbored the disease-causing variant KCNJ5 p.G151R.
MATERIALS AND METHODS
Clinical studies
The patient was recruited to an ongoing large study on the genetics of primary aldosteronism (NIH protocol 00-CH-0160) first approved by the Institutional Review Board (IRB) of the Eunice Kennedy Shriver National Institute for Child Health & Human Development, Division of Intramural Research (DIR). The patient signed an informed consent for all clinical and genetic studies.
Sanger sequencing
DNA was extracted from peripheral blood leukocytes, right and left adrenal fat and 11 adrenal nodules (5 left nodules, 6 right nodules) using DNA easy blood and tissue kit according to the manufacturer protocol (Qiagen, Hilden, Germany). Primers to amplify KCNJ5 were designed to amplify its hotspot region (comprising the most frequent disease-causing variants in KCNJ5—c.451G>A and c.503T>G). Primers used for the PCR reaction were as follows: forward—5′-CAGCGCTACATGGAGAAGAG-3′; reverse 5′-CCACGTTGATGTCTGTCTGG-3′. Annealing temperature of 60 °C was used for the PCR amplification. Classical bidirectional Sanger sequencing was performed using a Genetic Sequencer ABI3500 (Applied Biosystems, Grand Island) to identify the disease-causing variants in KCNJ5.
Mosaicism analysis by deep next generation sequencing
To determine the proportion of cells exhibiting the disease-causing variant in question, we amplified the specific region with targeted flanking primers. Each primer was composed of two parts: (i) the 3′ end was complementary to the targeted genomic sequence; and (ii) all primers had appended to their 5′ end either forward or reverse adaptor sequences to allow the addition of barcodes and Illumina sequencing adaptors. These appended sequences were: forward—5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG [locus-specific sequence]; and reverse—5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG [locus-specific sequence]. The locus specific sequences used were: forward 5′-AAACCTCAGTGGCTTCGTGT-3′ and reverse 5′-GAGGGTCTCCGCTCTCTTCT-3′. Amplicons generated from the sample DNA were then converted to sequenced libraries according to Illumina’s 16S Metagenomic Sequencing Library Preparation kit. Libraries were sequenced on an Illumina MiSeq using version 2 chemistry and generated 2 × 150-bp paired-end reads. Read pairs were trimmed with cutadapt, joined via “PEAR,” and aligned to expected amplicons with BWA. Variant frequencies were evaluated by counting bases at the variant position in aligned reads. Normalization was done using the WT sample result as “zero.” The experiment was done in technical triplicates and we tested one nodule and four germline DNA (left and right adrenal fat, peripheral blood and buccal swab) from the individual in the study. We also tested a germline DNA sample from the parents of the study individual and a germline DNA sample from an unaffected individual (wild type (WT) control).
Immunostaining
Deparaffinized sections of adrenal tumors were immuno-stained using antibodies against CYP11B2 (rabbit antihuman NBP2-1389, Novus Biologicals, Colorado, EUA). Routine Hematoxylin & Eosin staining was performed at Histoserv, Inc. Slides were scanned in a ×10 objective and pictures were taken using Keyence BZ-X710 (Osaka, Japan) microscope.
RESULTS
Clinical case
An eleven-year-old Caucasian male was initially seen at the National Institutes of Health Clinical Center in 1998 for early-onset severe hypertension. He was diagnosed with hypertension at the age of 5 years and was initially evaluated for renal causes of hypertension. He was empirically treated with propranolol, with modest improvement in his hypertension, and subsequently switched to hydrochlorothiazide but developed hypokalemia and nifedipine monotherapy was trialed thereafter. He was subsequently referred to the National Institutes of Health and diagnosed with PA. Laboratory studies demonstrated an initial serum aldosterone of 24 ng/dl (reference range 3–21 ng/dl), with repeat value of 45.1 ng/dl, and undetectable plasma renin activity (<0.2 ng/ml/hr).
He underwent confirmatory testing while on a high salt diet with intake of 109 mEq/day (equivalent to an oral salt load test). On the third day of the diet, his aldosterone level was 70.2 ng/dl and renin <0.2 ng/ml/hr. Twenty-four-hour urinary aldosterone was 23.5 mcg/24 hr. CT of the adrenal glands showed bilateral enlargement, consistent with BAH (Figure 1). He underwent adrenal vein sampling, with results consistent with bilateral disease. GRA was ruled out as his aldosterone levels failed to suppress on dexamethasone and sequencing for a chimeric CYP11B1/CYP11B2 gene was negative. He then underwent whole exome sequencing from peripheral DNA, which did not reveal a disease-causing variant (data not shown). His parents were phenotypically normal with negative genetic evaluations. He was started on spironolactone, but this caused gynecomastia. In 2004, he was transitioned from amiloride to eplerenone. Due to uncontrolled hypertension and worsening renal function, he underwent bilateral adrenalectomy in 2017. Histology showed that both adrenal glands had changes consistent with BAH with nodules measuring up to 3 cm in their greatest dimension (Supplementary Figure 1).
Figure 1.

Bilateral adrenal hyperplasia (arrows) on computed tomography.
Long suspected to have a de novo case of a genetic form of PA, due to lack of family history and early age of presentation; classified initially as FH-II, he agreed to undergo additional genetic testing.
Sanger sequencing analysis of tumor DNA samples revealed the presence of disease-causing variant KCNJ5 p.G151R in 11 different adrenal nodules
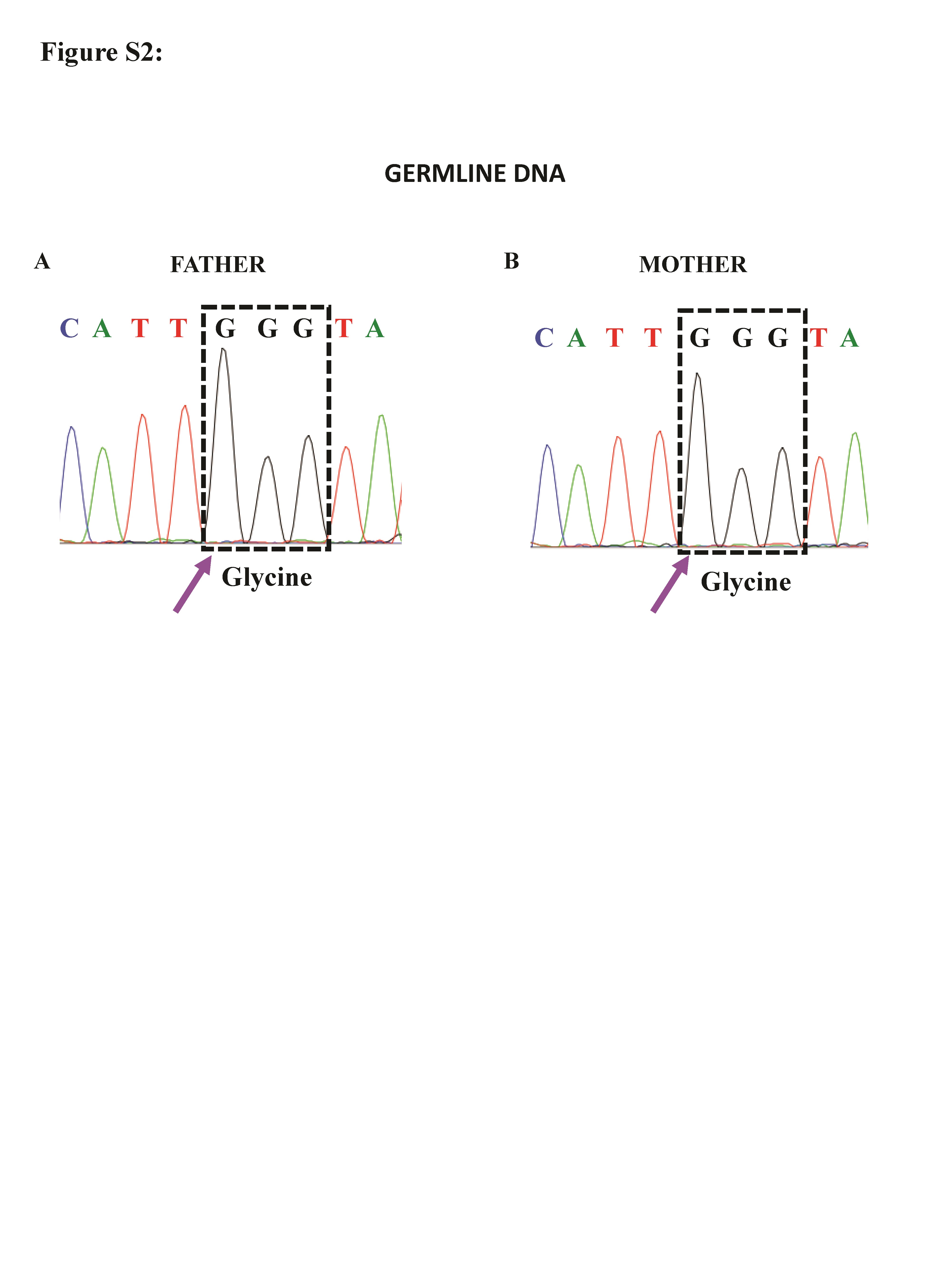
Screening for KCNJ5 hotspot region p.G151R and p.L168R was performed in peripheral blood leukocytes, right and left adrenal fat DNA, and 11 different adrenal nodules by Sanger sequencing. The c.451G>A KCNJ5 (p.G151R) defect was identified exclusively in the tumor samples; the mutant allele was present in all 11 nodules but not in the blood, nor in the adrenal fat samples (Figure 2). Genomic DNA from parents was analyzed and the KCNJ5 c.451G>A defect, which was not present in their DNA (Supplementary Figure 2).
Figure 2.

Electropherogram of part of KCNJ5 sequence including the position c.451 in the DNA. No mutant alleles were detected in peripheral blood leucocytes DNA (a) or adrenal fat (b). The c.451G>A KCNJ5 defect was detected in all the adrenal nodules analyzed, arrow denotes the location of the variant (c).
CYP11B2 is highly expressed in the KCNJ5-mutated adrenal tumors
CYP11B2 is highly expressed in APAs and therefore is a powerful histopathological tool to confirm excessive aldosterone production and localize aldosterone-producing cells in the adrenal gland.17 As we found the disease-causing variant in KCNJ5 in the nodules, we stained one section of a left and another of a right adrenal lesion for CYP11B2; our results showed high expression of CYP11B2 throughout the nodules (Figure 3).
Figure 3.

Representative histopathological analysis of two adrenal nodules of a KCNJ5 mosaic individual. Hematoxylin-eosin staining of a left adrenal nodule (a). Immunostaining for CYP11B2 of a left adrenal nodule (b). Hematoxylin-eosin staining of a right adrenal nodule (c). Immunostaining for CYP11B2 of a right adrenal nodule (d). Corresponding amplified images from the squares numbered with 1 and 2 in the lower magnification field can be seen in the adjacent images numbered as 1 and 2. Slides were scanned in a ×10 objective.
Deep sequencing analysis revealed mosaicism in KCNJ5
Considering the presence of the same KCNJ5 sequence variant in all the adrenal tumors and no detectable changes in the genomic DNA, we sought to determine the proportion of cells exhibiting the pathogenic variant in question and therefore investigated whether this individual could harbor mosaicism for a KCNJ5 defect. We performed “deep” next generation sequencing (NGS) of right and left adrenal fat tissue, peripheral blood leukocytes, buccal swab, and tumor DNA (nodule). Approximately 39% of the mutant allele was identified in the nodule sample tested, while in peripheral blood leukocytes’ DNA, we found 0.23% of the mutant and in other germline samples tested we found between 0.1% and 0.16% of the mutant allele (Table 1), suggesting the presence of very low levels of KCNJ5 mosaicism at the germline level. The average number of reads per sample was 1,433,999.
Table 1.
Frequency of variant KCNJ5 p.Gly151Arg detected by deep sequencing in different tissues of the individual in study and the parents
| Patient code | Tissue sample | Gene | cDNA | Protein | Total number of reads | Mutation frequency (%) |
|---|---|---|---|---|---|---|
| ADT102.01 (patient) | Right adrenal fat | KCNJ5 | c.451G>A | p.Gly151Arg | 1,652,034 | 0.16 |
| ADT102.01 (patient) | Left adrenal fat | KCNJ5 | c.451G>A | p.Gly151Arg | 1,393,092 | 0.11 |
| ADT102.01 (patient) | Blood | KCNJ5 | c.451G>A | p.Gly151Arg | 1,114,362 | 0.23 |
| ADT102.01 (patient) | Swab | KCNJ5 | c.451G>A | p.Gly151Arg | 1,397,124 | 0.10 |
| ADT102.01 (patient) | Nodule | KCNJ5 | c.451G>A | p.Gly151Arg | 985,195 | 38.783 |
| ADT102.02 (father) | Blood | KCNJ5 | WT | WT | 1,863,106 | 0.00 |
| ADT102.04 (mother) | Blood | KCNJ5 | WT | WT | 1,885,344 | 0.00 |
| Negative control (WT) | Blood | KCNJ5 | WT | WT | 1,181,736 | 0.00 |
Abbreviations: WT, wild type.
DISCUSSION
We describe the first patient with BAH caused by mosaicism for a KCNJ5 pathogenic defect. Although germline defects are expected to cause no more than 10% of all tumors,18 NGS has uncovered many cases of causative defects that are present in a few cells. In endocrine tumor syndromes, mosaicism for a postzygotic mutational event is causative of McCune Albright syndrome (MAS) and HIF2A-related polycythemia-paraganglioma syndrome.19–21 Mosaicism is also found in other tumor predisposition syndromes such as tuberous sclerosis. The implications of germline mosaicism for causative variants of tumor predisposition syndromes are immense in regard to genetic counseling for patients and their families, as well as for tumor surveillance which may affect the prognosis of the respective disorders. Germline mosaicism may explain why multiple affected offspring carrying an apparently de novo pathogenic variant may be born to unaffected parents.
Disease causing KCNJ5 defects are present in approximately 24–82% of individuals diagnosed with APA, a percentage that varies between centers and ethnicity.22 On the other hand, FH-III is extraordinarily rare.23 It is possible that few cases of mosaicism exist within large cohorts of patients with APA and/or BAH but their identification would require NGS and a search for low-level mosaicism, as was apparent in our case.24 Such patients may be those with multiple APAs and/or adrenocortical hyperplasia (BAH or other forms) upon histopathological evaluation of the excised tissue, which may be bilateral. Their identification is important not only to guide clinical care for the individual (since both adrenal glands may be affected) but also for the risk of transmission.
Is it possible that our patient’s disease was due to multiple de novo disease-causing variants of the same type, in concurrent adenomas? Figure 3 demonstrates that all our patient’s nodules were strongly positive for CYP11B2, as shown in other cases of PA9; however, this was also true for most of the surrounding tissue showing hyperplasia consistent with what has been described in FH-III.8 Although multiple de novo disease-causing variants have been identified in heterogeneously staining nodules including KCNJ5, CACNA1D, and ATP1A125 in the same patient, given our patient’s pattern of CYP11B2 staining, the identification of the KCNJ5 p.G151R allele in all 11 nodules derived from both adrenal glands, and the discovery of low-level mosaicism for the same defect in blood, makes this possibility very unlikely.
In conclusion, a patient previously thought to have FH-II due to an unknown defect was found to have BAH due to mosaicism for a “hot-spot” disease-causing variant in KCNJ5. The finding has implications for the studies of patients with aldosterone-producing lesions, the patients’ management, prognosis and their genetic counseling.
Supplementary Material
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
We are indebted to the NICHD’s IRP Molecular Genomics Core (Dr Steven Coon, Dr. James Iben, and Dr Li Tianwei). This work was supported by the Eunice Kennedy Shriver National Institute for Child Health & Human Development Division of Intramural Research (DIR; NIH Intramural Grant Z01-HD008920-01 to Dr C.A.S.).
DISCLOSURE
The authors declared no conflict of interest.
REFERENCES
- 1. Dutta RK, Söderkvist P, Gimm O. Genetics of primary hyperaldosteronism. Endocr Relat Cancer 2016; 23:R437–R454. [DOI] [PubMed] [Google Scholar]
- 2. Zennaro MC, Boulkroun S, Fernandes-Rosa F. Genetic causes of functional adrenocortical adenomas. Endocr Rev 2017; 38:516–537. [DOI] [PubMed] [Google Scholar]
- 3. Korah HE, Scholl UI. An update on familial hyperaldosteronism. Horm Metab Res 2015; 47:941–946. [DOI] [PubMed] [Google Scholar]
- 4. Monticone S, Bandulik S, Stindl J, Zilbermint M, Dedov I, Mulatero P, Allgaeuer M, Lee CC, Stratakis CA, Williams TA, Tiulpakov A. A case of severe hyperaldosteronism caused by a de novo mutation affecting a critical salt bridge Kir3.4 residue. J Clin Endocrinol Metab 2015; 100:E114–E118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scholl UI, Stölting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, Vichot AA, Jin SC, Loring E, Untiet V, Yoo T, Choi J, Xu S, Wu A, Kirchner M, Mertins P, Rump LC, Onder AM, Gamble C, McKenney D, Lash RW, Jones DP, Chune G, Gagliardi P, Choi M, Gordon R, Stowasser M, Fahlke C, Lifton RP. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet 2018; 50:349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lafferty AR, Torpy DJ, Stowasser M, Taymans SE, Lin JP, Huggard P, Gordon RD, Stratakis CA. A novel genetic locus for low renin hypertension: familial hyperaldosteronism type II maps to chromosome 7 (7p22). J Med Genet 2000; 37:831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xekouki P, Hatch MM, Lin L, Rodrigo de A, Azevedo M, de la Luz Sierra M, Levy I, Saloustros E, Moraitis A, Horvath A, Kebebew E, Hoffman DA, Stratakis CA. KCNJ5 mutations in the National Institutes of Health cohort of patients with primary hyperaldosteronism: an infrequent genetic cause of Conn’s syndrome. Endocr Relat Cancer 2012; 19:255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, Lolis E, Wisgerhof MV, Geller DS, Mane S, Hellman P, Westin G, Åkerström G, Wang W, Carling T, Lifton RP. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011; 331:768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fernandes-Rosa FL, Giscos-Douriez I, Amar L, Gomez-Sanchez CE, Meatchi T, Boulkroun S, Zennaro MC. Different somatic mutations in multinodular adrenals with aldosterone-producing Adenoma. Hypertension 2015; 66:1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, Penton D, Schack VR, Amar L, Fischer E, Walther A, Tauber P, Schwarzmayr T, Diener S, Graf E, Allolio B, Samson-Couterie B, Benecke A, Quinkler M, Fallo F, Plouin PF, Mantero F, Meitinger T, Mulatero P, Jeunemaitre X, Warth R, Vilsen B, Zennaro MC, Strom TM, Reincke M. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 2013; 45:440–444, 444e1. [DOI] [PubMed] [Google Scholar]
- 11. Scholl UI, Goh G, Stölting G, de Oliveira RC, Choi M, Overton JD, Fonseca AL, Korah R, Starker LF, Kunstman JW, Prasad ML, Hartung EA, Mauras N, Benson MR, Brady T, Shapiro JR, Loring E, Nelson-Williams C, Libutti SK, Mane S, Hellman P, Westin G, Åkerström G, Björklund P, Carling T, Fahlke C, Hidalgo P, Lifton RP. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 2013; 45:1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Campbell IM, Shaw CA, Stankiewicz P, Lupski JR. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet 2015; 31:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Forsberg LA, Gisselsson D, Dumanski JP. Mosaicism in health and disease—clones picking up speed. Nat Rev Genet 2017; 18:128–142. [DOI] [PubMed] [Google Scholar]
- 14. Spinner NB, Conlin LK. Mosaicism and clinical genetics. Am J Med Genet C Semin Med Genet 2014; 166C:397–405. [DOI] [PubMed] [Google Scholar]
- 15. King DA, Sifrim A, Fitzgerald TW, Rahbari R, Hobson E, Homfray T, Mansour S, Mehta SG, Shehla M, Tomkins SE, Vasudevan PC, Hurles ME; Deciphering Developmental Disorders Study . Detection of structural mosaicism from targeted and whole-genome sequencing data. Genome Res 2017; 27:1704–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qin L, Wang J, Tian X, Yu H, Truong C, Mitchell JJ, Wierenga KJ, Craigen WJ, Zhang VW, Wong LC. Detection and quantification of mosaic mutations in disease genes by next-generation sequencing. J Mol Diagn 2016; 18:446–453. [DOI] [PubMed] [Google Scholar]
- 17. Nanba K, Omata K, Else T, Beck PCC, Nanba AT, Turcu AF, Miller BS, Giordano TJ, Tomlins SA, Rainey WE. Targeted molecular characterization of aldosterone-producing adenomas in white Americans. J Clin Endocrinol Metab 2018; 103:3869–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Konnick EQ, Pritchard CC. Germline, hematopoietic, mosaic, and somatic variation: interplay between inherited and acquired genetic alterations in disease assessment. Genome Med 2016; 8:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buffet A, Smati S, Mansuy L, Ménara M, Lebras M, Heymann MF, Simian C, Favier J, Murat A, Cariou B, Gimenez-Roqueplo AP. Mosaicism in HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab 2014; 99:E369–E373. [DOI] [PubMed] [Google Scholar]
- 20. Salpea P, Stratakis CA. Carney complex and McCune Albright syndrome: an overview of clinical manifestations and human molecular genetics. Mol Cell Endocrinol 2014; 386:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, Popovic V, Stratakis CA, Prchal JT, Pacak K. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med 2012; 367:922–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murakami M, Yoshimoto T, Nakabayashi K, Nakano Y, Fukaishi T, Tsuchiya K, Minami I, Bouchi R, Okamura K, Fujii Y, Hashimoto K, Hata KI, Kihara K, Ogawa Y. Molecular characteristics of the KCNJ5 mutated aldosterone-producing adenomas. Endocr Relat Cancer 2017; 24:531–541. [DOI] [PubMed] [Google Scholar]
- 23. Monticone S, Tetti M, Burrello J, Buffolo F, De Giovanni R, Veglio F, Williams TA, Mulatero P. Familial hyperaldosteronism type III. J Hum Hypertens 2017; 31:776–781. [DOI] [PubMed] [Google Scholar]
- 24. Tamura A, Nishimoto K, Seki T, Matsuzawa Y, Saito J, Omura M, Gomez-Sanchez CE, Makita K, Matsui S, Moriya N, Inoue A, Nagata M, Sasano H, Nakamura Y, Yamazaki Y, Kabe Y, Mukai K, Kosaka T, Oya M, Suematsu S, Nishikawa T. Somatic KCNJ5 mutation occurring early in adrenal development may cause a novel form of juvenile primary aldosteronism. Mol Cell Endocrinology 2017; 441:134–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nanba K, Chen AX, Omata K, Vinco M, Giordano TJ, Else T, Hammer GD, Tomlins SA, Rainey WE. Molecular heterogeneity in aldosterone-producing adenomas. J Clin Endocrinol Metab 2016; 101:999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.