

Key Points
Question
Are rare pathogenic genetic variants associated with risk for sporadic moyamoya disease?
Findings
In a genetic association study including a 24-proband discovery cohort and an 84-proband validation cohort of non–East Asian patients with moyamoya disease, whole-exome sequencing identified rare, damaging variants in DIAPH1 (diaphanous-1), a key effector of actin remodeling in vascular cells and platelets. DIAPH1 was enriched in mural cells of the midgestational human brain and its coexpression network converges on cytoskeleton regulatory pathways.
Meaning
These results suggest DIAPH1 as a novel moyamoya disease risk gene and may implicate impaired vascular cell actin remodeling in moyamoya disease pathogenesis, with likely diagnostic and therapeutic ramifications.
Abstract
Importance
Moyamoya disease (MMD), a progressive vasculopathy leading to narrowing and ultimate occlusion of the intracranial internal carotid arteries, is a cause of childhood stroke. The cause of MMD is poorly understood, but genetic factors play a role. Several familial forms of MMD have been identified, but the cause of most cases remains elusive, especially among non–East Asian individuals.
Objective
To assess whether ultrarare de novo and rare, damaging transmitted variants with large effect sizes are associated with MMD risk.
Design, Setting, and Participants
A genetic association study was conducted using whole-exome sequencing case-parent MMD trios in a small discovery cohort collected over 3.5 years (2016-2019); data were analyzed in 2020. Medical records from US hospitals spanning a range of 1 month to 1.5 years were reviewed for phenotyping. Exomes from a larger validation cohort were analyzed to identify additional rare, large-effect variants in the top candidate gene. Participants included patients with MMD and, when available, their parents. All participants who met criteria and were presented with the option to join the study agreed to do so; none were excluded. Twenty-four probands (22 trios and 2 singletons) composed the discovery cohort, and 84 probands (29 trios and 55 singletons) composed the validation cohort.
Main Outcomes and Measures
Gene variants were identified and filtered using stringent criteria. Enrichment and case-control tests assessed gene-level variant burden. In silico modeling estimated the probability of variant association with protein structure. Integrative genomics assessed expression patterns of MMD risk genes derived from single-cell RNA sequencing data of human and mouse brain tissue.
Results
Of the 24 patients in the discovery cohort, 14 (58.3%) were men and 18 (75.0%) were of European ancestry. Three of 24 discovery cohort probands contained 2 do novo (1-tailed Poisson P = 1.1 × 10−6) and 1 rare, transmitted damaging variant (12.5% of cases) in DIAPH1 (mammalian diaphanous-1), a key regulator of actin remodeling in vascular cells and platelets. Four additional ultrarare damaging heterozygous DIAPH1 variants (3 unphased) were identified in 3 other patients in an 84-proband validation cohort (73.8% female, 77.4% European). All 6 patients were non–East Asian. Compound heterozygous variants were identified in ena/vasodilator-stimulated phosphoproteinlike protein EVL, a mammalian diaphanous-1 interactor that regulates actin polymerization. DIAPH1 and EVL mutant probands had severe, bilateral MMD associated with transfusion-dependent thrombocytopenia. DIAPH1 and other MMD risk genes are enriched in mural cells of midgestational human brain. The DIAPH1 coexpression network converges in vascular cell actin cytoskeleton regulatory pathways.
Conclusions and Relevance
These findings provide the largest collection to date of non–East Asian individuals with sporadic MMD harboring pathogenic variants in the same gene. The results suggest that DIAPH1 is a novel MMD risk gene and impaired vascular cell actin remodeling in MMD pathogenesis, with diagnostic and therapeutic ramifications.
This genetic association study analyzes genetic characteristics of patients with moyamoya disease.
Introduction
Moyamoya disease (MMD) is a primary cerebrovascular disorder characterized by progressive stenosis and eventual occlusion of the bilateral intracranial internal carotid arteries and their proximal branches.1,2 Resultant tissue ischemia leads to compensatory development of abnormally dilated and fragile collateral vessels that appear on cerebral angiography as a “puff of smoke” (Japanese, moyamoya3). The incidence of MMD is highest in the Japanese population4,5 and is 10 times lower in non–East Asian populations.6,7 Moyamoya disease is one of the most frequent causes of pediatric stroke.8 Current therapy consists of stroke prevention via platelet inhibition with aspirin and indirect or direct neurosurgical revascularization, with the attendant risks of morbidity.9The progressive nature of MMD requires early diagnosis to optimize selection of patients for surgery, which is the only currently effective disease treatment.
Gaps in our understanding of MMD pathogenesis impede the development of preventive, diagnostic, and therapeutic strategies. Pathologic changes in MMD include fragmentation of the inner elastic lamina and concentric fibrocellular thickening of the intima associated with increased proliferation of α-actin–positive vascular smooth muscle cells (VSMCs).10,11,12,13 Genetic factors play a substantial role; 10% of Japanese patients have affected first-degree relatives, as do 6% of patients in the US.14,15 Identification of 4 genes involved in familial monogenic MMD11,16,17,18,19,20 and several monogenic forms of moyamoya syndrome21,22,23,24,25 has implicated nitric oxide signaling (GUCY1A3), genomic stability (RNF213), actin polymerization (ACTA2), RAS signaling (with NF-1, Noonan, and Costello syndromes), and the NOTCH pathway (with Alagille syndrome), among others, in disease pathogenesis (eTable 1 in the Supplement). However, these rare pathogenic variants with large effect size account for less than 2% of MMD cases.19 Moreover, although contributing significantly to the genetic burden of MMD, the common variant risk factor RNF213 is represented predominantly in Japanese and other East Asian populations.16,26,27 Therefore, a significant proportion of MMD remains genetically undefined, especially among the European population.5,28
The sporadic nature of most MMD cases limits the utility of classic genetic approaches. Whole-exome sequencing (WES) of relatively small, well-phenotyped, trio (case-parent)-based cohorts has identified pathogenic de novo variants (DNVs) in sporadic cases of neurodevelopmental disease,29,30,31 including vein of Galen malformation and other arteriovenous malformations.17,32,33 We hypothesized that trio-based WES might uncover new, rare pathogenic gene variants with large effect size that increase the risk for sporadic MMD. We provide data herein suggesting that DIAPH1 is a new MMD risk gene for sporadic MMD in non–East Asian individuals.
Methods
All study procedures and protocols complied with human investigation committees, institutional review boards, and human research protection programs at Yale University School of Medicine, New Haven, Connecticut, and the University of Texas Health Science Center, Houston. Written informed consent for genetic studies was obtained from all participants. Participants did not receive financial compensation. This study followed the Strengthening the Reporting of Genetic Association Studies (STREGA) reporting guideline for genetic association studies.
Inclusion criteria of the Yale Center for Mendelian Genomics–based 24-patient discovery cohort (22 trios) and the University of Texas–based 84-patient validation cohort (29 trios) (eTable 2 in the Supplement) were identical and restricted to primary MMD.5 Cases were diagnosed by neurosurgical specialists using international consensus criteria.34 Patients and participating family members provided buccal swab samples (SK-2S DNA buccal swab kits, Isohelix), medical records, neuroimaging study results, and operative reports. Patient phenotypes are presented in eTable 3 in the Supplement. Representative neuroimaging is presented in Figure 1.
Figure 1. De Novo and Rare, Damaging Transmitted DIAPH1 Variants in Moyamoya Disease.
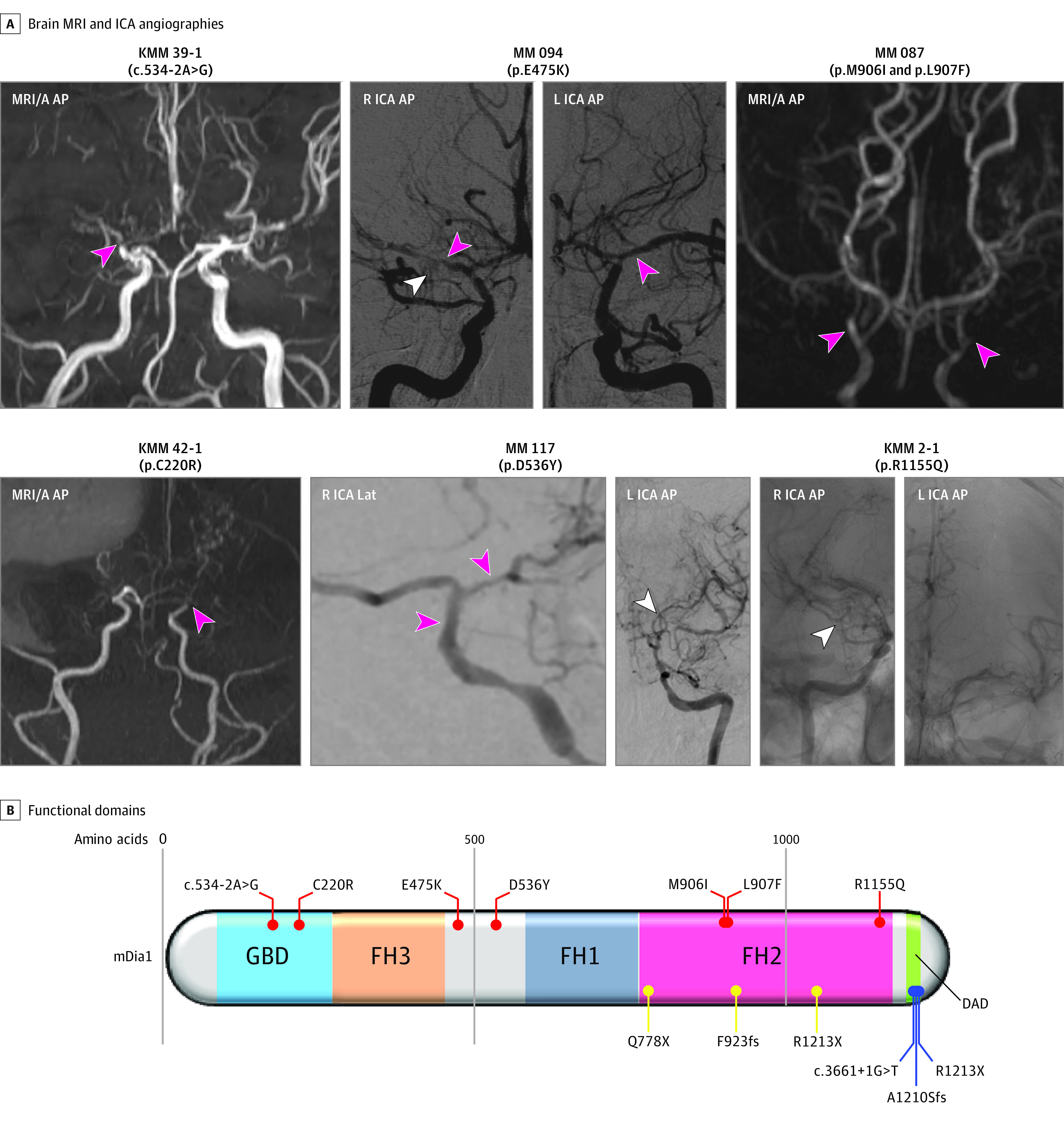
A, Brain magnetic resonance angiography (MRI/A) and carotid, digital subtraction catheter angiography (ICA) catheter angiography demonstrating severe narrowing of the bilateral intracranial internal carotid and middle cerebral arteries (pink arrowheads), along with the associated compensatory development of collateral vasculature comprising abnormally dilated and fragile moyamoya vessels (white arrowheads). B, Linear representation of functional domains (represented by dark rectangles) of mammalian Diaphaneous-1 (mDia1), encoded by DIAPH1, with mapped disease-associated variants. Shown are the C-terminal inhibitory diaphanous autoregulatory domain (DAD) (green) and the N-terminal Rho-GTPase-binding autoinhibitory domain (GBD/FH3) (blue/orange). Also depicted are the FH1 (gray) and F-actin binding and nucleating FH2 (pink) Formin domains. FH2 binds and nucleates F-actin. mDia1 variants are mapped onto the protein structure. Variants associated with moyamoya disease are in red. Variants associated with seizures, cortical blindness, and microcephaly syndrome (OMIM 616632) are in yellow. Variants associated with autosomal deafness with or without thrombocytopenia (OMIM 124900) are in blue. Note: both de novo mDia1 variants in moyamoya disease localize to the Rho-binding GBD. KMM/MM indicates proband identification; L, left; Lat, lateral; mDia1, mammalian diaphanous-1; and R, right.
Exon capture was performed using the Nimblegen v.2 exome capture reagent (Roche) in 3 participants, Nimblegen SeqxCap EZ MedExome Target Enrichment Kit (Roche) in 9 participants, or xGen Exome Research Panel v1 (IDT) in 56 participants, followed by 101 base pair paired-end sequencing using the Illumina NovaSeq platform (eTable 4 in the Supplement). Sequence reads were aligned to the human reference genome GRCh37/hg19 using BWA-MEM. Single-nucleotide variants and small indels were called using a combination of genome analysis toolkit HaplotypeCaller35,36 and Freebayes.37 Functional annotations were added using ANNOVAR38 with the dbNSFP39 database. Allele frequency annotations originated from the gnomAD (v.2.1.1) and Bravo databases.40,41 MetaSVM and combined annotation-dependent depletion, version 1.6 (CADD v1.6) algorithms were used to predict deleteriousness of missense variants (D-Mis, defined as MetaSVM-deleterious or combined annotation-dependent depletion, version 1.6 score ≥20).42,43 Inferred classes of loss-of-function (LoF) variants consisted of stop-gain, stop-loss, frameshift insertions/deletions, canonical splice site and start-loss variants. Loss-of-function and D-Mis variants were considered damaging.
De novo variants were called using TrioDeNovo.44 Candidate DNVs were further filtered based on the following criteria: (1) exonic or splice-site variants, (2) read depth of 10 in the proband and both parents; (3) minimum proband alternative read depth of 5; (4) proband alternative allele ratio greater than or equal to 28% if having less than 10 alternative reads or greater than or equal to 20% if having greater than or equal to 10 alternative reads, (5) alternative allele ratio in both parents greater than or equal to 3.5%, and (6) global minor allele frequency (MAF)≤4 × 10−4 in ExAC.
Rare heterozygous variants were identified as having a Bravo MAF≤5 × 10−4 and were classified as LoF or D-Mis by the previously described filtering criteria. Predicted LoF and D-Mis variants were further filtered for high quality using the following criteria: (1) Pass Genome Analysis Toolkit variant quality score recalibration, (2) read depth greater than or equal to 8 independent reads, and (3) genotype quality score greater than or equal to 20. Recessive genotypes and DNVs were excluded.
Recessive genotypes were filtered for rare (MAF≤1 × 10−3 in Bravo) homozygous and compound heterozygous variants that exhibited high-quality sequence reads (Pass Genome Analysis Toolkit variant quality score recalibration, GQ≥20, ≥4 total reads total for homozygous, and a minimum of 8 reads for compound heterozygous variants). Only LoF and D-mis were considered as potentially damaging. For probands whose parents’ WES data were not available, only homozygous variants were analyzed. Detailed methods regarding kinship analysis, ruling out duplicated samples, and principal component analysis are given in the eMethods in the Supplement.
After filtering using the aforementioned criteria for each type of variant, in silico visualization was performed to remove false-positive calls. Variants in the top candidate genes were further confirmed by Sanger sequencing (eFigure 1 in the Supplement).
Because the MMD trios were captured by 3 different reagents (ie, Nimblegen v.2, MedExome, and IDT), we took the union of all bases covered by different capture reagents and generated a browser-extensible data file representing a unified capture for all trios. We used bedtools (version 2.27.1) to extract sequences from the browser-extensible data file.45 A total of 1798 control trios consisting of unaffected siblings and parents of patients with autism spectrum disorder from the Simons Simplex Collection were analyzed in parallel.46 We then applied a sequence context-based method to calculate the probability of observing a DNV for each base in the coding region, adjusting for sequencing depth in each gene as described previously.47 The eMethods in the Supplement provides further details.
The burden of DNVs in MMD cases was determined using the denovolyzeR package (eMethods in the Supplement).48 To examine whether any gene contains more protein-damaging DNVs than expected, the expected number of protein-altering DNVs was calculated from the corresponding probability, adjusting for cohort size. The Poisson test was then used to compare the observed DNVs for each gene vs the expected number of DNVs. Because separate tests were performed for protein-damaging and LoF DNVs, the Bonferroni multiple-testing threshold was equal to 1.3 × 10−6 (α = .05/[2 tests ×19 347 genes]).
The gnomAD control cohort was filtered with identical criteria. A 2-tailed Fisher exact test was used to compare the number of total heterozygous damaging variants observed in DIAPH1, ACTA2, and RNF213 between 18 European MMD cases and non-Finnish European controls in the gnomAD (without disease-enriched TOPMed samples) database,40 regardless of the transmission pattern.
High-confidence MMD genes (monogenic causes of MMD) are provided in eTable 1 in the Supplement. Further clinical significance was determined by screening ClinVar database variants for those with predicted pathogenicity49
Polioudakis et al50 identified 16 prenatal cell types and associated marker genes in a single-cell RNA sequence atlas that maps the human midgestational brain (17-18 gestational weeks). We sought to test for enrichment of previously known and cohort-determined MMD genes in each of these cell types. Known MMD genes were compiled from the literature (eTable 1 in the Supplement). Candidate MMD risk genes from our cohort were defined using similar criteria from prior genomics studies as genes intolerant to LoF variants(pLI>0.9) with 1 or more LoF DNV and genes intolerant to missense variants (mis-Z>2) with 1 or more missense DNV (eTable 5 in the Supplement).29,31 Using an indicator-based binomial logistic regression model, we determined the significance of association between a gene’s status as an MMD gene and its identity as a cell-type marker for all genes expressed in 3 or more cells of the single-cell RNA-sequencing atlas, accounting for gene length and guanine-cytosine content as covariates. Enrichment significance was defined at the Bonferroni multiple-testing cutoff (α = .05/16 = 3.13 × 10−3).
The Betsholtz mouse brain vasculature scRNaseq database was used as an additional way to assess Diaph1 expression in cell types.51,52 The database was preprocessed by filtering for mitochondrial genes, library size, and cell doublets, followed by library normalization and log-transformation using scprep.53 Data were imputed using Markov Affinity-based graph imputation of cells.54 Optimal cluster number was determined by computing a silhouette score after principal component analysis linear-dimensionality reduction followed by kmeans clustering. Potential of heat-diffusion for affinity-based transition embedding,55 a nonlinear method of dimensionality reduction, was used for visualization. Gene-specific clusters were identified using mean difference in scprep. Marker genes were visualized to validate cluster identity (endothelial cells: Cdh5 and Pecam1, pericytes Pdgfrb and Cspg4, smooth muscle cells: Acta2 and Tagln, fibroblasts: Col1a1 and Col1a2, and oligodendrocytes: Plp1 and Cnp).
We then implemented k-nearest-neighbors conditional-density resampled estimate of mutual information (kNN-DREMI), which is a computational method successfully applied to study statistical dependency of gene expression in single-cell RNA sequencing data to identify genes associated with Diaph1 expression in mouse cerebrovascular cells.54,56,57 We determined the direction of relationship of the top Diaph1-scored genes (95th percentile) and performed hypergeometric enrichment analysis for the genes positively coregulated with Diaph1 expression using KEGG pathways from the molecular signatures database (MSigDB).58 Heatmaps were generated using pheatmap in R, version 4.0.2 (R Foundation for Statistical Computing). For visualization purposes, a random subset of 400 cells was used and visualized genes were restricted to those with a DREMI score greater than the 98th percentile. Columns were ordered by Diaph1 expression, whereas rows were hierarchically clustered.
Results
Of the 24 patients with MMD patients in our discovery cohort (22 trios) (eTable 2 in the Supplement), 14 (58.3%) were male and 18 (75.0%) were of European ancestry (eTable 6 in the Supplement). DNA was isolated and WES was performed.31 More than 93.7% of the targeted bases had 8 or more independent reads, and more than 89.7% had 15 or more independent reads (eTable 4 and eTable 7 in the Supplement).
We first screened for rare, damaging heterozygous variants (MAF≤5 × 10−4 in Bravo41) in genes previously implicated in MMD, including ACTA2, BRCC2, GUCY1A1, MTCP1, PCNT, RNF213, and YY1AP1 (eTable 1 in the Supplement). Three European probands carried different transmitted D-Mis variants (p.Cyc998Arg, p.Ile4717Thr, and p.Thr4845Lys) in highly conserved residues of RNF21359 (eTable 8 in the Supplement).11 Another European patient with MMD carried a transmitted D-Mis variant (p.Ile77Val) in ACTA2 (eTable 8 in the Supplement). To our knowledge, none of these variants found exclusively in White probands has been previously reported in patients with MMD. Orthogonal case-control analysis of these variants in 18 European probands with MMD vs non-Finnish European controls in the gnomAD database (without TOPMed samples) was significant in the case of RNF213 (odds ratio, 6.45; 2-tailed Fisher P = .01) but did not reach the multiple comparisons significance threshold for ACTA2 (Table 1).
Table 1. Case-Control Burden Analysis of All Rare Protein-Damage Variants for DIAPH1, ACTA2, and RNF213a.
| Gene | pLI Scoreb | mis_Zc | Yale MMD European Cases | Non-Finnish gnomAD-TOPMed (v2.1.1) | OR (95% CI) | P valued | ||
|---|---|---|---|---|---|---|---|---|
| Alleles, No. | Alleles, No. | |||||||
| Alternative | Reference | Alternative | Reference | |||||
| DIAPH1 | 0.92 | 1.65 | 3 | 33 | 1092 | 146 232 | 12.17 (2.39-38.89) | 2.4 × 10−3 |
| ACTA2 | 0.93 | 3.2 | 1 | 35 | 101 | 147 321 | 41.67 (1.01-254.7) | .02 |
| RNF213 | 0 | 2.3 | 3 | 33 | 2056 | 145 838 | 6.45 (1.26-20.58) | .01 |
Abbreviations: MMD, moyamoya disease; OR, odds ratio.
Rare variants (minor allele frequency, ≤5 × 10−4) protein-damaging mutations compared between 24 MMD cases from Yale Center for Mendelian Genomics and gnomAD-TOPMed controls.
pLI is a gene-wide constraint metric that estimates the probability of being intolerant to loss-of-function mutations.
mis_Z is a gene-wide constraint metric that estimates the probability of being intolerant to missense mutations.
A 2-tailed Fisher exact test was used to evaluate the genetic association by comparing frequencies of rare alleles between Yale Center for Mendelian Genomics MMD European cases and non-Finnish gnomAD (without disease-enriched TOPMed samples) and Simons simplex collection controls. The multiple-testing cutoff was 0.017 (α = .05/3).
Identification of Multiple DNVs and Damaging Variants
We next examined the contribution of DNVs to MMD risk. Our MMD cohort demonstrated a rate of 1.09 coding region DNVs per proband (eTable 5 in the Supplement), following the expected Poisson distribution and similar to previous results with the identical sequencing platform31 (eFigure 2 in the Supplement). We did not observe any significant enrichment of DNVs in any functional class (eTable 9 in the Supplement). However, 1 gene (DIAPH1) harbored 2 damaging heterozygous DNVs (c.534-2A>G and p.Cys220Arg) in unrelated European MMD probands, which is a very unlikely chance event (enrichment, 1372; 1-tailed Poisson P value = 1.1 × 10−6) (Table 2). In contrast, we identified no DNV in DIAPH1 among 1798 Simons simplex collection control trios.
Table 2. Characteristics of 6 Patients With Variants in DIAPH1.
| Proband ID | Ethnicity | Sex | Type | Class | Gene | Position (GRCh37) | AA change | Bravo WGS Freq a | gnomAD WES Freq | MetaSVM | CADD1.6 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Yale Center for Mendelian Genomics (24 MMD cases) | |||||||||||
| KMM_2-1 | European | Male | Transmitted from father | D-Mis | DIAPH1 | 5:140905715:C:T | p.R1155Q | 8.0 × 10−6 | 2.4 × 10−5 | T | 27.8 |
| KMM_39-1 | European | Male | DNV | Splicing | DIAPH1 | 5:140962861:T:C | c.534-2A>G | NA | NA | NA | 33 |
| KMM_42-1 | European | Female | DNV | D-Mis | DIAPH1 | 5:140961905:A:G | p.C220R | NA | NA | D | 28.2 |
| University of Texas Health Science Center at Houston (84 MMD cases) | |||||||||||
| MM 087 | African American/African | Female | Unphased | D-Mis | DIAPH1 | 5:140908798:G:A | p.L907F | 8.0 × 10−6 | NA | T | 23.9 |
| MM 087 | African American/African | Female | Unphased | D-Mis | DIAPH1 | 5:140908799:C:A | p.M906I | 8.0 × 10−6 | NA | T | 22.6 |
| MM 094 | European | Male | Transmitted from father | D-Mis | DIAPH1 | 5:140955835:C:T | p.E475K | 4.5 × 10−4 | 1.3 × 10−3 | T | 28.8 |
| MM 117 | African American/African | Female | Unphased | T-Mis | DIAPH1 | 5:140954569:C:A | p.D536Y | 8.0 × 10−6 | 4.1 × 10−6 | T | 15.99 |
Abbreviations: AA, amino acid; Bravo WGS Freq, Bravo whole-genome sequencing frequency; CADD1.6, combined annotated-dependent depletion; D-Mis, deleteriousness of missense variants; DNV, de novo variant; gnomAD WES Freq, Gnome aggregation database whole exome sequence frequency; MetaSVM, meta-analytic support vector machine; MMD, moyamoya disease; NA, not applicable.
This global frequency is based on 132 345 genomes in the Bravo database and is independent of ethnicity; Gnome Aggregation Database Whole Exome Sequence Frequency. This frequency is based on 125 748 exomes in the gnomAD database (v2.1.1) and is independent of ethnicity. Meta-analytic support vector machine is an ensemble score that predicts the tolerability of a variant. Combined annotated-dependent depletion is a validated tool for scoring the deleteriousness of single nucleotide deletions, insertions, or variations.
DIAPH1 (CCDS43374.1; RefSeq: NM_005219.4), encoding mammalian Diaphanous-1 (mDia1), is highly intolerant to LoF variant per gnomAD (pLI = 0.92) (Figure 1). Both DNVs map to the mDia1 guanosine triphosphatase (GTP)ase binding domain (GBD) domain, which mediates the interaction between activated GTP-bound RhoA and mDia160 (Figure 1 and Table 2). The c.534-2A>G alteration in the canonical acceptor splice site likely leads to exclusion of the 87 base pair exon 6, deleting 29 in-frame amino acids within the GBD domain. Arg substitution at the highly conserved Cys220 residue in mDia1 is predicted to disrupt tight helical packing of its GBD/FH3 domain required for RhoA-mDia1 interaction60 (eFigure 3 in the Supplement).
A transmitted heterozygous D-Mis (p.Arg1155Gln) mDia1 variant was detected in a third European patient with MMD. p.Arg1155Gln is ultrarare in Bravo (MAF = 8.0 × 10−6) and deleterious (CADD score, 27.8). The monomeric FH2 domains of mDia1 inhibit actin polymerization and bind filamentous actin (F-actin) in a 1:1 complex at micromolar concentrations.61 p.Arg1155Gln maps to the mDia1 FH2 domain (Figure 1 and Table 2). Gln substitution at the highly conserved Arg1155 residue is predicted to disrupt a hydrogen bond network and destabilize secondary structure of the FH2 domain required for F-actin binding (eFigure 3 in the Supplement).
An orthogonal case-control analysis of rare, damaging DIAPH1 variants in all 18 European MMD probands vs controls in the non-Finnish European gnomAD database (without TOPMed samples) provide additional support for DIAPH1 as an MMD risk gene (odds ratio, 12.17; 2-tailed Fisher exact test P = 2.4 × 10−3) (Table 1).
We prioritized DIAPH1 for further genomic validation because it was the only gene to harbor multiple damaging DNVs, showed variant enrichment in MMD cases vs controls, accounted for 12.5% of damaging variants in our discovery cohort, and has a biologically plausible mechanism given its role in RhoA-mediated actin dynamics in vascular cells and platelets.62 We therefore screened for rare, damaging DIAPH1 variants in a validation cohort of 84 MMD probands negative for known MMD gene pathogenic variants by WES.17
We identified 4 additional DIAPH1 protein-altering variants (3 unphased and 1 transmitted variant in 3 unrelated individuals with MMD (Figure 1, Table 2). In 1 patient, p.Met906Ile and p.Leu907Phe substitutions in the mDia1 FH2 domain were identified on the same allele. The p.Glu475Lys and p.Asp536Tyr D-mis variants in the linker region between the FH1 and FH3 domains were noted in 2 other unrelated MMD probands. All 4 DIAPH1 variants impact conserved residues and are predicted to significantly impair protein structure (eFigure 3 in the Supplement). These data lend support to the pathogenicity of these DIAPH1 variants in MMD and suggest haploinsufficiency as a genetic mechanism, with incomplete penetrance of a dominant trait in 2 cases.
Clinical Presentation of DIAPH1 Probands and Association With Thrombocytopenia
All patients with DIAPH1 variants presented with sporadic, bilateral MMD disease (Table 3). Both patients with DIAPH1 DNVs had severe disease (Suzuki grade IV or worse), and each presented with ischemic strokes, in contrast to only 22.2% of patients without DIAPH1 variants (P < .001). Patients with MMD and DIAPH1 variants who had available preoperative complete blood cell counts had thrombocytopenia (blood platelets, values <150 × 103/μL that required preoperative platelet transfusions (Table 3). The patient with the de novo DIAPH1 p.Cys220Arg variant developed preoperative nontraumatic bilateral subdural hemorrhages and the postoperative complication of subdural hemorrhage related to recalcitrant thrombocytopenia.
Table 3. Clinical Presentations of 6 Patients With Mutations in DIAPH1.
| Proband ID | Age at presentation, y | Presenting symptoms | Laterality | Suzuki grade | Infarct (yes/no), region | Other MMD/stroke sequelae | Treatments | Comorbidities |
|---|---|---|---|---|---|---|---|---|
| KMM 2-1 | 8 | Exertional headaches, ischemic stroke | Bilateral | Right: II-III | Yes, right frontal | None | Right pial synangiosis | Thrombocytopenia (platelets, 52 × 103/μL) |
| Left: I | ||||||||
| KMM 39-1 | 7 | Headaches, ischemic stroke | Bilateral | Right: IV | Yes, right aca-mca and mca-pca border zones | None | Right pial synangiosis | Behavioral problems (aggression), motor tics (EEG negative), thrombocytopenia (platelets, 96 × 103/μL) |
| Left: II-III | Left myo synangiosis | |||||||
| KMM 42-1 | 1 | Ischemic stroke, seizures | Bilateral | Bilateral: V-VI | Yes, numerous right-sided strokes | Seizures, precocious puberty (hypothalamic infarct), visual impairment, developmental delay | Bilateral pial synangiosis | Neonatal jaundice, syndactyly, thrombocytopenia (platelets, 62 × 103/μL) |
| MM 087 | 2 | Ischemic stroke | Bilateral | Data NA | Yes, right frontal | None | None | None |
| MM 094 | 26 | Ischemic stroke | Bilateral | Data NA | Yes, right frontal and left frontal | Difficulty with executive function, social skills, reading, and writing | Bilateral encephalo, duro, and arterio synangiosis | ADD, obesity, obstructive sleep apnea, asthma |
| MM 117 | 47 | Ischemic stroke, seizures | Bilateral | Right: IV | Yes, data NA | Hemianopsia, seizures | Data NA | Hypertension, left-ventricular hypertrophy, coronary artery disease, hypothyroidism |
Abbreviations: ADD, attention-deficit disorder; EEG, electroencephalogram; MMD, moyamoya disease; NA, not available.
SI conversion factor: To convert platelets to ×109/L, multiply by 1.
Compound Heterozygous Variants in EVL, a mDia1 Interactor and Actin Remodeler
To identify additional rare variants with large effect size that contribute to MMD, we evaluated recessive genotypes in 24 MMD trios. No genes harbored more than 1 recessive genotype (eTable 10 in the Supplement reports all damaging recessive genotypes). One patient harbored compound heterozygous D-mis variants (p.Gln236Arg and p.Arg363Lys) in EVL (pLI = 1.00), encoding the Ena/vasodilator-stimulated phosphoprotein (VASP)-like protein EVL. Similar to patients with DIAPH1 DNVs, the proband with the EVL variant had severe, bilateral MMD and thrombocytopenia. As with all Ena/VASP proteins, EVL has an N-terminal Ena/VASP homology EVH1 domain, a C-terminal EVH2 domain, and a central proline-rich domain. Both p.Gln236Arg and p.Arg363Lys map to EVL’s EVH2 domain, which mediates protein multimerization and binds F-actin in vitro.63
Enrichment of DIAPH1 and Other MMD Risk Genes in Mural Cells of the Midgestational Human Brain
To gain insight into the biological role of genes associated with MMD, we examined potential enrichment of known (eTable 1 in the Supplement) and cohort-determined (DIAPH1, CDH2, LRP2, MAP2K4, NOTCH3, TPR, and ZFC3H1) (eMethods and eTable 5 in the Supplement) MMD risk genes in the largest available single-cell RNA transcriptomic atlas of human midgestational brain development50 (Figure 2). Known MMD genes are enriched in pericytes (P = 1.3 × 10−3), and cohort-determined MMD risk genes are enriched in pericytes (P = 1.7 × 10−3), endothelial cells (P = 7.6 × 10−4), and interneurons of the prenatal caudate ganglionic eminence (P = 1.9 × 10−3). Enrichment of MMD genes in endothelial cells and pericytes suggests that MMD variants disrupt convergent pathways to affect mural cell biology.
Figure 2. Enriched Expression of DIAPH1 and Other Moyamoya Disease Risk Genes in Vascular Cells.
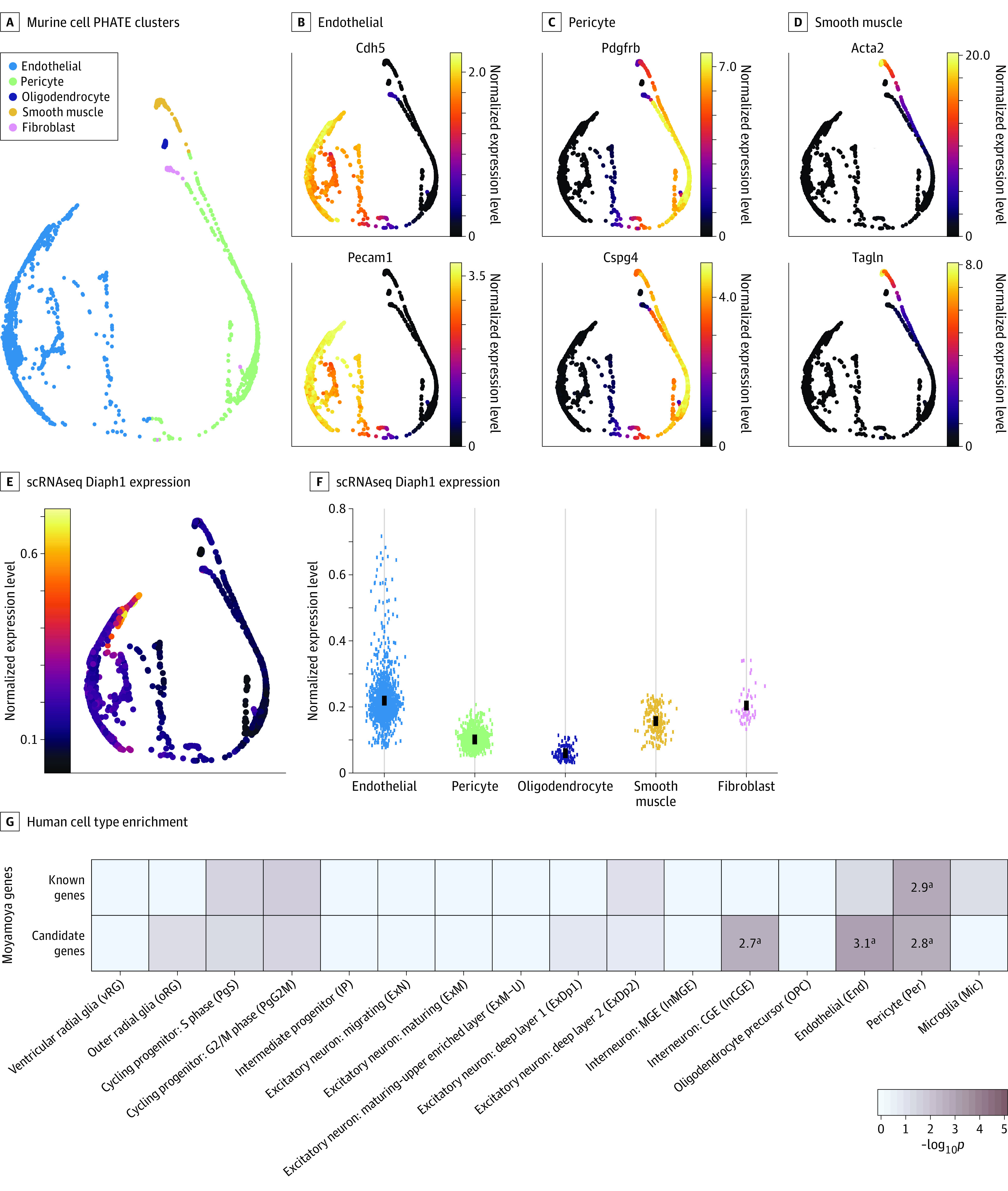
A-D, Cell type–specific clusters in mouse brain vascular cells using Betsholtz scRNaseq data. A, Potential of heat-diffusion for affinity-based transition embedding (PHATE) plot clusters of endothelial cells, pericytes, oligodendrocytes, smooth muscle cells, and fibroblasts. B-D, PHATE plots of 2 marker genes per cluster. B, Endothelial cells expressing Cdh5 and Pecam1. C Pericytes expressing Pdgfrb and Cspg4. D, Smooth muscle cells expressing Acta2 and Tagln. E, PHATE plot of cell type–specific expression of Diaph1. Expression maps best to endothelial cells. F, Diaph1 messenger RNA abundance in each cell cluster. G, Enrichment analysis across cell type markers of the midgestational human brain 69 for known human genes and cohort-determined candidate risk genes for moyamoya disease.
aRepresents significant enrichment at the Bonferroni multiple-testing cutoff (α = .05/16 = 3.13 × 10−3).
Genes Co-regulated With Diaph1 Expression in Vascular Cells Are Enriched for Roles in Actin Remodeling
The above results prompted us to study Diaph1 expression in mouse brain vascular cells in greater detail using Betsholtz scRNA sequencing data,51,52 as described in the Methods section. The top 3 cell types represented in the database were endothelial cells, pericytes, and smooth muscle cells (Figure 2A-D). Diaph1 expression was enriched in endothelial cells and fibroblasts (Figure 2E-F). kNN-DREMI analysis revealed that genes coexpressed with Diaph1 were enriched in an adherens junction (false discovery rate = 1.02 × 10−5) and regulation of actin cytoskeleton (false discovery rate = 3.27 × 10−4) (eFigure 4 in the Supplement), implicating Diaph1 in vascular organization and vascular tone regulation.
Discussion
We have identified a novel association of de novo and rare, transmitted damaging variants in DIAPH1 with the risk of MMD. The variant burden of DIAPH1 in MMD surpassed genome-wide significance thresholds, and variants were significantly increased in cases vs controls. The discovery of multiple additional, ultrarare, damaging DIAPH1 variants in an independent cohort of 84 probands supports our findings.
Previous work in monogenic familial forms of MMD has implicated impaired cytoskeleton regulation and increased fibrocellular proliferation of VSMCs in MMD pathogenesis.11,64 Pressure- and stretch-induced actin polymerization in VSMCs underlies blood vessel constriction,65 and agonist-induced actin polymerization mediates platelet surface adhesion, aggregation, and thrombus formation.66 These events are regulated by RhoA and other Rho family small GTPases, which cycle between inactive GDP-bound and active GTP-bound states.67 Dysregulation of Rho GTPases and their signaling effectors has been implicated in vascular cell (eg, vasospasm and stroke)68 and platelet (thrombocytopenia) diseases.69
DIAPH1 encodes the RhoA GTPase mDia1,61,70 which contains a C-terminal inhibitory diaphanous autoregulatory domain and an N-terminal Rho- GBD embedded within the larger diaphanous autoinhibitory domain.60,71 The binding of activated RhoA allows the mDia1 FH2 domain to bind F-actin and stimulate the addition of actin monomers at barbed ends.72 DIAPH1 pathogenic variants have been implicated in 2 other mendelian diseases. Heterozygous truncating DIAPH1 variants (including the recurrent variant p.Arg1213Ter) cause deafness 1 with or without thrombocytopenia (OMIM 124900)73,74; these variants eliminate the C-terminal autoinhibitory diaphanous autoregulatory domain and lead to increased mDia1 gain of function.74 In contrast, homozygous loss-of-function variants cause a syndrome characterized by seizures, cortical blindness, and microcephaly (OMIM 616632).75
Moyamoya disease–associated DIAPH1 variants are heterozygous and likely result in haploinsufficiency, with evidence of incomplete penetrance or potential somatic mosaicism in 1 of our trios. De novo variants localize to the GBD-binding domain and lead to more severe MMD and thrombocytopenia, the latter of which is present also in patients with DFNA1 with DIAPH1 variants. Notably, none of the probands with DIAPH1-variant MMD had deafness. This absence of deafness and the discovery of multiple DIAPH1 variants in a cohort of such modest size decrease the chance that DIAPH1 variants were responsible only for thrombocytopenia and simply coincident with MMD.
Moyamoya disease–associated DIAPH1 DNVs might result in loss of activation of mDia1 by abrogating its interaction with activated GTP-bound RhoA. Moyamoya disease–variants in other parts of the protein distal to the GBD domain but proximal to the autoinhibitory diaphanous autoregulatory domain also likely impair mDia1 function. For example, site-directed mutagenesis in the FH2 domain—the site of multiple mDia1 variants in MMD—abolishes bundling of actin filaments induced by a constitutively active mDia1 mutant.76 Phenotypic heterogeneity among patients with DIAPH1 variants likely reflects differences in variant zygosity (homozygous vs heterozygous), type (LoF vs D-mis), and domain localization of variants within the mDia1 protein. Determinants of incomplete penetrance and variable expressivity remain obscure but may include common genetic or environmental modifiers (eg, inflammatory or oxidative triggers) or stochastic elements.77
Moyamoya disease DIAPH1 variants impairing RhoA-mDia1–regulated actin remodeling may cause neointimal expansion and progressive narrowing of the bilateral internal carotid arteries.78 Consistent with this hypothetical mechanism, Rho-A-mDia1 regulates signal transduction of oxidative stress and inflammatory events that control VSMC migration, vascular remodeling, and pathological neointimal expansion.62 Moreover, DIAPH1 single-nucleotide variants are associated with increased susceptibility to ischemic stroke risk, especially small-artery occlusion subtypes.79 The next most frequently mutated gene among non–East Asian patients with MMD, ACTA2, encodes the smooth muscle–specific isoform of α-actin in VSMCs.80 ACTA2-mutant vascular stenosis is associated, in turn, with fibrocellular proliferation of intimal VSMCs.80 Impaired RhoA-mDia1–regulated actin dynamics could also help explain the thrombocytopenia seen in patients with MMD.81
Limitations
This study has limitations. The interpretation of our results is limited by our discovery cohort size. Furthermore, identified variants in DIAPH1 are not identical and therefore may have slightly different biological effects or carry different phenotype risk. Nonetheless, these results demonstrate the power of trio-based WES for identifying potential rare, large-effect pathogenic variants in rare disease.
Conclusions
Future WES of additional MMD trios and the modeling of discovered variants in appropriate model systems will be important topics of investigation that may yield new insight into the pathogenic mechanism of DIAPH1 in MMD. In addition, further research might foster the discovery of other new monogenic forms of disease and define clinically relevant MMD subtypes.
eFigure 1. Sanger Validation of Identified DIAPH1 Variants
eFigure 2. De Novo Variant Rate Closely Approximates Poisson Distribution in Primary MMD Cases and Controls
eFigure 3. Phylogenetic Conservation and Structural Implication of De Novo and Rare, Damaging Transmitted DIAPH1 Variants in Moyamoya Disease
eFigure 4. kNN-DREMI Diaph1-Associated Genes and KEGG Terms
eTable 1. High Confidence, Monogenic Causes of Primary MMD and Syndromes With Associated MM Pathology
eTable 2. Number of Studied Cases From Yale, UT Health, and Simons Simplex Collection (SSC)
eTable 3. Phenotypes for Primary Probands
eTable 4. Summary Sequencing Statistics for the Primary MMD and Control Cohorts
eTable 5. De Novo Variants in 22 Primary MMD Trios
eTable 6. Demographic Characteristics of Primary MMD Cases and Controls
eTable 7. Sequencing Metrics for All Reported Samples
eTable 8. Rare Transmitted/Unphased Variants in Known MMD Genes
eTable 9. De Novo Variant Enrichment Analysis for Each Functional Class in 22 Primary Moyamoya Case Trios
eTable 10. Damaging Recessive Variants in 26 MMD Cases
eMethods. Detailed Methods
eReferences
References
- 1.Takeuchi K, Shimizu K. Hypoplasia of the bilateral internal carotid arteries. Brain Nerv. 1957;9:37–43. [Google Scholar]
- 2.Jung YS, Wang W, Jun S, et al. Deregulation of CRAD-controlled cytoskeleton initiates mucinous colorectal cancer via β-catenin. Nat Cell Biol. 2018;20(11):1303-1314. doi: 10.1038/s41556-018-0215-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeuchi K, Shimizu K. Hypoplasia of the bilateral internalcarotid arteries. No To Shinkei. 1957(9):37-43. [Google Scholar]
- 4.Roder C, Nayak NR, Khan N, Tatagiba M, Inoue I, Krischek B. Genetics of moyamoya disease. J Hum Genet. 2010;55(11):711-716. doi: 10.1038/jhg.2010.103 [DOI] [PubMed] [Google Scholar]
- 5.Guey S, Tournier-Lasserve E, Hervé D, Kossorotoff M. Moyamoya disease and syndromes: from genetics to clinical management. Appl Clin Genet. 2015;8:49-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baba T, Houkin K, Kuroda S. Novel epidemiological features of moyamoya disease. J Neurol Neurosurg Psychiatry. 2008;79(8):900-904. doi: 10.1136/jnnp.2007.130666 [DOI] [PubMed] [Google Scholar]
- 7.Uchino K, Johnston SC, Becker KJ, Tirschwell DL. Moyamoya disease in Washington State and California. Neurology. 2005;65(6):956-958. doi: 10.1212/01.wnl.0000176066.33797.82 [DOI] [PubMed] [Google Scholar]
- 8.Lee S, Rivkin MJ, Kirton A, deVeber G, Elbers J; International Pediatric Stroke Study . Moyamoya disease in children: results from the International Pediatric Stroke Study. J Child Neurol. 2017;32(11):924-929. doi: 10.1177/0883073817718730 [DOI] [PubMed] [Google Scholar]
- 9.Ferriero DM, Fullerton HJ, Bernard TJ, et al. ; American Heart Association Stroke Council and Council on Cardiovascular and Stroke Nursing . Management of stroke in neonates and children: a scientific statement from the American Heart Association/American Stroke Association. Stroke. 2019;50(3):e51-e96. doi: 10.1161/STR.0000000000000183 [DOI] [PubMed] [Google Scholar]
- 10.Huang S, Guo ZN, Shi M, Yang Y, Rao M. Etiology and pathogenesis of moyamoya disease: an update on disease prevalence. Int J Stroke. 2017;12(3):246-253. doi: 10.1177/1747493017694393 [DOI] [PubMed] [Google Scholar]
- 11.Liu W, Morito D, Takashima S, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS One. 2011;6(7):e22542. doi: 10.1371/journal.pone.0022542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takagi Y, Kikuta K, Nozaki K, Hashimoto N. Histological features of middle cerebral arteries from patients treated for moyamoya disease. Neurol Med Chir (Tokyo). 2007;47(1):1-4. doi: 10.2176/nmc.47.1 [DOI] [PubMed] [Google Scholar]
- 13.Masuda J, Ogata J, Yutani C. Smooth muscle cell proliferation and localization of macrophages and T cells in the occlusive intracranial major arteries in moyamoya disease. Stroke. 1993;24(12):1960-1967. doi: 10.1161/01.STR.24.12.1960 [DOI] [PubMed] [Google Scholar]
- 14.Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. 2009;360(12):1226-1237. doi: 10.1056/NEJMra0804622 [DOI] [PubMed] [Google Scholar]
- 15.Fukui M, Kono S, Sueishi K, Ikezaki K. Moyamoya disease. Neuropathology. 2000;20(suppl):S61-S64. doi: 10.1046/j.1440-1789.2000.00300.x [DOI] [PubMed] [Google Scholar]
- 16.Kamada F, Aoki Y, Narisawa A, et al. A genome-wide association study identifies RNF213 as the first moyamoya disease gene. J Hum Genet. 2011;56(1):34-40. doi: 10.1038/jhg.2010.132 [DOI] [PubMed] [Google Scholar]
- 17.Pinard A, Guey S, Guo D, et al. The pleiotropy associated with de novo variants in CHD4, CNOT3, and SETD5 extends to moyamoya angiopathy. Genet Med. 2020;22(2):427-431. doi: 10.1038/s41436-019-0639-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hervé D, Philippi A, Belbouab R, et al. Loss of α1β1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am J Hum Genet. 2014;94(3):385-394. doi: 10.1016/j.ajhg.2014.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallace S, Guo DC, Regalado E, et al. Disrupted nitric oxide signaling due to GUCY1A3 mutations increases risk for moyamoya disease, achalasia and hypertension. Clin Genet. 2016;90(4):351-360. doi: 10.1111/cge.12739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miskinyte S, Butler MG, Hervé D, et al. Loss of BRCC3 deubiquitinating enzyme leads to abnormal angiogenesis and is associated with syndromic moyamoya. Am J Hum Genet. 2011;88(6):718-728. doi: 10.1016/j.ajhg.2011.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Warejko JK, Schueler M, Vivante A, et al. Whole exome sequencing reveals a monogenic cause of disease in ≈43% of 35 families with midaortic syndrome. Hypertension. 2018;71(4):691-699. doi: 10.1161/HYPERTENSIONAHA.117.10296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39(12):1488-1493. doi: 10.1038/ng.2007.6 [DOI] [PubMed] [Google Scholar]
- 23.Guo DC, Duan XY, Regalado ES, et al. ; University of Washington Center for Mendelian Genomics . Loss-of-function mutations in YY1AP1 lead to Grange syndrome and a fibromuscular dysplasia-like vascular disease. Am J Hum Genet. 2017;100(1):21-30. doi: 10.1016/j.ajhg.2016.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milewicz DM, Østergaard JR, Ala-Kokko LM, et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A. 2010;152A(10):2437-2443. doi: 10.1002/ajmg.a.33657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rauch A, Thiel CT, Schindler D, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319(5864):816-819. doi: 10.1126/science.1151174 [DOI] [PubMed] [Google Scholar]
- 26.Zhu B, Liu X, Zhen X, et al. RNF213 gene polymorphism rs9916351 and rs8074015 significantly associated with moyamoya disease in Chinese population. Ann Transl Med. 2020;8(14):851. doi: 10.21037/atm-20-1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park YS, An HJ, Kim JO, et al. The role of RNF213 4810G>A and 4950G>A variants in patients with moyamoya disease in Korea. Int J Mol Sci. 2017;18(11):E2477. doi: 10.3390/ijms18112477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu W, Senevirathna ST, Hitomi T, et al. Genomewide association study identifies no major founder variant in Caucasian moyamoya disease. J Genet. 2013;92(3):605-609. doi: 10.1007/s12041-013-0304-5 [DOI] [PubMed] [Google Scholar]
- 29.Furey CG, Choi J, Jin SC, et al. De novo mutation in genes regulating neural stem cell fate in human congenital hydrocephalus. Neuron. 2018;99(2):302-314.e4. doi: 10.1016/j.neuron.2018.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin SC, Lewis SA, Bakhtiari S, et al. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat Genet. 2020;52(10):1046-1056. doi: 10.1038/s41588-020-0695-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin SC, Dong W, Kundishora AJ, et al. Exome sequencing implicates genetic disruption of prenatal neuro-gliogenesis in sporadic congenital hydrocephalus. Nat Med. 2020;26(11):1754-1765. doi: 10.1038/s41591-020-1090-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duran D, Zeng X, Jin SC, et al. Mutations in chromatin modifier and ephrin signaling genes in vein of Galen malformation. Neuron. 2019;101(3):429-443. doi: 10.1016/j.neuron.2018.11.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang M, Ding X, Zhang Q, et al. Exome sequencing of 112 trios identifies recessive genetic variants in brain arteriovenous malformations. J Neurointerv Surg. 2020;neurintsurg-2020-016469. doi: 10.1136/neurintsurg-2020-016469 [DOI] [PubMed] [Google Scholar]
- 34.Research Committee on the Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis; Health Labour Sciences Research Grant for Research on Measures for Intractable Diseases . Guidelines for diagnosis and treatment of moyamoya disease (spontaneous occlusion of the circle of Willis). Neurol Med Chir (Tokyo). 2012;52(5):245-266. doi: 10.2176/nmc.52.245 [DOI] [PubMed] [Google Scholar]
- 35.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297-1303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43(1110):11.10.1-11.10.33. doi: 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv:12073907. Preprint posted online July 12, 2012.
- 38.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37(3):235-241. doi: 10.1002/humu.22932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karczewski KJ, Francioli LC, Tiao G, et al. ; Genome Aggregation Database Consortium . The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taliun D, Harris DN, Kessler MD, et al. . Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. bioRxiv. 2019:563866. doi: 10.1101/563866 [DOI] [PMC free article] [PubMed]
- 42.Dong C, Wei P, Jian X, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet. 2015;24(8):2125-2137. doi: 10.1093/hmg/ddu733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886-D894. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei Q, Zhan X, Zhong X, et al. A bayesian framework for de novo mutation calling in parents-offspring trios. Bioinformatics. 2015;31(9):1375-1381. doi: 10.1093/bioinformatics/btu839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841-842. doi: 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krumm N, Turner TN, Baker C, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015;47(6):582-588. doi: 10.1038/ng.3303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samocha KE, Robinson EB, Sanders SJ, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46(9):944-950. doi: 10.1038/ng.3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ware JS, Samocha KE, Homsy J, Daly MJ. Interpreting de novo variation in human disease using denovolyzeR. Curr Protoc Hum Genet. 2015;87:7.25.1-7.25.15. doi: 10.1002/0471142905.hg0725s87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062-D1067. doi: 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Polioudakis D, de la Torre-Ubieta L, Langerman J, et al. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron. 2019;103(5):785-801.e8. doi: 10.1016/j.neuron.2019.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He L, Vanlandewijck M, Mäe MA, et al. Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci Data. 2018;5:180160. doi: 10.1038/sdata.2018.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vanlandewijck M, He L, Mäe MA, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554(7693):475-480. doi: 10.1038/nature25739 [DOI] [PubMed] [Google Scholar]
- 53.Chen X, Burkhardt DB, Hartman AA, et al. MLL-AF9 initiates transformation from fast-proliferating myeloid progenitors. Nat Commun. 2019;10(1):5767. doi: 10.1038/s41467-019-13666-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Dijk D, Sharma R, Nainys J, et al. Recovering gene interactions from single-cell data using data diffusion. Cell. 2018;174(3):716-729.e27. doi: 10.1016/j.cell.2018.05.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moon KR, van Dijk D, Wang Z, et al. Visualizing structure and transitions in high-dimensional biological data. Nat Biotechnol. 2019;37(12):1482-1492. doi: 10.1038/s41587-019-0336-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dutrow EV, Emera D, Yim K, et al. The human accelerated region HACNS1 modifies developmental gene expression in humanized mice. bioRxiv. 2019. doi: 10.1101/2019.12.11.873075 [DOI]
- 57.Krishnaswamy S, Spitzer MH, Mingueneau M, et al. Systems biology: conditional density-based analysis of T cell signaling in single-cell data. Science. 2014;346(6213):1250689. doi: 10.1126/science.1250689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liberzon A. A description of the molecular signatures database (MSigDB) website. In: Stem Cell Transcriptional Networks. Springer; 2014:153-160. doi: 10.1007/978-1-4939-0512-6_9 [DOI] [PubMed] [Google Scholar]
- 59.Scholz B, Korn C, Wojtarowicz J, et al. Endothelial RSPO3 controls vascular stability and pruning through non-canonical WNT/Ca(2+)/NFAT signaling. Dev Cell. 2016;36(1):79-93. doi: 10.1016/j.devcel.2015.12.015 [DOI] [PubMed] [Google Scholar]
- 60.Rose R, Weyand M, Lammers M, Ishizaki T, Ahmadian MR, Wittinghofer A. Structural and mechanistic insights into the interaction between Rho and mammalian Dia. Nature. 2005;435(7041):513-518. doi: 10.1038/nature03604 [DOI] [PubMed] [Google Scholar]
- 61.Shimada A, Nyitrai M, Vetter IR, et al. The core FH2 domain of diaphanous-related formins is an elongated actin binding protein that inhibits polymerization. Mol Cell. 2004;13(4):511-522. doi: 10.1016/S1097-2765(04)00059-0 [DOI] [PubMed] [Google Scholar]
- 62.Touré F, Fritz G, Li Q, et al. Formin mDia1 mediates vascular remodeling via integration of oxidative and signal transduction pathways. Circ Res. 2012;110(10):1279-1293. doi: 10.1161/CIRCRESAHA.111.262519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bachmann C, Fischer L, Walter U, Reinhard M. The EVH2 domain of the vasodilator-stimulated phosphoprotein mediates tetramerization, F-actin binding, and actin bundle formation. J Biol Chem. 1999;274(33):23549-23557. doi: 10.1074/jbc.274.33.23549 [DOI] [PubMed] [Google Scholar]
- 64.Young AM, Karri SK, Ogilvy CS, Zhao N. Is there a role for treating inflammation in moyamoya disease?: a review of histopathology, genetics, and signaling cascades. Front Neurol. 2013;4:105. doi: 10.3389/fneur.2013.00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. 2002;16(1):72-76. doi: 10.1096/cj.01-0104hyp [DOI] [PubMed] [Google Scholar]
- 66.Aslan JE, McCarty OJ. Rho GTPases in platelet function. J Thromb Haemost. 2013;11(1):35-46. doi: 10.1111/jth.12051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moreno-Domínguez A, Colinas O, El-Yazbi A, et al. Ca2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J Physiol. 2013;591(5):1235-1250. doi: 10.1113/jphysiol.2012.243576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nunes KP, Rigsby CS, Webb RC. RhoA/Rho-kinase and vascular diseases: what is the link? Cell Mol Life Sci. 2010;67(22):3823-3836. doi: 10.1007/s00018-010-0460-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bros M, Haas K, Moll L, Grabbe S. RhoA as a key regulator of innate and adaptive immunity. Cells. 2019;8(7):E733. doi: 10.3390/cells8070733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shinohara R, Thumkeo D, Kamijo H, et al. A role for mDia, a Rho-regulated actin nucleator, in tangential migration of interneuron precursors. Nat Neurosci. 2012;15(3):373-380, S1-S2. doi: 10.1038/nn.3020 [DOI] [PubMed] [Google Scholar]
- 71.Otomo T, Otomo C, Tomchick DR, Machius M, Rosen MK. Structural basis of Rho GTPase-mediated activation of the formin mDia1. Mol Cell. 2005;18(3):273-281. doi: 10.1016/j.molcel.2005.04.002 [DOI] [PubMed] [Google Scholar]
- 72.Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593-627. doi: 10.1146/annurev.biochem.75.103004.142647 [DOI] [PubMed] [Google Scholar]
- 73.Lynch ED, Lee MK, Morrow JE, Welcsh PL, León PE, King MC. Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science. 1997;278(5341):1315-1318. doi: 10.1126/science.278.5341.1315 [DOI] [PubMed] [Google Scholar]
- 74.Stritt S, Nurden P, Turro E, et al. ; BRIDGE-BPD Consortium . A gain-of-function variant in DIAPH1 causes dominant macrothrombocytopenia and hearing loss. Blood. 2016;127(23):2903-2914. doi: 10.1182/blood-2015-10-675629 [DOI] [PubMed] [Google Scholar]
- 75.Al-Maawali A, Barry BJ, Rajab A, et al. Novel loss-of-function variants in DIAPH1 associated with syndromic microcephaly, blindness, and early onset seizures. Am J Med Genet A. 2016;170A(2):435-440. doi: 10.1002/ajmg.a.37422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ishizaki T, Morishima Y, Okamoto M, Furuyashiki T, Kato T, Narumiya S. Coordination of microtubules and the actin cytoskeleton by the Rho effector mDia1. Nat Cell Biol. 2001;3(1):8-14. doi: 10.1038/35050598 [DOI] [PubMed] [Google Scholar]
- 77.Timberlake AT, Choi J, Zaidi S, et al. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife. 2016;5:5. doi: 10.7554/eLife.20125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sakaguchi T, Yan SF, Yan SD, et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest. 2003;111(7):959-972. doi: 10.1172/JCI200317115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ren Z, Chen X, Tang W, et al. Association of DIAPH1 gene polymorphisms with ischemic stroke. Aging (Albany NY). 2020;12(1):416-435. doi: 10.18632/aging.102631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo DC, Papke CL, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84(5):617-627. doi: 10.1016/j.ajhg.2009.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bearer EL, Prakash JM, Li Z. Actin dynamics in platelets. Int Rev Cytol. 2002;217:137-182. doi: 10.1016/S0074-7696(02)17014-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eFigure 1. Sanger Validation of Identified DIAPH1 Variants
eFigure 2. De Novo Variant Rate Closely Approximates Poisson Distribution in Primary MMD Cases and Controls
eFigure 3. Phylogenetic Conservation and Structural Implication of De Novo and Rare, Damaging Transmitted DIAPH1 Variants in Moyamoya Disease
eFigure 4. kNN-DREMI Diaph1-Associated Genes and KEGG Terms
eTable 1. High Confidence, Monogenic Causes of Primary MMD and Syndromes With Associated MM Pathology
eTable 2. Number of Studied Cases From Yale, UT Health, and Simons Simplex Collection (SSC)
eTable 3. Phenotypes for Primary Probands
eTable 4. Summary Sequencing Statistics for the Primary MMD and Control Cohorts
eTable 5. De Novo Variants in 22 Primary MMD Trios
eTable 6. Demographic Characteristics of Primary MMD Cases and Controls
eTable 7. Sequencing Metrics for All Reported Samples
eTable 8. Rare Transmitted/Unphased Variants in Known MMD Genes
eTable 9. De Novo Variant Enrichment Analysis for Each Functional Class in 22 Primary Moyamoya Case Trios
eTable 10. Damaging Recessive Variants in 26 MMD Cases
eMethods. Detailed Methods
eReferences