

Abstract
B-Cell lymphoma 2 (BCL-2) regulates cell death in humans. In this study, combined multiscale in silico approaches and in vitro studies were employed. A small-molecule library that includes more than 210 000 compounds was used. The predicted therapeutic activity value (TAV) of the compounds in this library was computed with the binary cancer quantitative structure–activity relationships (QSAR) model. The molecules with a high calculated TAV were used in 26 individual toxicity QSAR models. As a result of this screening protocol, 288 nontoxic molecules with high predicted TAV were identified. These selected hits were then screened against the BCL-2 target protein using hybrid docking and molecular dynamics (MD) simulations. The interaction energies of identified compounds were compared with two known BCL-2 inhibitors. Then, the short MD simulations were carried out by initiating the best docking poses of 288 molecules. Average MM/GBSA energies were computed, and long MD simulations were employed to selected hits. The same calculations were also applied for two known BCL-2 inhibitors. Moreover, a five-site (AHRRR) structure-based pharmacophore model was constructed, and this model was used in the screening of the same database. On the basis of hybrid data-driven ligand identification study, final hits were selected and used in in vitro studies. Based on results of the time-resolved fluorescence resonance energy transfer (TR-FRET) analysis, further filtration was carried out for the U87-MG cell line tests. MTT cell proliferation assay analysis results showed that selected three potent compounds were significantly effective on glioma cells.
Keywords: binary QSAR modeling, virtual screening, BCL-2, text mining, colorimetric biochemical in vitro assay, TR-FRET assay
Cancer-related deaths account for about one-eighth of all deaths worldwide. Cancer is defined by the uncontrolled growth and the proliferation of unusual cells.1 The risk of developing cancer has doubled in the last 30 years.2 While there were more than 17 million new cancer cases in the world in 2018, the total number of deaths was about 9.6 million.2 Although the rate of cancer-related deaths has fallen dramatically in recent years, it remains the most important health problem worldwide.3 In addition to targeted cancer treatments,4 hyperthermia,5 immunotherapy,6 antidotes/toxicity modifiers,7 resistance modulation,8 and alternative formulations;9 even though they have serious side effects radiotherapy and surgery are still leading the treatments of this disease.10 Since a tumor poses a serious threat to human health, it is important to find effective and specific antitumor drugs. Although surgery and radiation are used to treat locally limited cancers, drug therapy is required to kill metastatic cancer cells.11
B-Cell lymphoma 2 (BCL-2) proteins are vital in controlling cell death. Overexpression of BCL-2 proteins is usually directly related to various types of cancer. Excessive expression of BCL-2 in chronic lymphocytic leukemia (CLL) cells was found to cause resistance to chemotherapy and tumor cell survival.
It is not clear how BCL-2 proteins affect apoptotic pathways, but it is estimated that these proteins form important homodimers and heterodimers by entering various protein–protein interactions for their biological functions. Whether a cell is prone to apoptosis depends on the heterodimer or homodimer form of the BCL-2 family genes. The BCL-2 family consists of two opposing groups: proapoptotic and antiapoptotic members. If proapoptotic proteins are high in the cell, then the cell is prone to apoptosis.12 Proapoptotic members are BAD, BAX, BID, BCL-Xs, BAK, BIM, PUMA, and NOXA. These proteins are located in the cytosolic domain. Cytochrome c and apoptosis inducing factor (AIF) induce apoptosis by increasing secretion. Antiapoptotic members are MCL-1, BCL-XL, and BCL-2. These proteins are also located in the outer membrane of the mitochondria, the endoplasmic reticulum, and the nuclear membrane. They provide pore formation and regulate ion transport. In particular, they control the Ca2+ concentration in the cell. They also inhibit apoptosis by blocking the release of AIF and cytochrome c with the precursor forms of caspases.12−14
Apoptosis occurs in two ways: the intrinsic (mitochondrial) pathway and the extrinsic pathway. Caspases are involved in both signal paths.15−17 Venetoclax (ABT-199), a small, bioavailable FDA-approved inhibitor, is particularly effective and safe in the treatment of CLL and is effective at the beginning of acute myeloid leukemia (AML). Navitoclax (ABT-263) was discovered as a BCL-2 inhibitor with high affinity (<1 nM) to BCL-2, BCL-W, and BCL-XL but not to myeloid cell leukemia-1 (MCL-1) or BCL-2 related protein A1.18 However, studies showing its activity as a chemotherapeutic agent on solid tumors is limited.19 Therefore, novel, safe and potent BCL-2 inhibitors are needed. The identification of new scaffolds as potential BCL-2 inhibitors will be a step closer to finding appropriate effective therapy in different cancer types. The development of potent inhibitors to control the level of BCL-2 proteins has received much attention in recent years. Thus, recently in silico guided approaches were used to identify new inhibitors of BCL-2.20
The complexity and quantity of biological information coupled with the rapidly increasing computational resources and power, as well as the vision of more efficient therapy and drug design, have created an increasing interest in computational biology and molecular simulations. Models created by integrated molecular modeling and artificial intelligence approaches using preclinical and clinical data provide new tools to understand the dynamic and topological profiles of drug targets, thus allowing us to design new therapeutics and solutions.
Recently, in our research group, we have searched and identified new BCL-2 inhibitors with molecular-modeling-guided approaches and compared the success rates of the correct identification of the hit compounds using docking-based and molecular dynamics (MD)-based approaches.20 Therefore, in the current study, which is the continuation of our effort for the identification of novel hits against BCL-2, combined hybrid in silico multiscale molecular modeling approaches and time-resolved fluorescence resonance energy transfer (TR-FRET)-guided screening of a library of small molecules was employed. Here, we slightly changed our previously applied screening algorithm20 for the identification of new BCL-2 inhibitors. In this study, virtual screening of a library of molecules (Specs-SC-10 mg-Nov2018) containing more than 210 000 small compounds identified with text mining and integrated ligand- and target-based drug screening methodologies was conducted, and new candidate hit compounds have been proposed.
The indole fragment is an important biological subunit and many FDA-approved drugs including anticancer compounds involve indole or indole derivatives.20,21 An indole ring system can be applied to other ring systems as well, such as benzothiazole and benzimidazoles. These compounds are also reported to have anticancer properties.22,23 Therefore, a text mining technique was employed in this study, and the molecules that include indole or its analogs were considered. These filtered compounds were used in binary quantitative structure–activity relationships (QSAR) models for the calculation of their therapeutic activity value (TAV) as well as their pharmacokinetic profiles. Moreover, a target-based pharmacophore model (i.e., e-pharmacophore modeling) was constructed, and this model was also used in the screening of this database. Identified compounds from both analyses were then used in molecular docking calculations and MD simulations, and tightly binding compounds among the large data set were identified.
Finally, we identified 15 potent hit compounds against BCL-2 activity from two different approaches using combined ligand-driven and target-driven based in silico methods. Selected hits among these compounds were initially tested by TR-FRET assay, and potent compounds validated by these methods were used in cell-line-based in vitro tests. Potent, effective, and safe BCL-2 inhibitors with new scaffolds were found in the current work by employing hierarchical and rigorous filtering methods.
Materials and Methods
Binary QSAR Models
Successful drug design requires a better understanding of the effect of the drug on the studied disease at the molecular level. Before the investigation of protein–ligand interactions, we aimed to filter out compounds using ligand-based approaches. For this aim, a binary QSAR-based platform called MetaCore/MetaDrug (MC/MD) from Clarivate Analytics (https://portal.genego.com) was used. MC/MD estimates the predicted values of therapeutic activity, pharmacokinetics, and absorption, distribution, metabolism, and excretion (ADME)/toxicity results for the investigated compounds using binary QSAR models. In MC/MD, the predicted TAVs are normalized to 0 and 1 (i.e., while 0 shows inactive prediction, 1 indicates active molecules). The details of MC/MD were explained in our previous papers.20,24 More than 210 000 ligands used from Specs-SC small-compound library were prepared in a text file (IUPAC format) by MarvinSketch.25 This text file was scanned to determine indol-based structures using an in-house Python-based text-mining script, and selected compounds were screened in the MC/MD “cancer QSAR” model. The identified active compounds by MC/MD were further filtered with 26 toxicity QSAR models.
The cancer QSAR model used has following statistical results: training set, 886; test set, 167; sensitivity, 0.89; specificity, 0.83; accuracy, 0.86; MCC, 0.72. Finally, we identified 288 compounds that show high therapeutic activity against cancer and without any toxicity prediction.
Ligand Preparation
Identified compounds were prepared with the OPLS-2005 force field26 using the LigPrep module27 of the Maestro Molecular Modeling Suit. The Epik module28 has been used at the physiological pH for the prediction of ionization states. In ligand preparation, all possible stereoisomers/tautomers for each ligand were also produced.
Protein Preparation
The structure of the BCL-2 protein was retrieved from RCSB Protein Data Bank (PDB ID 4LXD).29 The Prime module30 of Maestro was used in the determination and fixing of missing side chains or backbones and loops. Water molecules around the cocrystallized compound within 5.0 Å were used in simulations, and others were removed. The PROPKA and OPLS2005 force field were used for ionization states and structural optimization, respectively.31
Molecular Docking Calculations
Glide, which is a grid-based docking program, was used to perform the molecular docking studies.32 The Glide scoring function33 was used in the calculations of docking scores of each screened compound at the BCL-2 target. The binding pocket residues were considered by residues within a 10.0 Å vicinity of the cocrystallized ligand. All compounds were screened at the active site of the BCL-2 using Glide/SP (standard precision)34,35 protocol, and 100 docking poses were requested for each tested compound.36,37
MD Simulations
MD simulations were employed for the complexes retrieved from the top docking poses, and up to 100 ns of production time was carried out by Desmond version 4.9 to analyze the time-dependent changes of structural and dynamical properties.38 Protein–ligand complex structures were placed in periodic orthorhombic boxes including the TIP3P water model,39 and the box sizes “buffer size” option was set to 10.0 Å in all three directions.40 To neutralize systems, Na+/Cl– ions were used, and to mimic the physiological conditions, 0.15 M NaCl solution was added. Simulations were conducted in a constant temperature and pressure environment (NPT ensemble). A 2.0 fs time step was used in the integration steps. A Nose–Hoover thermostat41 and Martyna–Tobias–Klein methods42 were used for constant temperature and pressure of the constructed simulations boxes at 310 K and 1 bar, correspondingly.
Molecular Mechanics/Generalized Born Surface Area (MM/GBSA)
Complexes of protein/ligand trajectories during the MD simulations were investigated by MM/GBSA to predict the binding free energies of screened hit compounds. The MM/GBSA studies were carried out by Schrodinger’s Prime module.43 The trajectories throughout the MD simulations were extracted at every 10 ps.30 A VSGB solvation model44 and OPLS-2005 force field26 were used for the refinement of poses within 3.0 Å from the ligand.
E-Pharmacophore-Based Database Screening
Ligand-based and target-based methodologies are important approaches in in silico drug design and development studies. In the e-pharmacophore-based database screening approach, the advantages of ligand-based and target-driven based approaches were combined for a fast screening of a large database of compounds.45,46 With the e-pharmacophore approach, pharmacophore sites can be obtained from available solved protein–ligand complexes. Pharmacophore groups can be identified from the bioactive conformer of a cocrystallized compound preserving a maximum of seven pharmacophore features as default. In this study, we used a well-known BCL-2 inhibitor Navitoclax analog. Hydrogen-bond acceptor (A), hydrogen-bond donor (D), aromatic ring (R), positive ionizable (P), and negative ionizable (N) are the pharmacophore properties in PHASE pharmacophore modeling. The e-pharmacophore hypothesis can be generated using a potent compound at the binding pocket of the target protein. Using the e-pharmacophore approach, pharmacophore sites were generated based on the conformation of Navitoclax analog in the BCL-2 active site (i.e., cocrystallized ligand at the 4LXD PDB file). Pharmacophore hypothesis features were found to be hydrogen-bond acceptor (A), hydrophobic group (H), and three aromatic rings (R), (i.e., a five-site AHRRR hypothesis).
Assessment of BCL-2 Inhibition by TR-FRET
The inhibition of BCL-2 protein was measured in the presence of its ligands and inhibitory molecules by using the BCL-2 TR-FRET Assay kit (BPS Biosciences, catalog no. 50222). The assay protocol contains a sample including a terbium-labeled donor, dye-labeled acceptor, peptide ligand, and BCL-2 protein, and each inhibitor was incubated for 2 h. All samples and controls were worked in triplicate; the tested solution concentrations for the selected hit compounds were 1 × 10–9, 1 × 10–8, 1 × 10–7, 1 × 10–6, 1 × 10–5, and 1 × 10–4 M.
Fluorescence intensity was read at two different wavelengths (i.e., donor and acceptors emissions at 620 and 665 nm, respectively), and each fluorescent read was excited at 344 nm. Data analysis was conducted by the TR-FRET ratio (665 nm emission/620 nm emission). The percent inhibitory activity of studied hit compounds was computed by the following formula:
FRETs, FRETneg, and FRETp are sample FRET, negative control FRET, and positive control FRET, respectively. The IC50 value of each studied hit molecule was calculated by the TR-FRET BCL-2 binding inhibition assay. Absorbance and IC50 values were measured by dose–response–inhibition plots and nonlinear regression analysis on Graphpad Prism v.8.
Cell Culture Experiments and MTT Analysis
Colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was used to assess in vitro activity of inhibitory molecules on U87-MG glial tumor cells. U87-MG glioma cells were chosen to study BCL-2 inhibition due to their intrinsically high level of BCL-2 gene expression under normal conditions. Cells were cultured in DMEM medium (Biosera) supplemented with 10% FBS (Gibco) and 1X penicillin/streptomycin (Multicell). Since higher plate confluence levels would slow cell proliferation regardless of molecular therapy, we determined the number of cells is seeded to ensure that none of the cells reached more than 60% confluence level during the treatment. Molecules showing 50% and above inhibitory activity on TR-FRET assay were selected for cell proliferation experiments. Glial cells were seeded at a density of 5 × 102 cells into each well of 96-well cell culture plates and incubated at 37 °C in a humidified atmosphere with 5% of CO2. Molecules were obtained in powder format and dissolved in 1% DMSO. First, stock solutions of molecules were prepared at a 4 mM stock solution concentration and diluted to the studied concentrations by using DMEM cell media. Each molecule had different IC50 concentrations specified by TR-FRET inhibitory assay in the range of 1 nM to 100 μM. For in vitro assays on cell culture experiments, two concentrations of molecules (i.e., low and high concentrations) were tested, one log above and one log below the concentration of IC50 value. Cells were treated with molecules following 24 h of preincubation. At each 24 h interval, treated and untreated cells were subjected to 10 μL of 5 mg/mL MTT in 1X PBS and incubated at 37 °C for 2–4 h. Then, formazan crystals were solubilized with 100 μL of HCl-SDS solubilization buffer, and the absorbance was measured at 570 nm on a microplate reader (HIDEX Multimode reader). MTT experiments were carried out for 120 h with or without treatment, and all experiments were replicated at least three times.
Identification of Compounds Using Hybrid Virtual Screening Algorithm
This work aimed to conduct a virtual screening workflow for a small-molecule database (i.e., Specs-SC small-molecule library) that includes around 210 000 available ligands targeting the BCL-2 and to identify new pharmacophore groups. It is well-known that indoles and indole derivatives are crucial compounds that have been used as therapeutic compounds in many different biological problems, including cancer. Thus, in this study, to decrease the large database to a manageable number with specified fragments, a text-mining study was initially employed. Hence, all molecules used from Specs-SC library in two-dimensional (2D, .sdf file format) were converted to a IUPAC text file (.name format) with the program MarvinSketch.25 Selected compounds containing the “indol” expression (i.e., indoles, indole derivatives, isoindoles, indolons, etc.) in the IUPAC names were obtained. A total of 1735 molecules that showed higher predicted therapeutic activity values than the selected cutoff value (0.5) were subjected to toxicity tests for 26 different toxicity QSAR models. Thus, 288 nontoxic compounds with potent activity against cancer were selected from the database. The 2D molecular structures of the 288 selected molecules were prepared and converted to their corresponding low energy 3D conformers. After this process, the total number of molecules increased to 472 due to different protonation states, and stereochemistry.
Molecular Docking
To identify potent ligands against BCL-2, a grid-based docking program (Glide) was used. Before the screening of compounds from the library, the docking program is validated with the known binding pose of cocrystallized ligand. Thus, the cocrystallized navitoclax analog (4LXD) was docked with the used docking protocol Glide/SP, and RMSD of the cocrystallized pose with the docking pose was calculated as 1.86 Å, although the ligand has quite flexible fragments (Figure S1). The top docking-scored pose for each ligand was used in in MD simulations. The screening algorithm carried out for identifying the novel hits against BCL-2 was summarized as a flowchart in Figure S2.
MD Simulations
MD simulations were initiated for the top recorded docking pose for each of the 288 molecules and 2 known BCL-2 inhibitors (venetoclax and navitoclax analog). Initially, short (1 ns) simulations were employed for a total of 288 hit molecules and 2 approved BCL-2 inhibitors at the target binding site, and the resulting coordinates were collected separately in the trajectory files. Table S1 represents the docking scores of the 288 compounds.
The average binding free energies and Z scores were calculated using MM/GBSA method and these values were compared with positive control compounds. 25 ns MD simulations were carried out and average MM/GBSA binding free energies were recalculated for selected 41 hits and two reference molecules by Prime. Based on calculated average MM/GBSA scores throughout the MD simulations, 17 selected hit compounds were forwarded to longer (100 ns) MD simulations. These MD simulations of 100 ns were carried out for selected 17 hits and 2 positive controls, and on the basis of their corresponding MM/GBSA scores, 10 molecules were considered for further analysis (Figure S3).
To get an initial insight into the role of identified putative inhibitor hits on the BCL2 target, we structurally investigated each compound at the binding site to predict the impact of the ligand on the structure and dynamics of the BCL-2. For this aim, we compared the local interactions of the binding pocket residues with the ligand. In Figure S4, RMSD plots of 10 selected hits based on GBSA scores were provided. RMSD–time plots show that most of the studied systems have small structural changes based on the initial backbone coordinates of the target protein. Mol6 showed the highest increase in protein’s backbone RMSD throughout the simulations as shown in Figure S4. The average Protein RMSD value of the 10 selected hit molecules is about 4.0 Å (except Mol6). All complex systems structurally stable after 20 ns MD simulations.
Table 1 shows the molecular docking scores and average MM/GBSA scores of the studied hit compounds as well as reference molecules. Ligand RMSDs were also conducted along with the protein RMSD plots. Figure S5 represents the LigFitProt (i.e., change of ligand RMSDs based on protein backbone, thus a translational motion of the ligand during the simulations) plot of the 10 hits as well as reference known BCL-2 inhibitors. Table S2 shows the corresponding mean RMSD values. FDA-approved venetoclax and navitoclax analog’s LigFitProt average RMSD results were 2.051 and 3.358 Å, respectively. The average LigFitProt RMSD values of the selected hit compounds (i.e., Mol45, Mol126, Mol136, and Mol146) were high, indicating that these compounds have large translational motions at the binding pocket throughout the simulations. However, other hits have average LigFitProt RMSD values similar to those of the positive control compounds.
Table 1. 2D Structures, MM/GBSA Average Scores, MetaCore/MetaDrug-Predicted Therapeutic Activity (Th. Act.) Values and Docking Scores in Glide/SP of Selected 10 Hits from Target-Driven Based Approach and FDA-Approved Drugs Venetoclax and Navitoclax Analog.

The internal motions of the ligands throughout the simulations were also investigated using the LigFitLig RMSD plots (Figure S6 and Table S2). The average LigFitLig RMSDs of all of studied the molecules (except for Mol146) were less than 2.5 Å, indicating that the molecules did not make a significant internal rotational motion at the binding pocket.
During simulations, to measure the effect of the selected compounds on the structural/dynamical properties of the target protein, root-mean-square fluctuations (RMSF) values were also calculated. To observe the fluctuation regions of the target structure, RMSF of peptide backbone atoms of each amino acid residue in the complex analyzes was employed. High RMSF values indicate highly mobile fluctuating regions of the target protein examined during MD simulations (i.e., loop regions (residue numbers 30–45 region)). The backbone RMSF plot of protein when complexed with the identified hit compounds and positive controls throughout the MD simulations is shown in Figure S7. Residue numbers 30–45 have higher fluctuations in the Mol87 and Mol6.
E-Pharmacophore Approach
Virtual screening (VS) is a commonly used approach for the identification of novel therapeutic compounds for different biological problems. Ripphausen et al.47 have carried out a comprehensive literature survey of prospective VS applications. In this survey, it is found that there are around 3 times more target-driven approaches in VS studies compared to ligand-based approaches (i.e., QSAR).47 However, when the success rates are compared it is found that ligand-based techniques are more successful approaches for the correct identification of potent compounds.47 Thus, as a significant and successful screening strategy, in our current VS study strategy, the e-pharmacophore approach was also carried out and considered in the selection of hit compounds for the in vitro studies.
The crystal structure of target protein (4LXD) in complex with the well-known BCL-2 inhibitor navitoclax analog was used to produce the e-pharmacophore hypothesis. Then the e-pharmacophore was carried out to screen the small-molecule data set obtained from Specs-SC. Energy-optimized pharmacophores were found by mapping the energetic terms from the scoring feature of Glide’s extra precision (XP) protocol to atom centers. Next, pharmacophore sites were produced, and the Glide/XP energies were summarized from the atoms comprising each pharmacophore site. The putative sites were then ranked based on these calculated energies and the most desired regions were selected for the pharmacophore hypothesis.48
The initial number for the generation of pharmacophore sites was set to default (7 sites); however, as shown in Figure 1, a five-site pharmacophore hypothesis was generated. The final hypothesis consists of a hydrophobic group (H), three aromatic rings (R), and hydrogen bond acceptor group (A): five-site AHRRR hypothesis. The database of Specs-SC compounds was screened over this developed pharmacophore hypothesis. On the basis of alignment scores of each compound with this five-site pharmacophore model, as well as their corresponding Glide/XP docking scores, five compounds were selected for future studies. Longer (100 ns) MD simulations were carried out on the initial poses of the BCL-2/ligand complexes obtained from the docking calculations to investigate the structural stability of the identified hits at the binding pocket of the target protein within the nanosecond time scale. Table 2 shows docking scores and average MM/GBSA scores of identified 5 hits from e-pharmacophore modeling throughout the 100 ns MD simulations.
Figure 1.
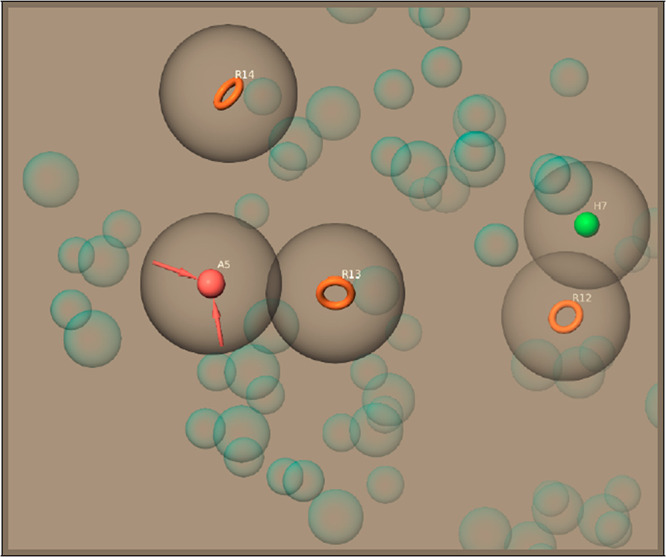
Hypothesis generated by e-pharmacophore method. Pharmacophore sites generated from crystal Protein Data Bank structure (4LXD) and well-known BCL-2 inhibitor navitoclax analogue preserving a maximum of seven pharmacophore features as default. A five-pharmacophore site hypothesis was generated.
Table 2. Specs ID, Molecule Number, 2D Structure, Docking Scores and MM/GBSA Average Scores of Identified 5 Compounds with E-Pharmacophore Hypothesis.

Ligand-Based and Structure-Based Virtual Screening
Some studies in the literature directly compare the success-rate results between pharmacophore-based virtual screening (PBVS) and docking-based virtual screening (DBVS). Steindl et al.49 compared the success rates of ligand-based and structure-based approaches for the identification of human Rhinovirus (HRV) coat protein inhibitors. They concluded that both screening procedures are valuable for the target of the HRV coat protein, which shows that the assets in the resulting hit list are enriched. In another study, Chen et al.50 compared the efficiency of the two VS approaches, namely, the PBVS and the DBVS approaches,51,52 and concluded that the PBVS method in the correct identification of actives from a set of compounds at the library is a more powerful technique compared to DBVS.50−58 The PBVS method is also used for postfiltering compounds selected through docking approaches.59 The work of Muthas et al.60 has shown that the final filtration with pharmacophores in their investigated targets improves enrichment rates compared to the docking-based approach alone. The pharmacophore-based screening technique is among the most effective tools that match the similarity of the 3D interaction pattern or pharmacophore model of druglike compounds in complexes with known ligands.61−65 Aboalhaija and Taha66 used a combination of pharmacophore modeling and QSAR studies to search the structural features required for potent BCL-2 inhibitors. They virtually screened the NCI chemical database and identified seven hit compounds against BCL-2. The in vitro results of these seven compounds leading to IC50 values between 2.6 and 38.7 μM.66 Wen et al.67 carried out QSAR-based virtual screening to identify potent BCL-2 specific ligands from the Specs-SC. They employed Random Forest (RF) classification and regression models for the identification of hit compounds and selected 12 compounds.67 The interactions of the selected compounds with BCL-2 were examined by surface plasmon resonance (SPR) binding assay, and 8 out of 12 compounds could directly bind to the BCL-2. However, only one compound showed significant cytotoxic effects on breast cancer cells (IC50, 5 μM).67 Yoou et al.66 used the protein chip method for the identification of fragment hits against BCL-2. They employed docking calculations for the 12 identified fragment hits.68 Using the molecular-docking-guided protein chip screening system, they derived a virtual compound with an important scaffold feature for the interaction of crucial residues at the BCL-2. Then, they tested the anticancer activity of 26 compounds that have similar structural scaffold features. Biological test results showed that three of these compounds have desired IC50 values ranging from 4.38 to 5.74 μM.68 Ramos et al.69 employed a high-throughput virtual screening study for the ZINC database that includes 642759 drug-like compounds. They screened these compounds against BCL-2 by docking calculations using AutoDock, and the top 1000 compounds were further screened at CDRUG which is a molecular fingerprint server for the prediction of potent anticancer compounds. Finally, they proposed five hit compounds.69
TR-FRET Analysis Confirms the BCL-2 Inhibition of Identified Compounds
Because of the advantages and highlighted high success rates of structure-based e-pharmacophore modeling, selected hit compounds by using this approach were used for their biological activities on cancer cells. TR-FRET analysis results showed inhibitory activity against the cancer cells for the following selected hits which showed at least 50% and above inhibitions at various molecule concentrations: AG-205/11944207, AM-879/14774006, AK-918/41759663, and AH-487/40936997. The performed TR-FRET assay represents a high success rate (i.e., 80%) of the selected hits which are obtained by the virtual screening of small molecule ligand database on the constructed e-pharmacophore model. IC50 values for tested molecules were listed in Table 3. The inhibitory concentrations ranged between 153.3 μM to 19 nM. Corresponding enzyme activity inhibition percentage values were changing between 55.61 and 95.57% at 100 μM (Table 3). Percent inhibitions of the selected active compounds were shown in Figure 2.
Table 3. IC50 and Percent Inhibition Values of Tested Compounds Obtained from In Silico Guided Virtual Screening Approaches.
| specs ID | IC50 value (μM) | percent inhibition (%) at 100 μM |
|---|---|---|
| AK-918/41759663 | 153.30 | 95.57 |
| AG-205/11944207 | 0.019 | 55.61 |
| AM-879/14774006 | 54.90 | 77.49 |
| AH-487/40936997 | 6.27 | 78.94 |
| AH-487/41490431 | NA | 36.11 |
Figure 2.

TR-FRET analysis confirms the inhibitory activity of identified hit molecules on BCL-2. The X-axis shows tested concentrations ranging 1 nM to 100 μM; the Y-axis shows the % inhibitory activity relative to samples without inhibitory molecules.
Inhibitory Molecules Restricted the Proliferation of U87-MG Glioma Cells
Selected hit molecules were evaluated in the U87-MG cell line following the determination of inhibitory concentration via TR-FRET analysis. On the basis of IC50 values, the acceptable inhibitory range was determined as high and low concentrations for each selected compound as detailed in the “Materials and Methods” section. High and low concentrations were 200 and 100 μM, 100 and 10 μM, 100 and 10 μM, and 10 and 1 μM for compounds AK-918/41759663, AG-205/11944207, AM-879/14774006, and AH-487/40936997, respectively.
Four selected active compounds were shown to be significantly effective on glioma cells by inhibiting and/or reducing cell proliferation. AK-918/41759663 and AG-205/11944207 showed the maximum inhibition effect by reducing the cell viability to less than 10% within 5 days. AM-879/14774006 also showed a significant reduction in cell viability. However, other selected inhibitory compound (AH-487/40936997) did not significantly reduce cell viability in less than 50% of cells. Only AK-918/41759663 showed effective cell proliferation reduction at both high (200 μM) and low (100 μM) concentrations. AG-205/11944207 and AM-879/14774006 have high efficacy at a high concentration (100 μM) but not at a low (10 μM) concentration, as expected. The cell viability of tested concentrations was shown in Figure 3. The promising active compound AH-487/40936997 (Mol2) based on their inhibitory potential shown by TR-FRET analysis, did not stably reduce cell proliferation. We suppose that inhibitor activity is correlated with a solubility of the compounds since inactive ligands were found to be partially soluble molecules in DMSO.
Figure 3.

MTT cell proliferation assay. Molecules having inhibition activity on U87-MG glioma cells were shown. Tested concentrations for each molecule were different (low and high concentrations) as determined in TR-FRET assay. Graphs were determined by comparing each treatment group with untreated control (without molecules), and error bars show the standard deviation.
Additionally, morphological changes within 4 days after treatment supported the quantitative results by massive cell detachments and the formation of apoptotic bodies under the microscope (Figure 4). To determine the dying cells, changes in the formation of tubular-shaped glial cells turning into a circular structure were observed.
Figure 4.

Microscopic evaluation of U87-MG cells for compounds AK-918/41759663, AG-205/11944207, AM-879/14774006, and AH-487/40936997. Cells were photographed and observed under microscope for 96 h. The DMSO group showed neat proliferation of cells as did the untreated group. Compound AK-918/41759663 showed a clear decrease starting from the first day at both concentrations (100 and 200 μM), while the effects of AG-205/11944207 and AM-879/14774006 on cell population are visible only at high concentrations above IC50 values (100 μM).
Although AH-487/40936997 showed a decreased effect on cell proliferation when compared with untreated cells, it exhibited no preventive impact on the U87-MG cell line. Hence, experimental results altogether suggest that three of the tested BCL-2 inhibitor compounds that showed an ability to bind to BCL-2 decreased cell proliferation and induced apoptotic cell death.
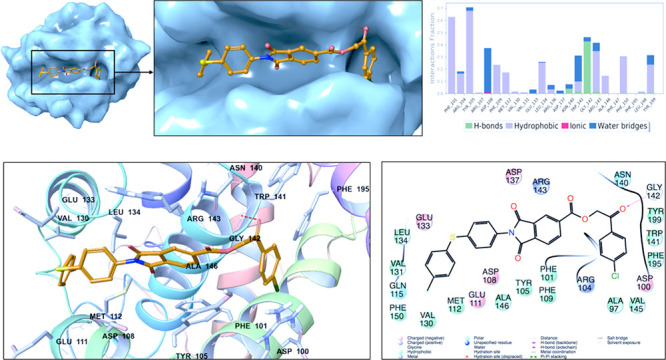
2D and 3D ligand interaction plots of the identified novel compounds at the catalytic site of BCL-2 are investigated in detail. Representative structures from MD simulations were used in 2D and 3D ligand interaction diagrams. Figure 5 displays the binding mode of Mol4 (AK-918/41759663) at the BCL-2 target protein. The important nonbonded chemical interactions between ligand and protein such as the hydrogen bond constructed with the GLY142 backbone (also observed in venetoclax) are conserved in the identified hit molecule Mol4. The conserved critical amino acids of BCL2 in the drug binding between Mol4 and venetoclax are also observed in PHE101, TYR105, and TYR199 which form hydrophobic interactions.
Figure 5.

(A) Representation of the binding mode of Mol-4 (Specs-SC ID AK-918/41759663) on the BCL-2 surface. (B) Ligand interaction diagram of Mol-4 throughout 100 ns molecular dynamics simulations. (C) Binding mode of Mol-4 in the active pocket of BCL-2. The protein is depicted as ribbons, and the compound is shown as sticks. (D) 2D ligand interactions with BCL-2 protein amino acid residues after MD simulations.
The statistical analysis for the protein–ligand contacts was conducted for the collected trajectories during each simulation, and it is found that the crucial amino acids (i.e., ARG143 and ASP108) which are needed for effective BCL2 inhibitors are mostly conserved throughout the MD simulations (Figure 5). Identified hit compound Mol1 (Specs-SC ID: AG-205/11944207) has mainly hydrophobic and hydrogen-bonding interactions with binding pocket residues. The interacting residues include PHE101, TYR105, TYR199, and ALA97 which form hydrophobic interactions (Figure 6). These residues have been also known to interact with BCL-2 for the FDA-approved drug venetoclax. Another crucial interaction was the hydrogen bond constructed with GLY142, which was also observed in venetoclax. The interaction of binding pocket residues ARG143 (forming hydrophobic and water bridge interactions), ASN140 (forming a hydrogen bond) and ARG104 (forming hydrophobic and water bridge interactions) were conserved during the MD simulations with Mol1. The ligand interaction diagrams of Mol2 (Specs-SC ID AH-487/40936997) and Mol5 (Specs-SC ID AM-879/14774006) which have high activity were also shown in Figures S8 and S9.
Figure 6.

(A) Representation of the binding mode of Mol-1 (Specs-SC ID AG-205/11944207) on the BCL-2 surface. (B) Ligand interaction diagram of Mol-1 throughout 100 ns molecular dynamics simulations. (C) Binding mode of Mol-1 in the active pocket of BCL-2. The protein is depicted as ribbons, and the compound is shown in sticks. (D) 2D ligand interactions with BCL-2 protein amino acid residues after MD simulations.
We used the latest state-of-the-art computational molecular modeling and dynamics approaches, which include advanced integrated text mining, virtual screening, and hybrid molecular modeling techniques, combined with in vitro binding and cell culture studies to identify the small-molecule inhibitors targeted to the BCL-2 binding site. On the basis of docking scores and interaction diagrams throughout the 100 ns MD simulations, 15 hit compounds (with high therapeutic activity and no toxicity) were identified. TR-FRET experiment results represented that hit compounds identified by the e-pharmacophore modeling has a high power for the correct identification of bioactive ligands from a large ligand database. The selected ligands were further evaluated in the U87-MG cell line, and among them three compounds were shown to be significantly effective on glioma cells by inhibiting and/or reducing cell proliferation.
While 5 compounds identified by the e-pharmacophore model screening were considered for the TR-FRET analysis, 4 of them were found active and among them 3 hits gave promising inhibitory activity results on glioma cells. Altogether, the success rate of the e-pharmacophore based method was 60%. This interesting result shows that although screening speed is much higher compared to the docking/MD-initiated method, since the success rate of e-pharmacophore-based screening is high; this method can be considered for the virtual screening of libraries in different biological problems.
MTT cell proliferation results showed that Mol4 (AK-918/41759663) and Mol1 (AG-205/11944207) showed the maximum inhibition effect. These compounds have IC50 values of 153.3 μM and 19 nM, respectively. AM-879/14774006 (Mol-5) also showed significant inhibition.
Results showed that the following residues are crucial for ligand binding: PHE101, TYR105, TYR199, and GLY142. Several hydrophobic interactions dominate the interactions. These crucial interactions are also observed in the FDA-approved drug venetoclax. When the scaffolds of active compounds were investigated it can be seen that they include following fragments: isoindolinecarboxylate, benzothiazole, and benzimidazole groups.
Consideration of these fingerprints together with indole analogs within the scaffolds of the designing of new compounds may improve activity against BCL-2. The identification of potent and safe small molecules as potential inhibitors of BCL-2 is a step closer to finding appropriate effective therapies for cancer. Our lead ligands identified from in silico guided screening can be used as a scaffold for further structural optimization and development, enabling further research in this promising field.
Acknowledgments
We thank Sena Gizem Suer for her help with the cell culture studies and the preparation of cell images. This study was supported by Bahçeşehir University, Scientific Research Projects Unit, Project No: BAP.2019-02.10.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00210.
Alignment of conformation of cocrystallized ligand obtained by X-ray and molecular docking, applied virtual screening workflow, MM/GBSA binding free energy analysis for the studied molecules at the binding pocket of BCL-2 throughout the MD simulations, protein RMSD graph of the Cα atoms of the BCL-2 throughout 100 ns MD simulations, LigFitProt and LigFitLig RMSD graphs for the hits and reference compounds, RMSF graphs for the 10 hits and reference compounds, 2D ligand interaction diagrams of Mol-2 and Mol-5, predicted docking scores for the 288 molecules, and average RMSD values of 10 hits and 2 known BCL2 inhibitors throughout the 100 ns MD simulations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hanahan D.; Weinberg R. A. (2011) Hallmarks of cancer: The next generation. Cell 144 (5), 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Siegel R. L.; Miller K. D.; Jemal A. (2015) Cancer statistics, 2015. Ca-Cancer J. Clin. 65 (1), 5–29. 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- Miller K. D.; Siegel R. L.; Lin C. C.; Mariotto A. B.; Kramer J. L.; Rowland J. H.; Stein K. D.; Alteri R.; Jemal A. (2016) Cancer treatment and survivorship statistics, 2016. Ca-Cancer J. Clin. 66 (4), 271–289. 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- Padma V. V. (2015) An overview of targeted cancer therapy. Biomed. 5 (4), 19. 10.7603/s40681-015-0019-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wust P.; Hildebrandt B.; Sreenivasa G.; Rau B.; Gellermann J.; Riess H.; Felix R.; Schlag P. (2002) Hyperthermia in combined treatment of cancer. Lancet Oncol. 3 (8), 487–497. 10.1016/S1470-2045(02)00818-5. [DOI] [PubMed] [Google Scholar]
- Upadhyaya M.; Spurlock G.; Majounie E.; Griffiths S.; Forrester N.; Baser M.; Huson S. M.; Gareth Evans D.; Ferner R. (2006) The heterogeneous nature of germline mutations in NF1 patients with malignant peripheral serve sheath tumours (MPNSTs). Hum. Mutat. 27 (7), 716. 10.1002/humu.9429. [DOI] [PubMed] [Google Scholar]
- Hasinoff B. B.; Aoyama R. G. (1999) Relative plasma levels of the cardioprotective drug dexrazoxane and its two active ring-opened metabolites in the rat. Drug Metab. Dispos. 27 (2), 265–268. [PubMed] [Google Scholar]
- Fracasso P. M.; Westerveldt P.; Fears C. A.; Rosen D. M.; Zuhowski E. G.; Cazenave L. A.; Litchman M.; Egorin M. J. (2000) Phase I study of paclitaxel in combination with a multidrug resistance modulator, PSC 833 (valspodar), in refractory malignancies. J. Clin. Oncol. 18 (5), 1124–1134. 10.1200/JCO.2000.18.5.1124. [DOI] [PubMed] [Google Scholar]
- Krishna R.; Mayer L. D. (1997) Liposomal doxorubicin circumvents PSC 833-free drug interactions, resulting in effective therapy of multidrug-resistant solid tumors. Cancer Res. 57 (23), 5246–5253. [PubMed] [Google Scholar]
- DeSantis C. E.; Lin C. C.; Mariotto A. B.; Siegel R. L.; Stein K. D.; Kramer J. L.; Alteri R.; Robbins A. S.; Jemal A. (2014) Cancer treatment and survivorship statistics, 2014. Ca-Cancer J. Clin. 64 (4), 252–271. 10.3322/caac.21235. [DOI] [PubMed] [Google Scholar]
- Scripture C. D.; Figg W. D. (2006) Drug interactions in cancer therapy. Nat. Rev. Cancer 6 (7), 546–558. 10.1038/nrc1887. [DOI] [PubMed] [Google Scholar]
- Adams J. M.; Cory S. (2001) Life-or-death decisions by the Bcl-2 protein family. Trends Biochem. Sci. 26 (1), 61–66. 10.1016/S0968-0004(00)01740-0. [DOI] [PubMed] [Google Scholar]
- Adrain C.; Martin S. J. (2001) The mitochondrial apoptosome: A killer unleashed by the cytochrome seas. Trends Biochem. Sci. 26 (6), 390–397. 10.1016/S0968-0004(01)01844-8. [DOI] [PubMed] [Google Scholar]
- Spierings D. C.; De Vries E. G.; Vellenga E.; Van Den Heuvel F. A.; Koornstra J. J.; Wesseling J.; Hollema H.; De Jong S. (2004) Tissue distribution of the death ligand TRAIL and its receptors. J. Histochem. Cytochem. 52 (6), 821–831. 10.1369/jhc.3A6112.2004. [DOI] [PubMed] [Google Scholar]
- Danial N. N.; Korsmeyer S. J. (2004) Cell Death: Critical Control Points. Cell 116 (2), 205–219. 10.1016/S0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Vieira H. L. A.; Boya P.; Cohen I.; El Hamel C.; Haouzi D.; Druillenec S.; Belzacq A. S.; Brenner C.; Roques B.; Kroemer G. (2002) Cell permeable BH3-peptides overcome the cytoprotective effect of Bcl-2 and Bcl-XL. Oncogene 21 (13), 1963–1977. 10.1038/sj.onc.1205270. [DOI] [PubMed] [Google Scholar]
- Smaili S. S.; Hsu Y.-T.; Youle R. J.; Russell J. T. (2000) Mitochondria in Ca2+ signaling and apoptosis. J. Bioenerg. Biomembr. 32 (1), 35–46. 10.1023/A:1005508311495. [DOI] [PubMed] [Google Scholar]
- Tse C.; Shoemaker A. R.; Adickes J.; Anderson M. G.; Chen J.; Jin S.; Johnson E. F.; Marsh K. C.; Mitten M. J.; Nimmer P.; Roberts L.; Tahir S. K.; Xiao Y.; Yang X.; Zhang H.; Fesik S.; Rosenberg S. H.; Elmore S. W. (2008) ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 68 (9), 3421–3428. 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Tan N.; Malek M.; Zha J.; Yue P.; Kassees R.; Berry L.; Fairbrother W. J.; Sampath D.; Belmont L. D. (2011) Navitoclax enhances the efficacy of taxanes in non-small cell lung cancer models. Clin. Cancer Res. 17 (6), 1394–1404. 10.1158/1078-0432.CCR-10-2353. [DOI] [PubMed] [Google Scholar]
- Tutumlu G.; Dogan B.; Avsar T.; Orhan M. D.; Calis S.; Durdagi S. (2020) Integrating Ligand and Target-Driven Based Virtual Screening Approaches With in vitro Human Cell Line Models and Time-Resolved Fluorescence Resonance Energy Transfer Assay to Identify Novel Hit Compounds Against BCL-2. Front. Chem. 8, 167. 10.3389/fchem.2020.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadha N.; Silakari O. (2017) Indoles as therapeutics of interest in medicinal chemistry: Bird’s eye view. Eur. J. Med. Chem. 134, 159–184. 10.1016/j.ejmech.2017.04.003. [DOI] [PubMed] [Google Scholar]
- Keri R. S.; Patil M. R.; Patil S. A.; Budagumpi S. (2015) A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur. J. Med. Chem. 89, 207–251. 10.1016/j.ejmech.2014.10.059. [DOI] [PubMed] [Google Scholar]
- Tahlan S.; Kumar S.; Kakkar S.; Narasimhan B. (2019) Benzimidazole scaffolds as promising antiproliferative agents: A review. BMC Chem. 13, 66. 10.1186/s13065-019-0579-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin K.; Durdagi S. (2021) Identifying new piperazine-based PARP1 inhibitors using text mining and integrated molecular modeling approaches. J. Biomol. Struct. Dyn. 39 (2), 681–690. 10.1080/07391102.2020.1715262. [DOI] [PubMed] [Google Scholar]
- ChemAxon . (2018) Marvinsketch 18.30.0.
- Banks J. L.; Beard H. S.; Cao Y.; Cho A. E.; Damm W.; Farid R.; Felts A. K.; Halgren T. A.; Mainz D. T.; Maple J. R.; Murphy R.; Philipp D. M.; Repasky M. P.; Zhang L. Y.; Berne B. J.; Friesner R. A.; Gallicchio E.; Levy R. M. (2005) Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 26 (16), 1752–1780. 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarala K. K.; Jamil K.; Devaraji V. (2014) Structure and putative signaling mechanism of protease activated receptor (PAR2) - A promising target for breast cancer. J. Mol. Graphics Modell. 53, 179–199. 10.1016/j.jmgm.2014.07.012. [DOI] [PubMed] [Google Scholar]
- Shelley J. C.; Cholleti A.; Frye L. L.; Greenwood J. R.; Timlin M. R.; Uchimaya M. (2007) Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 21 (12), 681–691. 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- Souers A. J.; Leverson J. D.; Boghaert E. R.; Ackler S. L.; Catron N. D.; Chen J.; Dayton B. D.; Ding H.; Enschede S. H.; Fairbrother W. J.; Huang D. C. S.; Hymowitz S. G.; Jin S.; Khaw S. L.; Kovar P. J.; Lam L. T.; Lee J.; Maecker H. L.; Marsh K. C.; Mason K. D.; Mitten M. J.; Nimmer P. M.; Oleksijew A.; Park C. H.; Park C. M.; Phillips D. C.; Roberts A. W.; Sampath D.; Seymour J. F.; Smith M. L.; Sullivan G. M.; Tahir S. K.; Tse C.; Wendt M. D.; Xiao Y.; Xue J. C.; Zhang H.; Humerickhouse R. A.; Rosenberg S. H.; Elmore S. W. (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19 (2), 202–208. 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Tripathi S. K.; Muttineni R.; Singh S. K. (2013) Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J. Theor. Biol. 334, 87–100. 10.1016/j.jtbi.2013.05.014. [DOI] [PubMed] [Google Scholar]
- (2011) Protein Preparation, version 2.5, Epik, Schrödinger, LLC, New York. [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. (2004) Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 47 (7), 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Raha K.; Merz K. M. (2004) A Quantum Mechanics-Based Scoring Function: Study of Zinc Ion-Mediated Ligand Binding. J. Am. Chem. Soc. 126 (4), 1020–1021. 10.1021/ja038496i. [DOI] [PubMed] [Google Scholar]
- Binkowski T. A.; Jiang W.; Roux B.; Anderson W. F.; Joachimiak A. (2014) Virtual high-throughput ligand screening. Methods Mol. Biol. 1140, 251–261. 10.1007/978-1-4939-0354-2_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X.-Y.; Zhang H.-X.; Mezei M.; Cui M. (2011) Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput.-Aided Drug Des. 7 (2), 146–157. 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subhani S.; Jayaraman A.; Jamil K. (2015) Homology modelling and molecular docking of MDR1 with chemotherapeutic agents in non-small cell lung cancer. Biomed. Pharmacother. 71, 37–45. 10.1016/j.biopha.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Roy S.; Kumar A.; Baig M. H.; Masařík M.; Provazník I. (2015) Virtual screening, ADMET profiling, molecular docking and dynamics approaches to search for potent selective natural molecules based inhibitors against metallothionein-III to study Alzheimer’s disease. Methods 83, 105–110. 10.1016/j.ymeth.2015.04.021. [DOI] [PubMed] [Google Scholar]
- (2011) Desmond, version 4.9, D. E. Shaw Research, New York. [Google Scholar]
- Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., and Hermans J. (1981) Interaction Models for Water in Relation to Protein Hydration, pp 331–342, Springer, Netherlands, Dordrecht. [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. (2006) Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49 (21), 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Hoover W. G. (1985) Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A: At., Mol., Opt. Phys. 31 (3), 1695–1697. 10.1103/PhysRevA.31.1695. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Tuckerman M. (2010) Constant pressure ab initio molecular dynamics with discrete variable representation basis sets. J. Chem. Phys. 133 (18), 184110. 10.1063/1.3499812. [DOI] [PubMed] [Google Scholar]
- Jacobson M. P.; Pincus D. L.; Rapp C. S.; Day T. J. F.; Honig B.; Shaw D. E.; Friesner R. A. (2004) A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins: Struct., Funct., Genet. 55 (2), 351–367. 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Li J.; Abel R.; Zhu K.; Cao Y.; Zhao S.; Friesner R. A. (2011) The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins: Struct., Funct., Genet. 79 (10), 2794–2812. 10.1002/prot.23106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salam N. K.; Nuti R.; Sherman W. (2009) Novel method for generating structure-based pharmacophores using energetic analysis. J. Chem. Inf. Model. 49 (10), 2356–2368. 10.1021/ci900212v. [DOI] [PubMed] [Google Scholar]
- Negi B.; Raj K. K.; Siddiqui S. M.; Ramachandran D.; Azam A.; Rawat D. S. (2014) In vitro antiamoebic activity evaluation and docking studies of metronidazole-triazole hybrids. ChemMedChem 9 (11), 2439–2444. 10.1002/cmdc.201402240. [DOI] [PubMed] [Google Scholar]
- Ripphausen P.; Nisius B.; Peltason L.; Bajorath J. (2010) Quo vadis, virtual screening? A comprehensive survey of prospective applications. J. Med. Chem. 53 (24), 8461–8467. 10.1021/jm101020z. [DOI] [PubMed] [Google Scholar]
- Salam N. K.; Nuti R.; Sherman W. (2009) Novel method for generating structure-based pharmacophores using energetic analysis. J. Chem. Inf. Model. 49 (10), 2356–2368. 10.1021/ci900212v. [DOI] [PubMed] [Google Scholar]
- Steindl T.; Langer T. (2005) Docking versus pharmacophore model generation: A comparison of high-throughput virtual screening strategies for the search of human rhinovirus coat protein inhibitors. QSAR Comb. Sci. 24, 470–479. 10.1002/qsar.200430929. [DOI] [Google Scholar]
- Chen Z.; Li H. L.; Zhang Q. J.; Bao X. G.; Yu K. Q.; Luo X. M.; Zhu W. L.; Jiang H. L. (2009) Pharmacophore-based virtual screening versus docking-based virtual screening: A benchmark comparison against eight targets. Acta Pharmacol. Sin. 30 (12), 1694–1708. 10.1038/aps.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Zhang K. Y. J. (2015) Hierarchical virtual screening approaches in small molecule drug discovery. Methods 71, 26–37. 10.1016/j.ymeth.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Kim D. (2012) Large-scale reverse docking profiles and their applications. BMC Bioinf. 13, S6. 10.1186/1471-2105-13-S17-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J. P.; Rees S. S.; Kalindjian S. B.; Philpott K. L. (2011) Principles of early drug discovery. Br. J. Pharmacol. 162 (6), 1239–1249. 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C. M.; Lim S. J.; Tong J. C. (2009) Recent advances in computer-aided drug design. Briefings Bioinf. 10 (5), 579–591. 10.1093/bib/bbp023. [DOI] [PubMed] [Google Scholar]
- Taft C. A.; da Silva V. B.; de Paula da Silva C. H. T. (2008) Current topics in computer-aided drug design. J. Pharm. Sci. 97 (3), 1089–1098. 10.1002/jps.21293. [DOI] [PubMed] [Google Scholar]
- Cummings M. D.; DesJarlais R. L.; Gibbs A. C.; Mohan V.; Jaeger E. P. (2005) Comparison of automated docking programs as virtual screening tools. J. Med. Chem. 48 (4), 962–976. 10.1021/jm049798d. [DOI] [PubMed] [Google Scholar]
- Kellenberger E.; Rodrigo J.; Muller P.; Rognan D. (2004) Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins: Struct., Funct., Genet. 57 (2), 225–242. 10.1002/prot.20149. [DOI] [PubMed] [Google Scholar]
- Warren G. L.; Andrews C. W.; Capelli A. M.; Clarke B.; LaLonde J.; Lambert M. H.; Lindvall M.; Nevins N.; Semus S. F.; Senger S.; Tedesco G.; Wall I. D.; Woolven J. M.; Peishoff C. E.; Head M. S. (2006) A critical assessment of docking programs and scoring functions. J. Med. Chem. 49 (20), 5912–5931. 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- McInnes C. (2007) Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 11 (5), 494–502. 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Muthas D.; Sabnis Y. A.; Lundborg M.; Karlén A. (2008) Is it possible to increase hit rates in structure-based virtual screening by pharmacophore filtering? An investigation of the advantages and pitfalls of post-filtering. J. Mol. Graphics Modell. 26 (8), 1237–1251. 10.1016/j.jmgm.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Braga R. C.; Andrade C. H. (2013) Assessing the Performance of 3D Pharmacophore Models in Virtual Screening: How Good are They?. Curr. Top. Med. Chem. 13 (9), 1127–1138. 10.2174/1568026611313090010. [DOI] [PubMed] [Google Scholar]
- Guner O.; Clement O.; Kurogi Y. (2004) Pharmacophore Modeling and Three Dimensional Database Searching for Drug Design Using Catalyst: Recent Advances. Curr. Med. Chem. 11 (22), 2991–3005. 10.2174/0929867043364036. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Langer T. (2005) LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 45 (1), 160–169. 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- Yang S. Y. (2010) Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discovery Today 15 (11–12), 444–450. 10.1016/j.drudis.2010.03.013. [DOI] [PubMed] [Google Scholar]
- Sanders M. P. A.; Barbosa A. J. M.; Zarzycka B.; Nicolaes G. A. F.; Klomp J. P. G.; De Vlieg J.; Del Rio A. (2012) Comparative analysis of pharmacophore screening tools. J. Chem. Inf. Model. 52 (6), 1607–1620. 10.1021/ci2005274. [DOI] [PubMed] [Google Scholar]
- Aboalhaija N. H.; Zihlif M. A.; Taha M. O. (2016) Discovery of new selective cytotoxic agents against Bcl-2 expressing cancer cells using ligand-based modeling. Chem.-Biol. Interact. 250, 12–26. 10.1016/j.cbi.2016.03.006. [DOI] [PubMed] [Google Scholar]
- Wen M.; Deng Z. K.; Jiang S. L.; Guan Y. Di; Wu H. Z.; Wang X. L.; Xiao S. S.; Zhang Y.; Yang J. M.; Cao D. S.; Cheng Y. (2019) Identification of a Novel Bcl-2 Inhibitor by Ligand-Based Screening and Investigation of Its Anti-cancer Effect on Human Breast Cancer Cells. Front. Pharmacol. 10, 391. 10.3389/fphar.2019.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoou M. S.; Cho S.; Choi Y. (2019) Molecular Docking-assisted Protein Chip Screening of Inhibitors for Bcl-2 Family Protein-protein Interaction to Discover Anticancer Agents by Fragment-based Approach. BioChip J. 13, 260–268. 10.1007/s13206-019-3306-4. [DOI] [Google Scholar]
- Ramos J.; Muthukumaran J.; Freire F.; Paquete-Ferreira J.; Otrelo-Cardoso A. R.; Svergun D.; Panjkovich A.; Santos-Silva T. (2019) Shedding light on the interaction of human anti-apoptotic Bcl-2 protein with ligands through biophysical and in silico studies. Int. J. Mol. Sci. 20, 860. 10.3390/ijms20040860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.