

Abstract
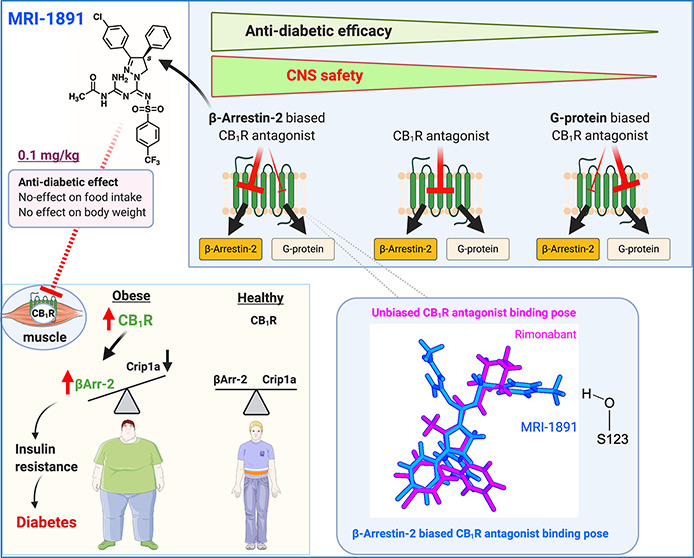
Seven-transmembrane receptors signal via G-protein- and β-arrestin-dependent pathways. We describe a peripheral CB1R antagonist (MRI-1891) highly biased toward inhibiting CB1R-induced β-arrestin-2 (βArr2) recruitment over G-protein activation. In obese wild-type and βArr2-knockout (KO) mice, MRI-1891 treatment reduces food intake and body weight without eliciting anxiety even at a high dose causing partial brain CB1R occupancy. By contrast, the unbiased global CB1R antagonist rimonabant elicits anxiety in both strains, indicating no βArr2 involvement. Interestingly, obesity-induced muscle insulin resistance is improved by MRI-1891 in wild-type but not in βArr2-KO mice. In C2C12 myoblasts, CB1R activation suppresses insulin-induced akt-2 phosphorylation, preventable by MRI-1891, βArr2 knockdown or overexpression of CB1R-interacting protein. MRI-1891, but not rimonabant, interacts with nonpolar residues on the N-terminal loop, including F108, and on transmembrane helix-1, including S123, a combination that facilitates βArr2 bias. Thus, CB1R promotes muscle insulin resistance via βArr2 signaling, selectively mitigated by a biased CB1R antagonist at reduced risk of central nervous system (CNS) side effects.
Keywords: biased antagonism, obesity, diabetes, insulin resistance, peripheral CB1R antagonist
The endocannabinoids anandamide (arachidonoyl ethanolamide, AEA) and 2-arachidonoyl glycerol (2-AG) are ubiquitous lipid mediators generated on demand from membrane phospholipid precursors in response to a rise in intracellular Ca2+ or metabotropic receptor activation. Endocannabinoids act on the same G-protein-coupled receptors (GPCR) that recognize the psychoactive ingredient of marijuana to produce a broad range of effects both in the brain and the periphery.1 The two main receptors involved are CB1 receptors (CB1R) that are highly expressed in the brain but also expressed at lower yet functional levels in most peripheral tissues, and CB2R, whose expression is more limited, primarily to cells of the immune and hematopoietic systems.1 The endocannabinoid/CB1R system (ECS) has emerged as a key regulator of lipid and carbohydrate metabolism.1 Activation of CB1R promotes energy conservation and inhibits energy expenditure, and an overactive ECS has been found to contribute to the development of visceral obesity and its metabolic consequences, commonly called the metabolic syndrome.2 Indeed, the CB1R antagonist rimonabant had shown promise as an antiobesity agent that also improved metabolic complications, including fatty liver, insulin resistance, and dyslipidemia,3,4 but ultimately, failed approval by the FDA due to unacceptable neuropsychiatric side effects.5 Dissociating therapeutic effects from unwanted side effects is a major challenge in drug development. In the case of the CB1R blockade, one way to achieve such separation is to limit the brain penetrance of the antagonist. In preclinical models of obesity/diabetes, such antagonists were found to be devoid of centrally mediated side effects while retaining metabolic efficacy.6,7 Another approach to selectively reduce side effects relies on biased signaling, as exemplified by μ-opioid agonists that do not recruit β-arrestin-2 to the receptor and, consequently, do not induce receptor internalization and the development of tolerance8 or respiratory depression,9 although the role of β-arrestin in opiate-induced respiratory depression and the functional selectivity of G-protein-biased μ-receptor agonists have recently been challenged.10 Another example is β-arrestin-biased angiotensin II receptor-1 agonists that do not increase blood pressure due to lack of G protein engagement but induce beneficial cardioprotective effects via β-arrestin signaling.9
CB1R signal mainly via Gi/o proteins, resulting in inhibition of adenylate cyclase and voltage-sensitive Ca2+ channels and activation of GIRK potassium channels and MAP kinases.11 Similar to other GPCRs, CB1R activation results in recruitment of β-arrestins, which not only can lead to receptor desensitization and internalization12 but also could contribute to CB1R signaling, such as the activation of p42/44 MAPK, which is partially mediated by β-arrestins.13 Although GPCR antagonists are more widely used than agonists as therapeutic agents, to date there has been no report of a biased GPCR antagonist,14 although a recently introduced dopamine D2 receptor ligand displayed D2R/β-arrestin antagonism and D2R/Giα agonism.15 In screening novel, peripherally restricted CB1R antagonist/inverse agonists, we identified a compound that is highly biased toward inhibiting CB1R-agonist-induced β-arrestin-2 recruitment compared to its ability to inhibit CB1R-agonist-induced GTPγS binding. We further show that CB1R in skeletal muscle signals via β-arrestin-2 to induce insulin resistance, whereas anxiety-like behaviors elicited by CB1R blockade in the brain are mediated entirely via G protein signaling. As a result, biased antagonism of CB1R signaling via β-arrestin-2 improves obesity-related insulin resistance without eliciting central nervous system (CNS)-mediated adverse behavioral effects.
Results
MRI-1891 Is a β-Arrestin-2-Biased Peripheral CB1R Antagonist
We modified the structure of the brain penetrant CB1R antagonist/inverse agonist ibipinabant16 in order to reduce its ability to cross the blood/brain barrier. The structurally modified compound (S)-MRI-1891 (referred to as MRI-1891) has increased total polar surface area and hydrogen bonding capacity relative to ibipinabant, predicting reduced brain penetrance (Figure 1a), while retaining subnanomolar CB1R binding affinity and >2000-fold CB1R/CB2R selectivity (Figure 1b). In functional assays, MRI-1891 displayed very high bias toward inhibiting CB1R-agonist-induced β-arrestin-2 (βArr2) recruitment (IC50: 21 pM) versus inhibiting CB1R-agonist-induced G protein activation, as monitored by GTPγS binding (IC50: 6 nM), with a potency ratio of 286. The concentration of the CB1R agonist CP-55,940 used in these assays corresponded to its EC80 for both G protein activation and βArr2 recruitment. In contrast to MRI-1891, the reference compound rimonabant was nonbiased (Figure 1c).
Figure 1.

(a) Chemical structure and physicochemical properties of (S)-MRI-1891 and its brain-penetrant parent compound SLV-319 (ibipinabant); (b) binding affinity of (S)-MRI-1891 to human CB1R and CB2R as determined by displacement of a radiolabeled cannabinoid agonist and crude membrane preparations from CHO-K1 cells stably transfected with hCB1R or hCB2R, as described in the Supporting Information, n = 3. (c) Inhibition of CB1R-agonist-induced GTPγS binding (dotted lines) and β-arrestin-2 recruitment (solid lines) by (S)-MRI-1891 (red) or rimonabant (blue), using hCB1R-CHO-K1 cell membrane (PerkinElmer, ES-110-M400UA) and PathHunter eXpress CNR1 CHO-K1 β-arrestin-2 assay, 93–0959E2CP0M, as described in the Supporting Information. Values represent mean ± SEM from 3–6 independent experiments. *, significant difference (P < 0.05) from IC50 values for inhibiting hCB1R-GTPγS signaling, as determined by t-test.
The reduced brain penetrance of MRI-1891 was confirmed by its reduced brain/plasma ratio (7%) (Figure 2a) relative to that of ibipinabant (22%)7 or rimonabant (100%, not shown) following acute oral doses of 3 mg/kg in male, wild-type mice. Accordingly, acute MRI-1891 treatment at doses of 1 or 10 mg/kg did not result in significant CB1R occupancy in the brain, as determined by CB1R positron emission tomography (PET, Figure 2b) and did not induce anxiogenic behavior even at the high dose of 30 mg/kg. By contrast, 3 mg/kg rimonabant caused significant brain CB1R occupancy and was highly anxiogenic (Figure 2c). There was a moderate increase in the brain/plasma ratio of MRI-1891 following 28 days of chronic administration at 3 mg/kg/day (Figure 2a). Brain CB1R occupancy was significant following a high (10 mg/kg/day) but not a low dose (1 mg/kg/day) regimen (Figure 2b). This “leakiness” of MRI-1891 may be related to its less efficient extrusion by ABC transporters than that of another peripheral CB1R antagonist that does not accumulate in the brain,17 as indicated by the smaller increase in brain levels of MRI-1891 compared to MRI-1867 in Mdr1a/b double-knockout mice compared to those in wild-type mice (Supplementary Figure 1). However, neither dose regimen was anxiogenic, whereas chronic treatment with rimonabant (3 mg/kg/day) induced strong anxiety (Figure 2c).
Figure 2.
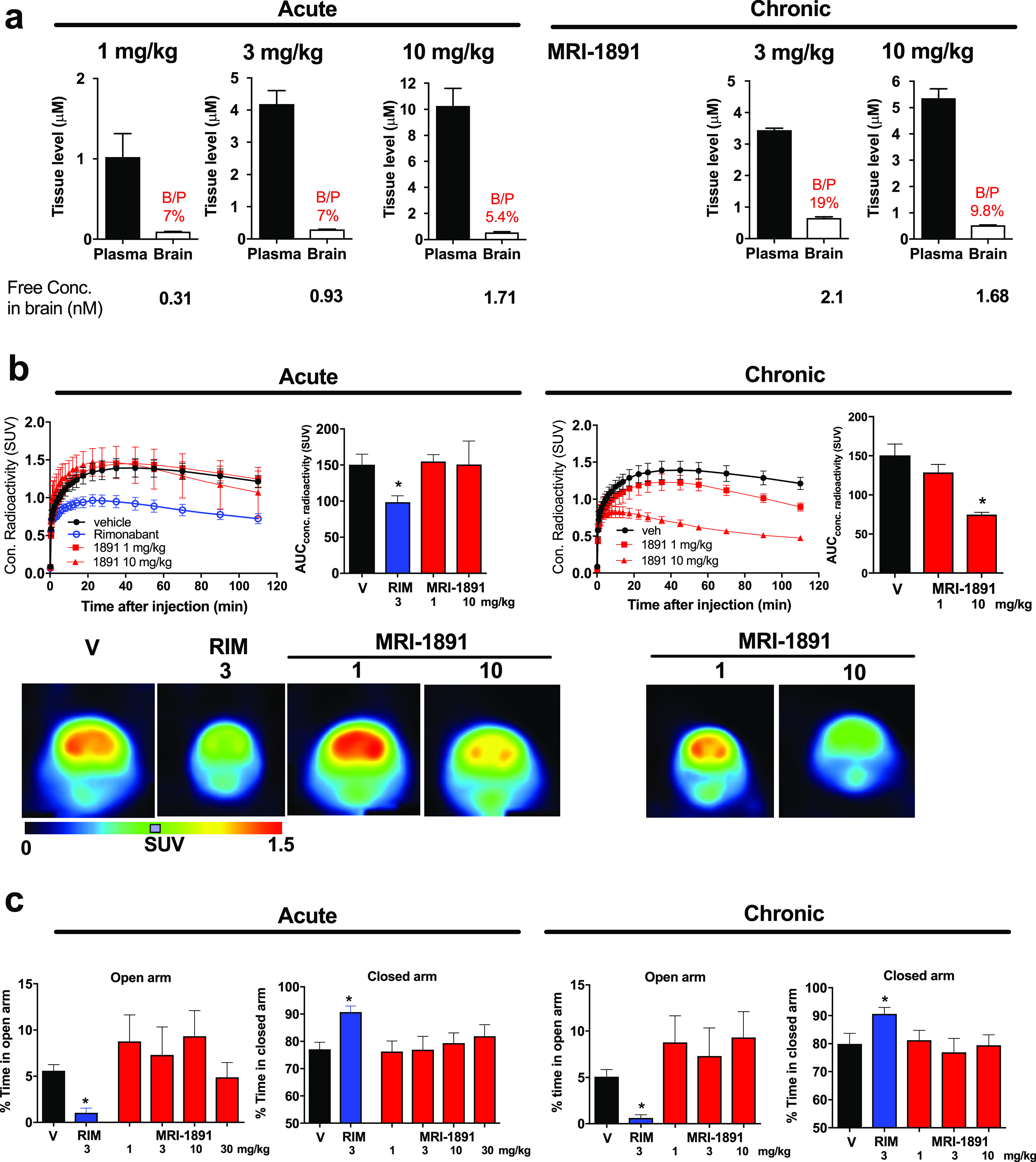
(a) Brain penetrance of (S)-MRI-1891 upon a single (acute) dose or 28 days of chronic oral dosing in lean control male C57Bl/6J mice. Drug levels in plasma and buffer-perfused brain were measured by LC/MS/MS 1 h after the last dose (plasma Cmax). Free concentration in brain was determined by equilibrium dialysis using crude membranes from the brain of CB1R-knockout (KO) mice as described and corresponded to 0.3% of total brain levels measured. (b) In vivo binding of (S)-MRI-1891 or rimonabant to mouse brain CB1R as assessed by displacement of a positron emission tomography (PET) radiotracer administered 1 h after acute dosing or 28 days of chronic oral administration of the CB1R antagonist, as described in the Supporting Information and in ref (7). Values represent mean ± SEM from 3 to 6 independent experiments. Scans from representative experiments are shown in the bottom. (c) Anxiogenic behavior induced by rimonabant, but not (S)-MRI-1891, as determined by the elevated plus maze test (see the Supporting Information). Columns and vertical bars represent mean ± SEM of 4–6 independent experiments.
To test whether similar doses of MRI-1891 could engage peripheral CB1R, we tested MRI-1891 for its ability to antagonize the CB1R-agonist-induced inhibition of upper gastrointestinal motility, as measured by the charcoal transit assay. As shown in Supplementary Figure 2a, the CB1R agonist arachidonoyl chloroethylamine (ACEA) at the maximally effective intraperitoneal dose of 10 mg/kg caused a 60% inhibition of upper GI motility in lean wild-type mice. This effect was antagonized in a dose-dependent manner by MRI-1891, with 3 mg/kg causing maximal and 1 mg/kg near-maximal antagonism. MRI-1891 increased upper GI motility at both 3 and 10 mg/kg in the absence of CB1R agonist indicating in vivo CB1R inverse agonism. These effects were similar to those seen with rimonabant (Supplementary Figure 2b). ACEA inhibited upper GI motility to the same extent in wild-type and βArr2-KO mice, indicating that CB1R inhibition of upper GI motility is βArr2-independent (Supplementary Figure 2c).
CB1R Blockade Induces Anxiety via G Protein Signaling
The absence of anxiogenic behavior despite significant brain CB1R occupancy following chronic dosing with 10 mg/kg/day MRI-1891 was puzzling, because a similar level of brain CB1R occupancy by rimonabant was associated with a strong anxiogenic response. This raised the possibility that anxiety induced by CB1R blockade results exclusively from signaling via G proteins and not via β-arrestin-2. We tested this hypothesis by analyzing the behavioral effects of central CB1R blockade in wild-type and βArr2-KO mice. Two behavioral responses to CB1R inverse agonists in mice are considered as good predictors of their neuropsychiatric side effects in humans: hyperambulatory activity as tested in drug-naïve animals and anxiety-like behavior in the elevated plus maze. A single oral dose of 3 mg/kg rimonabant elicited similar, long-lasting hyperambulatory activity in βArr2-KO mice and their wild-type littermates (Figure 3a) and also caused identical, strong anxiogenic responses in both strains, as indicated by near-complete shutdown of exploratory activity in the open arm and a parallel increase in time spent in the closed arm of the elevated plus maze paradigm (Figure 3b). These findings indicate that altered β-arrestin-2 signaling is not involved in these behavioral responses to central CB1R blockade.
Figure 3.
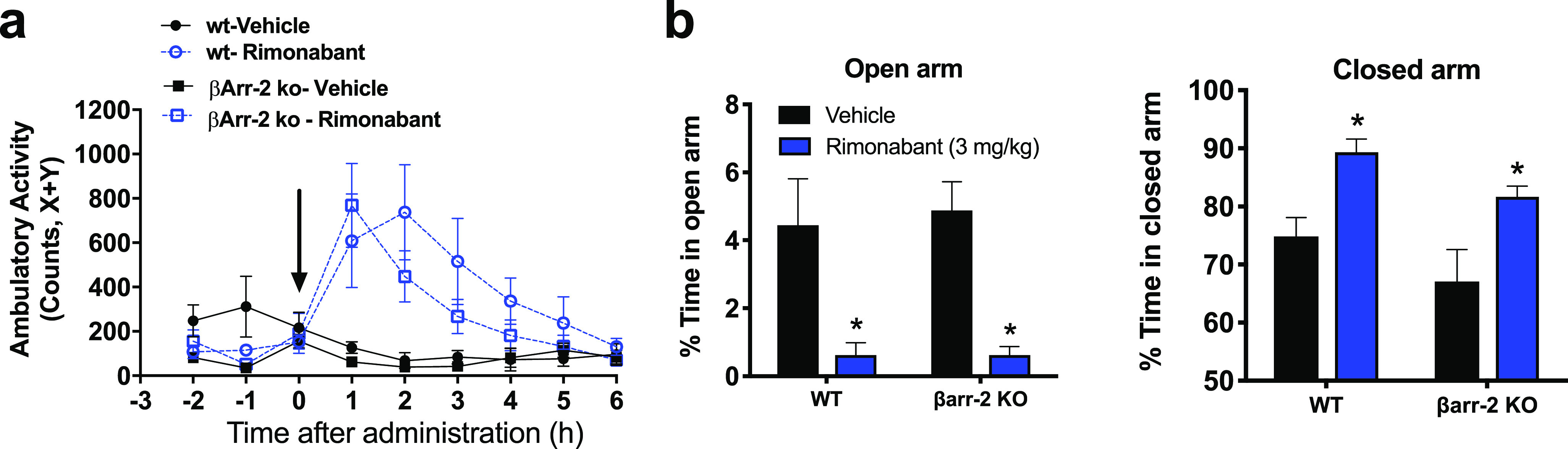
Behavioral effects of rimonabant mediated by brain CB1R are similar in wild-type and βArr2-KO mice. (a) Hyperambulatory activity induced by 3 mg/kg rimonabant in drug-naïve mice, as quantified by beam disruption in an x–y box; (b) anxiogenic effect of rimonabant as tested in the elevated plus maze. Points or columns and vertical bars represent mean ± SEM from 4–6 experiments; *, significant difference (P < 0.05) from corresponding vehicle-treated group, as determined by 2-way ANOVA followed by Dunnett’s multiple comparisons test.
We next examined the involvement of βArr2 signaling on the metabolic effects of CB1R blockade in mice with high-fat-diet-induced obesity/metabolic syndrome (DIO). In male, wild-type C57BL6/J mice kept on a high-fat diet for 14 weeks and then treated daily for 6 days with different oral doses of MRI-1891, MRI-1891 caused an acute and robust, dose-dependent decrease in food intake that returned to normal in 5–6 days, whereas a progressive decrease in body weight was maintained throughout the treatment period (Supplementary Figure 3a,b). MRI-1891 also reversed in a dose-dependent manner the obesity-induced hyperleptinemia (Supplementary Figure 3c), insulin resistance, and glucose intolerance, as quantified using the intraperitoneal insulin sensitivity and glucose tolerance tests (Supplementary Figure 3d,e). Obesity was also associated with marked hyperinsulinemia and modest hyperglycemia, which were also dose dependently mitigated by MRI-1891 (Supplementary Figure 3f,g).
CB1R Promotes Skeletal Muscle Insulin Resistance via βArr2
We next compared the effects of MRI-1891 in DIO wild-type and DIO βArr2-KO mice. Daily treatment with 3 mg/kg MRI-1891 reduced food intake and body weight nearly identically in the two strains (Figure 4a,b). The reversal of hyperleptinemia was also similar in the two strains (Figure 4c), which is compatible with resensitization to endogenous leptin being responsible for the appetite and weight reducing effects, as proposed earlier.7 In contrast, the marked hyperinsulinemia of wild-type DIO mice was completely reversed by 7 days of treatment with 3 mg/kg/day MRI-1891, whereas a more moderate level of hyperinsulinemia in βArr2-KO DIO mice was unaffected by the same MRI-1891 treatment regimen (Figure 4d). Similarly, wild-type DIO mice developed profound insulin resistance, which was completely reversed by MRI-1891 treatment, whereas βArr2-KO DIO mice remained insulin-sensitive, which was not significantly affected by MRI-1891 (Figure 4e).
Figure 4.

Effects of (S)-MRI-1891 treatment on food intake (a), body weight (b), plasma leptin (c), nonfasting plasma insulin (d), and insulin sensitivity (e) of male wild-type and βArr2-KO mice with high-fat diet induced obesity. Body weight at the start of treatment was 45.6 ± 0.4 g. Points or columns and vertical lines represent mean ± SEM from 8–10 animals. Intraperitoneal insulin sensitivity test (ipIST) was conducted as described in the Supporting Information. *, significant difference (P < 0.05) within the indicated groups, as determined by 2-way ANOVA followed by Dunnett’s multiple comparisons test. #, significant difference (P < 0.05) between wild-type and βArr-2 KO groups, as determined by 2-way ANOVA followed by Sidaks’s multiple comparisons test.
Tissue-specificity of glycemic control was analyzed using the hyperinsulinemic–euglycemic insulin clamp that also included 2-deoxyglucose infusion near the end of the clamp. Hepatic glucose production significantly increased in DIO versus lean wild-type mice, and this effect was attenuated by a single oral dose of 1 mg/kg MRI-1891 similarly in wild-type and βArr2-KO mice (Figure 5a). However, the obesity-induced reduction of glucose clearance was partially reversed by MRI-1891 in wild-type but not in βArr2-KO mice (Figure 5b), which was reflected by a similar differential effect of MRI-1891 on glucose infusion rate (Figure 5c). Further analysis indicated that the tissue responsible for the differential effect of MRI-1891 on glucose clearance was skeletal (soleus) muscle, in which the obesity-induced inhibition of 2-deoxyglucose uptake was reversed by a 1 mg/kg dose of MRI-1891 in wild-type mice but was unaffected by the same treatment in βArr2-KO mice (Figure 5d).
Figure 5.

Glycemic control of lean control mice and mice with high-fat diet induced obesity treated with vehicle or (S)-MRI-1891, as analyzed by hyperinsulinemic/euglycemic insulin clamps and described in the Supporting Information. Hepatic glucose production (a), glucose clearance or Rd (b), glucose infusion rate (c), and 2-deoxyglucose uptake into soleus muscle (d) were analyzed from data obtained from the clamp, as described in the Supporting Information. *, significant difference (P < 0.05) within the indicated groups, as determined by 2-way ANOVA followed by Dunnett’s multiple comparisons test. #, significant difference (P < 0.05) between wild-type and βArr-2 KO groups, as determined by 2-way ANOVA followed by Sidaks’s multiple comparisons test. Columns and vertical bars indicate mean ± SEM from 6–8 animals. For an explanation, see the text.
2-Deoxyglucose uptake was also tested in the absence of a hyperinsulinemic clamp, in wild-type, βArr2-KO, and βArr1-KO mice. In both wild-type and βArr1-KO mice, the high-fat-diet (HFD)-induced marked suppression of 2-deoxyglucose uptake into soleus muscle was reversed by a single dose of 1 mg/kg MRI-1891, whereas similar treatment of HFD-fed βArr2-KO mice again failed to affect muscle glucose uptake (Figure 6a), suggesting that CB1R signaling in skeletal muscle is βArr2-dependent but not βArr1-dependent. Because of the very high potency of MRI-1891 to inhibit CB1R-induced βArr2 signaling, we tested whether 7 days of treatment with a low dose of 0.1 mg/kg/day MRI-1891, which does not significantly affect body weight or food intake, can influence muscle glucose uptake. In wild-type DIO mice, such treatment caused a partial, but significant, reversal of the HFD-induced 2-deoxyglucose uptake into soleus muscle, whereas no such effect was evident in βArr2-KO DIO mice (Figure 6b).
Figure 6.

Analyses of the role of βArr2 in CB1R-induced, obesity-related muscle insulin resistance. (a) 2-Deoxyglucose was infused into anesthetized wild-type, βArr2-KO or βArr1-KO mice and its uptake measured in soleus muscle from lean mice or mice with diet-induced obesity 1 h following treatment with a single oral dose of 1 mg/kg (S)-MRI-1891 or vehicle. (b) 2-Deoxyglucose uptake measured as in panel (a), except that treatment with (S)-MRI-1891 was for 7 days at 0.1 mg/kg/day. (c) Insulin-induced Akt phosphorylation and its CB1R-mediated inhibition were analyzed in mock-transfected and βArr2-siRNA-transfected C2C12 myotubes. Each treatment was tested in duplicate aliquots of cells, analyzed by Western blot using β-actin as loading control, and quantified by densitometry. The level of βArr2 knockdown is illustrated by the bar graph on the right. Note that the inhibition of insulin-induced akt-phosphorylation by the CB1R agonist is inhibited by MRI-1891 and is absent in cells with βArr2 knockdown. (d) CB1R-mediated inhibition of insulin-induced Akt phosphorylation is absent in C2C12 myotubes with Crip1a overexpression and (e) is enhanced in myotubes with Crip1a knockdown. (f) High-fat diet-induced obesity results in downregulation of Crip1a expression in soleus muscle from wild-type but not from βArr2-KO mice. *, significant difference (P < 0.05) within the indicated groups, as determined by 2-way ANOVA followed by Dunnett’s multiple comparisons test. #, significant difference (P < 0.05) between the groups, as determined by 2-way ANOVA followed by Sidaks’s multiple comparisons test. Columns and vertical bars represent mean ± SEM from 8–10 animals.
The role of βArr2 in CB1R inhibition of insulin signaling was further analyzed in C2C12 mouse muscle myotubes. Exposure of cultured C2C12 cells with 100 nM insulin caused a robust increase in Akt-2 phosphorylation, which was inhibited in the presence of 5 μM CP-55940, a CB1R agonist. This inhibition was prevented by simultaneous exposure of the cells to 100 nM MRI-1891 or in cells with siRNA-mediated knockdown of βArr2 (Figure 6c).
Crip1a Regulates CB1R Signaling via βArr2 in Skeletal Muscle
It has been recently reported that Crip1a, a CB1R distal C-terminal associated protein,18 competes with β-arrestins for binding to CB1R distal and central C-terminal domains that could affect CB1R signaling via β-arrestins.19,20 We therefore overexpressed Crip1a in C2C12 cells and analyzed CB1R-mediated inhibition of insulin signaling in mock-transfected and Crip1a-transfected cells. As illustrated in Figure 6d, exposure of the mock-transfected control cells to 100 nM insulin triggered robust Akt-2 phosphorylation, which was significantly inhibited by the CB1R agonist CP-55940. Preincubation of the cells with 100 nM of MRI-1891 alone did not affect the insulin response but completely abrogated the inhibitory response to CP-55940. In sharp contrast, in cells transfected with Crip1a, CP-55940 failed to inhibit insulin-induced Akt-2 phosphorylation either in the absence or presence of MRI-1891. In contrast, siRNA-mediated Crip1a knock-down potentiated the inhibitory effect of CP-55940 on insulin-induced Akt-2 phosphorylation (Figure 6e).
The above findings suggest that Crip1a is a functional antagonist of endocannabinoid/CB1R/βArr2 signaling in skeletal muscle, and loss of this function in DIO mice may contribute to the CB1R-mediated insulin resistance. We therefore measured the expression of Cnrip1, the gene encoding Crip1a, in skeletal muscle and found that Cnrip1 is robustly downregulated in wild-type DIO compared to lean control mice, without a similar diet-induced change being detectable in βArr2-KO mice (Figure 6f).
Computational Study of MRI-1891/CB1R Interaction
To identify the residues of CB1R that give rise to β-arrestin biased antagonism by MRI-1891, molecular dynamics (MD) simulations were carried out using the X-ray structure of CB1R cocrystallized with taranabant.21 The MD simulations were performed with a set of conformers of MRI-1891 (cf. the Supporting Information) and with the unbiased antagonist rimonabant and ibipinabant as controls.
The simulations indicate that Arms 1 and 2 of MRI-1891 (Figures 7 and 8) are well-stabilized by aromatic residues deep in the binding pocket, as seen in the X-ray structures of CB1R bound to taranabant21 or rimonabant-like AM6538.22 The Cl atom of Arm 1 interacts electrostatically with several nonpolar H atoms, but most strongly with W279. Although all the MRI-1891 conformers (cf. the Supporting Information and Figure 8a) remained stable throughout the simulations and interconversions were observed, only one conformer (conf. 1) had optimal interactions with the receptor in the orientations shown, as determined by the number and frequency of favorable contacts (cf. Methods) and was consistent with the mutation data. In this mode, Arm 3 of MRI-1891 spans the region of Arm 3 of both taranabant and rimonabant (Figure 8b), whereas Arm 4 is oriented in the opposite direction, interacting with residues not in contact with either rimonabant or taranabant (Figures 7 and 8b).
Figure 7.

Left: persistent, statistically significant MRI-1891/CB1R interactions in the most favorable binding mode (conf. 1; cf. Figure 8 and in the Supporting Information). Residues in blue indicate electrostatic interactions (through nonpolar H) with Cl or F of MRI-1891; in green, N-terminal residues engaged in hydrophobic interactions. black residues with hydrophobic or nonpolar interactions; those in red indicate hydrogen bond interactions (W denotes water). MRI-1891 atoms: O (red), N (blue), C (light green), S (yellow), Cl and F (dark green), and H (white). Right: location of the CB1R residues interacting with MRI-1891 in the context of the receptor (extracellular and side views); numbers in brackets indicate the TMHs. Most persistent interactions with Arm 1 are colored blue (mainly TMHs 3, 5, and 6), with Arm 2 in yellow (TMHs 2 and 3), with Arm 3 in red (TMHs 1–3), and with Arm 4 in purple (mainly the N-terminal loop). The interactions of CF3 of Arm 3 with TMH1 and CH3 of Arm 4 with the N-term affect the movement of TMH1 (red arrow).
Figure 8.

(a) Left: general side view of the MRI-1891 binding mode (conf. 1; cf. Figure 7 and the Supporting Information) showing the positions of the four arms in the receptor. Right: all the conformers of MRI-1891 considered in this study; only conf. 1 in the binding mode of Figure 7 showed optimal and persistent interactions with the receptor throughout the simulations and is consistent with all the mutation studies. (b) Top: overlay of agonist AM841 and antagonist taranabant (left), as they appear in their relative positions in the crystal structures (5XR8 and 5U09, respectively); comparison with the structure of MRI-1891 (middle); the overlay of MRI-1891 (green) onto rimonabant (red) was done using the heavy atoms of the five-member ring as a common docking point (right). Each arm plays a distinct role and interacts with a different region of the receptor. MRI-1891 combines in a single scaffold the arms distribution of agonists and antagonists, a property that may be essential to impart biased property in general. Bottom: detail of the S123 (TMH1) position relative to the trifluoromethyl group of MRI-1891 and four nearby nonpolar residues on adjacent TMHs (snapshot of the simulation; I119 omitted for clarity).
The sulfonamide group of Arm 3 is stabilized by water entering the pocket, creating short chains that bridge these groups to the receptor. The CF3 group interacts electrostatically with several residues, particularly S123, whereas the aryl ring is well-packed against several nonpolar residues, including I119 (transmembrane helix-1 [TMH1]), F170 and F174 (TMH2), M384 (TMH7), and with I105 and M109 in the N-terminal segment, forming a relatively compact cluster. Mutations M384S, I105A, and M109A resulted in suppression of CP-55940-induced βArr2 recruitment, suggesting a key role of this cluster in CB1R agonism by CP-55940 (Supplementary Figure 4). These interactions involve residues at the top of the TMH1, i.e., where the N-terminal segment connects with the helix (Figure 7, right panel) and may help modulate the movement or stabilize the conformation of TMH1 (red arrow in Figure 7). Several cocrystals of CB1R reported recently23 suggest that conformational changes of TMH1 may be associated with differences in agonist/antagonist activity, presumably because of its proximity to TMH7, the movement of which affects the intracellular C-terminal helix, and possibly β-arrestin recruitment.24 We focused on S123 (TMH1) due to its interaction with the CF3 group of MRI-1891 (Figure 8b, lower right panel); Arm 3 of both ibipinabant and rimonabant do not interact with this residue. The S123A mutation resulted in a 4-fold reduction of inhibitory potency of MRI-1891, but not of rimonabant, toward βArr2 signaling (Figure 9c). However, the mutation did not alter either CB1R binding affinity (Figure 9a) or CB1R inhibitory potency toward G protein signaling (Figure 9b) of either MRI-1891 or rimonabant compared to that of wildtype.
Figure 9.

S123A mutation of hCB1R results in a selective decrease in the inhibitory potency of MRI-1891 against CB1R-agonist-induced βArr2 recruitment (c) but not G protein activation (b), without causing a similar change in the effects of rimonabant (right panels) or affecting the binding affinity of either compound (a). GTPγS binding and βArr2 recruitment in CHO cells stably transfected with wild-type and S123A mutant hCB1R were conducted using human CB1 receptor cDNA (hCNR1, NM_016083) in the pCI vector (Promega) for GTPγS and radioligand binding assays, and in the pCMV-hCNR1-PK vector (Eurofins/DiscoverX) for β-arrestin-2 recruitment assays via the PathHunter system as described in the Supporting Information. Points and vertical bars represent mean ± SEM from 8 independent experiments. Numbers indicate Kd values calculated using computerized curve fitting and the Cheng–Prusoff equation.
All the polar groups of Arm 4 are stabilized mainly by water, although the NH2 of guanidine also forms transient H-bonds with S383 of TMH7. Notably, the methyl group of Arm 4 interacts hydrophobically with several residues on the N-terminal segment, including F102, M103, and, more persistently, with F108 (Figure 7). These interactions appear to be critical for the biased activity of MRI-1891, since Arm 4 of ibipinabant (SLV319) lacks the acyl group (Figure 8b), which may account for the negligible bias of SLV319 (G/βArr ≈2).
Discussion
The present study provides the first example of a biased orthosteric GPCR antagonist. The CB1R antagonist/inverse agonist MRI-1891 is highly potent in suppressing CB1R-agonist-stimulated βArr2 recruitment with an IC50 of 21 pM and is about 300 times less potent in inhibiting CB1R-agonist-induced activation of G protein activation (IC50: 6 nM). Importantly, this bias results in functional selectivity, as we found that CB1R modulate anxiogenic behavior, body weight, appetite, and hepatic glucose production predominantly via G protein activation, whereas CB1R modulation of muscle insulin sensitivity is predominantly via βArr2 signaling. Furthermore, the cannabinoid receptor interacting protein 1a (Crip1a) competes with βArr2 for binding to CB1R in skeletal muscle where its expression is downregulated in DIO/insulin resistance, suggesting its involvement in the physiological control of insulin sensitivity.
Recent findings suggest that different conformations of GPCRs mediate agonist-induced G protein activation and β-arrestin recruitment25 and that a specific phosphorylation pattern at the C terminus of GPCRs, induced by GPCR kinases (GRKs), determines β-arrestin recruitment to GPCRs and regulates their intracellular functions.25,26 Another receptor domain, an Asp–Arg–Tyr (DRY) motif in the second intracellular loop is critical for G protein binding, but is also involved in β-arrestin recruitment.27 Biased agonists preferentially activate conformations linked to G protein or β-arrestin signaling, respectively. Although there is no published evidence for a biased orthosteric GPCR antagonist, pregnenolone was proposed to be a biased allosteric CB1R antagonist,28 based on its inability to compete with ligand binding to the CB1R, but its ability to inhibit certain behavioral responses to Δ9-tetrahydrocannabinol (THC) by selectively interfering with THC-induced ERK1/2MAPK phosphorylation without affecting the parallel inhibition of cAMP accumulation.28 However, others reported that pregnenolone displaced radiolabeled rimonabant from CB1R binding sites but failed to affect THC-induced ERK1/2 phosphorylation.29 Furthermore, as pregnenolone appears to bind to a domain distinct from the ligand binding pocket of CB1R,28 the receptor conformation it promotes is likely different from the conformation of the CB1R/MRI-1891 complex.
The MD simulations suggest that the presence of Arms 3 and 4 in a single molecular scaffold, with each arm playing a distinct and largely independent role (Figure 7), may be important to impart biased CB1R antagonism. In particular, Arm 4 is absent in the unbiased inverse agonists rimonabant and taranabant and too short in ibipinabant to interact effectively with the receptor. Moreover, the structure of MRI-1891 suggests that Arm 3 spans a region common to antagonists, and Arm 4 spans a region common to agonists, as illustrated in Figure 8b. This combination may affect G protein and βArr2 signaling pathways separately. There seems to be a direct and an indirect effect of the S123A mutation (Figures 8b and 9) on the structure/dynamics of CB1R and its interactions with MRI-1891 that explain the similar affinities of the wt and S123A mutant and their differences in potency. The CF3 group of Arm 3 engages S123 directly but also can develop electrostatic interactions with the nonpolar H atoms of A123 or other nonpolar residues nearby (Figure 7 and 8b). It is noted that Arm 3 of rimonabant does not interact with S123. Also, S123 is at the center of a group of nonpolar residues, namely I119 in TMH1, F170 and F174 in TMH2, and M384 in TMH7 (Figure 8b) that can form a relatively compact cluster once the methyl group of A123 is introduced. These interactions make TMH1 less flexible when compared to the wild-type CB1R, and consequently, the N-terminal segment becomes less effective in modulating the TMH1 movement in response to the action of Arm 3 or 4 (Figure 7). The mobility and adaptability of the N-terminal/TMH1 motif appears to be important for activity, judged by the differences observed in the crystal structure of the agonist-bound versus the antagonist-bound receptor.23 The X-ray structure of rimonabant-like22 and taranabant crystallized with CB1R21 show that this region changes significantly to accommodate Arm 3 of either ligand, with TMH1 moving away from the receptor core and the N-terminal tail pushed inwardly. It is plausible that the movement of TMH1 modulated by the N-terminal residues potentiates the βArr2 bias of MRI-1891, particularly via Arm 4. This interpretation is in line with the A123S mutant having a 4-fold lower inhibitory potency of MRI-1891 toward βArr2 signaling but not G-protein signaling, without affecting either parameter for rimonabant (Figure 9).
β-Arrestins not only play a key role in GPCR internalization and desensitization but also can serve as scaffolds for signaling complexes involved in various cellular responses.30 CB1R has affinity for βArr2 higher than that for βArr1. βArr2 has been implicated in agonist-induced internalization of CB1R,31 whereas βArr1 was shown to mediate CB1R-agonist-induced ERK1/2 phosphorylation.32 To analyze the functional consequences of biased orthosteric antagonism of CB1R by MRI-1891, we first sought to determine the involvement of β-arrestin signaling in CB1R-mediated behavioral and metabolic effects of endocannabinoids. The anxiogenic effect of rimonabant, thought to reflect a blockade of CB1R in the dopaminergic reward pathway,33 was identical in wild-type and βArr2-KO mice, suggesting the lack of significant βArr2 involvement in mediating this effect. This may also explain the lack of a similar anxiogenic response to chronic treatment of mice with 10 mg/kg/day of MRI-1891 that caused significant CB1R occupancy in the CNS, as revealed by CB1R PET studies. The free concentration of MRI-1891 in the brain following chronic treatment with daily doses of 10 mg/kg (1.7 nM) exceeded its CB1R binding Kd of 0.3 nM but remained below its IC50 of 6 nM for inhibiting CB1R-agonist-induced G protein activation, thus providing a therapeutic window. This finding supports the concept that functional inhibitory potency of the G protein pathway rather than binding affinity is the parameter that best predicts the neuropsychiatric side effects of CB1R antagonist/inverse agonists.
In contrast to the G protein signaling bias of CB1R in modulating anxiogenic behavior, the diabetogenic effect of CB1R in skeletal muscle, due to inhibition of insulin-induced glucose uptake, occurs via βArr2 signaling. First, whole-body glucose clearance and muscle 2-deoxyglucose uptake during a hyperinsulinemic clamp were significantly higher in both lean and obese βArr2-KO mice than in their respective wild-type littermates, indicating increased insulin sensitivity in the absence of βArr2. This is in agreement with a significant increase in glucose tolerance in skeletal-muscle-specific βArr2-KO mice34 but opposite an earlier report of decreased insulin sensitivity in global βArr2-KO mice.35 One possible reason for the discrepant results is that the genetic background of the mice used in the latter studies was different. Second, the HFD-induced suppression of 2-deoxyglucose uptake into skeletal muscle was largely reversed by MRI-1891 treatment in wild-type and βArr1-KO mice, whereas it remained unaffected in βArr2-KO mice. Third, in C2C12 mouse myoblasts, inhibition of insulin-induced Akt phosphorylation by the CB1R agonist CP-55940 was abrogated by either siRNA-mediated knockdown of βArr2 or by MRI-1891. The dominant role of βArr2 in CB1R signaling appears to be unique to skeletal muscle, as other metabolic effects of CB1R blockade in obese animals, including reductions in appetite, body weight, plasma leptin levels, and hepatic glucose production and increased glucose uptake into adipose tissue, were similar in wild-type and βArr2-KO mice. One consequence of the high signaling bias of MRI-1891 is its greater apparent in vivo potency for improving insulin sensitivity compared to its potency to induce other metabolic effects or unwanted CNS-mediated side effects, which has therapeutic implications for the treatment of insulin resistance and diabetes.
Finally, the present findings reveal a possible novel physiological function of cannabinoid receptor interacting protein 1a (Crip1a). Crip1a was previously demonstrated to compete with βArr2 for binding to the phosphorylated central or distal C-terminal peptides of CB1R19 and as a result can attenuate agonist-induced CB1R downregulation20 and cellular signaling.36 The present observation, that overexpression of Crip1a in C2C12 myotubes abrogates βArr2-mediated CB1R signaling whereas its knockdown potentiates it, is fully compatible with the above findings and suggests that Crip1a and, possibly, other CB1R interacting proteins such as SGIP1,37 are involved in glycemic control in diabetes and related metabolic disorders with altered energy balance.38 Indeed, the observed robust downregulation of CRIP1a expression in skeletal muscle of DIO compared to lean mice is compatible with a protective role of Crip1a in maintaining muscle insulin sensitivity.
Acknowledgments
The graphical abstract was generated in Biorender.com. This work utilized the high-performance computer capabilities of the Biowulf/HPC cluster at the NIH (http://hpc.nih.gov).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00048.
Supplementary figures as described in the text; chemical synthesis and characterization procedures; material and methods (PDF)
Author Present Address
# M.R.I.: Section on Medicinal Chemistry, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD 20892 USA
Author Present Address
∇ T.J.: INSERM Lipids, Nutrition, Cancer UMR1231, University of Burgundy and Franche-Comté, Dijon, France
Author Present Address
◆ R.C.: Section on Fibrotic Disorders, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD 20892 USA
Author Contributions
Z.L., N.J.C., C.N.Z., and R.C. conducted in vivo experiments and in vitro receptor binding and functional assays. R.C. conducted LC/MS/MS analyses of tissue levels of (S)-MRI-1891 and conducted behavioral and gut motility assays; T.J. performed ipGTT and ipIST. G.G. conducted and analyzed insulin clamp experiments; MRI designed, synthesized, and validated the structure of (S)-MRI-1891; H.L.P. generated site-directed mutations of hCNR1. J.M. assisted with insulin clamp experiments and proofread manuscript; J.W. provided research resources and reviewed the manuscript. J.-S.L. and R.B.I. conducted and analyzed CB1R PET studies; S.A.H. and Y.S.L. conducted and interpreted Molecular Dynamics simulations. G.K. and R.C. conceived the study and analyzed results; G.K. wrote manuscript.
Supported by intramural funds of NIAAA to G.K. S.A.H. and Y.S.L. were supported by the NIH Intramural Research Program through the Center for Information Technology. J.W. and J.M. received funds from the Intramural Research Program of the NIDDK.
The authors declare the following competing financial interest(s): G.K., R.C., and M.R.I. are listed as co-inventors on two US Government patents covering (S)-MRI-1891. The remaining authors declare no competing interests.
Supplementary Material
References
- Pacher P.; Batkai S.; Kunos G. (2006) The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 58 (3), 389–462. 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri C.; Di Marzo V. (2013) The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 17 (4), 475–490. 10.1016/j.cmet.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Van Gaal L. F.; Rissanen A. M.; Scheen A. J.; Ziegler O.; Rossner S. (2005) Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet 365 (9468), 1389–1397. 10.1016/S0140-6736(05)66374-X. [DOI] [PubMed] [Google Scholar]
- Despres J. P.; Golay A.; Sjostrom L. (2005) Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N. Engl. J. Med. 353 (20), 2121–2134. 10.1056/NEJMoa044537. [DOI] [PubMed] [Google Scholar]
- Le Foll B.; Gorelick D. A.; Goldberg S. R. (2009) The future of endocannabinoid-oriented clinical research after CB1 antagonists. Psychopharmacology (Berl) 205 (1), 171–174. 10.1007/s00213-009-1506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J.; Vemuri V. K.; Liu J.; Batkai S.; Mukhopadhyay B.; Godlewski G.; Osei-Hyiaman D.; Ohnuma S.; Ambudkar S. V.; Pickel J.; Makriyannis A.; Kunos G. (2010) Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J. Clin. Invest. 120 (8), 2953–2966. 10.1172/JCI42551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J.; Cinar R.; Liu J.; Godlewski G.; Wesley D.; Jourdan T.; Szanda G.; Mukhopadhyay B.; Chedester L.; Liow J. S.; Innis R. B.; Cheng K.; Rice K. C.; Deschamps J. R.; Chorvat R. J.; McElroy J. F.; Kunos G. (2012) Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 16 (2), 167–179. 10.1016/j.cmet.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groer C. E.; Tidgewell K.; Moyer R. A.; Harding W. W.; Rothman R. B.; Prisinzano T. E.; Bohn L. M. (2007) An opioid agonist that does not induce mu-opioid receptor--arrestin interactions or receptor internalization. Mol. Pharmacol. 71 (2), 549–557. 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin J. D.; Crombie A. L.; Soergel D. G.; Lark M. W. (2014) Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol. Sci. 35 (7), 308–316. 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Kliewer A.; Gillis A.; Hill R.; Schmiedel F.; Bailey C.; Kelly E.; Henderson G.; Christie M. J.; Schulz S. (2020) Morphine-induced respiratory depression is independent of beta-arrestin2 signalling. Br. J. Pharmacol. 177 (13), 2923–2931. 10.1111/bph.15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turu G.; Hunyady L. (2010) Signal transduction of the CB1 cannabinoid receptor. J. Mol. Endocrinol. 44 (2), 75–85. 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- Daigle T. L.; Kwok M. L.; Mackie K. (2008) Regulation of CB1 cannabinoid receptor internalization by a promiscuous phosphorylation-dependent mechanism. J. Neurochem. 106 (1), 70–82. 10.1111/j.1471-4159.2008.05336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibsen M. S.; Connor M.; Glass M. (2017) Cannabinoid CB1 and CB2 Receptor Signaling and Bias. Cannabis Cannabinoid Res. 2 (1), 48–60. 10.1089/can.2016.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. (2014) What is pharmacological ’affinity’? Relevance to biased agonism and antagonism. Trends Pharmacol. Sci. 35 (9), 434–441. 10.1016/j.tips.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Weiwer M.; Xu Q.; Gale J. P.; Lewis M.; Campbell A. J.; Schroeder F. A.; Van de Bittner G. C.; Walk M.; Amaya A.; Su P.; et al. (2018) Functionally Biased D2R Antagonists: Targeting the beta-Arrestin Pathway to Improve Antipsychotic Treatment. ACS Chem. Biol. 13 (4), 1038–1047. 10.1021/acschembio.8b00168. [DOI] [PubMed] [Google Scholar]
- Need A. B.; Davis R. J.; Alexander-Chacko J. T.; Eastwood B.; Chernet E.; Phebus L. A.; Sindelar D. K.; Nomikos G. G. (2006) The relationship of in vivo central CB1 receptor occupancy to changes in cortical monoamine release and feeding elicited by CB1 receptor antagonists in rats. Psychopharmacology (Berl) 184 (1), 26–35. 10.1007/s00213-005-0234-x. [DOI] [PubMed] [Google Scholar]
- Cinar R.; Iyer M. R.; Liu Z.; Cao Z.; Jourdan T.; Erdelyi K.; Godlewski G.; Szanda G.; Liu J.; Park J. K.; et al. (2016) Hybrid inhibitor of peripheral cannabinoid-1 receptors and inducible nitric oxide synthase mitigates liver fibrosis. JCI Insight 1 (11), e87336. 10.1172/jci.insight.87336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus J. L.; Liu Y.; Wallis K. T.; Egertova M.; Bhartur S. G.; Mukhopadhyay S.; Shi S.; He H.; Selley D. E.; Howlett A. C.; Elphick M. R.; Lewis D. L. (2007) CB1 cannabinoid receptor activity is modulated by the cannabinoid receptor interacting protein CRIP 1a. Mol. Pharmacol. 72 (6), 1557–1566. 10.1124/mol.107.039263. [DOI] [PubMed] [Google Scholar]
- Blume L. C.; Patten T.; Eldeeb K.; Leone-Kabler S.; Ilyasov A. A.; Keegan B. M.; O’Neal J. E.; Bass C. E.; Hantgan R. R.; Lowther W. T.; Selley D. E.; Howlett A. L. (2017) Cannabinoid Receptor Interacting Protein 1a Competition with beta-Arrestin for CB1 Receptor Binding Sites. Mol. Pharmacol. 91 (2), 75–86. 10.1124/mol.116.104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume L. C.; Leone-Kabler S.; Luessen D. J.; Marrs G. S.; Lyons E.; Bass C. E.; Chen R.; Selley D. E.; Howlett A. C. (2016) Cannabinoid receptor interacting protein suppresses agonist-driven CB1 receptor internalization and regulates receptor replenishment in an agonist-biased manner. J. Neurochem. 139 (3), 396–407. 10.1111/jnc.13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z.; Yin J.; Chapman K.; Grzemska M.; Clark L.; Wang J.; Rosenbaum D. M. (2016) High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 540 (7634), 602–606. 10.1038/nature20613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T.; Vemuri K.; Pu M.; Qu L.; Han G. W.; Wu Y.; Zhao S.; Shui W.; Li S.; Korde A.; et al. (2016) Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 167 (3), 750–762.e14. 10.1016/j.cell.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T.; Vemuri K.; Nikas S. P.; Laprairie R. B.; Wu Y.; Qu L.; Pu M.; Korde A.; Jiang S.; Ho J. H.; Han G. W.; Ding K.; Li X.; Liu H.; Hanson M. A.; Zhao S.; Bohn L. M.; Makriyannis A.; Stevens R. C.; Liu Z. J. (2017) Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 547 (7664), 468–471. 10.1038/nature23272. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kapur A.; Hurst D. P.; Fleischer D.; Whitnell R.; Thakur G. A.; Makriyannis A.; Reggio P. H.; Abood M. E. (2007) Mutation studies of Ser7.39 and Ser2.60 in the human CB1 cannabinoid receptor: evidence for a serine-induced bend in CB1 transmembrane helix 7. Mol. Pharmacol. 71 (6), 1512–1524. 10.1124/mol.107.034645. [DOI] [PubMed] [Google Scholar]
- Reiter E.; Ahn S.; Shukla A. K.; Lefkowitz R. J. (2012) Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197. 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich V. V.; Gurevich E. V. (2006) The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol. Ther. 110 (3), 465–502. 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyombolai P.; Toth A. D.; Timar D.; Turu G.; Hunyady L. (2015) Mutations in the ’DRY’ motif of the CB1 cannabinoid receptor result in biased receptor variants. J. Mol. Endocrinol. 54 (1), 75–89. 10.1530/JME-14-0219. [DOI] [PubMed] [Google Scholar]
- Vallee M.; Vitiello S.; Bellocchio L.; Hebert-Chatelain E.; Monlezun S.; Martin-Garcia E.; Kasanetz F.; Baillie G. L.; Panin F.; Cathala A.; Roullot-Lacarriere V.; Fabre S.; Hurst D. P.; Lynch D. L.; Shore D. M.; Deroche-Gamonet V.; Spampinato U.; Revest J. M.; Maldonado R.; Reggio P. H.; Ross R. A.; Marsicano G.; Piazza P. V. (2014) Pregnenolone can protect the brain from cannabis intoxication. Science 343 (6166), 94–98. 10.1126/science.1243985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajehali E.; Malone D. T.; Glass M.; Sexton P. M.; Christopoulos A.; Leach K. (2015) Biased Agonism and Biased Allosteric Modulation at the CB1 Cannabinoid Receptor. Mol. Pharmacol. 88 (2), 368–379. 10.1124/mol.115.099192. [DOI] [PubMed] [Google Scholar]
- Violin J. D.; Lefkowitz R. J. (2007) Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 28 (8), 416–422. 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Gyombolai P.; Boros E.; Hunyady L.; Turu G. (2013) Differential beta-arrestin2 requirements for constitutive and agonist-induced internalization of the CB1 cannabinoid receptor. Mol. Cell. Endocrinol. 372 (1–2), 116–127. 10.1016/j.mce.2013.03.013. [DOI] [PubMed] [Google Scholar]
- Flores-Otero J.; Ahn K. H.; Delgado-Peraza F.; Mackie K.; Kendall D. A.; Yudowski G. A. (2014) Ligand-specific endocytic dwell times control functional selectivity of the cannabinoid receptor 1. Nat. Commun. 5, 4589. 10.1038/ncomms5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horder J.; Harmer C. J.; Cowen P. J.; McCabe C. (2010) Reduced neural response to reward following 7 days treatment with the cannabinoid CB1 antagonist rimonabant in healthy volunteers. Int. J. Neuropsychopharmacol. 13 (8), 1103–1113. 10.1017/S1461145710000453. [DOI] [PubMed] [Google Scholar]
- Meister J.; Bone D. B. J.; Godlewski G.; Liu Z.; Lee R. J.; Vishnivetskiy S. A.; Gurevich V. V.; Springer D.; Kunos G.; Wess J. (2019) Metabolic effects of skeletal muscle-specific deletion of beta-arrestin-1 and −2 in mice. PLoS Genet. 15 (10), e1008424. 10.1371/journal.pgen.1008424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan B.; Zhao J.; Wu H.; Duan B.; Shu G.; Wang X.; Li D.; Jia W.; Kang J.; Pei G. (2009) Deficiency of a beta-arrestin-2 signal complex contributes to insulin resistance. Nature 457 (7233), 1146–1149. 10.1038/nature07617. [DOI] [PubMed] [Google Scholar]
- Smith T. H.; Blume L. C.; Straiker A.; Cox J. O.; David B. G.; McVoy J. R.; Sayers K. W.; Poklis J. L.; Abdullah R. A.; Egertova M.; Chen C. K.; Mackie K.; Elphick M. R.; Howlett A. C.; Selley D. E. (2015) Cannabinoid receptor-interacting protein 1a modulates CB1 receptor signaling and regulation. Mol. Pharmacol. 87 (4), 747–765. 10.1124/mol.114.096495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova A.; Techlovska S.; Dvorakova M.; Chambers J. N.; Kumpost J.; Hubalkova P.; Prezeau L.; Blahos J. (2016) SGIP1 alters internalization and modulates signaling of activated cannabinoid receptor 1 in a biased manner. Neuropharmacology 107, 201–214. 10.1016/j.neuropharm.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Trevaskis J.; Walder K.; Foletta V.; Kerr-Bayles L.; McMillan J.; Cooper A.; Lee S.; Bolton K.; Prior M.; Fahey R.; Whitecross K.; Morton G. J.; Schwartz M. W.; Collier G. R. (2005) Src homology 3-domain growth factor receptor-bound 2-like (endophilin) interacting protein 1, a novel neuronal protein that regulates energy balance. Endocrinology 146 (9), 3757–3764. 10.1210/en.2005-0282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.