

Summary
CHK1 is a protein kinase that functions downstream of activated ATR to phosphorylate multiple targets as part of intra-S and G2/M DNA damage checkpoints. Its role in allowing cells to survive replicative stress has made it an important target for anti-cancer drug discovery. Activation of CHK1 by ATR depends on their mutual interaction with CLASPIN, a natively unstructured protein that interacts with CHK1 through a cluster of phosphorylation sites in its C-terminal half. We have now determined the crystal structure of the kinase domain of CHK1 bound to a high-affinity motif from CLASPIN. Our data show that CLASPIN engages a conserved site on CHK1 adjacent to the substrate-binding cleft, involved in phosphate sensing in other kinases. The CLASPIN motif is not phosphorylated by CHK1, nor does it affect phosphorylation of a CDC25 substrate peptide, suggesting that it functions purely as a scaffold for CHK1 activation by ATR.
Keywords: DNA damage signaling, protein kinase mechanisms, checkpoints
Graphical abstract
Highlights
-
•
Novel crystal forms of the CHK1 kinase domain are reported
-
•
Nucleotide-bound CHK1 structure
-
•
Phosphorylated CLASPIN peptide-bound CHK1 structure
-
•
CHK1-CLASPIN interaction does not affect kinase kinetics
Day et al. report the structure of a phosphorylated CLASPIN-derived peptide bound to CHK1 and accompanying biochemistry that demonstrates this interaction does not directly affect the kinetics of the CHK1 kinase, suggesting that the role of the conserved kinase domain phosphate binding pocket is in recruitment or scaffolding of CHK1.
Introduction
Checkpoint kinase 1, or CHK1, plays a key role in the cellular response to DNA damage at different points in the cell cycle through its function in both the intra-S-phase checkpoint, which allows damage to be repaired to facilitate the accurate duplication of genomes, and the G2/M checkpoint, which prevents entry into mitosis of cells with damaged chromosomes (Smits and Gillespie, 2015). Among many other targets involved in these two checkpoint responses, CHK1 phosphorylates key residues in the three CDC25 phosphatases, resulting in their deactivation by subsequent degradation or export from the nucleus. This leads to cell-cycle arrest as the phosphatases can no longer activate cyclin-dependent kinase CDK1 or CDK2, the drivers of the cell cycle. In addition, CHK1 has other important functions in the S phase, including a role in origin firing, fork progression, stabilizing stalled replication forks, fork restart, and pausing the cell cycle under conditions of replication stress (Iyer and Rhind, 2017).
CHK1 consists of an N-terminal kinase domain (Chen et al., 2000) joined by a flexible linker region to a C-terminal kinase-associated (KA1) domain. As an attractive cancer drug discovery target, many crystal structures of the CHK1 kinase domain (CHK1-KD) have been determined bound to various fragments and inhibitors. In all structures of CHK1-KD, whether in apo form or with an inhibitor bound, the kinase domain is in an active conformation with an ordered activation loop, but with a slightly open lobe arrangement leading to the misalignment of the catalytic residues. CHK1 activation occurs when key residues in the linker region are phosphorylated by ATR (Niida et al., 2007). These disrupt intramolecular interactions between the kinase and the KA1 domain (Emptage et al., 2017), and mutations that disrupt the KA1 domain lead to constitutive activity (Gong et al., 2015). Additional autophosphorylation of the KA1 domain further activates the kinase (Gong et al., 2018), and there may be a role for further phosphorylation-driven intermolecular interactions with other factors.
CLASPIN was identified as an interactor with CHK1 in Xenopus extracts (Kumagai and Dunphy, 2000) and was required for the activation of CHK1 in response to DNA replication blocks and stalled replication forks, a function that is conserved in humans (Clarke and Clarke, 2005). The interaction between CHK1 and CLASPIN has also been demonstrated to play a role in the checkpoint response to UV-induced DNA damage (Jeong et al., 2003). CLASPIN is required for the ATR-dependent phosphorylation of CHK1 (Kumagai et al., 2004) leading to its activation, and furthermore, the ATR-dependent phosphorylation of CHK1 has been demonstrated to be directly stimulated by CLASPIN in an in vitro reconstituted system using recombinant human proteins (Lindsey-Boltz et al., 2009).
CLASPIN is predicted to be largely disordered (Figure S1A), forming a natively unfolded protein (Wright and Dyson, 2015) with three repeated linear motifs that mediate the interaction with CHK1 (Chini and Chen, 2006; Jeong et al., 2003; Kumagai and Dunphy, 2003). These sites in CLASPIN have been reported to be phosphorylated by CHK1 itself (Chini and Chen, 2006) but also through a CHK1-independent route (Bennett et al., 2008) that involves casein kinase 1 (CK1) (Meng et al., 2011). More recently an additional pathway has been described in which the three sites are phosphorylated by CDC7 (Yang et al., 2019). Previous studies implicated a site in CHK1-KD, centered around Arg-129, Thr-153, and Arg-162, required for binding to the three phosphorylation sites in CLASPIN in immunoblot experiments (Jeong et al., 2003).
Here we biochemically characterize the phosphorylation-mediated CLASPIN-CHK1 interaction and determine the crystal structure of a complex between CHK1-KD and a phosphorylated CLASPIN peptide. Our results reveal the molecular details of this interaction in the context of CHK1 activation and uncover a new role for an evolutionarily conserved phosphate binding pocket in the kinase domain, which is adapted to multiple functions in different kinase families.
Results
A single site in CHK1-KD can bind to the phosphorylated CLASPIN motifs
To characterize the interactions between CHK1 and the three repeated phosphorylation sites in CLASPIN (Figure 1A), we synthesized phosphopeptides corresponding to the three different repeats centered on pT916, pS945, and pS982, and measured their binding to CHK1-KD using fluorescence polarization (FP). All three peptides independently bound the kinase domain with affinities in the high nanomolar range (Figure 1B), with the tightest interaction between the kinase domain and the peptide encapsulating the pS945 site. The interaction was abrogated when treated with λ-phosphatase (Figure 1C), demonstrating that the interaction is indeed phosphorylation dependent. Mutations in residues that contribute to the binding of a sulfate ion in the original structure of the CHK1-KD, Arg-129 and Arg-162 (Chen et al., 2000), also abolished the interaction (Figure 1D).
Figure 1.

In vitro characterization of CHK1-CLASPIN interaction
(A) Schematic representation of CHK1 and CLASPIN and the sequences of the three repeated CHK1 binding motifs in CLASPIN. The underlined regions correspond to the sequences used to synthesize fluorescein-labeled peptides, and the sequence consensus is shown. See also Figure S1.
(B) Fluorescence polarization experiments show binding between a CHK1 construct consisting of only the kinase domain (CHK1-KD) and the three motifs in CLASPIN. All error bars are one standard error of the mean.
(C) Treatment with λ-phosphatase abolishes binding, confirming that the interaction is specific for the phosphorylated CLASPIN peptides.
(D) R129A and R162E mutations, which are in residues involved in sulfate binding in CHK1, also abolish interaction with the phosphorylated peptides, demonstrating that this is the only site responsible for binding to each of the individual motifs in CLASPIN.
Crystal engineering
Analysis of CHK1-KD structures deposited in the Protein Databank (PDB) reveals two different lattices in which the protein crystallizes (Figure 2A). However, attempts to either soak a phosphorylated CLASPIN peptide into crystals of CHK1-KD or co-crystallize a complex of CHK1-KD with a CLASPIN peptide were unsuccessful, yielding crystals of CHK1 alone or in complex with staurosporine in both of the previously observed crystal forms. Examination of the molecular packing (Figure 2B) in these crystal forms suggested that the region surrounding the putative phosphate binding site was potentially occluded by contacts with other molecules in the crystal lattice, and this was likely to interfere with binding of a CLASPIN motif in that region (confirmed once the structure of the complex had been determined). In an attempt to relieve this potential restriction, we made a point mutation changing Asp-10 to an Arg residue, distant from both the active site and the putative CLASPIN interaction interface, intended to disrupt a salt bridge contact in the lattice (Figure 2C), thereby destabilizing the predominantly observed crystal form and allowing new packing variants to be produced.
Figure 2.

CHK1 lattice engineering
(A) Summary of CHK1-KD structures in the PDB; cell dimensions are averages across the indicated number of structures in each case.
(B) The asymmetric unit of PDB entry 1NVR, representative of the predominant lattice, contains a single copy of the kinase domain, shown in yellow, with symmetry-related molecules shown here in gray. The phosphorylated CLASPIN binding site is marked with a sulfate ion in the structure, while the active site is occupied by the presence of staurosporine.
(C) Details for the salt bridge between Asp-10 and Arg-181 that is disrupted by the D10R mutation shown alongside its location with respect to the active site, marked by staurosporine, and putative CLASPIN interaction surface, marked with the sulfate ion.
Crystal structure of CHK1 bound to ATPγS
In an attempt to produce a CLASPIN-bound crystal structure of CHK1-KD, various combinations of nucleotides, inhibitors, and peptides were combined and set up in crystallization trials. One of these attempts, where the CLASPIN peptide and ATPγS were included, yielded a structure of the kinase domain bound to the nucleotide alone. The kinase domain is in a conformation similar to that seen in the two known crystal forms, with the nucleotide binding site, activation loop, and catalytic residues ordered and positioned in an active, but open, conformation (Figure 3A). There is clear density for the ATPγS with the ribose in a C3-endo conformation (Figure 3B). This is in contrast to a reported structure of an AMPPNP-bound complex in which the ribose was in the C5-endo conformation and the phosphate groups could not be seen (Chen et al., 2000). However, the coordinates and diffraction data for that structure have never been deposited in the PDB, so the reliability of that observation cannot be ascertained. In the ATPγS complex, two magnesium ions, chelated by residues Asn-135 and Asp-148, can be seen in the active site balancing the charge on the phosphate groups of the nucleotide. Whereas the phosphate groups can be unambiguously placed in this structure, the γ-phosphate is not positioned in the presumed correct location for catalysis, with the catalytic Asp-130 residue situated ∼8 Å from the bridging thioether of the ATPγS (Figure 3C). This is not dissimilar to CDK2, where structures of several activation states are known, including both an active open state (Russo et al., 1996), where the activation loop is ordered but the catalytic aspartate is similarly distant from the nucleotide as for this structure, and an active closed confirmation (Bao et al., 2011), representing the transition state of the enzyme. The conformation of CHK1 seen here may be due to constraints imposed by the crystal lattice or potentially represents a nucleotide-bound open conformation formed prior to substrate peptide binding, which might promote full closure of the active site to bring the aspartate residue into the correct position for catalysis to occur. A substrate peptide-bound structure would help to distinguish between these possibilities.
Figure 3.

Nucleotide-bound CHK1 structure
(A) Superposition of the ATPγS-bound CHK1 structure (yellow) with an apo CHK1 structure, 1IA8 (gray); both the overall arrangement of the N and C kinase lobes and the details of key active-site residues are identical between the two models.
(B) 2mFo-DFc map showing electron density at 2σ for ATPγS in the structure.
(C) Details of the distance between the catalytic Asp-130 residue and the γ-phosphate of the ATPγS.
Crystal structure of CHK1 bound to CLASPIN
A further trial, where the kinase domain was mixed with the CLASPIN peptide and the broad-spectrum ATP-competitive kinase inhibitor staurosporine, produced a structure in which both the inhibitor and the peptide were present. There are two independent copies of the kinase domain in the asymmetric unit of this crystal lattice, and while both of the CHK1 molecules are able to accommodate the CLASPIN peptide, in one of the chains the peptide is constrained by the lattice and likely adopts an artifactual conformation where the peptide interacts with multiple CHK1 molecules (Figure S2). The CLASPIN peptide bound to the other chain adopts a position and conformation situated between, and making interactions with, the C helix and the activation loop of a single kinase domain (Figure 4A), with density for the peptide visible in an omit map (Figure 4B).
Figure 4.

CLASPIN-bound CHK1 structure
(A) Structure of a CLASPIN pS945 peptide (purple) bound to CHK1-KD (yellow) with staurosporine bound in the active site. See also Figure S2.
(B) A 2mFo-DFc omit map showing density at 2σ for the CLASPIN peptide in the structure.
(C) Superposition of the CLASPIN pS945 CHK1 structure (yellow) with a staurosporine-bound CHK1 structure 1NVR (gray). The overall arrangements of the N and C kinase lobes are identical between the two models.
(D) Details of the interaction with the phosphate group of the bound CLASPIN peptide. Residues in the peptide are labeled in bold.
(E) Details of the hydrophobic interactions made between the kinase domain and the CLASPIN peptide.
(F) Fluorescence polarization experiments show that binding between the CLASPIN peptide and CHK1 can be abrogated by a mutation in the CLASPIN sequence. All error bars are one standard error of the mean.
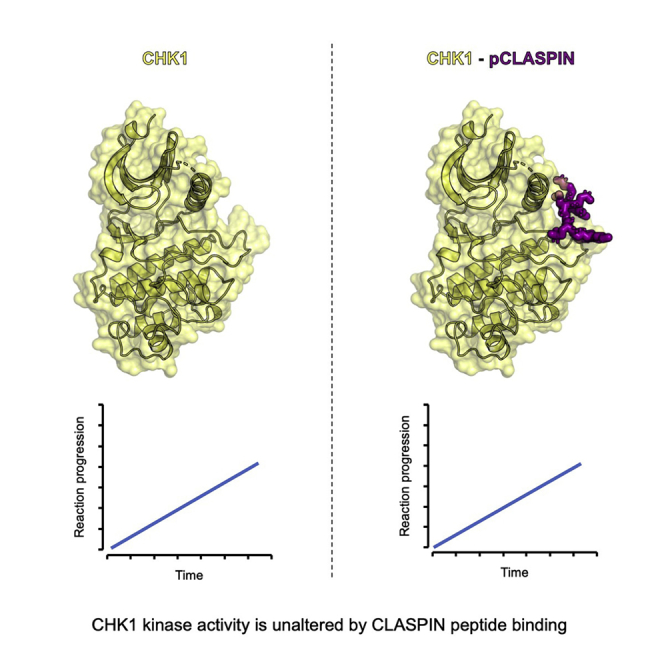
Notably, the overall positioning of the kinase domain lobes with respect to each other, and the active-site architecture, is the same as previously determined apo or inhibitor-bound structures (Figure 4C), with an overall root-mean-square deviation of 0.252 Å for all protein atoms from a staurosporine-bound structure (Zhao et al., 2002). This suggests that CLASPIN binding does not play a significant role in aligning the active-site residues into a catalytically active conformation.
The phosphorylated state of the CLASPIN peptide is recognized by the side chains of CHK1 Lys-54, Arg-129, Thr-153, and Arg-162, coordinating the phosphate group of CLASPIN pSer-945, with additional H-bond interactions between the main-chain carbonyl of CLASPIN pSer-945 and the side chain of CHK1 Arg-162 (Figure 4D). Specificity for the CLASPIN sequence beyond the phosphorylation is provided by a series of predominantly hydrophobic interactions (Figure 4E). Leu-943 of CLASPIN sits between the side chains of CHK1 Lys-53, Lys-54, and Cys-57, while CLASPIN Leu-940, Leu-941, and Phe-948 protrude into a hydrophobic groove on the surface of the kinase domain lined by the side chains of CHK1 Met-61, Val-154, Tyr-157, and Arg-162.
To validate the CLASPIN binding mode seen in the crystal structure, a peptide with a point mutation in the phenylalanine conserved between the three motifs, and shown from the crystal structure to be important for the interaction, was synthesized, and its binding to CHK1-KD was measured using FP. The mutation was found to significantly lower the affinity of the interaction in the FP assay (Figure 4F).
CLASPIN binding is uncoupled from CHK1 regulation
The position of the CLASPIN binding site on CHK1 that our study reveals is distant from the proposed CHK1-KA1 domain binding site based on models derived from the MARK1 kinase structure (Emptage et al., 2018) (Figure 5A), but in a similar location compared with the cyclin binding site in CDKs (Russo et al., 1996). This suggests that CLASPIN binding would be unaffected by the KA1 domain-regulated activation state of CHK1. To test this, we purified full-length CHK1, treated it with λ-phosphatase to remove any activating phosphorylation, and measured its affinity for the CLASPIN phosphopeptide in an FP assay (Figure 5B). As predicted, the CLASPIN peptide still bound KA1-autoinhibited CHK1, but with 2-fold lower affinity than for CHK1-KD, indicating some negative cooperativity between KA1-domain and CLASPIN binding. However, the relative weakness of the effect suggests that it is unlikely to be biologically significant.
Figure 5.

CHK1-CLASPIN interaction does not directly affect kinase activity
(A) Model for the interaction of the KA1 domain (orange) with the kinase domain (yellow) for CHK1 produced by superposing both the KA1 domain and the CLASPIN-bound kinase domain on the KA1-autoinhibited MARK1 kinase structure 6C9D. Fluorescence polarization experiments showing CLASPIN peptide interactions with full-length CHK1 or a CHK1 construct consisting of only the kinase domain are presented. The CLASPIN peptide binds with comparable affinity to both constructs.
(B) Fluorescence polarization experiments showing affinities of the CLASPIN pS945 peptide for full-length CHK1 or the CHK1 kinase domain alone. The presence of the KA1 regulatory region of CHK1 has a minimal effect on the affinity for the CLASPIN peptide. All error bars are one standard error of the mean.
(C) Fluorescence polarization experiments showing CDC25C substrate peptide interactions with a CHK1 construct consisting of only the kinase domain, in the presence or absence of the CLASPIN pS945 peptide. The presence of the CLASPIN peptide has no significant effect on the affinity of the kinase for the substrate peptide.
(D) Reaction rates for NADH-coupled assays showing ATP turnover by either full-length CHK1 or a CHK1 construct consisting of only the kinase domain in the presence of increasing concentrations of the CLASPIN pS945 peptide. Taken together with (C), these data show that CLASPIN binding has no allosteric effect on CHK1 kinase function. See also Figures S3A–S3D.
(E) NADH-coupled ATPase assay for CHK1-KD incubated with a CDC25C substrate, mutant S216A CDC25C substrate, and CLASPIN S945 substrate. These data show that the CLASPIN peptide itself is not a substrate for CHK1 kinase, contrary to previous suggestions (Chini and Chen, 2006). See also Figure S3E.
CLASPIN binding does not affect CHK1 activity
The coincidence of the CLASPIN binding site in CHK1 with cyclin binding to CDKs suggests that CLASPIN, like cyclins, might modulate the affinity of the kinase for its substrate and/or affect its catalytic activity. To test this, we used FP to determine the affinity of a CDC25C substrate peptide for CHK1-KD in the presence or absence of the pS945 CLASPIN peptide, but saw no significant difference (Figure 5C), suggesting that the CHK1-CLASPIN interaction does not modulate the affinity of CHK1-KD for substrate.
To determine whether CLASPIN binding has an allosteric effect on CHK1 kinase activity, we used an NADH-coupled ATPase assay to determine the rate of ATP turnover in the presence of a substrate peptide corresponding to the CHK1 phosphorylation site in CDC25C. Under the conditions tested, the rate of the reaction was 2.25 mol min−1 mol−1 for the full-length kinase and 7.73 mol min−1 mol−1 for the kinase domain alone. For both full-length CHK1 and CHK1-KD, the addition of the CLASPIN pS945 peptide had no effect on the rate of ATP turnover of the reaction in the presence of the CDC25C peptide (Figure 5D). To show that the ATP turnover measured was the result of the phosphorylation of Ser-216 in the CDC25C peptide, rather than some other activity, a CDC25C substrate peptide in which Ser-216 was replaced with an alanine was used, which showed no activity in the assay. We also looked at whether the CLASPIN peptide itself could be phosphorylated by CHK1, as has been suggested (Chini and Chen, 2006), but saw no stimulation of ATP turnover by CHK1 in the presence of the unphosphorylated S945 CLASPIN peptide (Figure 5E).
Discussion
The results presented here provide a reliable model for the nucleotide-bound state of CHK1-KD and reveal the molecular basis of the phosphorylation-dependent interaction between CHK1 and CLASPIN. Our data show that CLASPIN binding has no significant direct effect on the regulation or kinase activity of CHK1, but suggest that CLASPIN acts as a pure scaffold, facilitating the recruitment of CHK1 into a larger complex in which it can be activated through phosphorylation by ATR, downstream of DNA damage.
The interactions of the bound CLASPIN motif with CHK1 explain the pattern of small hydrophobic residues at positions −1, −2, −4, and −5 relative to the phosphorylated Ser/Thr, and a glycine at +1 and phenylalanine at +3, all of which are very strongly conserved in the three repeats of this motif that occur throughout the vertebrates. As our structural and biochemical data show that a single copy of this motif is sufficient for a high-affinity interaction with a unique site on CHK1, the apparent biological need for a triple tandem array of these motifs in CLASPIN is far from clear. Pull-down studies of Xenopus CLASPIN, in which the third repeat is less strongly conserved, indicated some possible cooperativity in CHK1 binding between the first two sites (Kumagai and Dunphy, 2003), favoring binding of multiple CHK1 molecules to a single CLASPIN. Focal accumulation of CHK1 at sites of ATR activation can be observed in cells following DNA damage (Bigot et al., 2019), and recruitment of multiple CHK1 molecules by each CLASPIN might be a means of generating a high concentration of CHK1 activity at sites of DNA damage.
Clustering of CHK1 molecules by CLASPIN could also play a role in the second stage of the CHK1 kinase activation process, which requires autophosphorylation at two consensus CHK1-phosphorylation motifs and at least one non-consensus site within the KA1 domain, subsequent to phosphorylation by ATR (Gong et al., 2018; Kasahara et al., 2010; Okita et al., 2012). Autophosphorylation mediated by phosphorylation-dependent clustering downstream of phosphorylation by ATM also underlies activation of the other checkpoint kinase, CHK2 (Ahn et al., 2002; Oliver et al., 2006). However, the two processes are mechanistically distinct, as CHK1 autophosphorylation occurs in an ancillary domain and may be partly intramolecular (Okita et al., 2012), while trans-autophosphorylation of CHK2 and related kinases occurs within the kinase domain itself and is strictly intermolecular (Oliver et al., 2007; Pike et al., 2008).
An alternative function for the tandem motifs of CLASPIN is as an internal scaffold, whereby prior phosphorylation of one motif that engages with the CLASPIN binding site on CHK1 facilitates binding of an adjacent unphosphorylated motif to the active site of CHK1 as a substrate, by increasing its local concentration. Phosphorylation of CLASPIN by CHK1 in vitro has been reported (Chini and Chen, 2006), but subsequent studies show that CHK1 is not required for CLASPIN phosphorylation in vivo (Bennett et al., 2008), where this function is fulfilled by CK1γ1 (Meng et al., 2011) and CDC7 (Yang et al., 2019). Our in vitro data show that an isolated unphosphorylated CLASPIN motif displays negligible affinity for CHK1 and no significant activity as a kinase substrate. Although we cannot completely rule out the possibility that CHK1 might phosphorylate a longer CLASPIN peptide spanning two or more motifs, where one is already phosphorylated, the lack of even a weak affinity or activity for the isolated unphosphorylated CLASPIN CHK1-binding motif argues against this.
CLASPIN utilizes a conserved phosphate binding pocket
The site occupied by the phosphorylated Ser/Thr in CLASPIN’s CHK1 binding motifs is a phosphate binding pocket that serves at least four different functions across the different kinases within which it is evolutionarily conserved (Figure 6). In all cases the residues that coordinate a bound phosphate group are highly conserved, but the rest of the binding site that provides the larger recognition process in each case is not. The most widely observed role for this site is to bind to the side chain of a phosphorylated threonine within the activation segment of the kinase and bridge it to the C helix within the N-terminal lobe (Endicott et al., 2012). Thus, for example, in CDK2, the side chains of Arg-50, Arg-126, and Arg-150 coordinate the phosphate group of pThr-160 of CDK2 (Figure 6B), thereby stabilizing the active conformation of the enzyme (Bao et al., 2011). The equivalent site in GSK3β (Arg-96, Arg-180, and Lys-205) functions in the specific recognition of “primed” substrates by coordinating the phosphate group on already phosphorylated residues within the substrate, positioned four residues downstream of the residue being phosphorylated, and thereby greatly enhances their affinity for the kinase and the efficiency of their phosphorylation (Dajani et al., 2001). This site is also used to regulate GSK3β kinase activity by facilitating binding of phosphorylated residues in inhibitory sequences, such as pSer-9 in the autoinhibitory N terminus of GSK3β itself, or pSer-1607 in low-density lipoprotein receptor-like protein 6 (LRP6) (Stamos et al., 2014) (Figure 6C), so that the upstream sequence occupies the active site and prevents substrate access. The structure of the CLASPIN-CHK1 complex described here (Figure 6D) adds a hitherto unknown role for this site, in providing a phosphorylation-dependent scaffold interaction that facilitates recruitment of the kinase to a specific cellular location.
Figure 6.

Varied roles of the conserved phosphate binding pocket
(A) Superposition of the kinase domains and interacting regions of CDK2 (pink), GSK3β (cyan), and CHK1 (yellow) showing location of the conserved phosphate binding pocket, marked with a phosphate group from the CHK1-CLASPIN structure presented here.
(B–D) Details of the interaction with phosphorylated residues in CDK2 (B), GSK3β (C), and CHK1 (D). The LRP6 inhibitor peptide bound to GSK3β is shown in pink and the CLASPIN scaffold peptide bound to CHK1 in purple.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| NEB® 5-alpha competent E. coli (high efficiency) | New England Biolabs | Cat#C2987H |
| Invitrogen™ MAX Efficiency™ DH10Bac competent cells | Fisher Scientific | Cat#10592663 |
| Chemicals, peptides, and recombinant proteins | ||
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | 000000005056489001; COEDTAF-RO ROCHE |
| Lambda protein phosphatase | New England Biolabs | Cat#P0753S |
| TURBO™ DNase | Fisher Scientific | Cat#10722687 |
| Staurosporine | New England Biolabs | Cat#9953 |
| ATPγS | Sigma-Aldrich | Cat#A1388 |
| PACT screen | Molecular dimensions | Cat#MD1-36 |
| CDC25C_210-227 | This paper (PPR Ltd) | Custom |
| CDC25C_S216A_210-227 | This paper (PPR Ltd) | Custom |
| Cy5-CDC25C | This paper (PPR Ltd) | Custom |
| Flu-CLASPIN_pT916 | This paper (PPR Ltd) | Custom |
| Flu-CLASPIN_pS945 | This paper (PPR Ltd) | Custom |
| Flu-CLASPIN_pS982 | This paper (PPR Ltd) | Custom |
| Flu-CLASPIN_F948A_pS945 | This paper (PPR Ltd) | Custom |
| CLASPIN_pS945 | This paper (PPR Ltd) | Custom |
| CLASPIN_S945 | This paper (PPR Ltd) | Custom |
| Critical commercial assays | ||
| Q5® site-directed mutagenesis kit | New England Biolabs | Cat#E0554S |
| Q5® high-fidelity 2X master mix | New England Biolabs | Cat#M0492L |
| Cellfectin II reagent | Fisher Scientific | Cat#10458833 |
| Deposited data | ||
| CHK1 kinase domain: ATPγS | This paper | PDB: 7AKM |
| CHK1 kinase domain: Staurosporine: CLASPIN pS945 | This paper | PDB: 7AKO |
| CHK1 kinase domain: Staurosporine, SO4 | (Zhao et al., 2002) | PDB: 1NVR |
| CHK1 kinase domain: SO4 | (Chen et al., 2000) | PDB: 1IA8 |
| CHK1 KA1 domain | (Emptage et al., 2017) | PDB: 5WI2 |
| MARK1 | (Emptage et al., 2018) | PDB: 6C9D |
| CDK2: Cyclin A | (Russo et al., 1996) | PDB: 1JST |
| GSK3: Axin | (Dajani et al., 2003) | PDB: 4NM7 |
| Experimental models: cell lines | ||
| Gibco® Sf9 cells | Thermofisher | Cat#11496015 |
| Sf-900 II SFM | Thermofisher | Cat#10902096 |
| Recombinant DNA | ||
| Plasmid: pFastBAC1 STREP-(3C)-CHK1-HIS | This paper | NA |
| Plasmid: pFastBAC1 STREP-(3C)-CHK1(1–290)-HIS | This paper | NA |
| Plasmid: pFastBAC1 STREP-(3C)-CHK1(1–290)_D10R-HIS | This paper | NA |
| Plasmid: pFastBAC1 STREP-(3C)-CHK1(1–290)_R129A_R162E-HIS | This paper | NA |
| Software and algorithms | ||
| Prism 6 for mac OS X (v6.0hr) | GraphPad Software |
https://www.graphpad.com RRID:SCR_002798 |
| SnapGene | GSL Biotech LLC |
http://www.snapgene.com/ RRID:SCR_015052 |
| PyMOL | Schrodinger LLC | PyMOL (RRID:SCR_000305) |
Resource availability
Lead contact
Further information and requests for resources and reagents should be direct to and will be fulfilled by the lead contact, LHP Laurence.pearl@sussex.ac.uk.
Materials availability
Plasmids generated in this study can be requested from the lead contact.
Data and code availability
Structure Factors and refined atomic coordinates have been deposited in the PDB with accession codes: PDB: 7AKM and PDB: 7AKO.
Experimental model and subject details
WT and mutant human CHK1 proteins were recombinantly expressed in SF9 insect cells. Cells were grown in 400 mL of Sf-900 II SFM media (Thermofisher, UK) at 27°C shaking at 140 RPM in 2 L bottles. Cells for expression were typically amplified by splitting at a density of 5 × 105 cells ml−1, once a density of 4 × 106 cells ml−1 had been reached, and infections with virus for expression were done once the cells had reached a density of 1.5 × 106 cells ml−1, with a multiplicity of infection of 5, before harvesting three days later.
Method details
Generation of bacmids and cell culture
Coding sequences for Strep and HIS tagged CHK1 full length and Kinase domain alone were synthesised and ligated into pFastBAC1 (Thermofisher, UK). Mutations were made using a Q5® Site-Directed Mutagenesis Kit (NEB, Hitchin, UK). Plasmids were transformed into DH10BAC cells (Fisher Scientific, UK) and bacmids were purified by standard procedures. Bacmids were transfected into SF9 cells (Thermofisher, UK) using Cellfectin II reagent (Fisher Scientific, UK), before viral amplification and protein expression.
Protein expression and purification
Recombinant full length CHK1, or kinase domain alone (CHK1-KD), was produced by infecting Sf9 cells with a baculovirus coding for a construct containing STREP-(3C)-CHK1-HIS or STREP-(3C)-CHK1(1–290)-HIS respectively. Cell pellets were re-suspended in lysis buffer containing 50mM HEPES pH 7.5, 200mM NaCl, 0.5mM TCEP, and supplemented with 10 U DNASE Turbo and protease inhibitor tablets, then disrupted by sonication, and the resulting lysate clarified by centrifugation at 40,000 x g for 60 min at 4°C. The supernatant was applied to a 5mL HiTrap TALON crude column (GE Healthcare, Little Chalfont, UK), washed first with buffer containing 50mM HEPES pH 7.5, 500mM NaCl, 0.5mM TCEP, followed lysis buffer supplemented with 10 mM 10mM imidazole, with any retained protein then eluted by application of the same buffer but now supplemented with 250mM imidazole. The eluted protein was diluted with lysis buffer before application to a 5mL HiTrap STREP column (GE Healthcare, Little Chalfont, UK), washed with lysis buffer and eluted using buffer supplemented with 2 mM Desthiobiotin.
For FP experiments, a Superdex 75 16/60 size exclusion column (GE Healthcare, Little Chalfont, UK) was used to purify the protein to homogeneity in 25mM HEPES pH 7.5, 200mM NaCl, 1mM EDTA, 0.25mM TCEP, 0.02% (v/v) Tween 20.
For crystallographic studies, the N-terminal STREP tag was removed by incubation with GST-3C protease (in house) for 12 hr at 4°C. A Superdex 75 16/60 size exclusion column (GE Healthcare, Little Chalfont, UK) was used to purify the kinase domain to homogeneity in 10mM HEPES pH 7.5, 200mM NaCl, 0.5mM TCEP.
Fluorescence polarisation experiments
Fluorescein-labelled (Flu-pT916 Flu-GGMDELLDLC(pT)GKFTSQ, Flu-pS945 Flu-GGMEELLNLC(pS)GKFTSQ, Flu-pS945_F948A Flu-GGMEELLNLC(pS)GKATSQ and Flu-pS982 Flu-GGMEEALALC(pS)GSFPTD) or CY5-labelled (Cy5-CDC25C-S216 Cy5-CGYGGLYRSPSMPENLNRPRLK) peptides (Peptide Protein Research Ltd, Bishops Waltham, UK) at a concentration of 100nM, or for the double peptide experiment 200 nM Cy5-CDC25C and 1000 nM CLASPIN pS945, were incubated at room temperature with increasing concentrations of CHK1-KD, or lambda phosphatase treated full length CHK1 in 25mM HEPES pH 7.5, 200mM NaCl, 1mM EDTA, 0.25mM TCEP, 0.02% (v/v) Tween 20 in a black 96-well polypropylene plate (VWR, Lutterworth, UK). Incubation with Lambda phosphatase (NEB, Hitchin, UK) supplemented with 1 mM MnCl2 was used to remove the phosphorylation on the peptides. Fluorescence polarisation was measured in a POLARstar Omega multimode microplate reader (BMG Labtech GmbH, Offenburg, Germany). Binding curves represent the mean of 3 independent experiments, with error bars of 1 standard deviation.
Crystallography
The pure CHK1-KD was mixed with a fifty molar excess of ATPγS (Sigma-Aldrich, Gillingham, UK) and a five molar excess of both a substrate peptide corresponding to residues 210–227 of CDC25 (GLYRSPSMPENLNRPRLK) and a pS945 CLASPIN peptide (MEELLNLC(pS)GKFT(pS)QD) (Peptide Protein Research Ltd, Bishops Waltham, UK) prior to concentration to 8 mg/mL for use in crystallisation trials. Crystals grew in condition PACT H11 (200 mM Sodium citrate tribasic dihydrate, 100 mM Bis-Tris propane pH 8.5 and 20% w/v PEG 3350) and following further optimisation of this condition single crystals were looped, soaked and cryoprotected in well solution supplemented with 30% (v/v) Ethylene Glycol before flash freezing in liquid nitrogen.
Staurosporin (NEB, Hitchin, UK) and the pS945 CLASPIN peptide (MEELLNLC(pS)GKFT(pS)QD) (Peptide Protein Research Ltd, Bishops Waltham, UK) were mixed with the pure CHK1-KD at two and five molar excess respectively prior to concentration to 8 mg/mL for use in crystallisation trials. Crystals grew in condition PACT F11 (200 mM Sodium citrate tribasic dihydrate, 100 mM Bis-Tris propane pH 6.5 and 20% w/v PEG 3350) and following further optimisation of this condition single crystals were looped, soaked and cryoprotected in well solution supplemented with 30% Ethylene Glycol and 10 mM peptide before flash freezing in liquid nitrogen. Data were collected on beamline I24 at the Diamond Synchrotron Lightsource and the structure was determined using PHASER (McCoy, 2017) to perform molecular replacement with PDB 1IA8 as a search model before refinement using the PHENIX software package (Headd et al., 2012). The structure was edited between refinements using COOT (Emsley et al., 2010) and the model quality and results of individual rebuilding/refinement cycles were evaluated using Molprobity (Chen et al., 2010) throughout the process.
Generation of protein structure figures
All figures were produced using PyMOL (v 2.2.2, Schrodinger LLC). For Figure 5A the CLASPIN bound kinase domain model from this paper, and the CHK1 KA1 domain structure (PDB 5WI2) were superposed on the KA1-autoinhibited MARK1 kinase structure (PDB 6C9D). For Figure 6A the kinase domains and interacting regions from CDK2 (PDB 1JST) and GSK3β-LRP6 complex (PDB 4NM7) were superposed onto the CHK1-CLASPIN structure.
ATPase assay
CHK1, to a concentration of 100 nM for the kinase domain or 200 nM for full length CHK1, was added to reaction mixtures containing Pyruvate Kinase and Lactic Dehydrogenase enzymes (Sigma-Aldrich, Gillingham, United Kingdom) with 0.010–0.016, and 0.015–0.023 units of activity respectively in 25 mM HEPES pH 7.5, 150 mM NaCl, 0.5mM TCEP, 0.5 mM PEP, 0.5 mM NADH, 1 mM ATP with 0.5 mM CDC25C (210–227) peptide (GLYRSPSMPENLNRPRLK) or CDC25C S216A (210–227) peptide (GLYRSPSMPENLNRPRLK) or CLASPIN S945 peptide (MEELLNLCSGKFTSQ) and with or without CLASPIN pS945 peptide (MEELLNLC(pS)GKFTSQ).(Peptide Protein Research Ltd, Bishops Waltham, UK) at different concentrations in a Thermoscientific 96 well UV microplate (Fisher scientific, Loughborough, UK). The plates were incubated at 25°C and absorbance at 340 nm was measured in a POLARstar Omega multimode microplate reader (BMG Labtech GmbH, Offenburg, Germany) at intervals of 30 s.
Quantification and statistical analysis
Protein disorder prediction
The amino acid sequence was submitted to the SPOT-Disorder-single online server (Hanson et al., 2019).
Fluorescence polarisation experiments
All data were fitted by non-linear regression, to a one site – specific binding model in Prism 6 for Mac OS X (v 6.0d, GraphPad Software) in order to calculate the reported disassociation constants (Kd).
RMSD calculations
Superposition and RMSD calculations were performed in PyMOL (v 2.2.2, Schrodinger LLC).
ATPase assay
All data were fitted by linear regression in Prism 6 for Mac OS X (v 6.0d, GraphPad Software) in order to calculate the reported rates. For Figure 5D the fitted data can be seen in Figure S3 along with residual plots for the fitted lines for this data and Figure 5E.
Acknowledgments
We thank Mark Roe for assistance with X-ray data collection and both Lihong Zhou and Raquel Arribas for help with baculovirus protein expression. We thank Dr. Neil Kad (University of Kent) for details of the NADH-coupled ATPase assay. We are grateful to Diamond Light Source Ltd. (Didcot, UK) for access to synchrotron radiation and to the Wellcome Trust for support for X-ray diffraction facilities at the University of Sussex. This work was supported by Cancer Research UK program grants C302/A14532 and C302/A24386 (A.W.O. and L.H.P.).
Author contributions
Conceptualization, M.D., A.W.O., and L.H.P.; methodology, M.D., A.W.O., and L.H.P.; validation, M.D., A.W.O., and L.H.P.; formal analysis, M.D., A.W.O., and L.H.P.; investigation, all authors; writing – original draft, M.D.; writing – review & editing, M.D., A.W.O., and L.H.P.; visualization, M.D.; supervision, L.H.P. and A.W.O.; funding acquisition, L.H.P. and A.W.O.
Declaration of interests
The authors declare no competing interests.
Published: March 30, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.str.2021.03.007.
Contributor Information
Antony W. Oliver, Email: antony.oliver@sussex.ac.uk.
Laurence H. Pearl, Email: laurence.pearl@sussex.ac.uk.
Supplemental information
References
- Ahn J.Y., Li X., Davis H.L., Canman C.E. Phosphorylation of threonine 68 promotes oligomerization and autophosphorylation of the Chk2 protein kinase via the forkhead-associated domain. J. Biol. Chem. 2002;277:19389–19395. doi: 10.1074/jbc.M200822200. [DOI] [PubMed] [Google Scholar]
- Bao Z.Q., Jacobsen D.M., Young M.A. Briefly bound to activate: transient binding of a second catalytic magnesium activates the structure and dynamics of CDK2 kinase for catalysis. Structure. 2011;19:675–690. doi: 10.1016/j.str.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett L.N., Larkin C., Gillespie D.A., Clarke P.R. Claspin is phosphorylated in the Chk1-binding domain by a kinase distinct from Chk1. Biochem. Biophys. Res. Commun. 2008;369:973–976. doi: 10.1016/j.bbrc.2008.02.154. [DOI] [PubMed] [Google Scholar]
- Bigot N., Day M., Baldock R.A., Watts F.Z., Oliver A.W., Pearl L.H. Phosphorylation-mediated interactions with TOPBP1 couple 53BP1 and 9-1-1 to control the G1 DNA damage checkpoint. Elife. 2019;8:e44353. doi: 10.7554/eLife.44353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Luo C., Deng Y., Ryan K., Register J., Margosiak S., Tempczyk-Russell A., Nguyen B., Myers P., Lundgren K. The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell. 2000;100:681–692. doi: 10.1016/s0092-8674(00)80704-7. [DOI] [PubMed] [Google Scholar]
- Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chini C.C., Chen J. Repeated phosphopeptide motifs in human Claspin are phosphorylated by Chk1 and mediate Claspin function. J. Biol. Chem. 2006;281:33276–33282. doi: 10.1074/jbc.M604373200. [DOI] [PubMed] [Google Scholar]
- Clarke C.A., Clarke P.R. DNA-dependent phosphorylation of Chk1 and Claspin in a human cell-free system. Biochem. J. 2005;388:705–712. doi: 10.1042/BJ20041966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajani R., Fraser E., Roe S.M., Yeo M., Good V.M., Thompson V., Dale T.C., Pearl L.H. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. EMBO J. 2003;22:494–501. doi: 10.1093/emboj/cdg068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajani R., Fraser E., Roe S.M., Young N., Good V., Dale T.C., Pearl L.H. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Emptage R.P., Lemmon M.A., Ferguson K.M., Marmorstein R. Structural basis for MARK1 kinase autoinhibition by its KA1 domain. Structure. 2018;26:1137–1143 e1133. doi: 10.1016/j.str.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emptage R.P., Schoenberger M.J., Ferguson K.M., Marmorstein R. Intramolecular autoinhibition of checkpoint kinase 1 is mediated by conserved basic motifs of the C-terminal kinase-associated 1 domain. J. Biol. Chem. 2017;292:19024–19033. doi: 10.1074/jbc.M117.811265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott J.A., Noble M.E., Johnson L.N. The structural basis for control of eukaryotic protein kinases. Annu. Rev. Biochem. 2012;81:587–613. doi: 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- Gong E.Y., Hernandez B., Nielsen J.H., Smits V.A.J., Freire R., Gillespie D.A. Chk1 KA1 domain auto-phosphorylation stimulates biological activity and is linked to rapid proteasomal degradation. Sci. Rep. 2018;8:17536. doi: 10.1038/s41598-018-35616-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong E.Y., Smits V.A.J., Fumagallo F., Piscitello D., Morrice N., Freire R., Gillespie D.A. KA1-targeted regulatory domain mutations activate Chk1 in the absence of DNA damage. Sci. Rep. 2015;5:10856. doi: 10.1038/srep10856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson J., Paliwal K.K., Litfin T., Zhou Y. SPOT-Disorder2: improved protein intrinsic disorder prediction by ensembled deep learning. Genomics Proteomics Bioinformatics. 2019;17:645–656. doi: 10.1016/j.gpb.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headd J.J., Echols N., Afonine P.V., Grosse-Kunstleve R.W., Chen V.B., Moriarty N.W., Richardson D.C., Richardson J.S., Adams P.D. Use of knowledge-based restraints in phenix.refine to improve macromolecular refinement at low resolution. Acta Crystallogr. D Biol. Crystallogr. 2012;68:381–390. doi: 10.1107/S0907444911047834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer D.R., Rhind N. The intra-S checkpoint responses to DNA damage. Genes (Basel) 2017;8:74. doi: 10.3390/genes8020074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S.Y., Kumagai A., Lee J., Dunphy W.G. Phosphorylated claspin interacts with a phosphate-binding site in the kinase domain of Chk1 during ATR-mediated activation. J. Biol. Chem. 2003;278:46782–46788. doi: 10.1074/jbc.M304551200. [DOI] [PubMed] [Google Scholar]
- Kasahara K., Goto H., Enomoto M., Tomono Y., Kiyono T., Inagaki M. 14-3-3gamma mediates Cdc25A proteolysis to block premature mitotic entry after DNA damage. EMBO J. 2010;29:2802–2812. doi: 10.1038/emboj.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A., Dunphy W.G. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell. 2000;6:839–849. doi: 10.1016/s1097-2765(05)00092-4. [DOI] [PubMed] [Google Scholar]
- Kumagai A., Dunphy W.G. Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1. Nat. Cell Biol. 2003;5:161–165. doi: 10.1038/ncb921. [DOI] [PubMed] [Google Scholar]
- Kumagai A., Kim S.M., Dunphy W.G. Claspin and the activated form of ATR-ATRIP collaborate in the activation of Chk1. J. Biol. Chem. 2004;279:49599–49608. doi: 10.1074/jbc.M408353200. [DOI] [PubMed] [Google Scholar]
- Lindsey-Boltz L.A., Sercin O., Choi J.H., Sancar A. Reconstitution of human claspin-mediated phosphorylation of Chk1 by the ATR (ataxia telangiectasia-mutated and rad3-related) checkpoint kinase. J. Biol. Chem. 2009;284:33107–33114. doi: 10.1074/jbc.M109.064485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J. Acknowledging errors: advanced molecular replacement with phaser. Methods Mol. Biol. 2017;1607:421–453. doi: 10.1007/978-1-4939-7000-1_18. [DOI] [PubMed] [Google Scholar]
- Meng Z., Capalbo L., Glover D.M., Dunphy W.G. Role for casein kinase 1 in the phosphorylation of Claspin on critical residues necessary for the activation of Chk1. Mol. Biol. Cell. 2011;22:2834–2847. doi: 10.1091/mbc.E11-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida H., Katsuno Y., Banerjee B., Hande M.P., Nakanishi M. Specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Mol. Cell. Biol. 2007;27:2572–2581. doi: 10.1128/MCB.01611-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita N., Minato S., Ohmi E., Tanuma S., Higami Y. DNA damage-induced CHK1 autophosphorylation at Ser296 is regulated by an intramolecular mechanism. FEBS Lett. 2012;586:3974–3979. doi: 10.1016/j.febslet.2012.09.048. [DOI] [PubMed] [Google Scholar]
- Oliver A.W., Knapp S., Pearl L.H. Activation segment exchange: a common mechanism of kinase autophosphorylation? Trends Biochem. Sci. 2007;32:351–356. doi: 10.1016/j.tibs.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Oliver A.W., Paul A., Boxall K.J., Barrie S.E., Aherne G.W., Garrett M.D., Mittnacht S., Pearl L.H. Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. EMBO J. 2006;25:3179–3190. doi: 10.1038/sj.emboj.7601209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike A.C., Rellos P., Niesen F.H., Turnbull A., Oliver A.W., Parker S.A., Turk B.E., Pearl L.H., Knapp S. Activation segment dimerization: a mechanism for kinase autophosphorylation of non-consensus sites. EMBO J. 2008;27:704–714. doi: 10.1038/emboj.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A.A., Jeffrey P.D., Pavletich N.P. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 1996;3:696–700. doi: 10.1038/nsb0896-696. [DOI] [PubMed] [Google Scholar]
- Smits V.A., Gillespie D.A. DNA damage control: regulation and functions of checkpoint kinase 1. FEBS J. 2015;282:3681–3692. doi: 10.1111/febs.13387. [DOI] [PubMed] [Google Scholar]
- Stamos J.L., Chu M.L., Enos M.D., Shah N., Weis W.I. Structural basis of GSK-3 inhibition by N-terminal phosphorylation and by the Wnt receptor LRP6. Elife. 2014;3:e01998. doi: 10.7554/eLife.01998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P.E., Dyson H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.C., Kato H., Shindo M., Masai H. Cdc7 activates replication checkpoint by phosphorylating the Chk1-binding domain of Claspin in human cells. Elife. 2019;8:e50796. doi: 10.7554/eLife.50796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Bower M.J., McDevitt P.J., Zhao H., Davis S.T., Johanson K.O., Green S.M., Concha N.O., Zhou B.B. Structural basis for Chk1 inhibition by UCN-01. J. Biol. Chem. 2002;277:46609–46615. doi: 10.1074/jbc.M201233200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structure Factors and refined atomic coordinates have been deposited in the PDB with accession codes: PDB: 7AKM and PDB: 7AKO.