

Abstract
While kinases have been attractive targets to combat many diseases including cancer, selective kinase inhibition has been challenging due to the high degree of structural homology in the active site, where many kinase inhibitors bind. We have previously discovered that 8-anilino-1-naphthalene sulfonic acid (ANS) binds an allosteric pocket in cyclin-dependent kinase 2 (Cdk2). Here, we detail the positive cooperativity between ANS and orthosteric Cdk2 inhibitors dinaciclib and roscovitine, which increase the affinity of ANS towards Cdk2 5- to 10-fold, and the relatively non-cooperative effects of ATP. We observe these effects using a fluorescent binding assay and HSQC NMR, where we noticed a shift from fast exchange to slow exchange upon ANS titration in the presence of roscovitine but not with an ATP mimic. The discovery of cooperative relationships between orthosteric and allosteric kinase inhibitors could further the development of selective kinase inhibitors in general.
Graphical Abstract
Since the advent of imatinib to treat chronic myeloid leukemia (CML), there has been interest in developing selective kinase inhibitors to treat a variety of disease states, most notably cancer. Despite this excitement, selective kinase inhibition has been challenging in many cases because most inhibitors are orthosteric and bind the same active site of the kinase as ATP, a region of the protein structurally conserved among kinases. In contrast, allosteric inhibitors have the promise to bind less conserved regions away from the active site of the protein. Allosteric inhibitors can potentially exploit the differences in these regions to confer selectivity among kinases. Already, the FDA has approved three allosteric inhibitors to treat various cancers – the MEK inhibitors trametinib, cobemetinib, and binimetinib for BRAF-mutated melanoma (1–3).
Although selective kinase inhibitors have been developed, such as the allosteric MEK inhibitors and imatinib, many patients still develop drug resistance (4, 5). Combination treatment of both an orthosteric and allosteric inhibitor targeting the same kinase shows preclinical promise to thwart cancer resistance (6, 7). For example, the addition of the orthosteric covalent inhibitor osimertinib improved the binding and inhibitory effects of allosteric inhibitor JBJ-04-125-02 towards mutant EGFR using pull-down and cell proliferation assays (6). The probability of a kinase mutating twice within a single cancer cell to evade inhibition by both inhibitors is small. Traditionally, orthosteric and allosteric inhibitors were assumed to bind their respective sites independently. However, there is growing literature evidence that small molecules binding to the active or allosteric sites can have profound global conformational effects on the kinase, likely affecting the binding of ligands at distinct sites (8, 9).
Cyclin-dependent kinase 2 (Cdk2) has been an attractive clinical target to treat cancer. In particular, many cancers with historically poor prognoses overexpress partner protein cyclins A and E, which bind and activate Cdk2 (10–13). Furthermore, selective Cdk2 inhibitors have promise as a therapeutic with relatively few side effects compared to many other cancer therapies, as Cdk2 knock-out mice are viable but sterile (14, 15). Because of this knock-out phenotype, Cdk2 is additionally a non-hormonal contraceptive target for both men and women, as it is necessary for meiosis but not mitosis. Previous efforts to target Cdk2 have focused on the development of orthosteric inhibitors, which, however, possessed off-target kinase activity, especially against indispensable cell cycle regulators like Cdk1 (16), due to the structural homology of their active sites with that of Cdk2 (17).
We have previously reported that two molecules of the commercial dye 8-anilino-1-naphthalene sulfonic acid (ANS) bind an allosteric pocket of Cdk2 (18). To date, ANS remains the only molecule confirmed by crystallography to bind this allosteric pocket of Cdk2, although previous docking studies and other indirect evidence suggests that more drug-like compounds could also bind this site (19). ANS has the environmentally sensitive property that its fluorescent yield is increased in hydrophobic environments, like the allosteric pocket of Cdk2. Taking advantage of this property, an ANS fluorescent binding assay was developed and ANS was found to have moderate affinity to Cdk2 (KD = 37 μM), consistent with its low inhibitory potential towards active (i.e., cyclin-bound and phosphorylated) Cdk2 (IC50 = 91 μM) (18). Additionally, co-crystallographic studies revealed that it is possible for certain ATP-site inhibitors (or another molecule of ANS) to bind the active site while ANS occupies the allosteric pocket (18). However, the extent of both positive and negative cooperativity between ANS and orthosteric inhibitors has never been established.
Allosteric kinase inhibitors have shown various cooperativity relationships with ATP and orthosteric inhibitors. For example, the affinity of FDA-approved allosteric MEK inhibitors increases when ATP is bound, but they have exhibited neither positive nor negative cooperativity with orthosteric inhibitors. Conversely, asciminib and JBJ-04-125-02 have enhanced inhibitory activity in the presence of orthosteric inhibitors, although a difference in affinity has never been quantified (6). In this paper, we show and quantify the positively cooperative relationship between the binding of orthosteric inhibitors (roscovitine and dinaciclib) and the allosteric inhibitor ANS in Cdk2. We also show that the affinity of ANS towards Cdk2 is unchanged and non-cooperative with ATP.
A crystal structure of ANS bound to Cdk2 reveals two ANS molecules bound to the allosteric pocket – the 1st deeply buried in the pocket and the 2nd more solvent-exposed (18). In another crystal structure, an additional molecule of ANS binds the active site under conditions of high ANS concentration (Figure 1) (18). This implies that the affinity of ANS for the active site is weaker than the affinity of ANS to the allosteric pocket. Additionally, ANS does not contain a known hinge-binding motif that is characteristic of active site inhibitors. To further elucidate the binding affinity of the three ANS molecules for Cdk2, we docked ANS into Cdk2 at each of the respective binding sites. Our studies predicted that the ANS molecule deep in the 1st allosteric pocket binds the strongest with a docking score of −10.2 kcal/mol, followed by relatively weaker affinities for the active site and 2nd allosteric sites with docking scores of −8.2 and −6.7 kcal/mol, respectively (Figure 1). Furthermore, the binding pose of ANS was well recapitulated for the 1st deep allosteric and active site ANS molecules (RMSD < 0.4 Å), while the binding pose of the phenyl ring of ANS could not be recapitulated for the 2nd solvent exposed allosteric ANS molecule. This is consistent with previous computational results that similarly suggested ANS binds the deeper 1st allosteric pocket with higher affinity than it does the 2nd solvent exposed allosteric site (19). Because ANS is predicted to bind strongest to the 1st allosteric site, it is likely the increased fluorescence observed upon ANS binding to Cdk2 is due to the binding of this ANS molecule. A saturation binding curve constructed using ANS fluorescence intensity was monophasic, consistent with a single binding site with a KD = 37 μM (18).
Figure 1.
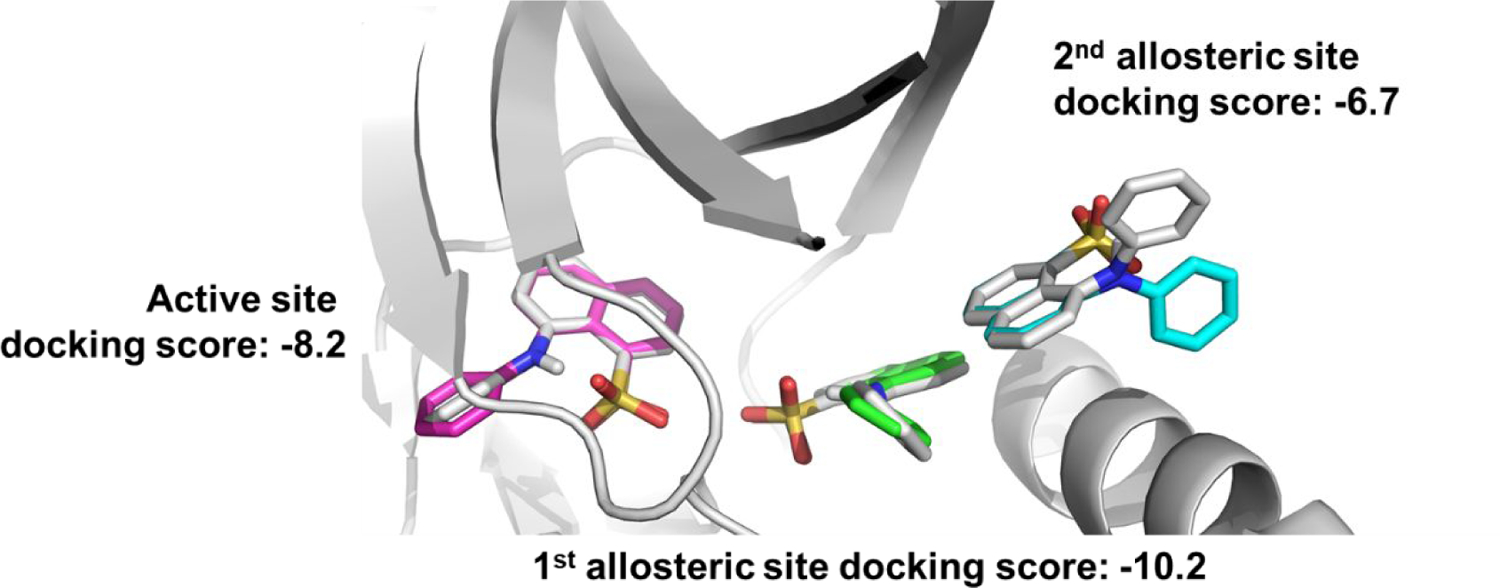
Docking ANS molecules into co-crystal structure with Cdk2 (gray, PDB ID: 3PXQ (18)). At high concentrations of ANS, 3 molecules of ANS are bound – two in allosteric sites (labeled 1st allosteric site and 2nd allosteric site) and another in the active site. The top docking scores and binding poses of each ANS molecule into its respective site (active site = magenta, 1st allosteric site = green, 2nd allosteric site = cyan) predicts different binding affinities of ANS for each site.
The ANS derivative p-Cl-ANS (Figure 2, panel A) has an improved environmentally-sensitive fluorescent nature compared to ANS due to an increase in both fluorescence intensity and affinity for Cdk2 (Figure 2, panel B) (18). The affinity of p-Cl-ANS was determined in saturation binding studies by the concentration-dependent fluorescence increase as was reported for ANS.(20) The KD of p-Cl-ANS for Cdk2 was 17 ± 3 μM (Figure 2, panel B), indicating an affinity about twice that reported for ANS (KD = 37 μM) (20). Nonspecific binding was determined in the presence of the orthosteric inhibitor SU9516 (50 M), which was established previously to noncompetitively block the specific binding of ANS to the allosteric site (20). In contrast to SU9516, which inhibits the binding of p-Cl-ANS, the orthosteric inhibitors roscovitine and dinaciclib stimulate the binding of p-Cl-ANS ~60–90% to the allosteric site determined in dose-response studies (Figure 2, panels C, D). This stimulation in p-Cl-ANS binding by both roscovitine and dinaciclib is completely blocked by the orthosteric inhibitor staurosporine, which was previously shown to have minimal effects on ANS binding (20). This confirms that roscovitine and dinaciclib bind to the ATP site. To determine the mechanism underlying this increase in binding, we conducted p-Cl-ANS saturation binding experiments in the presence of fixed concentrations of roscovitine and dinaciclib, which revealed that these ATP site ligands increase the affinity of p-Cl-ANS 5- to 10-fold to 3.4 ± 1.0 μM and 1.9 ± 0.8 μM, respectively (Figure 2, panel E). Therefore, the ATP site inhibitors roscovitine and dinaciclib enhance the binding of p-Cl-ANS by increasing its affinity to the ANS allosteric site, rather than by increasing the apparent number of binding sites. In contrast, both ATP and the non-hydrolyzable ATP site ligand adenylyl-imidodiphosphate (AMP-PNP) had little effect on the binding of p-Cl-ANS, suggesting that ATP itself is only weakly or not cooperative with p-Cl-ANS (Figure 2, panel F). The small apparent inhibition and stimulation of p-Cl-ANS by ATP and AMP-PNP at 1 mM, respectively, may indicate a weak interaction, as these effects were blocked by staurosporine. Based on the p-Cl-ANS fluorescent binding studies, ATP site inhibitors can be positively cooperative (roscovitine and dinaciclib), negatively cooperative (SU9516), or relatively non-cooperative (staurosporine) linked to the allosteric ANS site, whereas ATP and AMP-PNP are only weakly cooperative, if at all. However, overlaying the crystal structures of Cdk2 without inhibitor bound (apo-Cdk2), or bound to roscovitine, dinaciclib, staurosporine, or SU9516 did not show substantial differences in Cdk2 structure that would indicate why the affinity of p-Cl-ANS for Cdk2 would differ in the presence of these orthosteric inhibitors (Figure 3).
Figure 2.
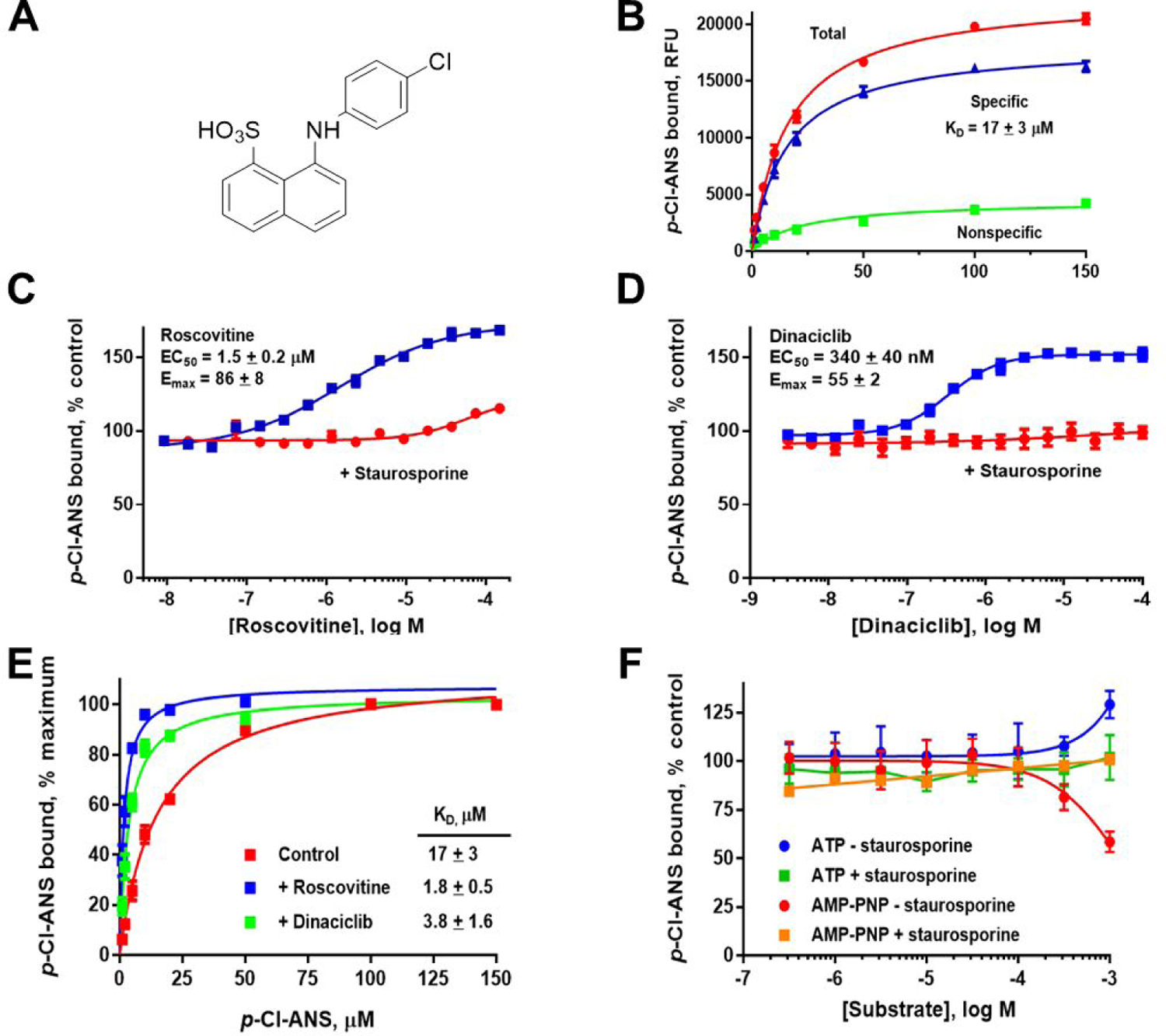
Cooperativity of orthosteric ligands and p-Cl-ANS as determined by fluorescent binding assay. A) The structure of p-Cl-ANS. B) p-Cl-ANS binds Cdk2 with an apparent KD of 17 ± 3 μM. C, D) Titrating in roscovitine or dinaciclib to Cdk2 in the presence of p-Cl-ANS increases the fluorescence of p-Cl-ANS, suggesting that these orthosteric inhibitors are positively cooperative with p-Cl-ANS binding. In contrast, increasing amounts of staurosporine had no effect on fluorescence, suggesting a non-cooperative binding relationship with p-Cl-ANS. E) The orthosteric inhibitors roscovitine and dinaciclib appear to increase the affinity of p-Cl-ANS to Cdk2 5- to 10-fold. F) ATP or AMP-PNP do not have much effect on the fluorescence of p-Cl-ANS bound to Cdk2, suggesting that ATP is relatively non-competitive and non-cooperative with p-Cl-ANS binding.
Figure 3.
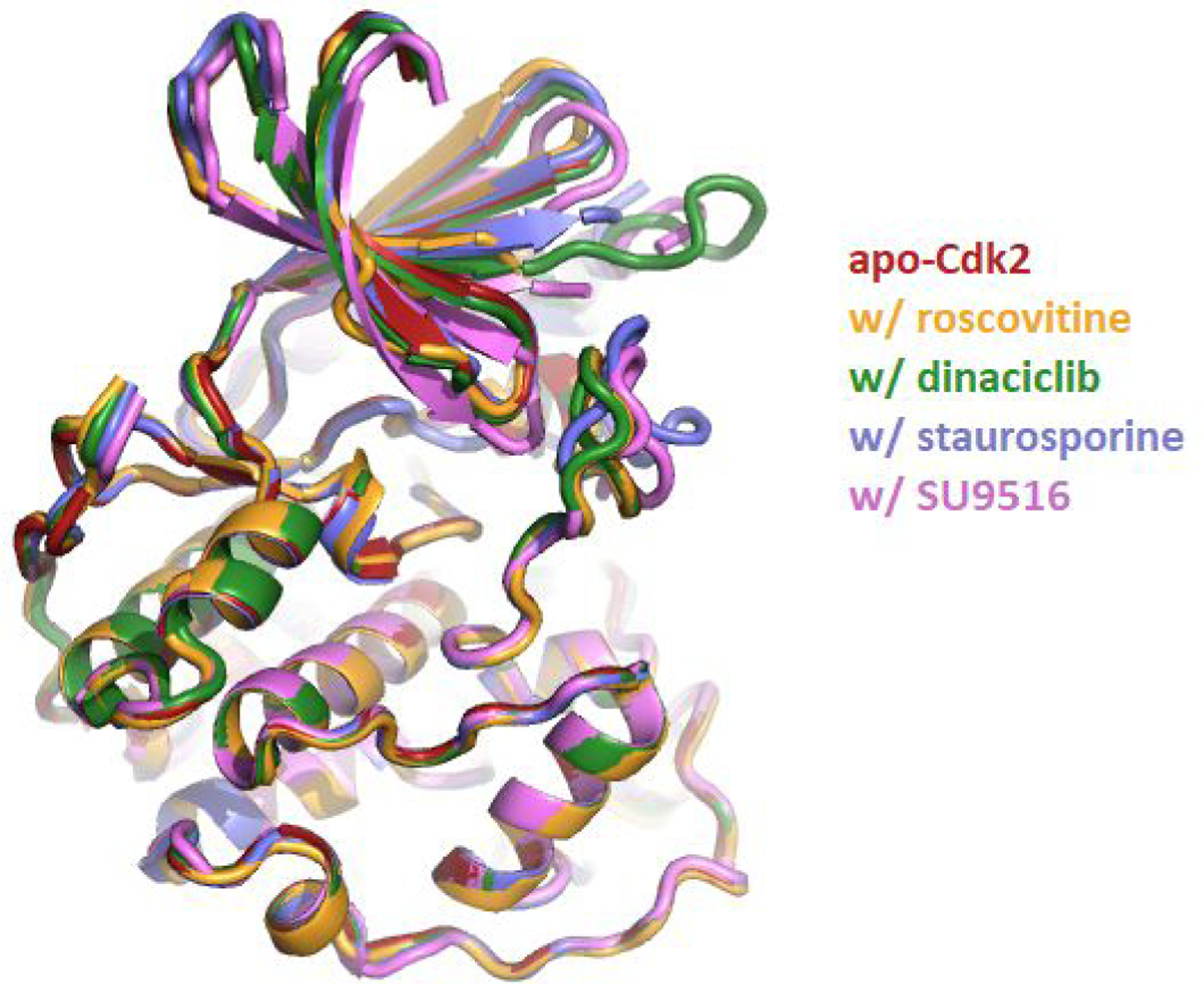
Crystal structure overlay of Cdk2 crystal structures. By color and PDB ID: apo-Cdk2 without inhibitor bound (red, PDB ID: 4EK3 (21)), bound to roscovitine (orange, PDB ID: 2A4L (22)), bound to dinaciclib (green, PDB ID: 4KD1 (23)), bound to staurosporine (blue, PDB ID: 4ERW (20)), and bound to SU9516 (violet, PDB ID: 3PY0 (18)). The RMSD between all five structures is less than 0.3 Å.
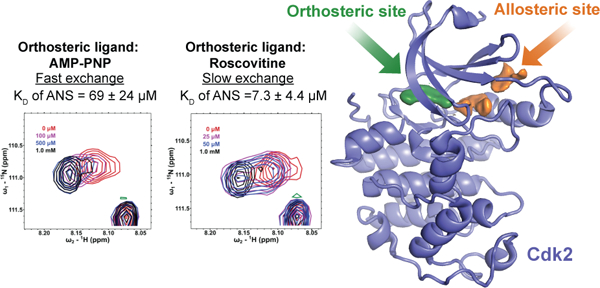
We needed to confirm that the observed positive binding cooperativity between p-Cl-ANS and orthosteric inhibitors was not the result of fluorescence interference from the orthosteric inhibitors themselves or a change in chemical environment/binding conformation of p-Cl-ANS. Spectral changes observed in HSQC NMR of Cdk2 afforded a non-fluorescent orthogonal means to examine the cooperativity between ANS and compounds that bind in the ATP-site. Titration of ANS into AMP-PNP bound to Cdk2 yielded chemical shift perturbations (CSP) indicative of fast exchange, suggesting relatively weak binding. Dose-response CSP analysis showed a KD of ANS towards Cdk2 in the presence of AMP-PNP ranging from 23–100 μM, depending on the residue examined (Figure 4, panel A; Supplementary Figure S1). Of 8 residues with well-resolved CSPs, the KD of ANS to Cdk2 was found to be 69 ± 24 μM, similar to the KD of ANS to apo-Cdk2 measured previously by fluorescence. This corresponded well with the observation that ATP binding does not affect the affinity of ANS towards Cdk2. However, in the presence of saturating amounts of the orthosteric inhibitor roscovitine, titration of ANS revealed slow exchange spectral changes, suggestive of tight binding. When ANS was added, a subsequent decrease in intensity of a parent peak and an increased intensity of a new peak was observed for the changes between spectra. A dose-response decay of the parent peak showed a KD of ANS for Cdk2 in the presence of roscovitine ranging from 3.2–16 μM, consistent with the 5- to 10-fold improvement in affinity observed by fluorescence in the p-Cl-ANS studies (Figure 4, panel B; Supplementary Figure S2). Of 6 well-resolved normalized decays examined with roscovitine bound, the average KD of ANS to Cdk2 was found to be 7.3 ± 4.4 μM. Together with the fluorescent data, the HSQC NMR data confirm the positive cooperativity between ANS and certain orthosteric inhibitors like roscovitine to Cdk2 and the lack of cooperative or competitive effects between ANS and ATP.
Figure 4.
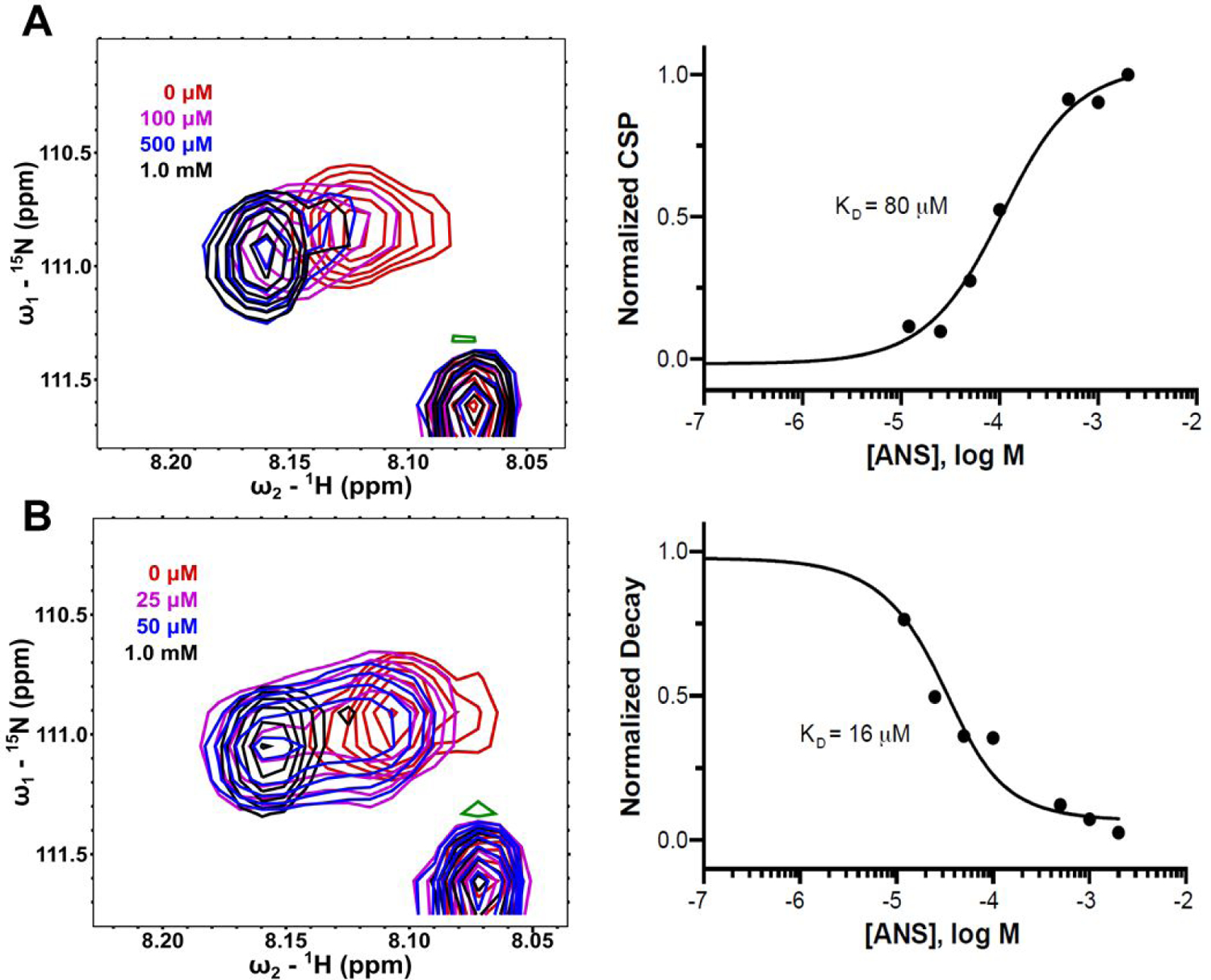
ANS titrations in HSQC NMR in the presence of various orthosteric ligands. The backbone nitrogen of a defined residue was examined to observe the fast and slow exchange regimes in the HSQC NMR spectra when ANS was titrated into Cdk2 in the presence of AMP-PNP (A) or roscovitine (B), respectively. For this particular residue, a 5-fold improvement in affinity was observed when roscovitine was bound. A zero-concentration data point is included in the fit but not displayed.
In conclusion, we observed and quantified for the first time a positive cooperative binding relationship between certain orthosteric inhibitors like roscovitine and dinaciclib and the allosteric inhibitor ANS in Cdk2 using two orthogonal biophysical techniques. Given the relatively weak affinity of p-Cl-ANS and ANS towards apo-Cdk2, administration of an orthosteric inhibitor with an allosteric inhibitor could produce a synergistic increase in inhibitory potency of weak allosteric inhibitors. Furthermore, the affinity of p-Cl-ANS and ANS towards Cdk2 did not appear to be affected by ATP binding, confirming that these analogs are allosteric inhibitors noncompetitive with ATP. We hypothesize that the enhanced affinity is likely due to unique changes in the dynamics of the kinase produced by each type of inhibitor resulting in cooperativity that is not captured by the static view of x-ray crystallography. This is consistent with previous molecular dynamics simulations that demonstrated an altered conformational landscape of Cdk2 in the presence of ANS compared to apo-Cdk2 (24). To further evaluate these dynamics, our use of HSQC NMR provides a unique context to get global framework to capture the dynamics of individual residue backbone atoms. The shift from a fast exchange regime in the presence of AMP-PNP to that of slow exchange in the presence of roscovitine is stark in confirming the cooperativity differences seen between the two in the fluorescent assay. The observed positive cooperativity relationship between orthosteric and allosteric inhibitors could aid selective Cdk2 inhibitor development in the future and selective kinase inhibitor development in general. Exploitation of these cooperative relationships could help confer a selectivity advantage in cases where allosteric and orthosteric inhibitors bind multiple kinases but bind better together for only particular kinases, such as in dual compound screens of known orthosteric and allosteric kinase inhibitors.
Supplementary Material
ACKNOWLEDGEMENTS
We thank E. Schönbrunn for providing purified Cdk2. We thank S. Jakkaraj for providing p-Cl-ANS. This work was supported by NIH/NICHD 5 U01 HD080431, NIH/NICHD 1 R61 HD099743, and NIH/NIGMS R01 GM121515. E. Faber was supported by NIH/NIGMS training grants T32 GM008244 and T32 GM008700 as well as by NIH/NCI fellowship F30 CA232303.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi
Full details of materials and methods, including purification of Cdk2, the p-Cl-ANS fluorescent binding assay, HSQC NMR ANS titrations, as well as Figures S1–S2 (PDF).
The authors declare no competing financial interest.
REFERENCES
- (1).Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandala M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, Sovak MA, Chang I, Choong N, Hack SP, McArthur GA, and Ribas A (2014) Combined vemurafenib and cobimetinib in BRAF-mutated melanoma, N. Engl. J. Med 371, 1867–1876. [DOI] [PubMed] [Google Scholar]
- (2).Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JM, Utikal J, Dreno B, Nyakas M, Middleton MR, Becker JC, Casey M, Sherman LJ, Wu FS, Ouellet D, Martin AM, Patel K, Schadendorf D, and Group MS (2012) Improved survival with MEK inhibition in BRAF-mutated melanoma, N. Engl. J. Med 367, 107–114. [DOI] [PubMed] [Google Scholar]
- (3).Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, Garbe C, Schadendorf D, Krajsova I, Gutzmer R, Chiarion-Sileni V, Dutriaux C, de Groot JWB, Yamazaki N, Loquai C, Moutouh-de Parseval LA, Pickard MD, Sandor V, Robert C, and Flaherty KT (2018) Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF -mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial, Lancet Oncol 19, 603–615. [DOI] [PubMed] [Google Scholar]
- (4).Milojkovic D, and Apperley J (2009) Mechanisms of resistance to imatinib and second-generation tyrosine inhibitors in chronic myeloid leukemia, Clin. Cancer Res 15, 7519–7527. [DOI] [PubMed] [Google Scholar]
- (5).Kakadia S, Yarlagadda N, Awad R, Kundranda M, Niu J, Naraev B, Mina L, Dragovich T, Gimbel M, and Mahmoud F (2018) Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma, Onco Targets Ther 11, 7095–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).To C, Jang J, Chen T, Park E, Mushajiang M, De Clercq DJH, Xu M, Wang S, Cameron MD, Heppner DE, Shin BH, Gero TW, Yang A, Dahlberg SE, Wong KK, Eck MJ, Gray NS, and Janne PA (2019) Single and dual targeting of mutant EGFR with an allosteric inhibitor, Cancer Discov 9, 926–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Eide CA, Zabriskie MS, Savage Stevens SL, Antelope O, Vellore NA, Than H, Schultz AR, Clair P, Bowler AD, Pomicter AD, Yan D, Senina AV, Qiang W, Kelley TW, Szankasi P, Heinrich MC, Tyner JW, Rea D, Cayuela JM, Kim DW, Tognon CE, O’Hare T, Druker BJ, and Deininger MW (2019) Combining the allosteric inhibitor asciminib with ponatinib suppresses emergence of and restores efficacy against highly resistant BCR-ABL1 mutants, Cancer Cell 36, 431–443 e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lake EW, Muretta JM, Thompson AR, Rasmussen DM, Majumdar A, Faber EB, Ruff EF, Thomas DD, and Levinson NM (2018) Quantitative conformational profiling of kinase inhibitors reveals origins of selectivity for Aurora kinase activation states, Proc. Natl. Acad. Sci. U. S. A 115, E11894–E11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pitsawong W, Buosi V, Otten R, Agafonov RV, Zorba A, Kern N, Kutter S, Kern G, Padua RA, Meniche X, and Kern D (2018) Dynamics of human protein kinase Aurora A linked to drug selectivity, Elife 7, e36656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Scaltriti M, Eichhorn PJ, Cortes J, Prudkin L, Aura C, Jimenez J, Chandarlapaty S, Serra V, Prat A, Ibrahim YH, Guzman M, Gili M, Rodriguez O, Rodriguez S, Perez J, Green SR, Mai S, Rosen N, Hudis C, and Baselga J (2011) Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients, Proc. Natl. Acad. Sci. U. S. A 108, 3761–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Porter PL, Malone KE, Heagerty PJ, Alexander GM, Gatti LA, Firpo EJ, Daling JR, and Roberts JM (1997) Expression of cell-cycle regulators p27Kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients, Nat. Med 3, 222–225. [DOI] [PubMed] [Google Scholar]
- (12).Karst AM, Jones PM, Vena N, Ligon AH, Liu JF, Hirsch MS, Etemadmoghadam D, Bowtell DD, and Drapkin R (2014) Cyclin E1 deregulation occurs early in secretory cell transformation to promote formation of fallopian tube-derived high-grade serous ovarian cancers, Cancer Res 74, 1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nakayama N, Nakayama K, Shamima Y, Ishikawa M, Katagiri A, Iida K, and Miyazaki K (2010) Gene amplification CCNE1 is related to poor survival and potential therapeutic target in ovarian cancer, Cancer 116, 2621–2634. [DOI] [PubMed] [Google Scholar]
- (14).Berthet C, Aleem E, Coppola V, Tessarollo L, and Kaldis P (2003) Cdk2 knockout mice are viable, Curr. Biol 13, 1775–1785. [DOI] [PubMed] [Google Scholar]
- (15).Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, and Barbacid M (2003) Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice, Nat. Genet 35, 25–31. [DOI] [PubMed] [Google Scholar]
- (16).Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, Tessarollo L, and Kaldis P (2012) Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration, Proc. Natl. Acad. Sci. U. S. A 109, 3826–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Asghar U, Witkiewicz AK, Turner NC, and Knudsen ES (2015) The history and future of targeting cyclin-dependent kinases in cancer therapy, Nat. Rev. Drug Discov 14, 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Betzi S, Alam R, Martin M, Lubbers DJ, Han H, Jakkaraj SR, Georg GI, and Schonbrunn E (2011) Discovery of a potential allosteric ligand binding site in CDK2, ACS Chem. Biol 6, 492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rastelli G, Anighoro A, Chripkova M, Carrassa L, and Broggini M (2014) Structure-based discovery of the first allosteric inhibitors of cyclin-dependent kinase 2, Cell Cycle 13, 2296–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Martin MP, Alam R, Betzi S, Ingles DJ, Zhu JY, and Schonbrunn E (2012) A novel approach to the discovery of small-molecule ligands of CDK2, Chembiochem 13, 2128–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kang YN, Stuckey JA Crystal structure of apo CDK2. 10.2210/pdb4EK3/pdb. [DOI]
- (22).De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, and Kim SH (1997) Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine, Eur J Biochem 243, 518–526. [DOI] [PubMed] [Google Scholar]
- (23).Martin MP, Olesen SH, Georg GI, and Schonbrunn E (2013) Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains, ACS Chem Biol 8, 2360–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pisani P, Caporuscio F, Carlino L, and Rastelli G (2016) Molecular dynamics simulations and classical multidimensional scaling unveil new metastable states in the conformational landscape of CDK2, PLoS One 11, e0154066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.