

ABSTRACT
The evolution of multidrug resistant pathogens and the diminishing supply of effective antibiotics are global crisis. Tiny Earth (TE) is undergraduate curriculum that encourage students to pursue science careers by engagement in authentic drug discovery research. Through the TE program, students identify environmental strains that inhibit other bacteria. Although these isolates may produce antibiotics based on the antagonistic phenotype, understanding the activity in regard to genome content remains elusive. Previously, we developed a transposon mutagenesis module for use with TE to identify genes involved in antibiotic production. Here, we extend this approach to a second semester undergraduate course to understand the origin of antagonism and genome diversity. Using a bioinformatics strategy, we identified gene clusters involved in activity, and with annotated genomes in hand, students were able to characterize strain diversity. Genomes were analyzed using different computational tools, including average nucleotide identity for species identification and whole genome comparisons. Because the focus of TE involves the evolution of drug resistance, predicted products in strains were identified and verified using a drug susceptibility assay. An application of this curriculum by TE members would assist in efforts with antibiotic discovery.
Keywords: Pseudomonas, antibiotic discovery, transposon mutagenesis, biosynthetic gene cluster, bioinformatics, citizen science, Tiny Earth
Students use a bioinformatics approach to characterize antagonistic strains.
INTRODUCTION
The evolution of antibiotic resistant bacteria is a global crisis (Robinson et al. 2016; CDC 2019), and in February 2017, a list of the top 12 bacteria for which new antibiotics are urgently needed was released by the WHO (Tacconelli et al. 2018). The main concerns included carbapenem-resistant strains of Acinetobacter baumannii, Pseudomonas aeruginosa andEnterobacteriaceae species. These are members of the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, A. baumannii, P. aeruginosa and Enterobacter species) that are recognized by the Infectious Disease Society of America as bacteria with the most significant risk to public health (Boucher et al. 2009). These disease-causing bacteria are responsible for the majority of nosocomial infections in the United States, and treatment options are dwindling due to their high levels of antibiotic resistance (Tommasi et al. 2015). Escalating the multidrug resistant (MDR) crisis is the shift of major pharmaceutical companies away from antibiotic discovery and synthesis (Pammolli, Magazzini and Riccaboni 2011; Scannell et al. 2012). As a result, sources of new antibiotics are depleted and bacteria are now resistant to the effects of most common drugs.
Tiny Earth (TE; https://tinyearth.wisc.edu/) was established to address the antibiotic crisis through a student crowdsourcing research initiative. During a semester-long lab course, students isolate bacteria from soil samples, characterize strains using phylogenetics and Basic Local Alignment Search Tool (BLAST) analysis, and test for antagonistic activity against safe relatives of the ESKAPE pathogens. TE efforts are well established across the United States and one of the goals of the program is to engage students in science, technology, engineering, and mathematics careers. Because of the success in education, the program has been implemented in high school, college and university curriculums, and as a consequence of this effort, a global collection of strains that exhibit inhibitory activity has been identified. To accommodate such a collection, a TE strain repository was recently developed for the storage of isolates and to archive curated metadata. This database provides an opportunity to further promote education and research through whole genome sequencing and analysis of these strains.
Although thousands of TE strains have been collected based on the antagonistic phenotype, understanding the activity with regard to genotype remains elusive to students. Previously, we developed a molecular approach for TE that utilizes transposon (Tn) mutagenesis and whole genome sequencing to identify genes involved in antagonistic activity (Davis et al. 2017). Here, we extend this strategy to bioinformatics to assist in understanding genome content. The Joint Genome Institute Integrated Microbial Genomes and Microbiomes (JGI IMG) is a computational system that permits bioinformatics analyses of genomes through a convenient and accessible interface (Markowitz et al. 2006, 2014). As of December 2019, IMG contains nearly 60 000 bacterial genomes that together contain 1.1 million biosynthetic gene clusters (BGCs) that are predicted to produce secondary metabolites such as antibiotics. Our strategy offers an opportunity for education in genomic research and also provides robust bioinformatics analyses for use with antagonistic strains by making use of the tens of thousands of bacterial genomes available through JGI IMG. With the ease of Pseudomonas strain isolation and its effective ability to antagonize strains accomplished in the first semester (Davis et al. 2017), we offer an educational platform that expands to a second semester of bioinformatics to understand genome diversity.
MATERIALS AND METHODS
Strain isolation, phylogenetics and transposon mutagenesis (first semester)
Soil samples were obtained from topsoil collected from the campus of Bowling Green State University (BGSU), OH, on 27 August 2015 (SWI36WT; Davis et al. 2017), 30 August 2016 (SWI44WT), 24 August 2017 (SWI6WT) and 16 January 2018 (SWI7WT). For growth conditions, antagonistic assays, 16S rRNA gene phylogenetic analysis and Tn mutagenesis, refer to Davis et al. (2017). All 16S rRNA gene sequences were submitted to GenBank under accession numbers MN399206–MN399335.
Genome sequencing and annotation
Genomic DNA from SWI36WT, SWI44WT, SWI6WT and SWI7WT was extracted using the Wizard Genomic DNA Purification Kit (Promega, Madison, Wisonsin, USA). PacBio sequencing was performed by the University of Delaware DNA Sequencing & Genotyping Center. Genomic DNA was sheared using g-tube to 20 kb fragments (Covaris). The PacBio libraries were prepared using standard PacBio protocol for 20 kb libraries (20 kb template preparation using BluePippin size-selection system). Each sample library was sequenced on a PacBio RS II instrument with one SMRT cell using P6-C4 chemistry with 6 h movie. Genomes were assembled using PacBio HGAP3 (Hierarchical Genome Assembly Process 3). Reads of inserts were filtered by quality 0.8 and read length 1 kb (Chin et al. 2013). All assemblies resolved into a single contig. The genomes may be accessed at JGI IMG under the taxon ID#, 2716884901, 2681813543, 2770939456 and 2791354873; and at GenBank under the accession numbers CP026675, CP026674, CP026676 and CP040930, for SWI36WT, SWI44WT, SWI6WT and SWI7WT, respectively. The genomes of the strains were annotated using JGI GOLD (Liolios et al. 2010) and GenBank.
Bioinformatics analyses (second semester)
The JGI IMG portal (https://img.jgi.doe.gov/) was utilized to perform bioinformatics. The species of SWIWT strains were identified using an average nucleotide identity (ANI) analysis. The SWI36WT, SWI44WT, SWI6WT and SWI7WT genomes were compared in a pairwise fashion to characterized Pseudomonas species available in the JGI database. For analysis of a few selected proteins with putative drug resistance functions, genomes were mined for loci involved in antibiotic resistance using the search function with the key words: lactam, aminoglycoside, chloramphenicol, macrolide, vancomycin and drug efflux pump. Pseudomonas aeruginosa PAO1 (JGI IMG genome ID # 637000218) was included for comparison to an ESKAPE pathogen. For whole genome comparison of SWI44WT and SWI6WT, the Artemis Comparison Tool was utilized (Carver et al. 2005), which is available through JGI IMG. DNA regions identified in SWI6WT that were absent in SWI44WT were defined by the presence of three or more adjacent genes present in one strain and not the other. PHASTER was utilized to identify the presence of a prophage (Arndt et al. 2016).
Antibiotic susceptibility test
The Kirby–Bauer disk diffusion susceptibility assay was used to test for antibiotic resistance (Hudzicki 2009). SWI36WT, SWI44WT, SWI6WT and SWI7WT were spread plated onto Muller Hinton agar (BD; Difco) medium from an overnight culture using a sterile cotton swab. Eight antibiotic disks (BD BBL) were placed over the spread plated strains: colistin (10 μg), carbenicillin (100 μg), chloramphenicol (30 μg), meropenem (10 μg), imipenem (10 μg), tobramycin (10 μg), ciprofloxacin (5 μg) and ceftazidime (30 μg). The diameters of the zones of inhibition were measured after 20 h of incubation. Strains were defined as resistant by the BD protocol if the zones of inhibition were less than 8 mm for colistin, 13 mm for carbenicillin, 12 mm for chloramphenicol, 13 mm for meropenem, 13 mm for imipenem, 12 mm for tobramycin, 15 mm for ciprofloxacin and 14 mm for ceftazidime.
RESULTS
The first semester: phyogenetics and antagonistic activity
As part of the TE curriculum (Fig. 1), soil-derived strains were isolated on the BGSU campus in August 2015 (Davis et al. 2017), August 2016, August 2017 and January 2018. The 16S rRNA gene of 130 isolates was amplified and sequenced, and a neighbor-joining phylogenetic tree was generated to assess the genetic diversity among the strains (Fig. 2). A NCBI nucleotide BLAST analysis (Mount 2007) identified bacteria at the genus level and confirmed that SWI36WT, SWI44WT, SWI6WT and SWI7WT belonged to the Pseudomonas genus. Other isolated strains included members from Citrobacter, Lysinibacillus, Paenibacillus and Serratia (Table S1, Supporting Information). All environmental isolates were tested by undergraduate students for antagonistic activity against non-pathogenic ‘safe’ relatives (SRs) of the ESKAPE pathogens (Acinetobacter baylyi, Bacillus subtilis, Enterobacter aerogenes and Escherichia coli), and the resulting data were overlaid to the phylogenetic tree (Fig. 2). From the 130 strains that were isolated, 47 exhibited antagonistic activity, of which 33 were Pseudomonas isolates and 13 of these inhibited multiple SRs. Since SWI44WT and SWI6WT inhibited all four SRs and SWI7 inhibited all except B. subtilis, graduate students determined their activity, outside of the lab class, against the ESKAPE pathogens and other disease-causing bacteria, including Listeria monocytogens and Bacillus cereus. SWI44WT and SWI6WT were able to inhibit E. faecium, K. pneumoniae, A. baumannii, E. cloacae and B. cereus. We utilized transposon mutagenesis previously described with SWI36WT (Davis et al. 2017) to identify loci in SWI44WT, SWI6WT and SWI7WT whose products may contribute to the inhibitory activity. About 10 000 mutants were screened for each strain and one loss-of-inhibition (LOI) mutant was identified in SWI7, two in SWI6 and six in SWI44. Due to semester constraints, the genomes of the wild-type strains were sequenced, annotated, and arbitrary PCR was performed after the semester end (Fig. 1). These data were used to identify loci involved in antagonistic activity and to characterize genome content.
Figure 1.
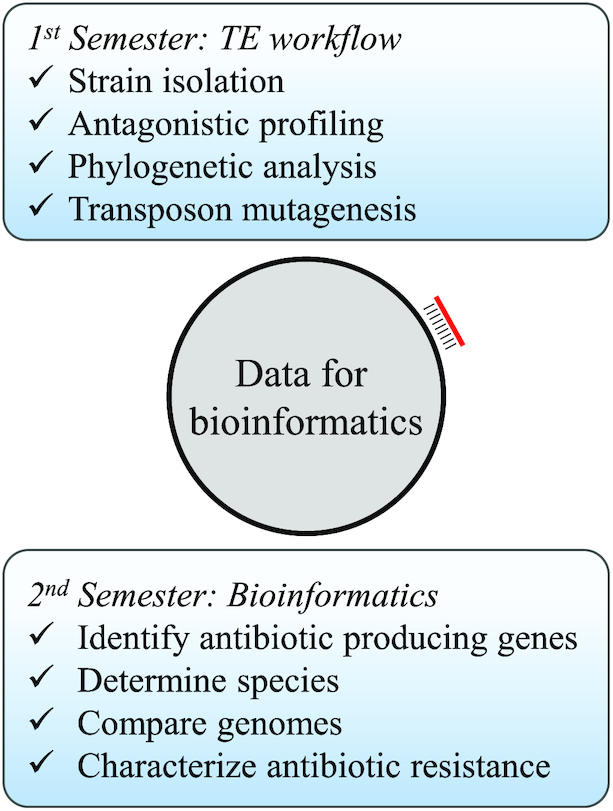
The expanded year-long TE workflow. The first and second semesters are boxed in light blue. Genome sequencing of the wild-type strains (gray circle) and arbitrary PCR (red line) of the transposon mutant are performed outside of the class. These data are used to expand TE to a second semester involving bioinformatics.
Figure 2.
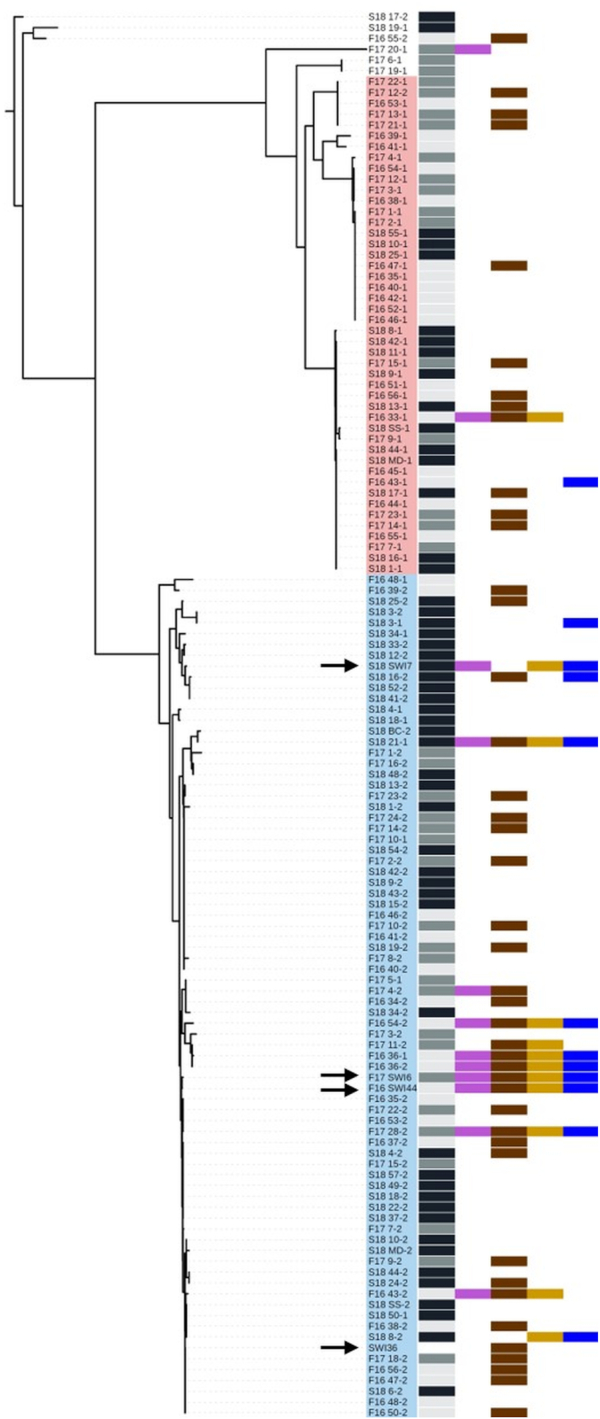
16S rRNA neighbor-joining phylogenetic tree of 130 TE soil-derived strains. Isolates were collected in September 2016, September 2017 and January 2018 and are depicted by light gray, dark gray and black bars, respectively. Among the 130 strains, 47 exhibited antagonistic activity of which 33 were pseudomonads and 13 of these inhibited multiple SRs. Antagonistic activity by the strains is indicated by the colored boxes: Acinetobacter baylyi (purple), Bacillus subtilis (brown), Enterobacter aerogenes (gold) and E. coli (blue). SWI36WT, SWI44WT, SWI6WT and SWI6WT are identified by black arrows, and the Bacillus and Pseudomonas clades were shaded pink and blue, respectively. The tree was rooted using the Serratia strain S18 17-2.
The second semester: bioinformatics
To expand the TE curriculum toward bioinformatics, we developed a second semester that utilizes a user friendly database for computational analyses (Fig. 1). The main goal was to understand antagonistic activity in regard to genome content. JGI IMG is a well-developed and continuously maintained database (Markowitz et al. 2006; Chen et al. 2017) that students can easily navigate and perform bioinformatic functions. All genomes submitted to NCBI are also available through JGI IMG, thus providing a comprehensive analysis of all currently sequenced genomes. Students registered as a user for JGI IMG and all bioinformatics assignments were performed using this computational platform unless otherwise stated. We demonstrate the utility of JGI IMG in an undergraduate lecture class through four assignments: (i) the identification of genes involved in antagonistic activity, (ii) species identification, (iii) genome comparison and (iv) drug resistance. Throughout the second semester, students worked on these tasks and generated a poster that they presented at the Ohio Branch American Society for Microbiology conference.
Assignment 1: identification of biosynthetic gene clusters
To identify the disrupted gene in SWI44LOI, SWI6LOI and SWI7LOI that were generated by Tn mutagenesis at the end of the first semester, students aligned the arbitrary PCR product to the annotated sequenced genome (Fig. 1) using the NCBI nucleotide BLAST sequence alignment function. Arbitrary PCR is a technique that allows amplification of the genomic sequence that flanks the Tn insert (Das et al. 2005), so alignment to the genome indicates the mutated gene. Locus coordinates were used to identify if the Tn inserted into a BGC. BGCs were computationally identified through JGI IMG annotation for each strain (Tables S2–S4, Supporting Information). These gene regions are predicted to encode secondary metabolites, so a Tn insertion in this region would suggest the encoded product is involved in antagonistic activity. In SWI44LOI and SWI6LOI, the Tn was found to disrupt a BGC. In SWI44, six LOI mutants were identified (Table S5, Supporting Information). With three of these mutants, the Tn inserted into a 42.0 kb BGC (JGI cluster 2716925891.Ga0172617 11.region2), which consists of 41 ORFs (Fig. 3A; Figure S8, Supporting Information) and is predicted to encode a thiopeptide. ORFs 10, 12 and 13 were mutated and predicted to encode a potassium-efflux system ancillary protein, an A-factor biosynthesis hotdog domain-containing protein and a NAD(P)-dependent dehydrogenase, respectively. The other three Tn insertions disrupted the gacS gene, which is a two-component system histidine kinase and is located 749 kb outside of the designated BGC. With SWI6WT, two LOI mutants were identified (Table S6, Supporting Information). One Tn disrupted the same BGC as in SWI44WT (2770957150.Ga0255654 11.region6), but in ORF 15, which is predicted to encode a NAD(P)-dependent dehydrogenase (Fig. 3B; Table S8, Supporting Information). The other mutation also occurred in the gacS gene as observed in SWI44LOI. The presence of multiple Tn insertions within a single BGC and gacS gene among two different strains suggests these loci encode the production of a secondary metabolite involved in antagonistic activity. One SWI7LOI mutant was identified (Table S7, Supporting Information) and predicted to be a hypothetical protein but was not located within in a BGC. The region around this locus was mined for a metabolite pathway (Fig. 3C), but there was no evidence of secondary metabolite production. This approach allows students to connect the antagonistic activity observed in the first semester to genes involved in the production of the active compound.
Figure 3.
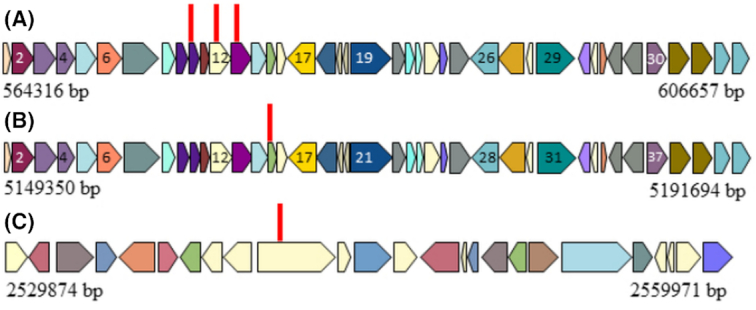
Tn mutated genes in SWI44WT, SWI6WT and SWI7WT that contribute to antagonistic activity. Tn mutants were identified in BGCs within (A) SWI44WT and (B) SWI6WT and were predicted to encode a thiopeptide. (C) The insertion in SWI7WT was not in a predicted BGC. The genes surrounding the mutated locus are shown, which differ from SWI44WT and SWI6WT. ORF numbers correspond to Table S8 (Supporting Information). Homologous ORFs are shaded the same color. Red vertical lines represent location of the Tn insert.
Assignment 2: species identification
Without genome sequence, species identification is difficult to perform because this involves technical lab experimentation such as multiple biochemical tests and whole genome hybridization. With sequenced genomes in hand, students performed an ANI analysis using the JGI IMG ANI function to determine species identification. Results showed that SWI44WT and SWI6WT were 99% similar to Pseudomonas taiwanensis DSM 21245 strain, suggesting these strains belong to the group P. taiwanensis (Table 1). SWI36WT was identified to be >98% similar to P. putida KT2440. Likewise, the ANI of SWI36WT to SWI44WT and SWI6WT was 85%, consistent with their status as different species. SWI7 was not closely related to any other Pseudomonas strain. Since the same BGC was identified in SWI44WT and SWI6WT and because the strains were similar based on ANI, a pairwise genome NCBI BLAST alignment was performed to determine genome similarity. Results revealed that SWI44WT and SWI6WT were 99% similar based on query coverage of 93% (SWI6WT vs SWI44WT) and 89% (SWI44WT vs SWI6WT). Thus, SWI44WT and SWI6WT strains were closely related based on genome content and of the same species while SWI36WT and SWI7WT were from a different group. These results presented a unique opportunity for students to compare closely related strains and to understand species based on genome content.
Table 1.
ANI analysis of SWI36WT, SWI44WT, SWI6WT and SWI7WT.
| Genome ID | Pseudomonas sp. | SWI36 | SWI44 | SWI6 | SWI7 |
|---|---|---|---|---|---|
| 637000218 | P. aeruginosa PAO1 | 77.38 | 77.46 | 77.48 | 76.87 |
| 637000219 | P. entomophila L48 | 85.34 | 85.24 | 85.22 | 82.76 |
| 637000220 | P. protegens Pf-5 | 79.31 | 79.44 | 79.30 | 78.63 |
| 637000221 | P. fluorescens Pf0-1 | 78.63 | 78.73 | 78.59 | 77.92 |
| 637000222 | P. putida KT2440 | 98.11 | 85.15 | 85.33 | 83.56 |
| 640427131 | P. mendocina ymp | 77.76 | 77.81 | 77.80 | 77.34 |
| 640427133 | P. stutzeri A1501 | 76.69 | 76.69 | 76.70 | 76.32 |
| 2508501074 | P. syringae pv. tomato DC300 | 77.39 | 77.48 | 77.45 | 76.83 |
| 2523533561 | P. taiwanensis DSM 21245 | 85.17 | 99.62 | 99.66 | 82.62 |
| 2681813543 | SWI36 | 100 | 85.12 | 85.59 | 83.68 |
| 2716884901 | SWI44 | 85.23 | 100 | 99.44 | 82.59 |
| 2770939456 | SWI6 | 85.59 | 99.44 | 100 | 82.63 |
| 2791354873 | SWI7 | 83.68 | 82.59 | 82.63 | 100 |
Assignment 3: genome comparison
To investigate genome similarity and differences between SWI6WT and SWI44WT, students performed a whole genome analysis using the Artemis Comparison Tool (ACT; Carver et al. 2005), which is also available through JGI IMG. Thirteen chromosomal regions were identified that were present only in SWI6WT (Fig. 4A; Table S9, Supporting Information). DNA regions in which three or more adjacent genes were present in one strain and not the other were of interest since these loci may give insight to gene gain or loss among the closely related strains. Seven of the thirteen regions encoded a predicted transposase or integrase suggesting lateral transfer of a gene region. These regions included but were not limited to genes with predicted functions involved in outer member components, transcriptional regulators, restriction enzymes, aminotransferases, efflux pumps and hypothetical proteins (Table S9, Supporting Information). Region 2 was 53 763 bp and PHASTER (Arndt et al. 2016) predicted a 39 193 bp prophage at genome coordinates 666 612–705 805 (Fig. 4B). A nucleotide BLAST analysis was performed using the prophage encoded region and results showed a 67% query coverage and 83% identity to the Pseudomonas putida strain PC2 genome yet only 31% coverage and 74% identity to Pseudomonas phage Phi3, the most similar phage, suggesting the identification of a novel prophage in strain SWI6WT. attL, attR and a tRNA were found near the phage boundary sites (Table S10, Supporting Information) and genes with functional similarity were grouped together suggesting a modular genome composition. Thus, the similarity between SWI44WT and SWI6WT provided a unique opportunity for students to identify differences likely due to lateral gene transfer among closely related strains.
Figure 4.

Genome comparison of strains SWI7WT, SWI44WT and SWI6WT. (A) ACT was utilized to visualize SWI genome similarity. SWI7WT, SWI44WT and SWI6WT were positioned on the top, middle and bottom of the alignment, respectively. The sequences are joined by colored bands that represent the matching genome regions. Red and blue bands represent DNA matches in the forward and reverse direction, respectively. DNA regions present in only SWI6WT compared to SWI44WT are numbered 1–13 on the SWI6 genome. (B) The prophage identified in SWI6WT is in region 2 and consists of 39 193 bp. Colored arrows pointing left or right represent ORFs transcribed in the forward or reverse direction, respectively. Gray filled arrows represent ORFs that were not predicted to be prophage associated. Colored ORFs were predicted to have prophage associated functions: red, site specific integrase; green, tail assembly; dark blue; lysis; light blue, DNA packaging; brown, capsid assembly; yellow, replication; purple, tRNA; and cream, unknown function. Numbers 1–41 correspond to ORFs in Table S10 (Supporting Information).
Assignment 4: drug resistance
Because the focus of TE involves the evolution of antibiotic resistance, we performed a search for genes predicted to be involved in drug resistance. We identified numerous loci with putative functions involved in enzymes that provide resistance to β-lactams, aminoglycosides, chloramphenicol, macrolides and vancomycin, and possibly other drugs through drug efflux pumps. In total, 34, 28, 28 and 26 putative genes involved in resistance were identified in SWI36WT, SWI44WT, SWI6WT and SWI7WT, respectively (Table S11, Supporting Information). Compared to an ESKAPE pathogen, P. aeruginosa PAO1 was found to encode 22 putative resistance genes, surprisingly less than the environmental strains. Based on these results, students predicted that the environmental SWIWT strains would exhibit MDR resistance. We tested eight antibiotics that differed based on compound classification and mode of action (Table 2). β-lactams and an aminoglycoside antibiotic were included since multiple genes were identified that conferred resistance to these drugs, in addition to chloramphenicol and ciprofloxacin; we also included the ‘last resort’ drug colistin. SWI36WT, SWI44WT and SWI6WT were resistant to the β-lactam carbenicillin, the aminoglycoside tobramycin, chloramphenicol and colistin. SWI7WT was resistant to carbenicillin, meropenem and chloramphenicol. These results show that environmental strains exhibit a MDR phenotype and have evolved mechanisms to combat a variety of antibiotics.
Table 2.
Antibiotic resistance of SWIWT strains.
| Resistance | ||||||
|---|---|---|---|---|---|---|
| Antibiotic | Classification | Inhibits | SWI36 | SWI44 | SWI6 | SWI7 |
| Carbenicillin | β-lactam | Cell wall synthesis | Yes | Yes | Yes | Yes |
| Ceftazidime | β-lactam | Cell wall synthesis | No | No | No | No |
| Imipenem | β-lactam | Cell wall synthesis | No | No | No | No |
| Meropenem | β-lactam | Cell wall synthesis | No | No | No | Yes |
| Chloramphenicol | Amphenicol | Protein synthesis | Yes | Yes | Yes | Yes |
| Tobramycin | Aminoglycoside | Protein synthesis | Yes | Yes | Yes | No |
| Ciprofloxacin | Quinolone | DNA replication | No | No | No | No |
| Colistin | Polypeptide | Membrane integrity | Yes | Yes | Yes | No |
DISCUSSION
Engaging undergraduate students in authentic research provokes interest in scientific careers and develops troubleshooting and critical thinking skills. Unfortunately, many students fulfil their undergraduate degree without the opportunity to engage in experiential learning. TE has been defined as inclusive Research Education Community (iREC)—a format that addresses a common scientific problem that is performed by multiple institutions and is supported by a centralized scientific structure (Hanauer et al. 2017). A similar education-research-based program is SEA-PHAGE, a well-defined iREC, which utilizes Mycobacterium bacteriophage for its research purpose and has been shown to be effective in science education (Anderson et al. 2011; Pope and Hatfull 2015). Recently, bacteriophage isolated from students in the SEA-PHAGE program was utilized to treat a Mycobacterium abscessus infection in a cystic fibrosis patient who did not respond to antibiotics (Dedrick et al. 2019). Thus, iREC programs are of importance in scientific discovery and education. The workflow presented here parallels that of SEA-PHAGE by integrating genome analysis into the research project. Due to semester constraints at most universities, the workflow was designed to span two semesters. The first semester involves utilizing Tn mutagenesis for the identification of LOI mutants (Davis et al. 2017). Required data linking the first and second semesters together involve arbitrary PCR of the mutant strains and whole genome sequencing of the respective wild-type isolate (Fig. 1). Both tasks can be performed by the Tiny Earth Genome Laboratory, which was developed to assist in the global application of this research endeavor (https://blogs.bgsu.edu/swigenomes/). The second semester involves using these data for the identification of genes involved in antagonistic activity and genome analyses.
For a proof of concept using our approach, strains from soil samples were isolated from the campus of BGSU and the proposed TE workflow was performed (Fig. 1). Students demonstrated that SWIWT strains were able to inhibit different SRs and pathogens (Fig. 2). Tn mutagenesis was performed on SWI44WT, SWI6WT and SWI7WT and their genomes were sequenced. For SWI44WT and SWI6WT, an identical BGC was identified and predicted to encode a thiopeptide (Fig. 3). Tn insertions were also found in a gene predicted to encode histidine sensor kinase (gacS gene), which is part of a two-component regulatory system involved in gene regulation. In Pseudomonas, the GacS sensor kinase has been shown to be involved in the regulation of secondary metabolites in Pseudomonas protegens Pf-5 (Corbell and Loper 1995) and the production of antagonistic factors by Pseudomonas fluorescens strains that inhibit plant (Laville et al. 1992) and fungal (Gaffney et al. 1994) pathogens. Given that the SWI44WT and SWI6WT had identical BGCs (Fig. 3), and were closely related based on the 16S rRNA gene (Fig. 2), students investigated if the strains were clonal. Bioinformatics analyses demonstrated that SWI44WT and SWI6WT were similar, but not identical in genome content based on ANI and ACT analyses while SWI7WT differed (Table 1 and Fig. 4), which provided students with an understanding of genetic diversity among similar and different strains. Because the evolution of antibiotic resistance is a main concept in SWI/TE, students sought to determine drug resistant profiles, so genomes were mined for resistance genes (Table S11, Supporting Information) and verified experimentally (Table 2). These results demonstrate that even among non-pathogenic strains, isolates have evolved a MDR phenotype; unfortunately, SWI36WT, SWI44WT and SWI6WT were resistant to colistin, a drug of last resort. Thus, the sequenced genomes provided a genetic understanding by establishing a link between phenotype and genotype both with antagonistic activity (and the identification of BGCs) and drug resistance (and the identification of resistance genes).
To culminate the second semester, students presented their work, in groups of two, at the Ohio Branch American Society for Microbiology conference. Posters were judged by faculty members and they scored the presentation on content, delivery, response and overall impression. The average poster presentation score from one (poor) to seven (outstanding) was a five (excellent), which reflected their engagement in the project considering this was the first authentic poster presentation at a scientific conference. This activity compiled a year-long course involving strain isolation, antagonistic activity, Tn mutagenesis and bioinformatics. In addition to educational purposes, the workflow presented here also provides important scientific data. For instance, the genes involved in antagonistic activity were identified, which can give valuable insight to compound prediction. Moreover, wild-type and respective LOI strains can be utilized by the Tiny Earth Chemical Hub (https://tinyearth.wisc.edu/chemistry-hub/) to extract the active compound using techniques such as high-performance liquid chromatography with prospects of antibiotic discovery. Furthermore, an implementation of this approach by SWI/TE members would provide a large collection of pseudomonads genomes that can be utilized for education and research purposes. Access to such a large genome collection would give a global insight to strain diversity within a bacterial group, the spread of drug resistance, and more importantly, the identification of diverse BGCs that would facilitate drug discovery by undergraduate students.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank members of TE for their continued support of the program, especially Jo Handelsman, Sarah Miller, Nicole Broderick and Kristen Butela. We also thank Bruce Kingham and Olga Shevchenko from the University of Delaware DNA Sequencing & Genotyping Center for their support in genome sequencing.
Conflicts of interest. None declared.
REFERENCES
- Anderson WA, Banerjee U, Drennan CL et al. Science education. Changing the culture of science education at research universities. Science. 2011;331:152–3. [DOI] [PubMed] [Google Scholar]
- Arndt D, Grant JR, Marcu A et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44:16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher HW, Talbot GH, Bradley JS et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. [DOI] [PubMed] [Google Scholar]
- Carver TJ, Rutherford KM, Berriman M et al. ACT: the artemis comparison tool. Bioinformatics. 2005;21:3422–3. [DOI] [PubMed] [Google Scholar]
- CDC. Antibiotic resistance threats in the United States. https://www.cdc.gov/media/releases/2019/p1113-antibiotic-resistant.html(30 January 2020, date last accessed).
- Chen IA, Markowitz VM, Chu K et al. IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017;45:507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Alexander DH, Marks P et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–9. [DOI] [PubMed] [Google Scholar]
- Corbell N, Loper JE A global regulator of secondary metabolite production in Pseudomonas fluorescens Pf-5. J Bacteriol. 1995;177:6230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Noe JC, Paik S et al. An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J Microbiol Methods. 2005;63:89–94. [DOI] [PubMed] [Google Scholar]
- Davis E, Sloan T, Aurelius K et al. Antibiotic discovery throughout the Small World Initiative: a molecular strategy to identify biosynthetic gene clusters involved in antagonistic activity. MicrobiologyOpen. 2017;6:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedrick RM, Guerrero-Bustamante CA, Garlena RA et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. 2019;25:730–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney TD, Lam ST, Ligon J et al. Global regulation of expression of antifungal factors by a Pseudomonas fluorescens biological control strain. Mol Plant Microbe Interact. 1994;7:455–63. [DOI] [PubMed] [Google Scholar]
- Hanauer DI, Graham MJ, Sea P et al. An inclusive Research Education Community (iREC): impact of the SEA-PHAGES program on research outcomes and student learning. Proc Natl Acad Sci. 2017;114:13531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudzicki J Kirby–Bauer disk diffusion susceptibility test protocol. ASM Laboratory Protocols. https://www.asm.org/Protocols/Kirby-Bauer-Disk-Diffusion-Susceptibility-Test-Pro(30 January 2020, date last accessed). [Google Scholar]
- Laville J, Voisard C, Keel C et al. Global control in Pseudomonas fluorescens mediating antibiotic synthesis and suppression of black root rot of tobacco. Proc Natl Acad Sci. 1992;89:1562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liolios K, Chen IM, Mavromatis K et al. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2010;38:D346–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, Korzeniewski F, Palaniappan K et al. The integrated microbial genomes (IMG) system. Nucleic Acids Res. 2006;34:D344–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, Chen IM, Palaniappan K et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42:D560–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount DW Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc. 2007;2007:pdb.top17. [DOI] [PubMed] [Google Scholar]
- Pammolli F, Magazzini L, Riccaboni M The productivity crisis in pharmaceutical R&D. Nat Rev Drug Discov. 2011;10:428–38. [DOI] [PubMed] [Google Scholar]
- Pope WH, Hatfull GF Adding pieces to the puzzle: new insights into bacteriophage diversity from integrated research-education programs. Bacteriophage. 2015;5:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TP, Wertheim HF, Kakkar M et al. Animal production and antimicrobial resistance in the clinic. Lancet. 2016;387:e1–3. [DOI] [PubMed] [Google Scholar]
- Scannell JW, Blanckley A, Boldon H et al. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11:191–200. [DOI] [PubMed] [Google Scholar]
- Tacconelli E, Carrara E, Savoldi A et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis. 2018;18:318–27. [DOI] [PubMed] [Google Scholar]
- Tommasi R, Brown DG, Walkup GK et al. ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov. 2015;14:529–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.