

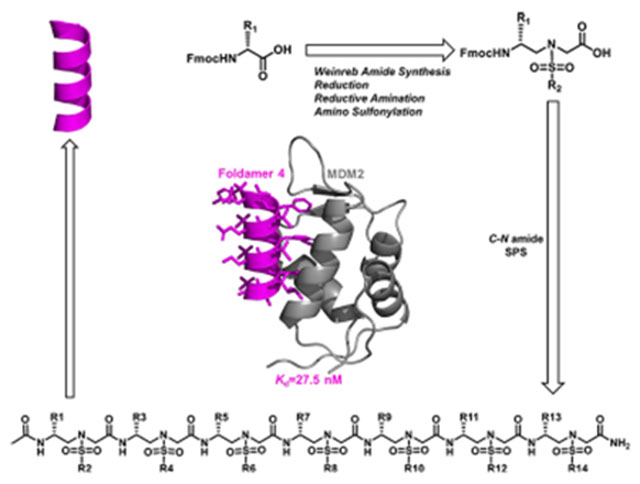
Abstract
Novel unprecedented helical foldamers have been effectively designed and synthesized. The homogeneous right-handed d-sulfono-γ-AApeptides represent a new generation of unnatural helical peptidomimetics, which have similar folding conformation to α-peptides, making them an ideal molecular scaffold to design α-helical mimetics. As demonstrated with p53-MDM2 PPI as a model application, the right-handed d-sulfono-γ-AApeptides reveal much-enhanced binding affinity compared to the p53 peptide. The design of d-sulfono-γ-AApeptides may provide a new and alternative strategy to modulate protein–protein interactions.
Graphical Abstract
INTRODUCTION
Foldamers1–5 have been increasingly explored for the development of potent and alternative bioactive molecules that may shape the future directions of drug discovery. Despite significant advancements in this endeavor,6–10 general strategies for addressing the enduring issues of disrupting protein–protein interactions (PPIs) remain to be improved. Among recent progresses, γ-AApeptides (oligomers of γ-substituted-N-acylated-N-aminoethyl amino acids), inspired by the backbone of chiral peptide nucleic acid, emerged as effective peptidomimetics that play important roles in chemical biology and biomedical sciences.11–14 Specifically, sulfono-γ-AApeptides, as proteolytically stable peptidomimetics, exhibit unusual folding stability by adopting a series of helical structures with a well-defined hydrogen bonding pattern (Figure 1C,D).15–19 Left-handed helical l-sulfono-γ-AApeptides, adopting a 14-H hydrogen bonding pattern and stabilized by both intramolecular hydrogen bonding and the curvature nature of sulfonamido moieties on the molecular backbone, have been successfully applied in targeting various α-helix-interacting proteins.20–22 However, the need to design a new class of unnatural helical peptidomimetics remains increasingly urgent, which may open windows for discovering new unnatural peptidomimetic inhibitors of PPIs. To this end, we envisioned that d-sulfono-γ-AApeptide right-handed helical foldamers, which are enantiomers of known left-handed l-sulfono-γ-AApeptides,15,20–22 should have more similar folding conformation to right-handed α-peptides, thereby making it easier and more straightforward to design α-helical mimetics (Figure 1A–F). Aside from mirroring helical conformation of l-sulfono-γ-AApeptides, d-sulfono-γ-AApeptides have all the advantageous attributes of left-handed l-sulfono-γ-AApeptides.
Figure 1.
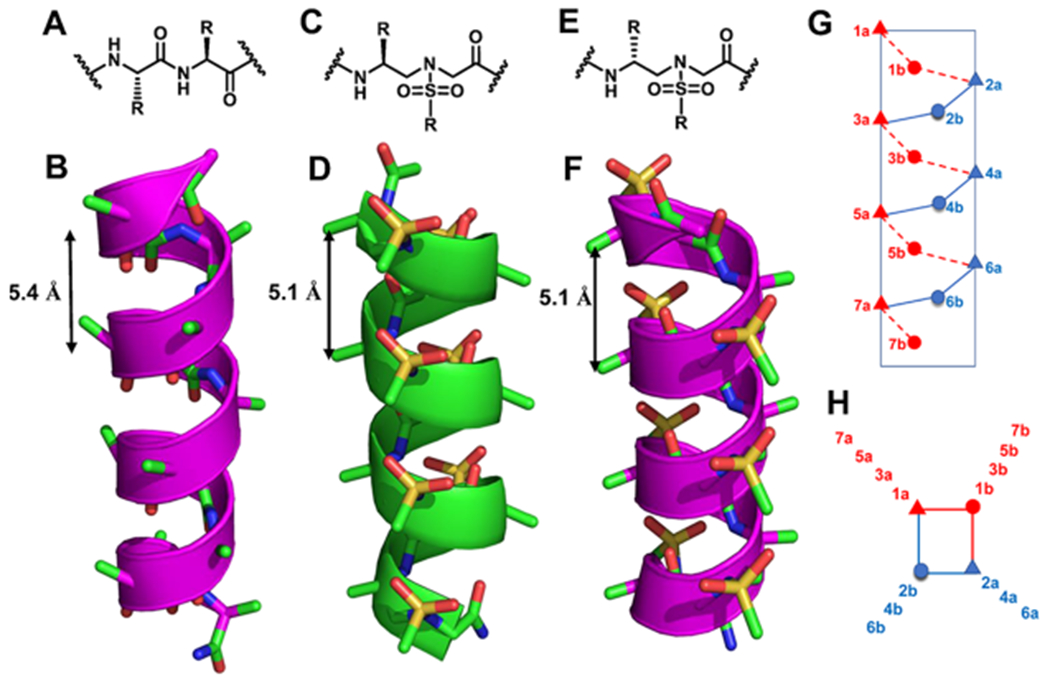
(A–F) Chemical and crystal structures of the α-peptides (A, B), chemical and crystal structures of homogeneous l-sulfono-γ-AApeptides (C, D),15 and chemical and modeled structures of homogeneous d-sulfono-γ-AApeptides (E, F). (G, H) Schematic representation of distribution of side chains from homogeneous d-sulfono-γ-AApeptides based on computational modeling. (G) Side view; (H) top view, helical wheel.
Although d-sulfono-γ-AApeptides have not previously reported, we speculated that the synthesis could be accomplished using the similar strategy for the synthesis of l-sulfono-γ-AApeptides.21 Therefore, different d-sulfono-γ-AApeptide building blocks could be designed and synthesized using Fmoc-protected D-amino acids as the starting materials (Scheme 1A,B).21 Subsequently, the sequences of d-sulfono-γ-AApeptide scaffolds could be synthesized (Scheme 1C).
Scheme 1.
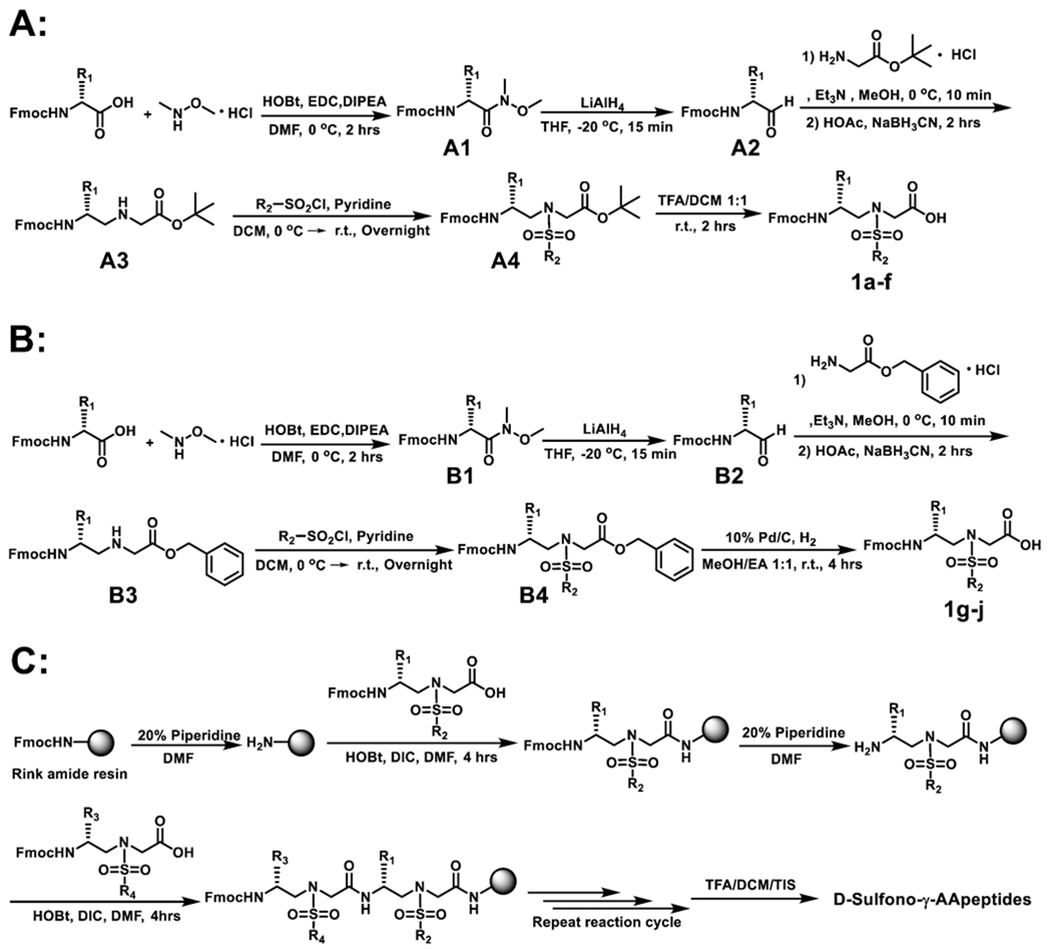
(A) Synthesis of Building Blocks 1a–f, (B) Synthesis of Building Blocks 1g–j, and (C) Solid-Phase Synthesis of d-Sulfono-γ-AApeptides
With the ability to access the sequences of the newly developed right-handed helical d-sulfono-γ-AApeptide scaffold, we questioned whether d-sulfono-γ-AApeptides could be designed to effectively disrupt PPIs. If so, it would offer an alternative strategy to generate potent helical peptidomimetics that inhibit various medicinally relevant PPIs. Herein, we report the first construction of homogeneous right-handed helical foldamers, using p53-MDM2 PPI as a model application.8,23–26
RESULTS AND DISCUSSION
The design of right-handed d-sulfono-γ-AApeptide inhibitors was straightforward. As shown in Figure 1G,H, the first d-sulfono-γ-AApeptide 1 we designed had chiral side chains at positions 2a, 4a, and 6a on the same face of the helical scaffold, and thus, this face was chosen to mimic the three critical residues Phe19, Trp23, and Leu 26 in p53 for binding with MDM2 (Scheme 2). The binding affinity of d-sulfono-γ-AApeptide 1 was determined by fluorescence polarization assays27 (Scheme 2 and Figure S2). P53 (16–29) was also included in the study for comparison (Scheme 2 and Figure S2).20,28 To our delight, d-sulfono-γ-AApeptide 1 bound to MDM2 with a Kd value of 220 nM, which is similar to the regular p53 peptide. This initial success demonstrates the potential of homogeneous right-handed d-sulfono-γ-AApeptides to mimic α-peptide helix:. The d-sulfono-γ-AApeptide 2 was created by changing Phe to a Trp group, which did not dramatically change the binding affinity. As expected, the replacement of Leu26 with bulkier residues in d-sulfono-γ-AApeptides 3 and 4 improved the binding affinity with Kd values of 110 nM (2-fold improvement) and 27.5 nM (8-fold improvement), respectively (Figure 3A, Figure S2, and Scheme 2). Interestingly, the binding activity of peptide 4 is comparable to that of the lead left-handed l-sulfono-γ-AApeptide (Kd = 26 nM),20 which is among the most potent peptidomimetic foldamers that bind p53. Due to the three critical residues contributing to the bulk of the binding energy,29 d-sulfono-γ-AApeptide 5 still exhibited good binding affinity. A d-sulfono-γ-AApeptide with only methyl side chains was also synthesized (Scheme 2, 6) for comparison. The importance of the key residues was manifested by 6 (Scheme 2), which lacks the three critical residues at positions 2a, 4a, and 6a, resulting in complete loss of the binding potency with MDM2. Subsequently, the p53 α-helix mimicry by right-handed d-sulfono-γ-AApeptide 4 was also assessed by direct comparison with the regular p53 and left-handed l-sulfono-γ-AApeptides (Figure 2) by computational modeling, which further suggests that d-sulfono-γ-AApeptides are ideal for mimicry of α-helix.
Scheme 2.
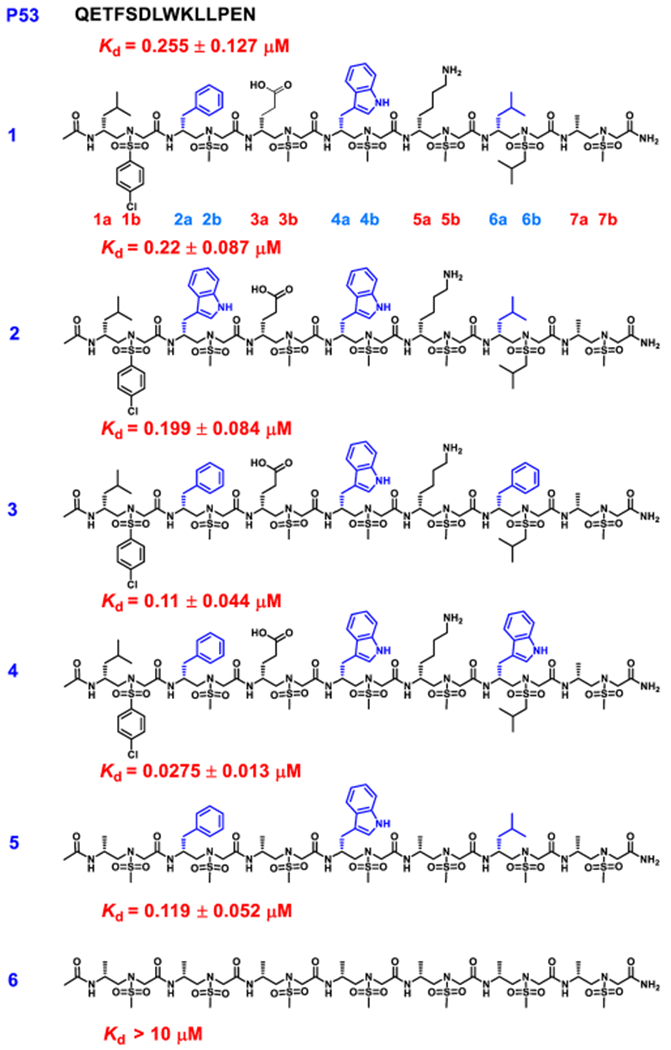
Structures of d-Sulfono-γ-AApeptides Investigated for the Disruption p53-MDM2 Interactiona
aThe side chains mimicking Phe19, Trp23, and Leu26 in p53 are shown in blue.
Figure 3.
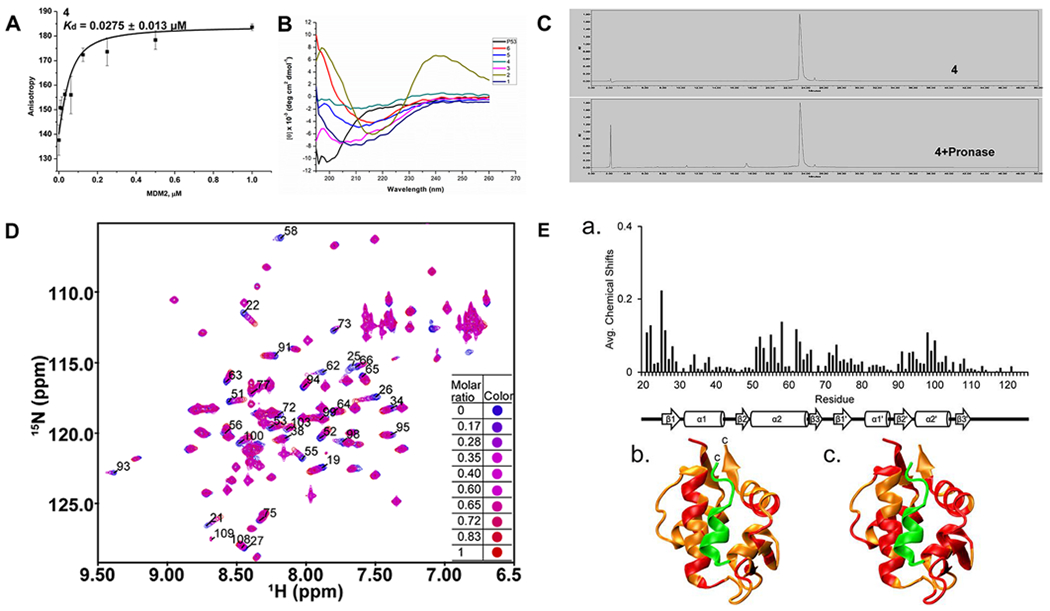
(A) Kd data of sulfono-γ-AApeptide 4 to MDM2. (B) CD spectra of p53 and sulfono-γ-AApeptides (100 μM) measured at room temperature in PBS buffer. (C) Analytic HPLC trace of 4 before and after incubation with pronase (0.1 mg/mL) in 100 mM pH 7.8 ammonium bicarbonate buffer at 37 °C. (D) Overlay of 15N HSQC spectra of MDM2 before (blue resonances) and after (red resonances) the addition of 4. (E) Chemical shift mapping of 4 bound to MDM217–125:(a) Average chemical shift changes in ppm (parts per million) of the MDM2 p53 binding domain (residues 17–125) when bound to 4. A 2° structure of MDM217–125 is shown below the graph. (b) Ribbon structure of MDM217–125 (orange) and p53TAD (green, residues 15–29) (PDB: 1YCR). Residues colored red have chemical shifts above the average of 0.032 ppm when bound to 4. (c) Ribbon structure of MDM217–125 (orange) and p53TAD15–29 (green) (PDB: 1YCR). Residues colored red have chemical shifts above the average of 0.032 ppm when bound to p53TAD15–29.
Figure 2.
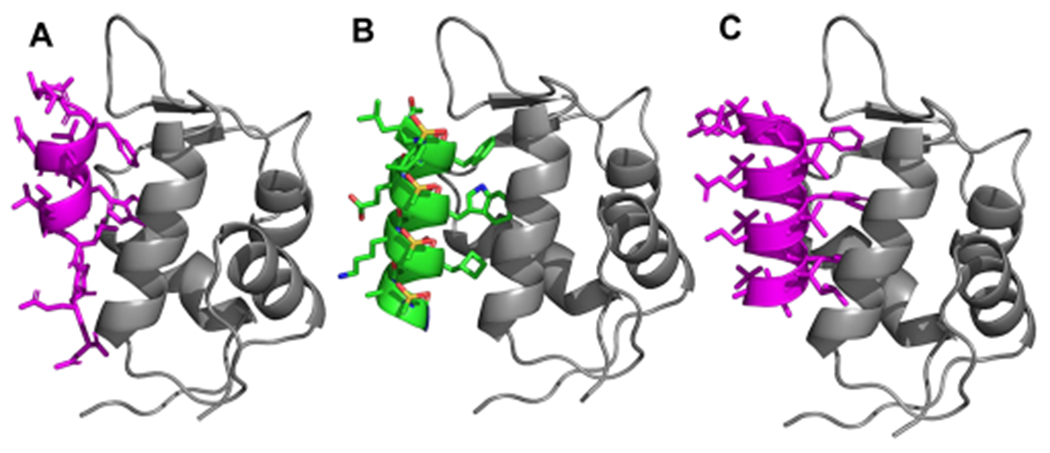
(A) Crystal structure of interaction of p53 with MDM2 (PDB: 1YCR). (B) Modeling of lead homogeneous l-sulfono-γ-AApeptide. (C) Designed homogeneous d-sulfono-γ-AApeptide 4 interaction with MDM2. P53 and homogeneous d-sulfono-γ-AApeptide are shown as magenta cartoon, homogeneous l-sulfono-γ-AApeptide is shown as green cartoon, and MDM2 is shown as gray cartoon.
We subsequently carried out circular dichroism (CD) spectroscopic studies to compare the helical propensity of regular p53 (16–29) and homogeneous d-sulfono-γ-AApeptides 1–6 in solution (Figure 3B). The CD studies were performed in phosphate-buffered saline (PBS) buffer between 190 and 260 nm. d-sulfono-γ-AApeptides 1–6 displayed very similar CD signatures. They all showed strong negative cotton effects between 205 and 215 nm, which is just opposite to the CD signature of left-handed l-sulfono-γ-AApeptides,15 suggesting that the d-sulfono-γ-AApeptides adopt similar right-handed helical conformations to α-peptides. d-sulfono-γ-AApeptide 2 is somewhat different from other analogues with the red shift and a positive maximum around 240 nm, possibly due to the aggregation of the sequence in solution.
To get a robust assessment of proteolytic susceptibility, we performed a peptide degradation assay of d-sulfono-γ-AApeptide 4 using pronase. Pronase is a mixture of broad scope endopeptidases and exopeptidases isolated from Streptomyces griseus, which is commonly used to assess the protease resistance of peptides.21,30 The assay was conducted by incubating 0.1 mg/mL lead peptide 4 with 0.1 mg/mL pronase in 100 mM ammonium bicarbonate buffer (pH 7.8) at 37 °C for 24 h. The stability was analyzed by HPLC-MS (Figure 3C). Compared with the complete degradation of regular p53, our d-sulfono-γ-AApeptide 4 showed no detectable degradation, demonstrating a remarkable stability toward enzymatic degradation, augmenting their potential in future biological applications.
Nuclear magnetic resonance (NMR) spectroscopy was then used to determine if 4’s binding site on MDM217–125 was similar to the binding site for the p53 transactivation domain (TAD). We measured the amide proton and nitrogen chemical shift changes of a uniformly 15N-lableled sample of MDM217–125 after a stoichiometric equivalent amount of 4 was added. Figure 3D shows an overlay of the 15N HSQC spectra of MDM2 before (blue resonances) and after (red resonances) the addition of 4. Figure 3E-a shows the average chemical shift changes in ppm for the amide proton and nitrogen resonances in MDM217–125. The average chemical shift changes for all the detectable MDM217–125 resonances when bound to 4 was 0.032 ppm. Figure 3E-b,c shows the structure of MDM217–125 in orange bound to p53TAD15–29 in green.31 Figure 3E-b shows MDM217–125 residues with chemical shift changes greater than 0.032 ppm when bound to 4 (colored red); 34 residues had chemical shift changes above the average. Figure 3E-c shows MDM217–125 residues with chemical shifts greater than 0.032 ppm when bound to p53TAD1–73 (colored red); 57 residues had chemical shift changes above the average. MDM217–125 β1 had shifts in the presence of 4 and p53TAD1–73, as well as β3′. α1, β1′, β2′, and α2′ had more residues with shifts above the 0.032 ppm threshold in the presence of p53TAD1–73 than 4; β3 had shifts only in the presence of 4 while α1′ and β2 had shifts only in the presence of p53TAD1–73. α2 had shifts in the presence of both 4 and p53TAD1–73. For 4, most of the large chemical shift changes were localized to the N-terminal half of α2. For p53TAD1–73, most of the large chemical shift changes were localized to the C-terminal half of α2. Based on these results, it appears that the binding site for 4 is similar to p53TAD1–73 but not identical. We are currently using this data to optimize peptide design.
In summary, we have introduced a new generation of homogeneous sulfono-γ-AApeptide helical foldamers, right-handed d-sulfono-γ-AApeptides. The helical conformation of d-sulfono-γ-AApeptides is similar to that of α-peptides, making it an ideal molecular scaffold to design α-helical mimetics. As demonstrated with their proof-of-concept application for the mimicry of p53 for the recognition of MDM2, right-handed d-sulfono-γ-AApeptides reveal a comparable binding potency to that of the existing left-handed l-sulfono-γ-AApeptides. This study opens the door to further applications of sulfono-γ-AApeptide helical foldamers for discovering potent unnatural peptidomimetic inhibitors of protein–protein interactions.
EXPERIMENTAL SECTION
General Information for d-Sulfono-γ-AApeptide Block Synthesis.
Fmoc-protected d-amino acids and fluorescein isothiocyanate (FITC) were purchased from Chem-Impex (Wood Dale, IL). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide, 1-hydroxybenzotriazole wetted with no less than 20 wt % water (HOBt), and N,N-diisopropylethylamine (DIPEA) were purchased from Oakwood Chemical (Estill, SC). Other chemicals and all solvents were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher and used without further purification. Sorbtech TLC plates (silica gel w/UV254) were used for thin layer chromatography. ICN silica gel (60 Å, 230–400 mesh, 32–63 μm) was employed for flash chromatography. 1H NMR spectra were obtained at 400 or 600 MHz using TMS as the internal standard. 13C NMR spectra were obtained at 100 or 150 MHz using TMS as the internal standard. The multiplicities, including singlet (s), doublet (d), doublet of doublets (dd), triplet (t), quartet (q), and multiplet (m), are reported. High-resolution mass spectra were obtained on an Agilent 6220 using electrospray ionization time-of-flight. d-Sulfono-γ-AApeptide building blocks were synthesized following previously reported methods.20–22
1a–f Were Synthesized Using the Following General Procedure A (Scheme 1). Weinreb Amide (A1) Synthesis.
To a dried 1 L round-bottom flask were added Fmoc-protected d-amino acids (6 g) and 200 mL of DMF. The mixture was stirred in an ice bath to which 1-hydroxybenzotriazole (1.2 equiv), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (1.2 equiv), and N,N-diisopropylethylamine (1.2 equiv) were added. After being stirred for 15 min at the same temperature, N,O-dimethylhydroxyl-amine hydrochloride (1.2 equiv) and N,N-diisopropylethylamine (1.2 equiv) were added and the solution mixture was stirred for another 2 h at 0 °C. After completion, 200 mL of H2O was added to the solution and then extracted with ethyl acetate (150 mL × 3). The organic layer was combined and washed with 1 N HCl (200 mL × 3), saturated NaHCO3 (200 mL × 1), and brine (200 mL × 1), dried over anhydrous sodium sulfate, and concentrated under reduced pressure to afford the desired Weinreb amide, which was used for the next step without further purification.
A2 Synthesis.
To a solution of the Weinreb amide (1 equiv) in anhydrous THF (150 mL) at −20 °C was added LiAlH4 (1.1 equiv). After being stirred for 15 min at the same temperature, the reaction was quenched by the addition of 1 N HCl (200 mL). After THF was evaporated, the solvent mixture was extracted three times with ethyl acetate, which was combined. The organic layer was washed with brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to afford the aldehyde, which was used in the next step without further purification.
A3 Synthesis.
A solution of A2 (1 equiv) in 150 mL of methanol was stirred in an ice bath for 10 min. Then, a solution of Gly-OtBu-HCl (1.1 equiv) and triethylamine (1.1) in 20 mL of methanol was added, and the solution was stirred at 0 °C for 10 min, followed by the addition of acetic acid (1.5 equiv) and sodium cyanoborohydride (2 equiv). The reaction was allowed to continue for 2 h. Then, the solvent was removed in vacuo. The resulting slurry was added to saturated sodium bicarbonate solution (150 mL) and extracted with ethyl acetate (100 mL × 3). The organic layer was combined, dried over sodium sulfate, and concentrated in vacuo. The pure product A3 was purified by flash column chromatography with ethyl acetate/hexane (1:1).
A4 Synthesis.
A solution of A3 (1 equiv) and pyridine (10 equiv) in 150 mL of dichloromethane was stirred in an ice bath. R2-SO2Cl (2 equiv) was then added. The solution was allowed to stir at 0 °C for 1 h and continued at room temperature overnight. After completion, the solvent was removed by vacuum. Flash chromatography using dichloromethane/ethyl acetate (10:1) gave the pure product A4. The slurry A4 was used directly in the next reaction.
1a–f Synthesis.
The slurry A4 was treated with 50 mL of 1:1 dichloromethane/trifluoroacetic acid for 2 h at room temperature. The solvent was then removed by air blowing. Pure 1a–f were obtained as colorless solids after flash column purification with 1:1 hexane/ethyl acetate.
1g–j Were Synthesized Using the Following General Procedure B (Scheme 1).
B1, B2, B3, and B4 were synthesized, following the above procedure A (Scheme 1). B4 in 150 mL of 1:1 methanol/ethyl acetate mixture was added to 10% Pd/C and hydrogenated at atmospheric pressure and room temperature. After completion, the solvent was evaporated, and the residue was purified by flash chromatography with hexane/ethyl acetate (1:1) as the eluent to give 1g–j as colorless foam solids.
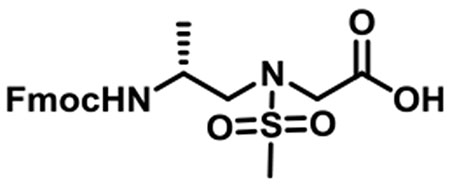
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)propyl)-N-(methylsulfonyl)glycine (1a).
1a was synthesized according to the general synthetic procedure A; overall yield = 26% (2.16 g); colorless solid; 1H NMR (400 MHz, DMSO-d6): δ 7.79 (d, J = 7.20 Hz, 2H), 7.61 (d, J = 6.80 Hz, 2H), 7.33 (t, J = 6.80 Hz, 2H), 7.25 (t, J = 7.20 Hz, 2H), 7.15 (d, J = 8.00 Hz, 1H), 4.20–4.30 (m, 2H), 4.14 (d, J = 6.00 Hz, 1H), 3.94 (s, 2H), 3.69–3.72 (m, 1H), 3.08–3.19 (m, 2H), 2.88 (s, 3H), 0.99 (d, J = 6.00 Hz, 3H). 13C{1H} NMR (100 MHz, DMSO-d6): δ 171.2, 156.0, 144.2, 141.1, 128.0, 127.4, 125.5, 120.4, 65.6, 52.4, 48.9, 47.2, 45.8, 18.6. HRMS (ESI) m/z: [M + H]+ calcd for C21H24N2O6S, 432.1355; found, 433.1419.
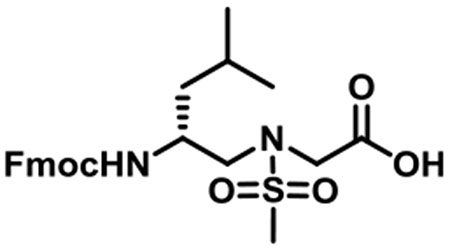
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentyl)-N-(methylsulfonyl)glycine (1b).
1b was synthesized according to the general synthetic procedure A; overall yield = 37% (3.35 g); colorless solid; 1H NMR (400 MHz, DMSO-d6): δ 7.78 (d, J = 7.60 Hz, 2H), 7.59–7.61 (m, 2H), 7.32 (t, J = 7.20 Hz, 2H), 7.21–7.26 (m, 2H), 7.06 (d, J = 9.20 Hz, 1H), 4.24–4.32 (m, 2H), 4.13 (t, J = 6.40 Hz, 1H), 3.91 (s, 2H), 3.63–3.68 (m, 1H), 3.18 (dd, J = 14.80, 5.20 Hz, 1H), 3.04 (q, J = 14.40, 8.80 Hz, 1H), 2.85 (s, 3H), 1.43–1.51 (m, 1H), 1.10–1.24 (m, 2H), 0.76 (q, J = 10.80, 6.80 Hz, 6H). 13C{1H} NMR (100 MHz, DMSO-d6): δ 171.1, 156.3, 144.3, 144.2, 141.1, 127.9, 127.4, 125.5, 120.4, 65.4, 51.9, 48.8, 48.1, 47.2, 41.2, 24.6, 23.6, 21.9. HRMS (ESI) m/z: [M + H]+ calcd for C24H30N2O6S, 474.1825; found, 475.1881.
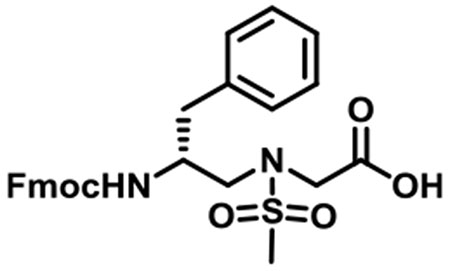
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-phenylpropyl)-N-(methylsulfonyl)glycine (1c).
1c was synthesized according to the general synthetic procedure A; overall yield = 39% (3.11 g); colorless solid; 1H NMR (400 MHz, DMSO-d6): δ 7.83 (d, J = 7.60 Hz, 2H), 7.58 (d, J = 7.20 Hz, 2H), 7.37 (t, J = 7.60 Hz, 2H), 7.26–7.30 (m, 3H), 7.21 (s, 1H), 7.20 (s, 2H), 7.11–7.14 (m, 1H), 4.14–4.23 (m, 2H), 4.06–4.11 (m, 1H), 3.93–4.02 (m, 2H), 3.86–3.90 (m, 1H), 3.37 (dd, J = 14.40, 5.60 Hz, 1H), 3.20 (q, J = 14.40, 5.60 Hz, 1H), 2.93 (s, 3H), 2.88 (t, J = 4.80 Hz, 1H), 2.58 (q, J = 13.60, 10.00 Hz, 1H), 2.01 (s, 1H). 13C{1H} NMR (100 MHz, DMSO-d6): δ 171.3, 156.1, 144.3, 144.2, 141.1, 139.1, 129.6, 128.5, 128.0, 127.5, 126.4, 125.6, 120.5, 65.7, 51.8, 51.6, 49.0, 47.1, 37.9. HRMS (ESI) m/z: [M + H]+ calcd for C27H28N2O6S, 508.1668; found, 509.1732.
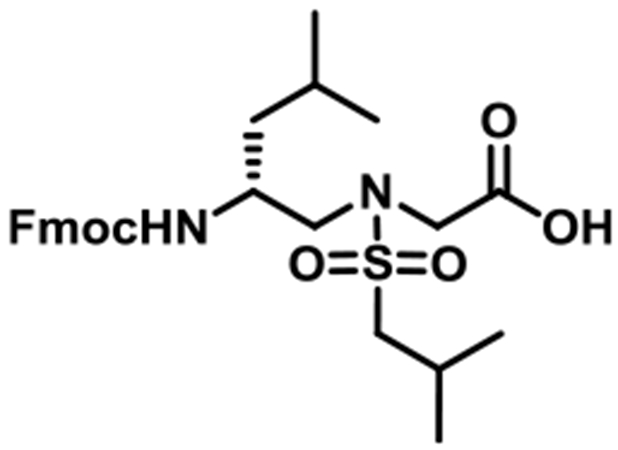
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentyl)-N-(isobutylsulfonyl)glycine (1d).
1d was synthesized according to the general synthetic procedure A; overall yield = 26% (2.291 g); colorless solid; 1H NMR (400 MHz, DMSO-d6): δ 7.80 (d, J = 7.20 Hz, 2H), 7.61 (d, J = 4.80 Hz, 2H), 7.33 (t, J = 7.20 Hz, 2H), 7.22–7.27 (m, 2H), 7.09 (d, J = 9.20 Hz, 1H), 4.29 (t, J = 10.40 Hz, 1H), 4.23 (t, J = 6.80 Hz, 1H), 4.13 (t, J = 6.80 Hz, 1H), 3.92 (s, 2H), 3.63–3.66 (m, 1H), 3.20 (dd, J = 14.40, 4.80 Hz, 1H), 3.07 (q, J = 14.40, 8.80 Hz, 1H), 2.85–2.95 (m, 2H), 1.97–2.07 (m, 1H), 1.47–1.49 (m, 1H), 1.21–1.25 (m, 2H), 0.90 (t, J = 4.40 Hz, 6H), 0.77 (q, J = 11.20, 6.40 Hz, 6H). 13C{1H} NMR (100 MHz, DMSO-d6): δ 171.2, 156.2, 144.3, 144.2, 141.1, 127.9, 127.4, 125.5, 120.4, 65.5, 59.3, 51.8, 48.5, 48.0, 47.2, 41.3, 24.6, 23.6, 22.5, 22.0. HRMS (ESI) m/z: [M + H]+ calcd for C27H36N2O6S, 516.2294; found, 517.2355.
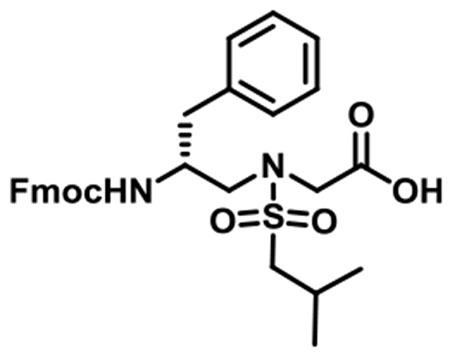
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-phenylpropyl)-N-(isobutylsulfonyl)glycine (1e).
1e was synthesized according to the general synthetic procedure A; overall yield = 29% (2.48 g); colorless solid; 1H NMR (400 MHz, DMSO-d6): δ 7.79 (d, J = 7.60 Hz, 2H), 7.54 (d, J = 7.60 Hz, 2H), 7.33 (t, J = 7.20 Hz, 2H), 7.22–7.66 (m, 3H), 7.14–7.20 (m, 4H), 7.08 (t, J = 3.20 Hz, 1H), 4.03–4.16 (m, 3H), 3.93 (t, J = 20.40 Hz, 2H), 3.82–3.85 (m, 1H), 3.34 (dd, J = 14.40, 5.20 Hz, 1H), 3.18 (q, J = 14.40, 8.40 Hz, 1H), 2.87–2.96 (m, 2H), 2.82 (dd, J = 13.60, 4.00 Hz, 1H), 2.54 (q, J = 13.60, 10.00 Hz, 1H), 1.98–2.08 (m, 1H), 0.91 (t, J = 5.60 Hz, 6H). 13C{1H} NMR (100 MHz, DMSO-d6): δ 171.3, 156.1, 144.2, 144.1, 141.1, 139.0, 129.5, 128.4, 128.0, 127.4, 126.4, 125.6, 120.4, 65.7, 59.3, 51.7, 51.5, 48.8, 47.1, 37.9, 24.6, 22.6. HRMS (ESI) m/z: [M + H]+ calcd for C30H34N2O6S, 550.2138; found, 551.2195.
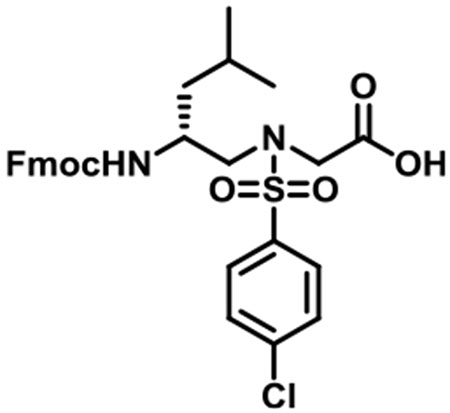
(R)-N-(2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentyl)-N-((4-chlorophenyl)sulfonyl)glycine (1f).
1f was synthesized according to the general synthetic procedure A; overall yield = 35% (3.4 g); colorless solid; 1H NMR (400 MHz, DMSO-d6): δ 12.72 (brs, 1H), 7.79 (d, J = 7.20 Hz, 2H), 7.72 (d, J = 8.40 Hz, 2H), 7.59 (d, J = 7.20 Hz, 2H), 7.52 (d, J = 8.40 Hz, 2H), 7.32 (t, J = 7.20 Hz, 2H), 7.23 (q, J = 13.60, 6.80 Hz, 2H), 7.01 (d, J = 8.80 Hz, 1H), 4.22–4.26 (m, 1H), 4.10–4.18 (m, 2H), 3.97 (q, J = 25.60, 18.40 Hz, 2H), 3.60 (brs, 1H), 3.36 (brs, 2H), 3.18 (dd, J = 14.40, 5.60 Hz, 1H), 3.07 (dd, J = 14.00, 8.00 Hz, 1H), 1.41–1.44 (m, 1H), 1.07–1.22 (m, 2H), 0.75 (d, J = 6.40 Hz, 3H), 0.71 (d, J = 6.40 Hz, 3H). 13C{1H} NMR (100 MHz, DMSO-d6): δ 170.3, 156.1, 144.3, 144.1, 141.1, 138.9, 138.0, 129.6, 129.3, 128.0, 127.4, 125.5, 120.4, 65.4, 52.4, 48.8, 48.0, 47.2, 41.1, 24.5, 23.7, 21.8. HRMS (ESI) m/z: [M + H]+ calcd for C29H31ClN2O6S, 570.1591; found, 571.1652.
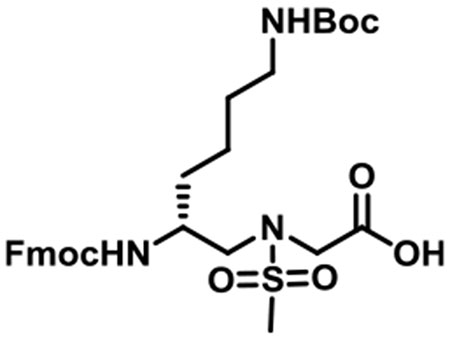
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-6-((tert-butoxycarbonyl)amino)hexyl)-N-(methylsulfonyl)glycine (1g).
1g was synthesized according to the general synthetic procedure B; overall yield = 25% (1.91 g); colorless solid; 1H NMR (400 MHz, DMSO-d6): δ 7.83 (d, J = 7.60 Hz, 2H), 7.65 (d, J = 7.60 Hz, 2H), 7.37 (t, J = 7.60 Hz, 2H), 7.27 (t, J = 6.80 Hz, 2H), 7.12 (d, J = 8.80 Hz, 1H), 6.68 (d, J = 4.80 Hz, 1H), 4.27–4.34 (m, 2H), 4.18 (t, J = 6.80 Hz, 1H), 3.96 (t, J = 18.80 Hz, 2H), 3.61 (brs, 1H), 3.24 (dd, J = 14.80, 5.20 Hz, 1h), 3.11 (q, J = 14.40, 8.80 Hz, 1H), 2.90 (s, 3H), 2.86 (d, J = 6.00 Hz, 2H), 1.38–1.42 (m, 2H), 1.33 (s, 9H), 1.17–1.28 (m, 4H). 13C{1H} NMR (100 MHz, DMSO-d6): δ 171.2, 156.4, 156.0, 144.3, 141.2, 128.0, 127.4, 125.6, 120.5, 77.7, 65.6, 51.6, 50.0, 48.8, 47.3, 31.9, 29.8, 28.7, 23.2. HRMS (ESI) m/z: [M + H]+ calcd for C29H39N3O8S, 589.2458; found, 590.2509.
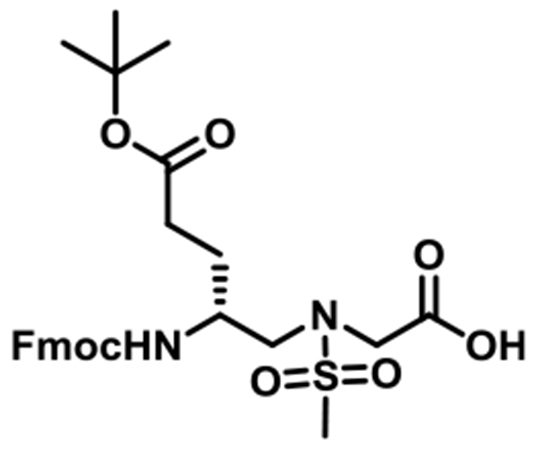
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-5-(tert-butoxy)-5-oxopentyl)-N-(methylsulfonyl)glycine (1h).
1h was synthesized according to the general synthetic procedure B; overall yield = 32% (2.46 g); colorless solid; 1H NMR (400 MHz, DMSO-d6): δ 7.79 (d, J = 7.60 Hz, 2H), 7.61 (dd, J = 7.20, 2.80 Hz, 2H), 7.33 (t, J = 7.20 Hz, 2H), 7.25 (t, J = 7.60 Hz, 2H), 7.10 (d, J = 8.80 Hz, 1H), 4.22–4.30 (m, 2H), 4.14 (t, J = 6.80 Hz, 1H), 3.91 (s, 2H), 3.60 (brs, 1H), 3.22 (dd, J = 14.40, 5.60 Hz, 1H), 3.08 (q, J = 14.40, 8.40 Hz, 1H), 2.87 (s, 3H), 2.04–2.17 (m, 2H), 1.66–1.68 (m, 1H), 1.39–1.46 (m, 1H), 1.31 (s, 9H). 13C{1H} NMR (100 MHz, DMSO-d6): δ 172.2, 171.1, 156.4, 144.3, 141.1, 128.0, 127.4, 125.5, 120.5, 79.9, 65.6, 51.2, 49.3, 48.6, 47.2, 31.7, 28.1, 27.5. HRMS (ESI) m/z: [M + H]+ calcd for C27H34N2O8S, 546.2036; found, 547.2094.
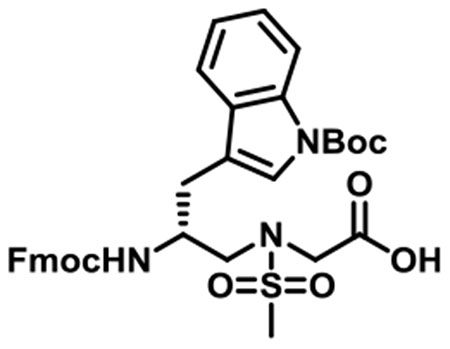
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(1-(tert-butoxycarbonyl)-1H-indol-3-yl)propyl)-N-(methylsulfonyl)-glycine (1i).
1i was synthesized according to the general synthetic procedure B; overall yield = 29% (2.216 g); colorless solid; 1H NMR (600 MHz, DMSO-d6): δ 12.86 (brs, 1H), 7.96 (d, J = 7.80 Hz, 1H), 7.78 (d, J = 7.80 Hz, 2H), 7.61 (d, J = 7.80 Hz, 1H), 7.49 (d, J = 3.60 Hz, 1H), 7.48 (s, 2H), 7.29–7.34 (m, 3H), 7.24 (t, J = 7.80 Hz, 1H), 7.14–7.19 (m, 3H), 4.11–4.17 (m, 2H), 4.08 (t, J = 6.60 Hz, 1H), 3.99 (d, J = 7.20 Hz, 2H), 3.42 (dd, J = 15.00, 6.00 Hz, 2H), 3.24 (q, J = 14.40, 8.40 Hz, 1H), 2.91 (s, 3H), 2.70 (q, J = 14.40, 9.60 Hz, 1H), 1.50 (s, 1H), 1.46 (s, 9H). 13C{1H} NMR (150 MHz, DMSO-d6): δ 169.3, 154.2, 142.2, 142.1, 139.1, 139.0, 128.9, 125.9, 125.3, 123.5, 123.4, 122.6, 121.9, 120.8, 118.4, 115.7, 113.0, 81.7, 63.8, 49.9, 48.1, 47.1, 45.0, 37.4, 26.0, 25.6. HRMS (ESI) m/z: [M + H]+ calcd for C34H37N3O8S, 647.2301; found, 648.2360.
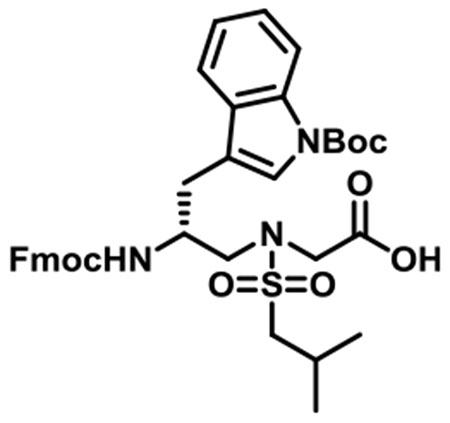
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(1-(tert-butoxycarbonyl)-1H-indol-3-yl)propyl)-N-(isobutylsulfonyl)-glycine (1j).
1j was synthesized according to the general synthetic procedure B; overall yield = 27% (2.11 g); colorless solid; 1H NMR (600 MHz, DMSO-d6): δ 7.96 (d, J = 7.80 Hz, 1H), 7.78 (d, J = 7.80 Hz, 2H), 7.63 (d, J = 7.80 Hz, 1H), 7.49 (t, J = 5.40 Hz, 3H), 7.36 (d, J = 9.00 Hz, 1H), 7.30–7.32 (m, 2H), 7.24 (t, J = 8.40 Hz, 1H), 7.15–7.19 (m, 3H), 4.13–4.17 (m, 1H), 4.06–4.11 (m, 2H), 4.00 (s, 1H), 3.98 (s, 1H), 3.44 (dd, J = 14.40, 5.40 Hz, 1H), 3.26 (q, J = 14.40, 8.40 Hz, 1H), 2.95 (dd, J = 13.80, 6.60 Hz, 1H), 2.88–2.92 (m, 2H), 2.71 (q, J = 15.00, 9.60 Hz, 1H), 2.00–2.07 (m, 1H), 1.50 (s, 1H), 1.46 (s, 9H), 0.91 (t, J = 6.60 Hz, 6H). 13C{1H} NMR (150 MHz, DMSO-d6): δ 171.4, 156.3, 144.2, 144.2, 141.2, 131.0, 128.0, 127.4 125.6, 125.6, 124.7, 124.0, 122.9, 120.5, 119.8, 117.8, 115.1, 83.8, 66.0, 59.4, 51.8, 50.3, 49.0, 47.2, 28.1, 24.7, 22.6, 22.6. HRMS (ESI) m/z: [M + H]+ calcd for C37H43N3O8S, 689.2771; found, 690.2830.
General Information for Peptide Synthesis.20–22
The synthesis was conducted in the solid phase using Rink Amide-MBHA resin (0.646 mmol/g, 100 mg) as the solid support. The resin was first swelled in DMF for 5 min and then treated with 20% piperidine in DMF (2 mL) for 15 min. The procedure was repeated once to completely remove the Fmoc protecting group. The beads were washed with DCM (×3) and DMF (×3). The d-sulfono-γ-AApeptide building block (2 equiv), DIC (4 equiv), and HOBt (4 equiv) were dissolved in 2 mL of DMF for 5 min, and the solution was added to the resin. The reaction vessel was shaken for 4 h until the reaction was complete. The reaction cycles were repeated to assemble the desired sulfono-γ-AApeptide sequences. For the capped sequence, the N-terminus of the sequence was reacted with acetic anhydride (1 mL) in pyridine (2 mL) (15 min × 2), and then the resin was cleaved off with TFA/DCM/TIS (6 mL, 50:48:2, v/v/v) for 3 h. The solution was collected, and the resin was washed with DCM (3 mL × 2). The solution was combined and evaporated under air flow, and the crude product was subsequently analyzed (1 mL/min flow rate) and purified (16 mL/min flow rate) by a Waters HPLC system equipped with both analytic and preparative modules. The gradient eluting method was as follows: 5% of solvent B (0.1% TFA in acetonitrile) in A (0.1% TFA in water) was increased to 100% over 50 min. The desired d-sulfono-γ-AApeptides were collected and lyophilized on a Labconco lyophilizer, with purity determined to be >95% by analytical HPLC.
For the FITC-labeled d-sulfono-γ-AApeptide synthesis, after installation of the last d-sulfono-γ-AApeptide building block, the Fmoc protecting group was removed, followed by washing with DCM (×3) and with DMF (×3). Fmoc-β-Ala-OH (2 equiv), DIC (4 equiv), and HOBt (4 equiv) were dissolved in 2 mL of DMF, and then the solution was added to the resin. The reaction vessel was shaken for 2 h until the coupling reaction was done. The Fmoc group was removed, and FITC (2 equiv) in 2 mL of DMF and DIPEA (6 equiv) was added to the resin to react overnight. After washing with DMF (×3) and DCM (×3), the resin was cleaved using TFA/DCM/TIS (6 mL, 50:48:2, v/v/v) for 3 h. The pure FITC-labeled d-sulfono-γ-AApeptides (>95%) were obtained using the same abovementioned method by HPLC.
Fluorescence Polarization Assay.
The binding affinity (Kd) of regular p53 and the d-sulfono-γ-AApeptides was obtained by fluorescence polarization (FP). GST-MDM2-1-150 containing human MDM2 was expressed in Escherichia coli, as previously described by us. The FP assay was conducted by incubating MDM2 (0.0625–1 μM) with 50 nM FITC-labeled peptide in 1× PBS. Dissociation constants (Kd) were determined, as reported previously.20–22
Circular Dichroism.
Circular Dichroism (CD) spectra were obtained on an Aviv 215 circular dichroism spectrometer using a 1 mm path length quartz cuvette. Three times of independent experiments were conducted. In each experiment, 10 scans were averaged for each sample (100 μM).15
Enzymatic Stability Study.
Compound 4 (0.1 mg/mL) was incubated with 0.1 mg/mL pronase at 37 °C for 24 h in 100 mM ammonium bicarbonate buffer (pH 7.8). The solution was concentrated in a speed vacuum at a high temperature to remove both solvent and ammonium bicarbonate salt. The residue was dissolved in water/acetonitrile and analyzed by HPLC.20–22
15N-1H HSQC NMR of Lead Peptide 4 in Complex with MDM2.
Experiments for 200 μM MDM217–125 in the presence and absence of a stoichiometric equivalent amount of peptide 4 were carried out at 25 °C on a Varian VNMRS 800 MHz spectrometer with a triple resonance pulse field Z-axis gradient cold probe at 30 °C. 1H-15N heteronuclear single-quantum coherence spectroscopy experiments were performed on 15N-labeled samples in 90% H2O/10% D2O. Buffer for peptide 4 and MDM217–125 experiments were 50 mM NaH2PO4, 50 mM NaCl, 1 mM EDTA, 2 mM DTT, 5% DMSO, and 0.02% NaN3 at pH 6.8. Data were acquired in the 1H and 15N dimensions using 9689.92-Hz (t2) × 2430.26-Hz (t1) sweep widths and 1024 (t2) × 128 (t1) complex data points. Bound spectra were collected in a molar equivalent of peptide 4.20
Supplementary Material
ACKNOWLEDGMENTS
This work was generously supported by NIH 5R01AG056569 (J. Cai), NIH 9R01AI152416 (J. Cai), NIH2R01CA14124406-A1 (G.D. and J. Chen), and NIH1R01GM115556-01A1 (G.D.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c00996.
General information; copies of 1H and 13C NMR spectra for all d-sulfono-γ-AApeptide building blocks; characterization and purities of peptides; and fluorescence polarization assays (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.0c00996
The authors declare no competing financial interest.
Contributor Information
Peng Sang, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
Yan Shi, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
Pirada Higbee, Department of Cell Biology, Microbiology and Molecular Biology, University of South Florida, Tampa, Florida 33620, United States.
Minghui Wang, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
Sami Abdulkadir, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
Junhao Lu, Department of Molecular Oncology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612, United States.
Gary Daughdrill, Department of Cell Biology, Microbiology and Molecular Biology, University of South Florida, Tampa, Florida 33620, United States.
Jiandong Chen, Department of Molecular Oncology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Floida 33612, United States.
Jianfeng Cai, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
REFERENCES
- (1).Roy A; Prabhakaran P; Baruah PK; Sanjayan GJ Diversifying the structural architecture of synthetic oligomers: the hetero folda-mer approach. Chem. Commun. 2011, 47, 11593–11611. [DOI] [PubMed] [Google Scholar]
- (2).Guichard G; Huc I Synthetic foldamers. Chem. Commun. 2011, 47, 5933–5941. [DOI] [PubMed] [Google Scholar]
- (3).Horne WS; Gellman SH Foldamers with Heterogeneous Backbones. Acc. Chem. Res. 2008, 41, 1399–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Goodman CM; Choi S; Shandler S; DeGrado WF Foldamers as versatile frameworks for the design and evolution of function. Nat. Chem. Biol. 2007, 3, 252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hecht S; Huc I Foldamers: Structure, Properties, and Applications; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- (6).De Poli M; Zawodny W; Quinonero O; Lorch M; Webb SJ; Clayden J Conformational photoswitching of a synthetic peptide foldamer bound within a phospholipid bilayer. Science 2016, 352, 575–580. [DOI] [PubMed] [Google Scholar]
- (7).Hamuro Y; Schneider JP; DeGrado WF De Novo Design of Antibacterial β-Peptides. J. Am. Chem. Soc. 1999, 121, 12200–12201. [Google Scholar]
- (8).Kritzer JA; Lear JD; Hodsdon ME; Schepartz A Helical β-Peptide Inhibitors of the p53-hDM2 Interaction. J. Am. Chem. Soc. 2004, 126, 9468–9469. [DOI] [PubMed] [Google Scholar]
- (9).Simon RJ; Kania RS; Zuckermann RN; Huebner VD; Jewell DA; Banville S; Ng S; Wang L; Rosenberg S; Marlowe CK Peptoids: a modular approach to drug discovery. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 9367–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang D-W; Zhao X; Hou J-L; Li Z-T Aromatic Amide Foldamers: Structures, Properties, and Functions. Chem. Sci. 2012, 112, 5271–5316. [DOI] [PubMed] [Google Scholar]
- (11).Shi Y; Teng P; Sang P; She F; Wei L; Cai J γ-AApeptides: Design, Structure, and Applications. Acc Chem. Res 2016, 49, 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Teng P; Shi Y; Sang P; Cai J γ-AApeptides as a New Class of Peptidomimetics. Chem. Eur J. 2016, 22, 5458–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shi Y; Challa S; Sang P; She F; Li C; Gray GM; Nimmagadda A; Teng P; Odom T; Wang Y; van der Vaart A; Li Q; Cai J One-Bead–Two-Compound Thioether Bridged Macrocyclic γ-AApeptide Screening Library against EphA2. J. Med. Chem. 2017, 60, 9290–9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shi Y; Parag S; Patel R; Lui A; Murr M; Cai J; Patel NA Stabilization of lncRNA GAS5 by a Small Molecule and Its Implications in Diabetic Adipocytes. Cell Chem. Biol. 2019, 26, 319–330.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shi Y; Yin G; Yan Z; Sang P; Wang M; Brzozowski R; Eswara P; Wojtas L; Zheng Y; Li X; Cai J Helical Sulfono-γ-AApeptides with Aggregation-Induced Emission and Circularly Polarized Luminescence. J. Am. Chem. Soc 2019, 141, 12697–12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Teng P; Gray GM; Zheng M; Singh S; Li X; Wojtas L; van der Vaart A; Cai J Orthogonal Halogen-Bonding-Driven 3D Supramolecular Assembly of Right-Handed Synthetic Helical Peptides. Angew. Chem., Int. Ed. 2019, 58, 7778–7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).She F; Teng P; Peguero-Tejada A; Wang M; Ma N; Odom T; Zhou M; Gjonaj E; Wojtas L; van der Vaart A; Cai J De Novo Left-Handed Synthetic Peptidomimetic Foldamers. Angew. Chem., Int. Ed. 2018, 57, 9916–9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Teng P; Niu Z; She F; Zhou M; Sang P; Gray GM; Verma G; Wojtas L; van der Vaart A; Ma S; Cai J Hydrogen-Bonding-Driven 3D Supramolecular Assembly of Peptidomimetic Zipper. J. Am. Chem. Soc. 2018, 140, 5661–5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Teng P; Ma N; Cerrato DC; She F; Odom T; Wang X; Ming L-J; van der Vaart A; Wojtas L; Xu H; Cai J Right-Handed Helical Foldamers Consisting of De Novo d-AApeptides. J. Am. Chem. Soc. 2017, 139, 7363–7369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sang P; Shi Y; Lu J; Chen L; Yang L; Borcherds W; Abdulkadir S; Li Q; Daughdrill G; Chen J; Cai J α-Helix-Mimicking Sulfono-γ-AApeptide Inhibitors for p53–MDM2/MDMX Pro-tein–Protein Interactions. J. Med. Chem. 2020, 63, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sang P; Zhang M; Shi Y; Li C; Abdulkadir S; Li Q; Ji H; Cai J Inhibition of β-catenin/B cell lymphoma 9 protein–protein interaction using α-helix–mimicking sulfono-γ-AApeptide inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 10757–10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sang P; Zhou Z; Shi Y; Lee C; Amso Z; Huang D; Odom T; Nguyen-Tran V; Shen W; Cai J The Activity of Sulfono-γ-AApeptide Helical Foldamers That Mimic GLP-1. Sci. Adv. 2020, 6, eaaz4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Grison CM; Miles JA; Robin S; Wilson AJ; Aitken DJ An α-Helix-Mimicking 12,13-Helix: Designed α/β/γ-Foldamers as Selective Inhibitors of Protein—Protein Interactions. Angew. Chem., Int. Ed. 2016, 55, 11096–11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bautista AD; Appelbaum JS; Craig CJ; Michel J; Schepartz A Bridged β3-Peptide Inhibitors of p53-hDM2 Complexation: Correlation between Affinity and Cell Permeability. J. Am. Chem. Soc. 2010, 132, 2904–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yin H; Lee G-I; Park HS; Payne GA; Rodriguez JM; Sebti SM; Hamilton AD Terphenyl-Based Helical Mimetics That Disrupt the p53/HDM2 Interaction. Angew. Chem., Int. Ed. 2005, 44, 2704–2707. [DOI] [PubMed] [Google Scholar]
- (26).Wang Z; Song T; Feng Y; Guo Z; Fan Y; Xu W; Liu L; Wang A; Zhang Z Bcl-2/MDM2 Dual Inhibitors Based on Universal Pyramid-Like α-Helical Mimetics. J. Med. Chem 2016, 59, 3152–3162. [DOI] [PubMed] [Google Scholar]
- (27).Knight SMG; Umezawa N; Lee H-S; Gellman SH; Kay BK A Fluorescence Polarization Assay for the Identification of In-hibitors of the p53–DM2 Protein–Protein Interaction. Anal. Biochem. 2002, 300, 230–236. [DOI] [PubMed] [Google Scholar]
- (28).Gilkes DM; Chen J Distinct Roles of MDMX in the Regulation of p53 Response to Ribosomal Stress. Cell Cycle 2007, 6, 151–155. [DOI] [PubMed] [Google Scholar]
- (29).Murray JK; Gellman SH Targeting protein–protein interactions: Lessons from p53/MDM2. Pept. Sci. 2007, 88, 657–686. [DOI] [PubMed] [Google Scholar]
- (30).Hook DF; Bindschädler P; Mahajan YR; šebesta R; Kast P; Seebach D The Proteolytic Stability of ‘Designed’ β-Peptides Containingα-Peptide-Bond Mimics and of Mixedα, β-Peptides: Application to the Construction of MHC-Binding Peptides. Chem. Biodiversity 2005, 2, 591–632. [DOI] [PubMed] [Google Scholar]
- (31).Kussie PH; Gorina S; Marechal V; Elenbaas B; Moreau J; Levine AJ; Pavletich NP Structure of the MDM2 Oncoprotein Bound to the p53 Tumor Suppressor Transactivation Domain. Science 1996, 274, 948–953. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.